
Abstract
Inherited retinal dystrophies (IRDs) affect 1 in 3000 individuals worldwide and are genetically heterogeneous, with over 270 identified genes and loci; however, there are still many identified disorders with no current genetic etiology. Whole exome sequencing (WES) provides a hypothesis-free first examination of IRD patients in either a clinical or research setting to identify the genetic cause of disease. We present a study of IRD in ten families from Alberta, Canada, through the lens of novel gene discovery. We identify the genetic etiology of IRDs in three of the families to be variants in known disease-associated genes, previously missed by clinical investigations. In addition, we identify two potentially novel associations: LRP1 in early-onset drusen formation and UBE2U in a multi-system condition presenting with retinoschisis, cataracts, learning disabilities, and developmental delay. We also describe interesting results in our unsolved cases to provide further information to other investigators of these blinding conditions.
Similar content being viewed by others


Introduction
Inherited retinal dystrophies (IRDs) affect ~1 in 3000 individuals and account for a large proportion of untreatable blinding disorders, however many conditions remain without a genetic explanation [1]. IRDs affect the light detecting cells of the retina, rod and/or cone photoreceptors, and may present as either non-syndromic or as a multisystem disorder (i.e. Bardet-Biedl syndrome). Advanced technologies such as next generation sequencing (NGS) have significantly improved our ability to both identify novel associations in family-based studies and provide genetic diagnoses in patients with IRDs. Currently, the proportion of IRD patients that receive a genetic diagnosis from a panel is variable (37–74%) [2]. Whole exome sequencing (WES) provides a hypothesis free first approach to identification of the genetic etiology of a condition, or as a research tool to identify novel genes and associations. The workflow for filtering and interpreting results of WES needs to be understood and appreciated, as not all investigations will result in the identification of previously documented variations in disease-associated genes. We report our experience with WES in Alberta, Canada using a cohort of ten families with heritable ocular disease selected for potential novel gene discovery. Through this study, we solved three families, which either confirmed or altered diagnoses, and found two potentially novel associations: variants in the low-density lipoprotein receptor related protein 1 (LRP1) gene in patients with macular drusen formation and in the ubiquitin conjugating enzyme E2 (UBE2U) gene in a pedigree of retinal dystrophy with associated systemic defects. In addition, we described potential associations in our unsolved cases, which will assist further investigations into these conditions.
Materials & methods
Family selection
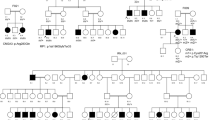
Families and individuals were selected based on clinical diagnoses, availability of DNA/phenotypic information from family members, and previous negative clinical/research genetic testing. In total, ten families were selected for our WES study and signed consent obtained. Clinical data on affected family members are listed in Table 1; pedigrees are presented in Fig. 1. This study was approved by the University of Alberta Human Ethics Office (Pro00045377). All procedures abide by the Declaration of Helsinki. Whole exome sequencing five micrograms of whole genomic DNA isolated from either saliva or blood from family members were sent to the Beijing Genomics Institute (BGI, Beijing, China) or DNALink (South Korea) for WES. BGI used the Agilent B5 (50 M) exome capture kit and was run on the BGISEQ-500 (Avg coverage of 100x), DNALink used the SureSelect Exome V5 QXT and was run on Illumina Hiseq2500 (Avg Coverage of 96x). WES filters were set for appropriate mode of inheritance, moderate and high impact variants (missense, splice, and nonsense variants), and a minor allele frequency (MAF) adjusted for rarity of the condition (<0.01 for rare conditions, <0.02 for more common presentations). Filtered gene lists were compared with a list of known retinal disease genes (RetNet https://sph.uth.edu/retnet/, Accessed May 31, 2020). Families who were not solved using this methodology were moved to a novel discovery pipeline, segregation analysis where possible, and in silico analyses (SIFT/PROVEAN [3, 4], PolyPhen-2 [5], MutationTaster [6], SplicePort [7], Human Splicing Finder [8]) for any variants. All variants and phenotypic data listed in this paper were submitted to the Leiden Open Variation Database (LOVD; https://www.lovd.nl/, submitted and accessed January 22nd, 2021).

Ten families were selected based on clinical diagnoses, availability of DNA/phenotypic information from family members, and previous negative clinical/research genetic testing. Shaded individuals indicate affected retinal conditions/syndromes further highlighted in Table 1.
