
Abstract
GEMIN5 exerts key biological functions regulating pre-mRNAs intron removal to generate mature mRNAs. A series of patients were reported harboring mutations in GEMIN5. No treatments are currently available for this disease. We treated two of these patients with oral Coenzyme Q10 (CoQ10), which resulted in neurological improvements, although MRI abnormalities remained. Whole Exome Sequencing demonstrated compound heterozygosity at the GEMIN5 gene in both cases: Case one: p.Lys742* and p.Arg1016Cys; Case two: p.Arg1016Cys and p.Ser411Hisfs*6. Functional studies in fibroblasts revealed a decrease in CoQ10 biosynthesis compared to controls. Supplementation with exogenous CoQ10 restored it to control intracellular CoQ10 levels. Mitochondrial function was compromised, as indicated by the decrease in oxygen consumption, restored by CoQ10 supplementation. Transcriptomic analysis of GEMIN5 patients compared with controls showed general repression of genes involved in CoQ10 biosynthesis. In the rigor mortis defective flies, CoQ10 levels were decreased, and CoQ10 supplementation led to an improvement in the adult climbing assay performance, a reduction in the number of motionless flies, and partial restoration of survival. Overall, we report the association between GEMIN5 dysfunction and CoQ10 deficiency for the first time. This association opens the possibility of oral CoQ10 therapy, which is safe and has no observed side effects after long-term therapy.
Similar content being viewed by others


Introduction
GEMIN5 is a highly conserved multifunctional protein with different RNA and protein targets. It is a cytosolic protein that is involved in assembling small RNA-protein complexes that then transit to the nucleus to participate in splicing of pre-mRNAs to generate mature mRNAs [1, 2]. Furthermore, abnormal interactions between GEMIN5 and spliceosome proteins, such as the survival motor neuron complex, may explain the neurological phenotype observed in patients with GEMIN5 mutations [3]. A series of patients have been reported to harbor genetic variants in GEMIN5, associated with two distinct phenotypes: a milder form characterized by a motor-predominant developmental delay, ataxia, and cerebellar atrophy, and a more severe infantile form resembling spinal muscular atrophy with a potentially fatal outcome [4,5,6,7,8]. Currently, no treatments are available for patients with GEMIN5 mutations.
In 2006, we described a case with ataxia and mitochondrial dysfunction attributed to a primary coenzyme Q10 (CoQ10) deficiency observed in a muscle biopsy of a patient with ataxia and mitochondrial dysfunction [9]. Subsequently, a second case with similar symptoms and CoQ10 deficiency was identified. Both cases improved with oral CoQ10. No mutations in CoQ10 biosynthesis genes were found with targeted Sanger sequencing of individual genes. Much later, using whole exome sequencing (WES), we identified the same GEMIN5 missense variant (c.3046 C > T; p.Arg1016Cys) in both individuals, who were both compound heterozygotes carrying two different truncating variants [4]. Notably, both patients exhibited a sustained beneficial response to CoQ10 treatment during an initial 2-year follow-up period [10]. To our knowledge, no demonstrated or previously published links between the GEMIN5 gene and CoQ10 biosynthesis or mitochondrial dysfunction have been reported thus far.
With this background, we aim to report the long-term follow-up of two patients harboring mutations in the GEMIN5 gene leading to an ataxic phenotype treated with oral CoQ10. Moreover, molecular evidence of the relationship between CoQ10 deficiency and GEMIN5 gene mutations is provided, both in fibroblasts from two individuals treated with oral CoQ10, an additional individual with a GEMIN5-related disease who could not be treated due to a fatal outcome, as well as in a fly model of GEMIN5 deficiency.
Subjects and methods
Complete subject and method descriptions are available in the online supplementary material.
Subjects
We describe three patients with GEMIN5-related disorders, two of whom (P1 and P2) were treated with CoQ10. Patient three (P3) was not treated with CoQ10 due to an early and fatal outcome. He was previously reported in Kour et al. [4] (case 4 from family 3), and the clinical description is stated in the supplementary text.
Methods
Clinical explorations
Neurological assessment of ataxia was performed using the International Cooperative Ataxia Rating Scale (ICARS) and the scale for the assessment and rating of ataxia (SARA) [11].
Neuroimaging studies: Brain MRIs were performed in 1.5T MRI scanners (Philips Ingenia, Philips Healthcare, Cleveland, Ohio, USA, and Signa Excite, General Electric, Milwaukee, Wisconsin, USA). The acquisition protocol included sagittal 3D-T1, axial or coronal FSE T2, axial or coronal FLAIR T2, and axial DWI.
