
Abstract
Aims or purpose
Glaucoma is the leading cause of vision loss and blindness in the world. Elucidating the pathogenesis of glaucoma and developing effective treatment should be the priority. Inflammation and oxidative stress play essential roles in glaucoma pathogeneisis. Kaempferol is a natural flavonol and has anti-inflammatory and anti-oxidative activities. In this study, we explored the potential effects of kaempferol on acute glaucoma.
Methods
We established the retinal ischemia-reperfusion (I/R) mice model and administrated kaempferol to I/R mice. We monitored the retina thickness change, retinal ganglion cell (RGC) death, caspase-8 and caspase-3 activation, NLRP1/NLRP3 inflammasomes activation, pro-inflammatory cytokines production, and activations of NF-κB and MAPKs signaling pathways.
Results
Kaempferol prevented retina thickness change and RGC death in I/R mice. The activations of caspase-8, caspase-3, and NLRP1/NLRP3 inflammasome activation were inhibited by kaempferol. Kaempferol prevented pro-inflammatory cytokines productions in I/R mice. The activation of NF-κB and JNK signaling pathways was also inhibited by Kaempferol in I/R mice.
Conclusion
Kaempferol attenuated retinal ganglion cell death by suppressing NLRP1/NLRP3 inflammasomes and caspase-8 via inhibiting NF-κB and JNK pathways in acute glaucoma.
Similar content being viewed by others

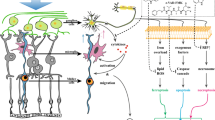

Introduction
Glaucoma is the leading cause of worldwide irreversible vision loss and blindness [1]. The damage to the optic nerve and the progressive degeneration of retinal ganglion cells (RGCs) are two major characteristics of glaucoma [2]. Although the pathogenesis of glaucoma is not fully elucidated, high intraocular pressure (IOP), increased oxidative stress, inflammation, and glutamate neurotoxicity have been implicated in glaucoma pathogenesis [3].
Trabecular meshwork blockage allows the build-up of aqueous humor and results in a rapid increase in IOP. Elevated IOP causes retinal ischemic reperfusion (I/R) injury. I/R contributes to cellular damage and causes RGC death glaucoma [4]. Oxidative stress is a key step in the progression of glaucoma and is connected to RGC apoptosis [5]. RGC apoptosis is an important component of glaucoma pathophysiology. Apoptosis is a process of programmed cell death requires activities of caspases (cysteine aspartyl-specific proteases) family [6]. Caspase-8 is the initiator caspase which triggers cell apoptosis and Caspase-3 is the effector caspase in apoptosis [7]. In glaucoma, the NF-κB signaling pathway is activated and results in the production of pro-inflammatory cytokines, which cause RGC death in retina [8]. Interestingly, NF-κB inhibitors have been shown to protect neurons against damage in the retina, suggesting that prevention of NF-κB activation would be a potential candidate to treat retinal denegation.
Inflammasomes are implicated in several I/R diseases. In I/R diseases, NOD-like receptor (NLR) families are activated and result in the processing of pro-IL-1β into it mature forms in a caspase-1-dependent manner. NLRP3 inflammasome has been reported to act as a sensor to detect the injury and then amplify the damage signal, leading to subsequent RGC loss [9]. In contrast, inhibiting NLRP3 inflammasome activation protected diabetic retinopathy, suggesting inhibition of NLRP3 inflammasome activation would be another potential candidate to treat retinal denegation [10].
Although much effort has been devoted to explore effective therapies to attenuate RGC death, there are still no practically effective drugs [11]. The mechanisms which are related to RGC death and glaucoma pathogenesis could be potential targets of effective therapies [12]. Kaempferol is a natural flavonol found in vegetables and fruits. Kaempferol has been reported to modulate signaling transduction pathways involved in apoptosis, angiogenesis, inflammation, oxidative stress, and metastasis [13]. Kaempferol has been shown to have anti-inflammatory activity targeting the NF-κB pathway [14], suppressing the mitogen-activated protein kinase (MAPK) signaling pathway [15], and NLRP3 inflammasome activation [16]. The anti-oxidative stress activities of kaempferol have also been reported [17]. As NF-κB and NLRP3 inflammasome plays essential role in glaucoma, the anti-inflammation activities of kaempferol by targeting NF-κB and NLRP3 suggested potential effects of kaempferol on glaucoma. Here, we explored the effect of kaempferol on acute glaucoma and reported that kaempferol prevented RGC death and inhibited NLRP1/NLRP3 inflammasome, NF-κB and JNK2 singling pathways.
