
Abstract
GEMIN5 exerts key biological functions regulating pre-mRNAs intron removal to generate mature mRNAs. A series of patients were reported harboring mutations in GEMIN5. No treatments are currently available for this disease. We treated two of these patients with oral Coenzyme Q10 (CoQ10), which resulted in neurological improvements, although MRI abnormalities remained. Whole Exome Sequencing demonstrated compound heterozygosity at the GEMIN5 gene in both cases: Case one: p.Lys742* and p.Arg1016Cys; Case two: p.Arg1016Cys and p.Ser411Hisfs*6. Functional studies in fibroblasts revealed a decrease in CoQ10 biosynthesis compared to controls. Supplementation with exogenous CoQ10 restored it to control intracellular CoQ10 levels. Mitochondrial function was compromised, as indicated by the decrease in oxygen consumption, restored by CoQ10 supplementation. Transcriptomic analysis of GEMIN5 patients compared with controls showed general repression of genes involved in CoQ10 biosynthesis. In the rigor mortis defective flies, CoQ10 levels were decreased, and CoQ10 supplementation led to an improvement in the adult climbing assay performance, a reduction in the number of motionless flies, and partial restoration of survival. Overall, we report the association between GEMIN5 dysfunction and CoQ10 deficiency for the first time. This association opens the possibility of oral CoQ10 therapy, which is safe and has no observed side effects after long-term therapy.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
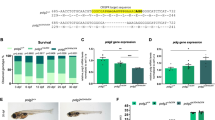
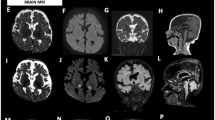

Data availability
Data about the basic research experiments are available upon reasonable request.
Code availability
Some of the data generated or analyzed during this study can be found within the published article and its supplementary files. Data of WES from patients is available from the corresponding author upon reasonable request.
References
Battle DJ, Kasim M, Yong J, Lotti F, Lau CK, Mouaikel J, et al. The SMN complex: an assembly machine for RNPs. Cold Spring Harb Symp Quant Biol. 2006;71:313–20.
Pellizzoni L, Yong J, Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–9.
Workman E, Kalda C, Patel A, Battle DJ. Gemin5 binds to the survival motor neuron mRNA to regulate SMN expression. J Biol Chem. 2015;290:15662–9.
Kour S, Rajan DS, Fortuna TR, Anderson EN, Ward C, Lee Y, et al. Loss of function mutations in GEMIN5 cause a neurodevelopmental disorder. Nat Commun. 2021;12:2558.
Saida K, Tamaoki J, Sasaki M, Haniffa M, Koshimizu E, Sengoku T, et al. Pathogenic variants in the survival of motor neurons complex gene GEMIN5 cause cerebellar atrophy. Clin Genet. 2021;100:722–30.
Francisco-Velilla R, Embarc-Buh A, Del Cano-Ochoa F, Abellan S, Vilar M, Alvarez S, et al. Functional and structural deficiencies of Gemin5 variants associated with neurological disorders. Life Sci Alliance. 2022; 5:e202201403.
Rajan DS, Kour S, Fortuna TR, Cousin MA, Barnett SS, Niu Z, et al. Autosomal recessive cerebellar atrophy and spastic ataxia in patients with pathogenic biallelic variants in GEMIN5. Front Cell Dev Biol. 2022;10:783762.
Ibrahim N, Naz S, Mattioli F, Guex N, Sharif S, Iqbal A, et al. A biallelic truncating variant in the TPR domain of GEMIN5 associated with intellectual disability and cerebral atrophy. Genes (Basel). 2023;14:707.
Artuch R, Brea-Calvo G, Briones P, Aracil A, Galvan M, Espinos C, et al. Cerebellar ataxia with coenzyme Q10 deficiency: diagnosis and follow-up after coenzyme Q10 supplementation. J Neurol Sci. 2006;246:153–8.
Pineda M, Montero R, Aracil A, O’Callaghan MM, Mas A, Espinos C, et al. Coenzyme Q(10)-responsive ataxia: 2-year-treatment follow-up. Mov Disord. 2010;25:1262–8.
Potashman MH, Mize ML, Beiner MW, Pierce S, Coric V, Schmahmann JD. Ataxia rating scales reflect patient experience: an examination of the relationship between clinician assessments of cerebellar ataxia and patient-reported outcomes. Cerebellum. 2022;22:1257–73.
Montero R, Sanchez-Alcazar JA, Briones P, Hernandez AR, Cordero MD, Trevisson E, et al. Analysis of coenzyme Q10 in muscle and fibroblasts for the diagnosis of CoQ10 deficiency syndromes. Clin Biochem. 2008;41:697–700.
Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc. 2012;7:1235–46.
Fernandez-Ayala DJ, Guerra I, Jimenez-Gancedo S, Cascajo MV, Gavilan A, Dimauro S, et al. Survival transcriptome in the coenzyme Q10 deficiency syndrome is acquired by epigenetic modifications: a modelling study for human coenzyme Q10 deficiencies. BMJ Open. 2013;3:e002524.