Laboratory studies
Clinical laboratory analysis
CoQ10 analysis in plasma, fibroblasts and muscle samples from patients were performed as previously reported [12], using reverse phase HPLC with electrochemical detection (Coulochem II, ESA, USA). CoQ10 from plasma, fibroblast, and muscle was extracted with n-hexane, evaporated under nitrogen stream, and dissolved in ethanol. 50 μL was injected onto the HPLC, and CoQ10 (ubiquinone and ubiquinol) was separated in a nucleosyl C-18 column (25 cm, Teknokroma) with a mobile phase consisting of 55/45 methanol/ethanol (v/v) plus 20 mmol/L of lithium perchlorate).
Genomics and bioinformatics: Genomic DNA from patient one was sequenced and analyzed, together with that of her father, by Whole Exome Sequencing (WES). Patients 2 and 3 were sequenced as trio WES.
Experiments in fibroblasts and fly model
The CoQ10 forms 13C-CoQ10 and 13C-DMQ10 quantification was performed with an UPLC coupled to mass spectrometry analysis. Activities of succinate dehydrogenase (Complex II), ubiquinol-cytochrome c oxidoreductase (Complex III), succinate-cytochrome c reductase (Complex II + III), and citrate synthase (CS) were measured in fibroblasts’ lysates by spectrophotometric assays as previously described [13]. Oxygen consumption, acidification rate, and mitochondrial bioenergetics were studied using a Seahorse XF24 Extracellular Flux Analyzer (Agilent). Fibroblast protein expression analysis was analyzed by western blot. Antibodies used in this study are listed in Supplementary Table 1.
Transcriptional analysis
RNA extraction, probe synthesis, and hybridization with Clariom D arrays for human samples (Affymetrix-ThermoFisher) were performed in triplicate as described [14].
Drosophila methods
Flies were maintained in a standard medium with supplements. RNAi line to knock down rigor mortis gene (rig; cg30149) was obtained from Vienna Drosophila RNAi Centre (VDRC#107777KK). The climbing and motionless assays were performed with 40–50-day-old flies. The lifespan assay was duplicated with 150 flies of either sex for each treatment.
Statistical analysis
All data are presented as mean ± SD. Statistical analysis was performed using GraphPad Prism version 9.3.1 for Windows (GraphPad Software) with 2-tailed unpaired t-test. Differences between groups were considered statistically significant at P < 0.05. For analysis of survival, a log-rank (Mantel-Cox) and Gehan-Breslow-Wilcoxon tests were performed. P < 0.05 is regarded as statistically significant.
Results
Neurological and neuroradiological outcome after CoQ10 treatment
Neurological outcome from P1 and P2, comparing the clinical symptoms and signs from the first visit at our hospital and after 20 and 13 years of follow-up under CoQ10 treatment, are detailed in Supplementary Table 2.
Patient one (P1)
A 32-year-old woman previously reported in 2006, when she was 12 years old [9]. At that time, supplementation with oral CoQ10 at 20 mg/kg/day was started because the muscle biopsy findings showed a CoQ10 deficiency, and expected mitochondrial respiratory chain dysfunction. She showed a remarkable clinical improvement, with the main symptoms related to cerebellar dysfunction disappearing, and the ICARS scores decreasing after 16 months of CoQ10 treatment, as previously reported [9]. In addition, the Wechsler Intelligence Scale for Children (WISC-R) was performed, which demonstrated no progression of her cognitive impairment, showing borderline intellectual functioning. Twenty years later, the neurological exam shows an ICARS total score of 10 points, with mainly mild posture and gait and involvement in the kinetic subdomains (Supplementary Videos 1 (pretreatment), and 2 (postreatment)). She has a university degree and leads an independent life with a full-time remote job. Brain MRI showed progressive atrophy of the cerebellar cortex and vermis as well as T2 hyperintensity of the dentate nuclei (Supplementary Fig. 1). She is currently taking CoQ10 at a dose of 10 mg/kg/day with no side effects. However, she reports worsening of tremor and gait when she has poor therapeutic compliance. Treatment compliance has been good, as plasma CoQ10 levels showed an average value of 6.5 µmol/L (normal values: 0.4-1.1 µmol/L).