Materials and methods
Retinal ischemia-reperfusion (IR) mice model
Six- to eight-week C57BL/6 female mice were housed in a 10–14-h light-to-dark-cycle environment. The temperature was kept at 22 ± 3 °C. Mice were anesthetized by intraperitoneally injecting 4.3% chloral hydrate with 10 ml/kg concentration. Pupils were dilated with 1% tropicamide, and corneas were anesthetized with 0.5% tetracaine hydrochloride eye drops topically. To increase the intraocular pressure (IOP), the anterior chamber of the right eye was cannulated with a 30-gauge needle and balance salt solution (Tono-Pen XL) was added to maintain at 70 mmHg for 60 min. The left eye was untreated and used as a control. After 60 min, the needle was withdrawn gradually and tobramycin was applied to avoid bacterial infection. Mice were killed at 48 h after the procedure.
Kaempferol administration
Kaempferol was purchased from Sigma-Aldrich (St. Louis, MO, USA). After induction of I/R injury, mice were randomly divided. Normal mice without treatment of ischemia-reperfusion and kaempferol were used as a control group. PBS was used as a vehicle control and I/R mice in vehicle control group were fed with 2 µl of PBS. For I/R + kaempferol group, I/R mice were administered intragastrically with kaempferol 100 mg/kg body weight. Mice were killedand eyes were enucleated 48 h after administration.
Histological examination
Histological examination was performed as described previously [18]. Mice eyes were enucleated, and then the whole eyes were fixed with 4% paraformaldehyde overnight. Next day samples were embedded in paraffin. Four 4-mm-thick sections through the optic nerve of each eye were cut, and sample slices of 10 µm per slide were further produced. Every twentieth slice was selected for tissue staining for hematoxylin and eosin (HE) staining. Total retinal thickness (from inner to outer limiting membrane) was measured in four adjacent areas within 1 mm distance to the optic nerve center using Axiovision Microscopy software (Carl Zeiss MicroImaging Inc., Oberkochen, Germany).
Fluoro-gold (FG) labeling of retinal ganglion cells (RGC)
FG labeling of RGC was performed as described previously [19]. Briefly, mice were allowed 7 days for retrograde transport of FG before kill to ensure proper RGC labeling. Mice were anesthetized by i.p. injection of a mixture of 100 mg/kg ketamine and 10 mg/kg xylazine and then placed in a stereotactic apparatus (Stoelting). In total, 1 μL of 4% FG (Invitrogen, Carlsbad, CA, USA) diluted in saline (weight/volume) was injected into both superior colliculi. Forty-eight hours after reperfusion, FG-positive RGCs were quantitated with a fluorescent microscope AxioImager (Carl Zeiss, Germany) after retinal flat mount preparation. Survived RGCs (gold dots) were counted by using Image Pro Plus (Version 6.0; Media Cybernetics).
Western blot
The whole retina was isolated from the eyeball and then lysed in RIPA buffer to make the homogenates. In some experiment, nuclear proteins were isolated using NE-PER™ nuclear and cytoplasmic extraction reagents (Thermo Fisher, Waltham, MA, USA). The whole-protein concentrations were measured using Bradford Protein Assay (Bio-Rad, Hercules, CA, USA). Total 25 µg protein was loaded on 12% SDS-PAGE. After transfer, membranes were blocked by 5% non-fat milk (Bio-Rad, USA) for 1 h at room temperature and then incubated with different primary antibodies: anti-active Caspase-3 (Abcam, Cambridge, MA, USA), anti-cleaved Caspase-8 (Cell Signaling, Danvers, MA, USA), anti-NALP1 (Cell Signaling, USA), anti-NALP3 (Cell Signaling, USA), anti-ASC (Cell Signaling, USA), anti-Capase-1p20 (Santa Cruz, Dallas, TX, USA), anti-β actin (Sigma, USA), anti-p-JNK2 (Thermo Fisher, USA), anti-JNK2 (Cell Signaling, USA), anti-p-ERK1/2 (Cell Signaling, USA), anti-ERK1/2 (Millipore, Billerica, MA, USA), anti-p-p38 (Cell Signaling, USA), anti-p38 (Cell Signaling, USA), anti-p-IKKαβ (Sigma, MO, USA), anti-IKKα (Abcam, USA), anti-NF-κB p65 (Thermo Fisher, USA), and anti-lamin B (Abcam, USA). The primary antibodies were incubated at 4 °C for overnight. Next day, HRP-conjugated corresponding secondary antibodies were incubated at room temperature for 1 h. After wash, chemiluminescent substrate (Thermo Fisher Scientific, Waltham, MA) was used to detect the bands. GS-900™ Calibrated Densitometer and Image Lab (Bio-Rad, Hercules, CA, USA) were used for quantitation following manufacturer’s instructions.