Clinical investigations
The age at clinical diagnosis, best corrected visual acuity (BCVA), fundus findings and ocular history, spectral domain ocular coherence tomography (SD-OCT) were reviewed and documented based on the availability. Multifocal electroretinography (mfERG) was recorded with the Espion system (Diagnosys, Lowell MA USA) using DTL electrodes according to ISCEV standards (www.ISCEV.org). Full field ERGs (ffERG) were recorded with the UTAS system (LKC, Gaithersburg, MD USA).
Results
Solved families
Family C137 — choroidal atrophy (Fig. 1)
The proband of family C137 presented at age 72 with chorioretinal atrophy (Table 1). While the peripheral retina was relatively preserved, bilateral macular depigmentation and moderate vascular attenuation were noted. Visual acuity was counting fingers (CF) in the left eye (Oculus Sinister; OS), and 20/800 in the right eye (Oculus Dexter; OD). Clinical investigation led to molecular investigations for choroideremia. Full sequencing of the coding region of CHM revealed no variants, and Western analysis showed normal REP1 protein expression in lymphoblastoid cells (data not shown). WES Results: WES analyses were compared with a list of known retinal genes and revealed a heterozygous variant within RPE65 (c.1430G > A: p.(D477G)). No other variants of interest were noted.
Family M59 — retinitis pigmentosa (Fig. 1)
Two males of Family M59 were diagnosed in early adolescence with retinitis pigmentosa (RP) characterized by night blindness, and peripheral vision loss (Table 1). OCT scan for II-1 showed irregularity of the outer retina with extensive atrophy of the ellipsoid zone and deposits at the level of the Bruch’s membrane-RPE complex (Supplementary Fig. 3B). Panel testing for autosomal recessive RP (Asper Biotech, Tartu Estonia) and X-linked genes (eyeGENE®) were negative. WES Results: WES revealed compound heterozygous variations within BBS1 (c.1169 T > G: p.(M390R) and c.1040del: p.(M347fs)) in both affected males (Table 2). Only the c.1040del variant was noted in the unaffected sister. Follow-up of this family revealed that both brothers suffered from mild learning disabilities, and the youngest brother had postaxial polydactyly on his right foot that was surgically removed. Neither brother was obese or had kidney problems. Considering the new phenotypic data and our genetic result, the diagnosis was altered from nonsyndromic RP to Bardet-Biedl Syndrome (BBS; OMIM 209900).
Family M53 — macular dystrophy (Fig. 1)
The proband of family M53 was initially diagnosed with a cone-dystrophy. At age 12, he had BCVA 20/400 vision in both eyes with reduced colour vision, hyper-reflective flecks in the macula. A ffERG revealed a reduced photopic b-wave. The photopic flicker response was within normal limits as was the scotopic series. The amplitude of the response of the mfERG was reduced, indicating loss of central retinal function. By age 19, a maculopathy was more apparent, with features of degeneration seen with fundus autofluorescence imaging. OCT across the macula showed thinning and atrophy of the ellipsoid zone (Supplementary Fig. 3A). WES Results: WES revealed a single previously known recessive pathogenic variant c.4469 G > A:p.(C1490Y) in ABCA4 in Stargardt macular dystrophy (OMIM 248200) [9]. This variant was heterozygous in the mother, and not present in the unaffected father. A second variant was found through a larger study of ABCA4 deep-intronic variants by our collaborators and was noted as c.4539 + 2028 C > T: p.(R1514Lfs*36) [10].