Patient two (P2)
He is a fifteen-year-old proband, the first-born male from non-consanguineous parents. He was referred to the neuropediatric department due to delayed motor development and tremor at 20 months of age. The neurological examination revealed microcephaly (−2.4 SD), mild axial hypotonia, and marked cerebellar signs, including slurred speech, slight limb dysmetria, and postural axial tremor. He could stand in a natural position without support, but walking was not achieved until later. The electrophysiological findings revealed a significant amplitude reduction suggestive of an axonal motor-sensory polyneuropathy, and brain MRI showed atrophy of the cerebellar cortex and vermis and T2 hyperintensity of the dentate nuclei (Supplementary Fig. 1). Muscle biopsy analysis showed a moderate CoQ10 deficiency. Treatment with oral CoQ10 at 30 mg/kg/day was started at 26 months of age. After 6 months of treatment, we noticed a significant improvement in the neurological exam, with decreased tremor and a significant improvement in fine motor skills. At 3.6 years of age, he developed mild pyramidal signs in the lower limbs (slight increase in distal muscle tone of the lower limbs, brisk deep tendon reflexes, and Babinski sign), which have remained stable to date. At the age of 5 years old, he began to walk with support, reaching unaided walking at 13 years old. At 15 years of age, the neurological exam showed slight dysarthria, rapid alternating movements and oscillating movement, and tremor with moderate amplitude on finger-nose testing, which was more marked in the left hand. The finger-finger test and heel shin test showed mild bilateral dysmetria. He could stand for more than 10 s without support in a natural position but not with feet together. Gait speed was slightly reduced with a broad base, and he could not walk with feet in a tandem position. He had increased distal limb tone and tendon reflexes. His score on the Scale for the Assessment and Rating of Ataxia (SARA) was 10 points. A neuropsychological test using the Wechsler Intelligence Scale (Fourth Edition (WISC-IV)) at the age of 9 years yielded a full-scale intelligence quotient (IQ) of 68, suggesting mild intellectual disability. The patient’s cognitive impairment showed no sustainable progression in the current evaluation. Head circumference normalized to −1.0 SD compared with the baseline situation. Brain MRI showed mild progression (Supplementary Fig. 1). He continues treatment with oral CoQ10 at 10 mg/kg/day with good compliance and an average CoQ10 value of 5.2 µmol/L (normal values: 0.4-1.1 µmol/L).
Laboratory results
Clinical laboratory analysis
Screening of inborn errors of metabolism, including mitochondrial biomarkers in blood and urine, was strictly normal over the course of the disease in P1 and P2 (not done in P3). However, due to clinical suspicion of a primary CoQ10 deficiency associated with ataxia, muscle and skin biopsies were performed in P1 and P2. In the muscle, a CoQ10 deficiency relative to citrate synthase activity, a surrogate of mitochondrial mass and number, was observed (P1: 1.16; P2: 0.72. Control range: 3.50–5.21 nmol CoQ10/citrate synthase units), along with a reduction in the activity of mitochondrial respiratory chain Complexes II + III, an activity highly dependent on CoQ [15]. In P1, the analysis of mitochondrial biomarkers performed in the muscle biopsy (COX staining, mitochondrial network, electron microscopy findings) supported the involvement of mitochondria, as previously reported [9].
Genetic diagnosis was reached by WES approach. Both patients were compound heterozygotes for mutations at the GEMIN5 gene: P1 bears a truncating mutation p.Lys742* (NM_015465.5: c.2224A > T), and the missense mutation p.Arg1016Cys (NM_015465.5: c.3046C > T). P2 has the same missense variant (p.Arg1016Cys), in combination with a truncating variant p.Ser411Hisfs*6 (NM_015465.5: c.1231_1232del). All variants were confirmed by Sanger sequencing. In P1 only her father was studied, since the mother’s sample was unavailable due to her sudden death by cardiac arrest. Both variants were identified in P2 and his parents. Both truncating mutations are predicted to trigger the Nonsense Mediated Decay (NMD) mechanisms and are considered null alleles, and both are classified as pathogenic according to the American College of Medical Genetics criteria [16]. The shared missense variant, p.Arg1016Cys is found in the general population with a minimum allele frequency (MAF) of 0.00448 (with 5 individuals being homozygotes, gnomAD v2.1, accessed April 2023). In silico impact prediction algorithms mostly classified this variant as damaging (CADD v1.6: 27.1, DANN 0.9992, SIFT, Polyphen2, etc.). This variant is classified by ACMG criteria as variant of unknown significance (VUS) but favoring pathogenicity by the in vitro functional analysis performed here (PS4, PM3, PP3, BS1). The in silico visualization of this variant shows that it affects a loop connecting two alfa-helices at the dimerization domain (Supplementary Fig. 2). P3 was previously reported in Kour et al. [4] (case 4, family 3), and he harbored the homozygous GEMIN5 missense mutation p.His923Pro (c.2768A > C). No mutations in mitochondrial genes or in CoQ10 biosynthesis related genes were detected (data not shown).
Functional studies: experiments in fibroblasts and fly model
Functional studies were conducted in fibroblasts from P1, P2, and P3. The total amount of CoQ10 was significantly lower in the three patients’ fibroblasts when compared with controls (Human Dermal Fibroblasts: HDF; Fig. 1A, Supplementary Fig. 3A), confirming the findings previously observed in muscle biopsies. Fibroblasts supplementation with exogenous CoQ10 restores CoQ10 to intracellular CoQ10 control levels except for P3 (the most severely affected patient), which shows only a slight increase. A defect of CoQ10 biosynthesis was identified in the patients’ fibroblasts after measuring the 13C-pHB incorporation into CoQ10 by LC-MS analysis (Fig. 1B, Supplementary Fig. 3B, C). The LC-MS analysis also detected the accumulation of the CoQ10 intermediate DMQ10, the substrate of the NADH-dependent hydroxylase COQ7, a component of the CoQ10 biosynthetic complex [17], but only in P1 fibroblasts.