Quantitative real-time PCR (RT-PCR)
RNeasy mini kit (Qiagen, Valencia, CA, USA) was used to isolate total RNA of the retina. The subsequent cDNA was synthesized by using the Quatitect reverse transcription kit (Qiagen, USA). Real-time PCR was performed using SYBR Green Master Mix (Qiagen, USA) following manufacturer’s instructions. Samples were normalized to internal control β-actin. Primer sequences used for RT-PCR were as follows: tumor necrosis factor α (TNF-α) forward primer: 5′-CGTGGAACTGGCAGAAGAGG-3′ and reverse primer: 5′-CTGCCACAAGCAGGAATGAG-3′, iNOS forward primer: 5′-ACAACAGGAACCTACCAGCTCA-3′ and reverse primer: 5′-GATGTTGTAGCGCCTGTGTGTCA-3′, interleukin 6 (IL-6) forward primer: 5′-CCAGAAACCGCTATGAAGTTCC-3′ and reverse primer: 5′-GTTGGGAGTGGTATCCTCTGTGA-3′, interleukin 1β (IL-1β) forward primer: 5′-GTTCCCATTAGACAACTGCACTACAG-3′ and reverse primer: 5′-GTCGTTGCTTGGTTCTCCTTGTA-3′, cyclooxygenase 2 (COX-2) forward primer: 5′-CCAGATGATATCTTTGGGGAGAC-3′ and reverse primer: 5′-CTTGCATTGATGGTGGCTG-3′, β-actin forward primer: 5′-GGCGGACTATGACTTAGTTG-3′ and reverse primer: 5′-AAACAACAA TGTGCAATCAA-3′.
Statistical analysis
All data were expressed as mean ± SD and one-way ANOVA was used for difference analysis. The difference was considered as significant when p < 0.05.
Results
Kaempferol inhibited IOP-induced retinal thickness change and prevented RGC death
The retina layers contain ganglion cell layer (GCL), inner nuclear layer (INL), inner plexiform layer (IPL), outernuclear layer (ONL), and outer plexiform layer (OPL). First, we analyzed the thickness change in control, I/R and I/R + kaempferol groups of mice. As shown in Fig. 1a, I/R mice had decreased retinal thickness when compared with normal mice, indicating elevated IOP caused retinal thickness change. In contrast, administration of kaempferol prevented the decreasing of retinal thickness in I/R mice and the retinal thickness of I/R + kaempferol mice was significantly higher than that of I/R mice, indicating kaempferol ameliorated the IOP-induced pathology. The retinal thickness was correlated to RGC death [20]. We further analyzed the RGC death among these three groups of mice. As shown in Fig. 1b, using FG labeling, we found there was obvious less RGC cells in I/R mice than in normal mice, indicating IOP-induced cell death in mice. Once I/R mice were treated with kaempferol, there was no obvious cell loss in I/R mice. After quantitation, kaempferol-treated I/R mice had significantly higher RGC survival rate than that I/R mice. Thus, our data suggested that kaempferol prevented IOP-induced cell death.