Partially solved
Family M73 — macular schisis and hearing loss (Fig. 1)
The 11-year-old proband of Family M73 presented with bilateral macular schisis and sensorineural hearing loss (SNHL). When diagnosed at age of 11, his BCVA was 20/150 on the right side and 20/40 on the left side. The descriptions for his fundus (Supplementary Fig. 2A) and OCT scan (Supplementary Fig. 3G) are included in Table 1. Chromosomal analysis and testing for known hearing loss genes were negative. ERG revealed only a residual cone flicker response, thus ruling out X-linked juvenile retinoschisis. A blood sample was obtained from the proband and a lymphoblastoid cell line was established via Epstein-Barr transduction. Given the genetic results in this family (described below), a possible peroxisomal biogenesis disorder (Heimler syndrome, OMIM 616617) was investigated. WES Results & cDNA Analysis: WES identified a heterozygous variant in PEX6, c.1802G > A: p.(R601Q) previously associated with Heimler Syndrome, a mild peroxisomal biogenesis disorder (OMIM 616617) [11, 12]. No nail or dental abnormalities were noted, and blood pipecolic/plasmalogen levels were normal. cDNA was isolated from a lymphoblastoid cell line, and the coding region of PEX6 was sequenced; no second coding or splicing variant was noted (data not shown). All variants can be found in Supplementary Table 1. As Falkenberg et al. [13] suggested an allelic expression imbalance can occur in the presence of a common 3’-UTR single-nucleotide polymorphism (SNP, rs144286892), we tested this specific region of the PEX6 3’-UTR. Sanger sequencing confirmed the proband is heterozygous for this SNP, which is in agreement with the allelic expression imbalance theory (Supplementary Fig. 1). However, the father (I-1) who passed the c.1802 G > A variant to the proband was also a heterozygote for rs144286892 and had no reported vision or hearing problems. This leaves this case partially solved.
Putatively novel findings
Family M54 and M70 — macular drusen (Fig. 1)
The proband of Family M54 was a 44-year-old female who presented with marked bilateral symmetrical macular drusen (Fig. 2B). The retina was otherwise normal, with no signs of degeneration. No other family members were affected, leading to a presumed recessive mode of inheritance. Family M70 presented with one affected female with similar bilateral macular drusen at age 36. OCT scan also showed numerous hyporeflective elevations of the retinal pigment epithelium in the central macula consistent with drusen (Supplementary Fig. 2E). The proband’s mother and sister had no retinal phenotype and a recessive mode of inheritance was considered. The father was unavailable for examination or for a DNA sample. WES Results: WES presumed that the appearance of drusen represented a heritable macular disorder with a relatively higher MAF ( < 0.02) as the trait is not rare in later age groups. Analysis of family M54 revealed compound heterozygous variations in LRP1 ([c.650 C > T]; [c.9628 G > C], Table 2). Compound heterozygous variants were also found in CPAMD8 ([c.1030 G > A];[c.5305 C > A]). Compound heterozygous variants within LRP1 were also noted within the proband of Family M70 ([c.2910 G > A];[c.11930 C > T]). In silico analyses can be found in Table 2, and all variants are listed in Supplementary Table 1.

A Normal fundus photo (top) and normal optical coherence tomography (OCT; bottom). B Fundus photo (top) of the proband from family M54 illustrating multiple large, confluent drusen in the central macula (white arrow) and nasal retina. OCT (bottom) shows numerous sub-RPE deposits in the macula consistent with drusen (white arrows). (C) and (D) Hypothesis of LRP1 involvement in MD: as extracellular matter accumulates in Bruch’s membrane, LRP1 endocytoses and destroys this matter. As LRP1 is mutated, this process slows, or is unable to bind particular ligands, leading to accumulated extracellular material, and drusen formation.
Family M72 — congenital cataracts, hearing loss, retinoschisis (Fig. 1)
The affected mother and three affected offspring of Family M72 (sons age 3 and 11, daughter age 7) were referred with dominantly inherited learning disabilities, facial dysmorphism, congenital cataracts, and congenital hearing loss (Table 1). OCT scan from II-1 showed splitting between the inner nuclear and the outer plexiform layer, consistent with retinoschisis (Fig. 4B). WES Results Analysis assumed a dominant mode of inheritance. WES showed no variants in known disease-related genes, and revealed several novel variants of interest, namely, variants in STUM c.62 A > G: p.(D21G) and UBE2U c.122 A > C: p.(E41A). In silico analyses of the STUM variant indicated that the variant was likely benign, though analyses predicted the UBE2U p.(E41A) variant to be pathogenic (Table 2). See Supplementary Table 1 for all identified variants.