A Total CoQ10 levels were measured by HPLC in GEMIN5 patients’ fibroblasts total homogenates. Cells were supplemented with 100 µM of CoQ10 prepared with BSA as vehicle. Non-treated cells were supplemented only with BSA at the same final concentration for 48 h. Results correspond to the average of 7 measurements for human dermal fibroblasts (HDF), 4 for P1, 2 for P2, and 5 for P3. (Statistical analysis was performed by Unpaired two-tailed t-test with the corresponding control). B Incorporation of 13C-4-HB to the quinone extract in 24 h as 13C-CoQ10 and 13C-DMQ10 in at least three biological replicates, measured in the patients’ fibroblasts. Cells were supplemented with 10 µM 13C-4-HB dissolved in water (Unpaired two-tailed t-test with the corresponding control cells). (****p < 0.0001, ***p < 0.001 **p < 0.01 and *p < 0.05).
The mitochondrial function in the patients’ fibroblasts was compromised, as indicated by the decrease in oxygen consumption rate (OCR), which was restored by CoQ10 supplementation. The restoration of OCR was complete in P1 fibroblasts and partial in P2 and P3 fibroblasts (Fig. 2A–F). We also measured succinate-DCPIP reductase (Complex II), decyl-ubiquinonol-cytochrome c reductase (Complex III) and succinate-cytochrome c reductase (Complex II + III) of the respiratory chain (Fig. 2G–I). When all activities were normalized using the citrate synthase activity as a mitochondrial mass marker, a significant decrease in Complex II + III was detected only in P1 fibroblasts (Fig. 2I). However, mitochondrial mass in patients’ fibroblasts was significantly decreased, as shown by citrate synthase activity measurement (Fig. 2J) and VDAC expression (Fig. 2K), indicating a significant decrease of mitochondrial respiratory chain activities in the analyzed patients’ fibroblasts, P1 and P3 (Fig. 2G–I).

A–C Oxygen consumption rate (OCR) was measured in patient (P1-3) and control (HDF) fibroblasts. Cells were supplemented with 100 µM of CoQ10 prepared with BSA as vehicle. Non-treated cells were supplemented only with BSA at the same final concentration for 48 h. Arrows indicate the addition of individual inhibitors. Results correspond to the average of six biological replicates ± SD. OL (oligomycin 0.4 μM); FCCP (Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone 0.1 μM); Rot (rotenone 0.1 μM); AntA (antimycin A 0.25 μM). D–F Quantitative data are plotted as the average ± SD (Two-way ANOVA with Sidak’s multiple comparison test). G–I Single (Complex II and III) and combined (Complex II + III) activities of the complexes were measured in fibroblasts homogenates from patients and controls. Results correspond to the average of at least three biological replicates ± SD, normalized by the citrate synthase activity (left axis) or normalized by protein content (right axis) (Unpaired two-tailed t-test). J Citrate synthase activity measured in fibroblast homogenates from patients and controls. Results correspond to the average of at four biological replicates ± SD. K Western blot analysis of VDAC protein levels in whole fibroblast lysates. A stain-free fluorescence signal was used as a loading control. Data correspond to the average of three normalized wells from patients’ fibroblasts expressed as relative expression ± SD. (Unpaired two-tailed t-test). (****p < 0.0001, ***p < 0.001 **p < 0.01 and *p < 0.05).
The transcriptomic analysis from P1 and P3 fibroblasts compared with controls (3 independent cell cultures) shows a general repression of genes involved in CoQ10 biosynthesis. The selected statistical criteria were fold change ≥2 or ≤−2, P ≥ 0.05 and false discovery rate (FDR) ≥ 0.05. The data description corresponding to each analysis (HDF versus each patient fibroblast and both samples in a patients’ pool) can be seen in Supplementary Fig. 4A. Principal components analysis clearly separated all samples, including control and patients’ fibroblasts, into three groups (Supplementary Fig. 4B). This separation was further confirmed by the hierarchical clustering analysis (Supplementary Fig. 4C). Most of genes responsible for CoQ10 biosynthesis were repressed in the patients’ pool (Supplementary Fig. 5) and in both patients’ fibroblasts, P1 and P3. However, the repression was remarkable in P3 fibroblasts, both in the number of genes repressed and its statistical significance (Supplementary Fig. 5).
GEMIN5 protein expression analysis by Western blot revealed a dramatic alteration in the protein pattern of fibroblasts from patients P1 and P3. Unlike the control, which showed a single band, both patients displayed additional bands of larger and smaller sizes (Fig. 3A). Furthermore, the analysis indicated significant alterations in the expression of proteins involved in CoQ10 synthesis, namely COQ3, COQ4, and COQ7 (Fig. 3B, C). Additionally, a decrease in the expression of subunits of mitochondrial respiratory chain complexes encoded by mtDNA, such as COX2 and ATP6, was observed, along with reduced levels of the mitochondrial factors TFAM (P1) and TUFM, which are involved in the transcription and translation of mtDNA-encoded genes (Fig. 3D, E).