The effect of kaempferol on IOP-induced RGC death. a Hematoxylin and eosin staining of retinal cross-sections showed difference of RGCs and the mean of the retinal tissue thickness among different groups 48 h after reperfusion. GCL ganglion cell layer, INL inner nuclear layer, IPL inner plexiform layer, ONL outernuclear layer, OPL outer plexiform layer. b FG labeling showed that the number of surviving cells in the RGC layer after IOP-induced injury compared with controls. N = 5 mice. Data are shown as mean ± SD. **P < 0.01 vs. Ctrl group (one-way ANOVA analysis)
Kaempferol prevented caspase-3 and caspase-8 activation
Elevated IOP induced RGC apoptosis through activating caspase-3 and caspase-8 [21]. As kaempferol inhibited RGC death, we continued to determine the effects of kaempferol on caspase-3 and caspase-8 activation by western blot. Consistent to previous reports, we found there was obvious increased active caspase-3 (Fig. 2a) and active caspase-8 (Fig. 2b) in I/R mice. Kaempferol significantly decreased protein levels of both active caspase-3 and caspase-8, indicating inhibitions of caspase-3 and caspasae-8 activation.

The effect of kaempferol on caspase-8 and caspase-3 activations in the retina of IOP-induced mice. a Western blot detected expression of active caspase-8 protein. b Western blot detected expression of active caspase-3 protein. N = 5 mice. Data are shown as mean ± SD. **P < 0.01 vs Ctrl group (one-way ANOVA analysis)
Kaempferol prevented NLRP1/ NLRP3 inflammation activation
NLRP1/NLRP3 inflammasome activation was implicated in glaucoma [19]. IOP induced the elevated expression of inflammasome components NLRP1 (Fig. 3a), NLRP3 (Fig. 3b), ASC (Fig. 3c), and active caspase-1 (Fig. 3d). Administration of kaempferol significantly decreased the levels of all four proteins, indicating kaempferol prevented NLRP1/NLRP3 inflammasome activation.

The effect of kaempferol on the activation of NLRP3 inflammasome in the retina of IOP-induced mice. a–d Western blot detected expression of NLRP1, NLRP3, ASC and caspase-1. N = 5 mice. Data are shown as mean ± SD. *P < 0.05, **P < 0.01 vs. Ctrl group (one-way ANOVA analysis)
Kaempferol inhibited inflammatory cytokines production and oxidative stress
Both inflammatory cytokines and oxidative stress played important roles in the process of glaucoma [22, 23]. We analyzed the effects of kaempferol on inflammatory cytokines production and oxidative markers in I/R mice retina by RT-PCR. We found that there were increased mRNA levels of interleukin 1β (IL-1β) (Fig. 4a), tumor necrosis factor α (TNF-α) (Fig. 4b), interleukin 6 (IL-6) (Fig. 4c), inducible nitric oxide synthase (iNOS) (Fig. 4d), and cyclooxygenase 2 (COX-2) in I/R mice retina (Fig. 4e). In contrast, kaempferol significantly decreased the mRNA levels of all five proteins, indicating kaempferol not only inhibited inflammatory cytokines production but also attenuated oxidative stress.

The effect of kaempferol on the productions of pro-inflammatory cytokines in the retina of IOP-induced mice. a–e qPCR detected expression of IL-1β, TNF-α, IL-6, iNOS, and Cox-2. N = 5 mice. Data are shown as mean ± SD. *P < 0.05, **P < 0.01 vs. Ctrl group (one-way ANOVA analysis)
Kaempferol inhibited activations of NF-κB and JNK signaling pathways
Both NF-κB and MAPK signaling pathways were responsible for inflammation response and implicated in glaucoma [24]. We further explored the effects of kaempferol on NF-κB and MAPK signaling pathway activations. In I/R mice, the MAPK signaling pathways were activated as we detected increased levels of phosphorylated-JNK2 (p-JNK2) (Fig. 5a), phosphorylated ERK1/2 (p-ERK1/2) (Fig. 5b), phosphorylated-p38 (p-p38) (Fig. 5c). Administration of kaempferol in I/R mice significantly decreased levels of p-JNK2 but not p-ERK1/2, p-p38, indicating that kaempferol inhibited IOP induced activation of the JNK2 signaling pathway. Similarly, we detected increased level of phosphorylated IKKα/β (p-IKKα/β) (Fig. 5d) and nuclear NF-κB p65 (Fig. 5e) in the retina of I/R mice, indicating IOP also induced NF-κB activation. The kaempferol treatment significantly decreased levels of both p-IKKα/β and nuclear NF-κB p65, indicating kaempferol inhibited IOP induced activation of NF-κB.