Unsolved families
Family M68 — macular dystrophy with normal fundus (Fig. 1)
The proband of family M68 presented with 20/40 vision in both eyes with −3 dioptres of myopia. A ffERG noted normal cone function and a mild reduction in the pure rod response. A mfERG was reduced in comparison to normal reflecting a retinopathy predominantly affecting the macula but not affecting overall cone function. Gradually the myopia progressed to −7 in both eyes and vision dropped to 20/150 (corrected). The fundus showed mild temporal pallor of the disc with normal SD-OCT imaging. He was labelled clinically as having occult macular dystrophy. WES Analysis: Further analysis indicated rare or novel variants within eight genes (Table 3). We noted heterozygous variants in two genes known to cause congenital stationary night blindness (CSNB): GRM6 (c.2092 C > G: p.(L698V)) and TRPM1 (c.3958 G > A: p.(E1320K)). These variants segregated in a ‘digenic’ fashion, and only the TRPM1 variant was present in the unaffected sister. A heterozygous variant c.1148del in CNGB3 was also noted though a second variant in CNGB3 was not discovered through WES.
Family M69 — macular dystrophy (Fig. 1)
The parents of family M69 were first cousins and an autosomal recessive mode of inheritance was prioritized. The two affected offspring (male 32, female 30) presented with a macular dystrophy. OCT scan from II-2 showed atrophy of the outer retina and absence of Bruch’s membrane in the central fovea (Supplementary Fig. 3D). While the brother provided a DNA sample and was not available for examination, he has been described as significantly near-sighted at a young age requiring the use of corrective lenses. WES Analysis: WES comparisons to retinal disease genes identified heterozygous variations in four genes and further analysis identified variants in seven additional genes (Table 3).
Family M71 — cataracts and retinal detachment (Fig. 1)
Family M71 exhibited a highly-penetrant, autosomal dominant condition, which spanned three generations with multiple affected individuals affecting primarily males (Fig. 1). The proband was born with left microphthalmia and retinal detachments were noted shortly after birth. When last examined, he had high myopia with bilateral cortical cataracts, which were removed in 2013 and revealed a previously unrecognized retinopathy (Supplementary Fig. 3F). A diagnosis of Wagner syndrome was considered. WES Analysis: WES analysis was carried out on four members (affected father, two affected brothers, and unaffected mother) and assumed dominant inheritance (Fig. 1). No variations were noted in genes known to cause Wagner syndrome or other retinal detachments (ATOH7, TSPAN12, LRP5, or NDP) or in genes known to cause ocular disease. WES identified 72 variations shared between all three affected individuals. Of these, novel variants were noted in nine genes (Table 3). All identified variants in family members of M71 can be found in Supplementary Table 2.
Discussion
WES provides a cost-effective analysis for clinical investigation in families with IRDs as well as research studies aiming to identify novel retinal disease genes. Our study used WES to study 10 families from Alberta, Canada, chosen from a database of ophthalmology patients (IMM) based on the likelihood of identifying novel genetic associations in retinal and ocular disease. Below we discuss these results in context of our genetic and clinical findings. We also describe potential genetic associations in our unsolved cases that provide insights for other investigators of these blinding conditions.
Solved cases
Choroidal atrophy
Clinical review of this patient indicated a likely diagnosis of choroideremia, however, molecular testing of CHM/REP1 precluded this diagnosis. Our results identified a RPE65 variant (c.1430 G > A: p.(D477G)) previously shown to cause choroidal atrophy [14]. Though RPE65 variants cause recessive Leber congenital amaurosis, this variant has a unique effect and is the only RPE65 variant known to cause a choroideremia-like appearance. Functional studies of the p.(D477G) variant have shown that the protein variant facilitates mono- and di-ubiquitination of the RPE65 protein [15], and another showed that c.1430 G > A leads to aberrant RNA splicing [16]. We suggest that cases of choroideremia with negative CHM sequencing should be tested for this RPE65 variant.
Bardet-Biedl syndrome
Our analysis identified two variants in BBS1 (c.1169 T > G: p.(M390R) and c.1040del: p.(M347fs)). The affected males were initially diagnosed as non-syndromic RP but the presence of known BBS1 variants, as well as additional family information (presence of post-axial polydactyly and learning disabilities), altered our diagnosis to Bardet-Biedl Syndrome, a multi-system ciliopathy. The c.1040del variant was previously reported in a large study of BBS1 [17]. In addition, the p.(M390R) variant has been reported to cause a wide spectrum of phenotypes from non-syndromic RP to severe BBS [18] when inherited in a recessive manner. This suggests that genetic modifiers or mutational burden influences phenotypic presentation, a phenomenon that has been documented to impact presentation in ciliopathies such as BBS [19, 20].