Western blot analysis in fibroblasts lysates of (A) GEMIN5, (B) COQ proteins (COQ3, COQ4, COQ5, and COQ7), and (C) some subunits of the MRC complexes (NDUFA9 (CI), SDHA (CII), QCR2 (CIII), COX2 (CIV)) levels and mtDNA factors (TFAM and TUFM). D and E Quantification (average of at least three normalized wells) expressed as relative expression ± SD) (Unpaired two-tailed t-test; ****p < 0.0001, ***p < 0.001, **p < 0.01 and *p < 0.05; each patient against the control, HDF). Loading normalization was performed using the Stain Free gel system.
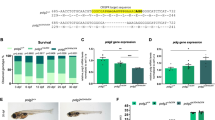
Additionally, we assessed the CoQ10 values in the rigor mortis defective fly model, which involves using RU-486 as an inducer of siRNA targeting the orthologue of the human GEMIN5 gene [4]. RU-486, also known as mifepristone, is a steroidal antiprogestogen typically used to bring about a medical abortion during pregnancy, but here the use is activating the fly GEMIN5 gene silencing. Details of RU-486 action can be found in the Supplementary text. D. melanogaster produces several isoforms of CoQ, with CoQ9 being the most abundant [18]. All CoQ levels were significantly decreased in the rigor mortis defective fly model (Fig. 4A, Supplementary Fig. 6A). Moreover, CoQ10 supplementation in rigor mortis-silenced adult flies partially restores intracellular CoQ8 and CoQ9 levels and completely restores CoQ10 levels (Fig. 4A, Supplementary Fig. 6A). It also increases the performance of adult flies in the climbing assay (Fig. 4B) and reduces the number of motionless flies (Fig. 4C, D). Additionally, CoQ10 supplementation partially restores the survival of male rigor mortis-silenced flies (Supplementary Fig. 6B). However, no effects were observed in females because the silencing induced by 20 µM RU-486 did not produce the same mortality as in males (Supplementary Fig. 6B).

A Male and female flies obtained after the cross of rigor mortis driver and UAS/GAL4 transgene were fed with normal food, RU-486 or RU-486 plus 0.1 mg/mL CoQ10 supplemented food. After 30 days from eclosion, flies were collected to quantify the amount of CoQ molecules present in flies (CoQ8, CoQ9, and CoQ10). Data are expressed as the average of 4 extractions performed with 30 flies ± SD. Total flies analyzed, 360. Unpaired two-tailed test, assuming gaussian distribution and assuming a similar SD. B Quantification of climbing assay in flies collected after 30 days from eclosion. Data correspond to the average ± SD of five independent experiments. Total flies analyzed, 300. Unpaired two-tailed test, assuming gaussian distribution and assuming a similar SD. C Motionless assay. Flies collected after 30 days from eclosion were subjected to motionless assay. Data correspond to the average ± SD of five independent experiments. Total flies analyzed, 300. Unpaired two-tailed test, assuming gaussian distribution and assuming a similar SD. D Picture corresponding to climbing and motionless assays.
Regarding flies’ development, CoQ10 supplementation significantly improves the number of adult flies that eclose from pupae compared to rigor mortis-silenced flies (Table 1). Rigor mortis silencing induced by the presence of RU-486 in food reduces the number of flies eclosed after larval development. This effect is initially independent of the RU-486 concentration, ranging from 1 nM to 100 nM. However, at a higher concentration of 300 nM, RU-486 induces a complete developmental arrest, and no flies are produced from eggs. CoQ10 supplementation at the permissive concentration of RU-486 (1 nM to 100 nM) restores the number of adults eclosed to a control level.
Discussion
A new syndrome caused by recessive mutations in GEMIN5 has been recently delineated [4, 7]. Here, we report on the clinical evolution of two patients with the milder phenotype of this condition, consisting of developmental delay, ataxia, and cerebellar atrophy. We conducted a long-term follow-up study of the two cases treated with CoQ10 when the genetic basis of their disease remained elusive. This is the first report to establish an association between GEMIN5 mutations and CoQ10 deficiency in patients and in cellular and animal models. This association opens a new therapeutic approach for this condition.
To date, autosomal recessive mutations in GEMIN5 have been identified in 39 patients and have been associated with a heterogeneous clinical spectrum that ranges from global developmental delay with cerebellar atrophy of early infantile onset to spastic ataxia syndrome with cerebellar atrophy that presents in adulthood [4,5,6,7,8]. The clinical features of our patients were similar to those recently reported and were characterized by late infantile-onset motor developmental delay and ataxic syndrome with static course [7].