Yubero D, Montero R, Artuch R, Land JM, Heales SJ, Hargreaves IP. Biochemical diagnosis of coenzyme q10 deficiency. Mol Syndromol. 2014;5:147–55.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Danhauser K, Herebian D, Haack TB, Rodenburg RJ, Strom TM, Meitinger T, et al. Fatal neonatal encephalopathy and lactic acidosis caused by a homozygous loss-of-function variant in COQ9. Eur J Hum Genet. 2016;24:450–4.
Liu J, Wu Q, He D, Ma T, Du L, Dui W, et al. Drosophila sbo regulates lifespan through its function in the synthesis of coenzyme Q in vivo. J Genet Genomics. 2011;38:225–34.
Emmanuele V, Lopez LC, Berardo A, Naini A, Tadesse S, Wen B, et al. Heterogeneity of coenzyme Q10 deficiency: patient study and literature review. Arch Neurol. 2012;69:978–83.
Potgieter M, Pretorius E, Pepper MS. Primary and secondary coenzyme Q10 deficiency: the role of therapeutic supplementation. Nutr Rev. 2013;71:180–8.
Beaudin M, Matilla-Duenas A, Soong BW, Pedroso JL, Barsottini OG, Mitoma H, et al. The classification of autosomal recessive cerebellar ataxias: a consensus statement from the society for research on the cerebellum and ataxias task force. Cerebellum. 2019;18:1098–125.
Wang Y, Hekimi S. The efficacy of coenzyme Q(10) treatment in alleviating the symptoms of primary coenzyme Q(10) deficiency: a systematic review. J Cell Mol Med. 2022;26:4635–44.
Francisco-Velilla R, Embarc-Buh A, Abellan S, Del Cano-Ochoa F, Ramon-Maiques S, Martinez-Salas E. Phosphorylation of T897 in the dimerization domain of Gemin5 modulates protein interactions and translation regulation. Comput Struct Biotechnol J. 2022;20:6182–91.
Shi Y, Mowery RA, Ashley J, Hentz M, Ramirez AJ, Bilgicer B, et al. Abnormal SDS-PAGE migration of cytosolic proteins can identify domains and mechanisms that control surfactant binding. Protein Sci. 2012;21:1197–209.
Ripolone M, Ronchi D, Violano R, Vallejo D, Fagiolari G, Barca E, et al. Impaired muscle mitochondrial biogenesis and myogenesis in spinal muscular atrophy. JAMA Neurol. 2015;72:666–75.
Acsadi G, Lee I, Li X, Khaidakov M, Pecinova A, Parker GC, et al. Mitochondrial dysfunction in a neural cell model of spinal muscular atrophy. J Neurosci Res. 2009;87:2748–56.
Miller N, Shi H, Zelikovich AS, Ma YC. Motor neuron mitochondrial dysfunction in spinal muscular atrophy. Hum Mol Genet. 2016;25:3395–406.
Acknowledgements
We are indebted to the patients and families with GEMIN5 mutations.
Funding
This research was funded by Instituto de Salud Carlos III (PI20-00340 and PI20/00541) (co-funded by European Regional Development Fund “A way to make Europe”), the Centre for Biomedical Research on Rare Diseases (CIBERER), the U.S. National Institute of Health (NIH), the National Institute of Neurological Disorders and Stroke (NINDS) R01NS134215 and Carmen de Torres grants (Institut de Recerca Sant Joan de Déu). CIBERER is an initiative from the Health Institute Carlos III.
Author information
Authors and Affiliations
Contributions
All authors critically reviewed the manuscript. RA, CSO, AGC, UP, and PN contributed to the experimental design. NJP, NR, MdMOC, PJML, CIO, JM, AN, DNdB, DSR and MP contributed to the clinical data collection, analysis, and evaluation. RU, DY, FP. performed the genetic data analysis. MVCA, ASC, DMFA, EGD, CO, AJPF, CSO, PN, RU, LS, TRF, and NJP conducting experiments, acquiring data, analyzing data, providing reagents. RA, CSO, NJP, RU, CO, MVCA drafted the manuscript and elaborated the main figures and tables.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All patients provided the informed consent for the genetic studies and skin biopsies collection. For supplementary videos, consent from patient 1 (P1) was obtained. For patient 2 (P2), the family refused to provide informed consent to publish the video. The study was conducted following the Declaration of Helsinki, and the ethical committee of the institution of Sant Joan de Déu Hospital has approved it.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cascajo-Almenara, M.V., Juliá-Palacios, N., Urreizti, R. et al. Mutations of GEMIN5 are associated with coenzyme Q10 deficiency: long-term follow-up after treatment. Eur J Hum Genet 32, 426–434 (2024). https://doi.org/10.1038/s41431-023-01526-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-023-01526-2
This article is cited by
-
Artificial intelligence â the next generation of sequencing?
European Journal of Human Genetics (2024)