The effect of kaempferol on the MAPKs and NF-κB activation in the retina of IOP-induced mice. a–e Western blot detected expression of p-JNK, p-ERK, p-p38, p-IKKs, and p65. N = 5 mice. Data are shown as mean ± SD. *P < 0.05, **P < 0.01 vs. Ctrl group (one-way ANOVA analysis)
Discussion
Glaucoma is a family of neurodegenerative diseases characterized by the loss of retinal ganglion cells (RGC), which ultimately leads to visual loss. Thus, developing treatments to prevent RGC death and preserve RGC in glaucoma should be the priority [12].
Several rodent models were established for glaucoma studies. As elevated intraocular pressure (IOP) is strongly associated with the progression of glaucoma, rodent models of glaucoma that involve optic nerve damage mediated through ocular hypertension are most commonly used [25]. Retinal ischemia-reperfusion (I/R) is a pathophysiological process contributing to cellular damage in glaucoma, and rodent models of I/R injury are providing significant insights into mechanism and treatment strategies for human I/R injury [26]. Upon I/R mice model, we found there was RGC loss 48 h after I/R. I/R mice model is a common used model to explore RGC survival and underlying mechanism, and investigate potential treatment to prevent glaucoma. In current study, we identified that kaempferol prevented I/R-induced retina thickness change by inhibiting RGC death. In addition, we further elucidated that kaempferol inhibited activation of caspase-8, capase-3 and NLRP1/NLRP3 inflammasome, prevented I/R-induced inflammatory cytokine production and oxidative stress by suppressing JNK and NF-κB pathways. Our data strongly suggested that kaempferol could be used as a potential treatment for glaucoma.
The pathophysiology of glaucoma is not well understood while it is believed that multiple factors contribute to it [27]. The elevated IOP results in RGC apoptosis and the correlation of elevated IOP level and RGC axon loss has been reported [28]. In current study, we found that kaempferol prevented RGC loss by inhibiting the caspase-8 and caspase-3 activation, indicating that kaempferol inhibited RGC apoptosis. The anti-apoptosis activity of kaempferol has been reported. Ruiz et al. reported that kaempferol inhibited caspase-3 activation and apoptosis in the vascular smooth muscle [29].
Both inflammatory cytokines and oxidative stress contributed to pathogenesis of glaucoma. With identified anti-oxidative and anti-inflammation activities in several other diseases models, kaempferol was explored for anti-oxidative and anti-inflammatory effects in I/R mice. We found that the pro-inflammatory cytokines, iNOS and COX-2 levels were significantly decreased in retina of kaempferol-treated I/R mice, indicating the anti-inflammatory and anti-oxidative activities of kaempferol also existed in acute glaucoma model. As oxidative stress caused apoptosis in RGC [30], our data indicated that the anti-oxidative activity of kaempferol contributed to its ability to inhibit RGC apoptosis.
A recent publication described the involvement of inflammasome in acute glaucoma [19]. Using the I/R mice model, Chi et al. reported that in acute glaucoma, TLR4 lead to increased caspase-8 expression and resulted in increased IL-1β expression and RGC death via the caspase-1-dependent pathway, which involved NLRP1/NLRP3 inflammasomes. Inhibition of caspase-8 activation significantly attenuates RGC death by downregulating the activation of NLRP1 and NLRP3. Our findings demonstrated that I/R-induced NLRP1/NLRP inflammasome and caspase-1 activation while kaempferol treatment prevented the inflammasome and caspase-1 activation and downstream IL-1β production.
The downstream signaling of TLR includes mitogen-activated protein kinases (MAPKs), interferon regulatory factors (IRFs) and NF-κB [31]. Both NF-κB and MAPK signaling pathways have been implicated in glaucoma [24, 32]. Our findings demonstrated that in I/R mice, NF-κB p65 nuclear translocation and phosphorylation of MAPK JNK2, p38, and ERK were upregulated, indicating NF-κB and MAPK signaling pathways were activated during glaucoma. Kaempferol has been reported to regulate both MAPKs and NF-κB signaling pathways [15]. Chen et al. demonstrated that in a LPS-induced acute lung injury mice model, kaempferol attenuated pulmonary edema and significantly reduced LPS-induced pro-inflammatory cytokine production by inhibiting MAPKs and NF-κB signaling pathways. Our data demonstrated that in I/R mice, kaempferol also prevented NF-κB p65 nuclear translocation and JNK but not ERK or p38 phosphorylation, suggesting that kaempferol exerted its anti-glaucoma role via interfering with the NF-κB and JNK pathways.