CoQ10 replacement therapy resulted in subjective and clinically observed improvements in both patients, with improvement in cerebellar dysfunction symptoms. In P1, the ICARS scale score remained stable throughout the disease. In P2, the SARA scale could not be applied at baseline (26 months old), but it remained stable and mildly affected at 11 and 14 years. CoQ10 deficiency is one of the few treatable causes of ataxia, and symptom improvement following replacement has been observed in several patients with autosomal-recessive cerebellar ataxias (ARCA) [19,20,21,22]. However, since there are few long-term studies, it is unclear how long these benefits might be sustained or whether they improve the natural history of the disease [7]. Nevertheless, due to the very limited number of patients and the lack of a natural history study of the disease, we cannot assert that the initial improvement and stability over time were related to the CoQ10 supplementation or the underlying etiology. Further clinical trials are necessary to validate our findings.
While the small number of previously published cases does not allow us to establish a robust phenotype-genotype correlation, it is striking to note that both cases reported here have a milder form of the disease and share the same missense variant. A few healthy individuals homozygous for the missense variant have been identified in the general population, suggesting a very mild effect of the mutation on protein function. The presence of this allele in combination with a severe allele (such as a truncating variant) lead to a milder phenotype, as also noticed in the three patients carrying this same mutation in Rajan et al. [7]. This variant is very close to the previously reported p.Asp1019Glu, also found in compound heterozygosis in a family presenting with the milder form of this condition (Family 12 in Kour et al. [4]). The in silico structural analysis of the previously described missense variants shows that most lie within an alpha-helix or a beta-sheet and probably affect the protein folding. The few mutations affecting loops lie on the WD40 repeats. Mutations p.Arg1016Cys, p.Asp1019Glu, and p.His923Pro, on the other hand, are found in a loop outside the WD40 repeats at the TPR-like dimerization module [6, 23], and thus, their effect on the protein structure is likely less severe, as suggested by the results of the functional experiments done here. In patients’ fibroblasts, GEMIN5 expression produces a triple band, where the middle band corresponds to the expected size of 170 kDa found in controls. Mutations in the TPR-like domain can produce conformational changes that induce a higher stabilization of some dimers (top band) or can induce non-completely denatured monomers (lower band) exhibiting a higher SDS-PAGE mobility [24].
Although a direct relationship was not reported between the canonical function of GEMIN5 and the respiratory metabolism, data obtained from patients’ fibroblast support mitochondrial dysfunction, including CoQ10 synthesis defects. A decrease in total CoQ10 in fibroblast was detected and affected the CoQ10 synthesis rate. The decrease in the OCR and the activity of some mitochondrial respiratory chain complexes support mitochondrial dysfunction. Previous studies have reported impaired mitochondrial biogenesis in muscle samples from patients with spinal muscular atrophy (SMA), mainly deficiency in mitochondrial respiratory complexes I, II, and IV, deficiency in cytochrome-c oxidase (COX) histochemical staining, and reduced levels of the respiratory chain subunits COX1, COX2 and COX4 [25]. In NSC-34 cells, the SMN siRNA knock-down drives a significant SMN protein decrease and ATP production 48 h after the transfection [26]. In ∆7 SMA mice model, motor neurons show a decrease in OCR compared to controls and midbrain neurons from the same model [27]. In fibroblasts from patients with defective GEMIN5, the fact that mitochondrial mass measured as citrate synthase activity and the VDAC expression were decreased rules out that GEMIN5 mutations specifically caused CoQ10 deficiency, which was probably the effect of a general repression of mitochondrial function. GEMIN5 may regulate mitochondrial metabolism, and it is plausible that the CoQ10 synthesis pathway is another pathway regulated by GEMIN5, in addition to those previously reported [6]. COQ genes and proteins expression data support this statement. The transcriptomic analysis showed that some COQ genes are repressed in patients’ fibroblasts. This repression is higher in P3 fibroblasts, the patient most severely affected of the three analyzed here. A similar effect was observed in COQ protein expression; some components of the CoQ10 biosynthetic complex (Q-synthome) were repressed in the patient’s fibroblast, together with reduced levels of some respiratory chain subunits encoded by mtDNA such as COX2 and ATP6.
GEMIN5 patients analyzed clinically in this study showed an improvement after supplementing with CoQ10. Oral CoQ10 supplementation significantly increased levels in those patients’ fibroblasts, partially restoring the OCR in P2 and P3 fibroblasts and completely in P1. The fly model corroborates those findings, the rigor mortis (GEMIN5 fly ortholog) silenced flies (rig) showed a general decrease of all CoQ molecules synthesized in flies, and the supplementation with CoQ10 partially restores the CoQ10 and CoQ9 levels without affecting CoQ8. Moreover, CoQ10 produces a significant impact in adult flies, improving climbing activity, reducing the motionless flies, and increasing lifespan in males. However, the CoQ10 effect could be muted in flies relative to humans, as CoQ10 is not the most abundant CoQ in this organism. This role corresponds to CoQ9, which moderately increases after CoQ10 supplementation [18]. A better effect was detected during embryo development; in rigor mortis silenced conditions, CoQ10 duplicates the number of adult flies eclosed from pupae. Since the number of eggs deposited after the crosses is similar and all pupae produced an adult fly, the defect caused by rigor mortis silencing must be produced on larval development, which is restored by CoQ10 supplementation. The positive effect of CoQ10 supplementation is associated with the silencing action of RU-486 in the rigor mortis flies but not with its possible toxicity since CoQ10 does not induce positive changes in the development of fly stocks that do not carry the GAL4 inducer (cg30149 and cg14437) or that do not carry the silencing cassette (w1118;tub-GS-GAL4).