Stargardt macular dystrophy
We identified a known pathogenic recessive variant in ABCA4 (c.4469 G > A: p.(C1490Y)) in the proband of family M53. This prompted a review of clinical data, as well as a search for the second variant, which ultimately identified a deep intronic variant through a collaborator c.4539 + 2028 C > T: p.(R1514Lfs*36) [10]. Combined, this data led us to diagnose Stargardt macular dystrophy, an adolescent-onset maculopathy. This case illustrates the importance of identifying heterozygous variants in WES data for recessive conditions, which may act as ‘guides’ for further analysis.
Peroxisomal biogenesis
WES analysis of family M73 identified a single variant in PEX6, c.1802G > A: p.(R601Q). This variant has been previously associated with a mild peroxisomal biogenesis disorder called Heimler syndrome [12], characterized by macular schisis, SNHL, and dental/nail abnormalities. Although the proband of this family presented with SNHL and macular schisis, no nail or dental abnormalities were identified and normal pipecolic/plasmalogen were noted in blood. These findings indicate that this is either a mild presentation of a peroxisomal disorder, or that our finding is coincidental. cDNA analysis showed no splicing abnormalities; however, this analysis was carried out in EBV transformed lymphoblast cells and may not contain the appropriate PEX6 isoform. It is possible that a second variant is controlling expression of the PEX6 gene through a regulatory sequence, which may be detectable through targeted or NGS.
Putatively novel findings
Heritable macular drusen (Families M54 & M70)
Drusen are subretinal deposits of lipids and proteins that are a major risk factor for age-related macular degeneration (AMD), though the exact relationship between drusen and photoreceptor death is not clear. We report two pedigrees with early-onset (<40 years of age) bilateral, macular drusen and compound heterozygous variants in LRP1: Family M54 with [c.650 C > T];[c.9628 G > C] and M70 with [c.2910 G > A];[c.11930 C > T]. One study showed that the c.650 C > T variant may activate a cryptic microRNA binding site, leading to altered expression of LRP1. Although the c.2910 G > A:p.(S970 = ) variant is synonymous, in silico splice predictions (SplicePort and Human Splice Finder) for the c.2910 G > A variant predict that this likely creates an exonic splice suppressor and alters an exonic splice enhancer near the variation. LRP1 is an intriguing candidate gene in the pathology of drusen formation. First, it has direct interaction with many components of drusen and proteins associated with MD, such as amyloid-beta [21], APOE, complement factors, and components of lipid metabolism. Second, LRP1 is expressed in the retinal pigment epithelium (RPE). The RPE provides nutrients to the neural retina, and the basal RPE is the site of drusen formation. Third, LRP1 provides a fascinating link between the three pathologies involved in age-related MD: lipid metabolism, complement pathway, and extracellular matrix homeostasis (Fig. 3). We hypothesize that dysfunction of LRP1 protein leads to accumulation of extracellular material over time due to altered endocytosis kinetics, resulting in drusen. Due to its multiple roles in MD-related pathways and our interesting genetic results, we hypothesize that LRP1 plays a role in drusen formation. Further studies of this protein and its relationship to drusen formation and MD are necessary.

The genetics of MD/dystrophy can be broken into three biological processes: (1) ECM homeostasis and remodeling (2) Lipid metabolism (3) Complement system. Proteins implicated in macular dystrophies or MD are highlighted in blue. LRP1 (red) through interaction with known proteins, provides a fascinating bridge between these three processes. Adapted from Kortvely and Ueffing [36].
Novel syndrome with cataracts, hearing loss, and intellectual delays (Family M72)
To the best of our knowledge, Family M72 presents a novel dominantly inherited condition. WES identified two novel variants: c.62 A > G: p.(D21G) in STUM, and c.122 A > C: p.(E41A) in UBE2U. Of interest, UBE2U has been reported to regulate RNF168 [22], an E2-ubiquitin conjugating enzyme that has been associated with the Radiosensitivity Immunodeficiency Dysmorphic features and Learning difficulties (RIDDLE) syndrome (OMIM 611943). Clinical assessment by a medical geneticist (OC) indicated this family shared some systemic dysmorphisms (short stature, small head circumference, low weight, hypertelorism) and behavioral/learning disabilities, similar to RIDDLE syndrome patients. We predict that the variant leads to an abnormal interaction between variant UBE2U and RNF168 and leads to a RIDDLE syndrome-like phenotype in our patients (Fig. 4). The affected mother of this family also developed breast cancer at age 31. The RNF168 system is involved with the repair of DNA damage and has a physical interaction with BRCA1, the most common cause of genetic breast cancer [23]. The diagnosis of breast cancer in our patient may be unrelated but is an interesting observation in the context of the UBE2U variant.