In conclusion, our current study provided the compelling evidence that kaempferol prevented RGC death by inhibiting the activation of caspase-8, caspase-3, and NLRP1/NLRP3 inflammasomes, preventing the production of pro-inflammatory cytokines through inhibiting NF-κB and JNK pathways.
Summary
What was known before
-
Glaucoma is the leading cause of vision loss and blindness in the world. Elucidating the pathogenesis of glaucoma and developing effective treatment should be the priority. Inflammation and oxidative stress play essential roles in glaucoma pathogenesis. Kaempferol is a natural flavonol and has anti-inflammatory and anti-oxidant activities.
What this study adds
-
Kaempferol attenuated retinal ganglion cell death by suppressing NLRP1/NLRP3 inflammasomes and caspase-8 via inhibiting NF-κB and JNK pathways in acute glaucoma.
References
Resnikoff S, Pascolini D, Etya’ale D, Kocur I, Pararajasegaram R, Pokharel GP, et al. Global data on visual impairment in the year 2002. Bull World Health Organ. 2004;82:844–51.
Sacca SC, Gandolfi S, Bagnis A, Manni G, Damonte G, Traverso CE, et al. The outflow pathway: a tissue with morphological and functional unity. J Cell Physiol. 2016;231:1876–93.
Kimura A, Namekata K, Guo X, Noro T, Harada C, Harada T. Targeting oxidative stress for treatment of glaucoma and optic neuritis. Oxid Med Cell Longev. 2017;2017:2817252.
Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. 2014;311:1901–11.
Erb C, Heinke M. Oxidative stress in primary open-angle glaucoma. Front Biosci (Elite Ed). 2011;3:1524–33.
Farkas RH, Grosskreutz CL. Apoptosis, neuroprotection, and retinal ganglion cell death: an overview. Int Ophthalmol Clin. 2001;41:111–30.
Guo L, Moss SE, Alexander RA, Ali RR, Fitzke FW, Cordeiro MF. Retinal ganglion cell apoptosis in glaucoma is related to intraocular pressure and IOP-induced effects on extracellular matrix. Invest Ophthalmol Vis Sci. 2005;46:175–82.
Halder SK, Matsunaga H, Ishii KJ, Ueda H. Prothymosin-alpha preconditioning activates TLR4-TRIF signaling to induce protection of ischemic retina. J Neurochem. 2015;135:1161–77.
Feng L, Liu X. NLRP3 inflammasome in retinal ganglion cell loss in optic neuropathy. Neural Regen Res. 2016;11:1077–8.
Hao J, Zhang H, Yu J, Chen X, Yang L. Methylene blue attenuates diabetic retinopathy by inhibiting NLRP3 inflammasome activation in STZ-induced diabetic rats. Ocul Immunol Inflamm. 2018. https://doi.org/10.1080/09273948.2018.1450516
Tian K, Shibata-Germanos S, Pahlitzsch M, Cordeiro MF. Current perspective of neuroprotection and glaucoma. Clin Ophthalmol. 2015;9:2109–18.
Xu Y, Yang B, Hu Y, Lu L, Lu X, Wang J, et al. Wogonin prevents TLR4-NF-kappaB-medicated neuro-inflammation and improves retinal ganglion cells survival in retina after optic nerve crush. Oncotarget. 2016;7:72503–17.
Chen AY, Chen YC. A review of the dietary flavonoid, kaempferol on human health and cancer chemoprevention. Food Chem. 2013;138:2099–107.
Kadioglu O, Nass J, Saeed ME, Schuler B, Efferth T. Kaempferol Is an anti-inflammatory compound with activity towards NF-kappaB pathway proteins. Anticancer Res. 2015;35:2645–50.
Chen X, Yang X, Liu T, Guan M, Feng X, Dong W, et al. Kaempferol regulates MAPKs and NF-kappaB signaling pathways to attenuate LPS-induced acute lung injury in mice. Int Immunopharmacol. 2012;14:209–16.
Lim H, Min DS, Park H, Kim HP. Flavonoids interfere with NLRP3 inflammasome activation. Toxicol Appl Pharmacol. 2018;355:93–102.