In conclusion, we report the association between GEMIN5 dysfunction and CoQ10 deficiency for the first time. This association opens the possibility of a therapeutic approach for this condition, consisting of oral CoQ10 administration. In the two cases reported here, CoQ10 therapy demonstrated subjective clinical improvement, and after long-term follow-up, the status of the patients remained stable with some improvements, although MRI results did not improve. All the experiments carried out in fibroblasts and the rigor mortis fly model confirmed the CoQ10 deficiency observed in patients, the mechanistic relationship between GEMIN5 mutations and CoQ10 deficiency, and more importantly, the correction of different mitochondrial functions after CoQ10 supplementation. Our report opens up new therapeutic approaches to GEMIN5-related ataxia syndrome and needs further validation with larger patient numbers.
Data availability
Data about the basic research experiments are available upon reasonable request.
Code availability
Some of the data generated or analyzed during this study can be found within the published article and its supplementary files. Data of WES from patients is available from the corresponding author upon reasonable request.
References
Battle DJ, Kasim M, Yong J, Lotti F, Lau CK, Mouaikel J, et al. The SMN complex: an assembly machine for RNPs. Cold Spring Harb Symp Quant Biol. 2006;71:313–20.
Pellizzoni L, Yong J, Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–9.
Workman E, Kalda C, Patel A, Battle DJ. Gemin5 binds to the survival motor neuron mRNA to regulate SMN expression. J Biol Chem. 2015;290:15662–9.
Kour S, Rajan DS, Fortuna TR, Anderson EN, Ward C, Lee Y, et al. Loss of function mutations in GEMIN5 cause a neurodevelopmental disorder. Nat Commun. 2021;12:2558.
Saida K, Tamaoki J, Sasaki M, Haniffa M, Koshimizu E, Sengoku T, et al. Pathogenic variants in the survival of motor neurons complex gene GEMIN5 cause cerebellar atrophy. Clin Genet. 2021;100:722–30.
Francisco-Velilla R, Embarc-Buh A, Del Cano-Ochoa F, Abellan S, Vilar M, Alvarez S, et al. Functional and structural deficiencies of Gemin5 variants associated with neurological disorders. Life Sci Alliance. 2022; 5:e202201403.
Rajan DS, Kour S, Fortuna TR, Cousin MA, Barnett SS, Niu Z, et al. Autosomal recessive cerebellar atrophy and spastic ataxia in patients with pathogenic biallelic variants in GEMIN5. Front Cell Dev Biol. 2022;10:783762.
Ibrahim N, Naz S, Mattioli F, Guex N, Sharif S, Iqbal A, et al. A biallelic truncating variant in the TPR domain of GEMIN5 associated with intellectual disability and cerebral atrophy. Genes (Basel). 2023;14:707.
Artuch R, Brea-Calvo G, Briones P, Aracil A, Galvan M, Espinos C, et al. Cerebellar ataxia with coenzyme Q10 deficiency: diagnosis and follow-up after coenzyme Q10 supplementation. J Neurol Sci. 2006;246:153–8.
Pineda M, Montero R, Aracil A, O’Callaghan MM, Mas A, Espinos C, et al. Coenzyme Q(10)-responsive ataxia: 2-year-treatment follow-up. Mov Disord. 2010;25:1262–8.
Potashman MH, Mize ML, Beiner MW, Pierce S, Coric V, Schmahmann JD. Ataxia rating scales reflect patient experience: an examination of the relationship between clinician assessments of cerebellar ataxia and patient-reported outcomes. Cerebellum. 2022;22:1257–73.
Montero R, Sanchez-Alcazar JA, Briones P, Hernandez AR, Cordero MD, Trevisson E, et al. Analysis of coenzyme Q10 in muscle and fibroblasts for the diagnosis of CoQ10 deficiency syndromes. Clin Biochem. 2008;41:697–700.
Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc. 2012;7:1235–46.
Fernandez-Ayala DJ, Guerra I, Jimenez-Gancedo S, Cascajo MV, Gavilan A, Dimauro S, et al. Survival transcriptome in the coenzyme Q10 deficiency syndrome is acquired by epigenetic modifications: a modelling study for human coenzyme Q10 deficiencies. BMJ Open. 2013;3:e002524.