A OCT from an unaffected individual through the macula. B OCT scan from II-1 shows splitting between the inner nuclear and the outer plexiform layer, consistent with retinoschisis (white arrows). C UBE2U has previously been shown to interact with the RIDDLE syndrome protein RNF168. D We hypothesize that the variant in UBE2U identified in Family M72, causes a lack of physical interaction with RNF168 and other interactors, leading to a RIDDLE-like syndrome.
Unsolved families
Macular dystrophy with normal fundus (Family M68)
WES analysis yielded no obvious results to pinpoint the genetic etiology of this retinopathy. Of interest was the heterozygous CNGB3 c.1148del variant, previously associated with achromatopsia, an autosomal recessive cone photoreceptor disease. However, we excluded the likelihood of a second CNGB3 variant as the electrophysiological results make a diagnosis of achromatopsia unlikely; however, the testing revealed a slightly depressed b-wave, indicating a dysfunction between photoreceptors and the interneurons. We identified two heterozygous variants in genes that cause CSNB: GRM6 (c.2092 C > G: p.(L698V)) and TRPM (c.3958 G > A: p.(E1320K)). Van Genderen et al. (2009) suggested that these two proteins directly interact and that TRPM1 is channel-gated by the GRM6 signaling pathway [24]. We postulate that digenic inheritance of these variants is the potential cause of this inherited retinopathy, though further segregation or functional testing is needed to confirm this.
Macular dystrophy (Family M69)
The two affected individuals shared variants in many genes not previously associated with IRD (Table 3), however a direct cause is not apparent. The most interesting candidate gene is CROCC, which encodes rootletin protein, a core component of the ciliary rootlet [25]. Knockout of rootletin leads to loss of the ciliary rootlet and photoreceptor degeneration in mice [26, 27]. Our reported patient variants may underlie the patient phenotype due to a fragile photoreceptor cilium, which could hinder light detection and phototransduction.
Cataracts and retinal detachment (Family M71)
Three affected individuals across two generations presented with retinal detachments. WES revealed no variants in known cataract or retinal detachment-associated genes. We observed novel variations in nine potential candidate genes identified by WES (Table 3). Pathogenicity predictor programs and gene expression data further narrowed this to three genes: ELAVL2, WTIP, and ATG2B. Embryonic Lethal Abnormal Vision-Like 2 (ELAVL2) is a neuron-specific RNA binding protein that regulates transcript expression during neuronal development [28, 29]. ELAVL2 is expressed early during retinal development, coincident with the differentiation of retinal neurons [30, 31]. Another notable gene is Wilms Tumour Interacting Protein (WTIP), which is important for cell–cell and cell–extracellular matrix adhesion in the kidney [32]. There is also evidence that WTIP associates with basal bodies of cilia [33]. WTIP expression and function have not been investigated in the eye, but it may play a role in adhesion of the retina to the extracellular matrix and cell–cell adhesion in the lens. Autophagy Related 2B (ATG2B) is a key component of autophagosome biogenesis [34, 35]. The ATG complex is comprised of ATG2A and ATG2B, which are functionally redundant — therefore, single loss-of-function variants are unlikely to produce a phenotype. However, whether they have overlapping expression in the human eye is unknown. The functional consequence of the patient’s missense variant in ATG2B is unknown; however, impaired autophagosome development could result in accumulation of damaged organelles and abnormal proteins, leading to cellular dysfunction and death.
Conclusions
Our approach used WES to identify novel genes in IRDs and through this work we have identified two putatively novel associations in retinal disease: LRP1 in drusen formation and UBE2U in a novel syndrome. In our experience, WES can provide a valuable tool into the interrogation of particularly difficult to solve cases. In some instances, it can provide a starting point by identifying a single variant in a recessive condition. In other cases, the genetic information provided by WES can establish or change a clinical diagnosis highlighting the need for multi-disciplinary clinical investigations before genetic studies, as some phenotypic features may be missed by a single specialist. In addition, WES can provide a direction for further interrogations in more difficult cases by identification of potentially novel associations.
References
Wang X, Wang H, Sun V, Tuan HF, Keser V, Wang K, et al. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet. 2013;50:674–88.