Samhan-Arias AK, Martin-Romero FJ, Gutierrez-Merino C. Kaempferol blocks oxidative stress in cerebellar granule cells and reveals a key role for reactive oxygen species production at the plasma membrane in the commitment to apoptosis. Free Radic Biol Med. 2004;37:48–61.
Chi W, Chen H, Li F, Zhu Y, Yin W, Zhuo Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-kappaB pathway in acute glaucoma. J Neuroinflamm. 2015;12:137.
Chi W, Li F, Chen H, Wang Y, Zhu Y, Yang X, et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc Natl Acad Sci USA. 2014;111:11181–6.
McKernan DP, Caplis C, Donovan M, O’Brien CJ, Cotter TG. Age-dependent susceptibility of the retinal ganglion cell layer to cell death. Invest Ophthalmol Vis Sci. 2006;47:807–14.
Qu J, Wang D, Grosskreutz CL. Mechanisms of retinal ganglion cell injury and defense in glaucoma. Exp Eye Res. 2010;91:48–53.
Tong Y, Zhou YL, Zheng Y, Biswal M, Zhao PQ, Wang ZY. Analyzing cytokines as biomarkers to evaluate severity of glaucoma. Int J Ophthalmol. 2017;10:925–30.
Izzotti A, Bagnis A, Sacca SC. The role of oxidative stress in glaucoma. Mutat Res. 2006;612:105–14.
Zhou X, Li F, Kong L, Tomita H, Li C, Cao W. Involvement of inflammation, degradation, and apoptosis in a mouse model of glaucoma. J Biol Chem. 2005;280:31240–8.
Johnson TV, Tomarev SI. Rodent models of glaucoma. Brain Res Bull. 2010;81:349–58.
Hartsock MJ, Cho H, Wu L, Chen WJ, Gong J, Duh EJ. A mouse model of retinal ischemia-reperfusion injury through elevation of intraocular pressure. J Vis Exp. 2016;113:e54065.
Agarwal R, Gupta SK, Agarwal P, Saxena R, Agrawal SS. Current concepts in the pathophysiology of glaucoma. Indian J Ophthalmol. 2009;57:257–66.
Chauhan BC, Pan J, Archibald ML, LeVatte TL, Kelly ME, Tremblay F. Effect of intraocular pressure on optic disc topography, electroretinography, and axonal loss in a chronic pressure-induced rat model of optic nerve damage. Invest Ophthalmol Vis Sci. 2002;43:2969–76.
Ruiz E, Padilla E, Redondo S, Gordillo-Moscoso A, Tejerina T. Kaempferol inhibits apoptosis in vascular smooth muscle induced by a component of oxidized LDL. Eur J Pharmacol. 2006;529:79–83.
Maher P, Hanneken A. The molecular basis of oxidative stress-induced cell death in an immortalized retinal ganglion cell line. Invest Ophthalmol Vis Sci. 2005;46:749–57.
Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–5.
Mammone T, Chidlow G, Casson RJ, Wood JPM. Expression and activation of mitogen-activated protein kinases in the optic nerve head in a rat model of ocular hypertension. Mol Cell Neurosci. 2018;88:270–91.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lin, C., Wu, F., Zheng, T. et al. Kaempferol attenuates retinal ganglion cell death by suppressing NLRP1/NLRP3 inflammasomes and caspase-8 via JNK and NF-κB pathways in acute glaucoma. Eye 33, 777–784 (2019). https://doi.org/10.1038/s41433-018-0318-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-018-0318-6
This article is cited by
-
Anti-inflammatory Effects of Siponimod in a Mouse Model of Excitotoxicity-Induced Retinal Injury
Molecular Neurobiology (2023)
-
Natural Immunosuppressants as a Treatment for Chronic Insomnia Targeting the Inflammatory Response Induced by NLRP3/caspase-1/IL-1β Axis Activation: A Scooping Review
Journal of Neuroimmune Pharmacology (2023)
-
Spotlight on pyroptosis: role in pathogenesis and therapeutic potential of ocular diseases
Journal of Neuroinflammation (2022)
-
Nanoparticles for the treatment of glaucoma-associated neuroinflammation
Eye and Vision (2022)
-
Caspase-1-dependent inflammasomes mediate photoreceptor cell death in photo-oxidative damage-induced retinal degeneration
Scientific Reports (2020)