Yubero D, Montero R, Artuch R, Land JM, Heales SJ, Hargreaves IP. Biochemical diagnosis of coenzyme q10 deficiency. Mol Syndromol. 2014;5:147–55.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Danhauser K, Herebian D, Haack TB, Rodenburg RJ, Strom TM, Meitinger T, et al. Fatal neonatal encephalopathy and lactic acidosis caused by a homozygous loss-of-function variant in COQ9. Eur J Hum Genet. 2016;24:450–4.
Liu J, Wu Q, He D, Ma T, Du L, Dui W, et al. Drosophila sbo regulates lifespan through its function in the synthesis of coenzyme Q in vivo. J Genet Genomics. 2011;38:225–34.
Emmanuele V, Lopez LC, Berardo A, Naini A, Tadesse S, Wen B, et al. Heterogeneity of coenzyme Q10 deficiency: patient study and literature review. Arch Neurol. 2012;69:978–83.
Potgieter M, Pretorius E, Pepper MS. Primary and secondary coenzyme Q10 deficiency: the role of therapeutic supplementation. Nutr Rev. 2013;71:180–8.
Beaudin M, Matilla-Duenas A, Soong BW, Pedroso JL, Barsottini OG, Mitoma H, et al. The classification of autosomal recessive cerebellar ataxias: a consensus statement from the society for research on the cerebellum and ataxias task force. Cerebellum. 2019;18:1098–125.
Wang Y, Hekimi S. The efficacy of coenzyme Q(10) treatment in alleviating the symptoms of primary coenzyme Q(10) deficiency: a systematic review. J Cell Mol Med. 2022;26:4635–44.
Francisco-Velilla R, Embarc-Buh A, Abellan S, Del Cano-Ochoa F, Ramon-Maiques S, Martinez-Salas E. Phosphorylation of T897 in the dimerization domain of Gemin5 modulates protein interactions and translation regulation. Comput Struct Biotechnol J. 2022;20:6182–91.
Shi Y, Mowery RA, Ashley J, Hentz M, Ramirez AJ, Bilgicer B, et al. Abnormal SDS-PAGE migration of cytosolic proteins can identify domains and mechanisms that control surfactant binding. Protein Sci. 2012;21:1197–209.
Ripolone M, Ronchi D, Violano R, Vallejo D, Fagiolari G, Barca E, et al. Impaired muscle mitochondrial biogenesis and myogenesis in spinal muscular atrophy. JAMA Neurol. 2015;72:666–75.
Acsadi G, Lee I, Li X, Khaidakov M, Pecinova A, Parker GC, et al. Mitochondrial dysfunction in a neural cell model of spinal muscular atrophy. J Neurosci Res. 2009;87:2748–56.
Miller N, Shi H, Zelikovich AS, Ma YC. Motor neuron mitochondrial dysfunction in spinal muscular atrophy. Hum Mol Genet. 2016;25:3395–406.
Acknowledgements
We are indebted to the patients and families with GEMIN5 mutations.
Funding
This research was funded by Instituto de Salud Carlos III (PI20-00340 and PI20/00541) (co-funded by European Regional Development Fund “A way to make Europe”), the Centre for Biomedical Research on Rare Diseases (CIBERER), the U.S. National Institute of Health (NIH), the National Institute of Neurological Disorders and Stroke (NINDS) R01NS134215 and Carmen de Torres grants (Institut de Recerca Sant Joan de Déu). CIBERER is an initiative from the Health Institute Carlos III.
Author information
Authors and Affiliations
Contributions
All authors critically reviewed the manuscript. RA, CSO, AGC, UP, and PN contributed to the experimental design. NJP, NR, MdMOC, PJML, CIO, JM, AN, DNdB, DSR and MP contributed to the clinical data collection, analysis, and evaluation. RU, DY, FP. performed the genetic data analysis. MVCA, ASC, DMFA, EGD, CO, AJPF, CSO, PN, RU, LS, TRF, and NJP conducting experiments, acquiring data, analyzing data, providing reagents. RA, CSO, NJP, RU, CO, MVCA drafted the manuscript and elaborated the main figures and tables.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All patients provided the informed consent for the genetic studies and skin biopsies collection. For supplementary videos, consent from patient 1 (P1) was obtained. For patient 2 (P2), the family refused to provide informed consent to publish the video. The study was conducted following the Declaration of Helsinki, and the ethical committee of the institution of Sant Joan de Déu Hospital has approved it.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cascajo-Almenara, M.V., Juliá-Palacios, N., Urreizti, R. et al. Mutations of GEMIN5 are associated with coenzyme Q10 deficiency: long-term follow-up after treatment. Eur J Hum Genet 32, 426–434 (2024). https://doi.org/10.1038/s41431-023-01526-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-023-01526-2
This article is cited by
-
Artificial intelligence â the next generation of sequencing?
European Journal of Human Genetics (2024)