Bravo-Gil N, Méndez-Vidal C, Romero-Pérez L, González-Del Pozo M, Rodríguez-De La Ruá E, Dopazo J, et al. Improving the management of Inherited Retinal Dystrophies by targeted sequencing of a population-specific gene panel. Sci Rep. 2016;6:1–10.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688.
Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–7.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. Mutationtaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Dogan RI, Getoor L, Wilbur WJ, Mount SM. SplicePort-An interactive splice-site analysis tool. Nucleic Acids Res. 2007;35:W285–91.
Desmet FO, Hamroun D, Lalande M, Collod-Bëroud G, Claustres M, Béroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67.
Lewis RA, Shroyer NF, Singh N, Allikmets R, Hutchinson A, Li Y, et al. Genotype/phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet. 1999;64:422–34.
Khan M, Cornelis SS, Pozo-Valero M Del, Whelan L, Runhart EH, Mishra K, et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genet Med. 2020;22:1235–46.
Wangtiraumnuay N, Alnabi WA, Tsukikawa M, Thau A, Capasso J, Sharony R, et al. Ophthalmic manifestations of Heimler syndrome due to PEX6 mutations. Ophthalmic Genet. 2018;39:384–90.
Ratbi I, Falkenberg KD, Sommen M, Al-Sheqaih N, Guaoua S, Vandeweyer G, et al. Heimler syndrome is caused by hypomorphic mutations in the peroxisome-biogenesis genes PEX1 and PEX6. Am J Hum Genet. 2015;97:535–45.
Falkenberg KD, Braverman NE, Moser AB, Steinberg SJ, Klouwer FCC, Schlüter A, et al. Allelic expression imbalance promoting a mutant PEX6 allele causes zellweger spectrum disorder. Am J Hum Genet. 2017;101:965–76.
Santer R, Schneppenheim R, Dombrowski A, Gotze H, Steinmann B, Schaub J. Mutations in RPE65 cause Leber’s congenital amaurosis. Nat Genet. 1997;15:57–61.
Choi EH, Suh S, Sander CL, Hernandez CJO, Bulman ER, Khadka N, et al. Insights into the pathogenesis of dominant retinitis pigmentosa associated with a D477G mutation in RPE65. Hum Mol Genet. 2018;27:2225–43.
Li Y, Furhang R, Ray A, Duncan T, Soucy J, Mahdi R, et al. Aberrant RNA splicing is the major pathogenic effect in a knock-in mouse model of the dominantly inherited c.1430A>G human RPE65 mutation. Hum Mutat. 2019;40:426–43.
Mykytyn K, Nishimura DY, Searby CC, Beck G, Bugge K, Haines HL, et al. Evaluation of complex inheritance involving the most common Bardet-Biedl syndrome locus (BBS1). Am J Hum Genet. 2003;72:429–37.
Estrada-Cuzcano A, Koenekoop RK, Senechal A, De Baere EBW, De Ravel T, Banfi S, et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome. Arch Ophthalmol. 2012;130:1425–32.
Lindstrand A, Frangakis S, Carvalho CMB, Richardson EB, McFadden KA, Willer JR, et al. Copy-number variation contributes to the mutational load of bardet-biedl syndrome. Am J Hum Genet. 2016;99:318–36.
Davis EE, Katsanis N. The ciliopathies: a transitional model into systems biology of human genetic disease. Curr Opin Genet Dev. 2012;22:290–303.
Storck SE, Hartz AMS, Bernard J, Wolf A, Kachlmeier A, Mahringer A, et al. The concerted amyloid-beta clearance of LRP1 and ABCB1/P-gp across the blood-brain barrier is linked by PICALM. Brain Behav Immun. 2018;73:21–33.
Guo Y, An L, Ng H-M, Sy SMH, Huen MSY. An E2-guided E3 Screen Identifies the RNF17-UBE2U pair as regulator of the radiosensitivity, immunodeficiency, dysmorphic features, and learning difficulties (RIDDLE) syndrome protein RNF168. J Biol Chem. 2017;292:967–8.
Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–34.
van Genderen MM, Bijveld MMC, Claassen YB, Florijn RJ, Pearring JN, Meire FM, et al. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am J Hum Genet. 2009;85:730–6.
Yang J, Liu X, Yue G, Adamian M, Bulgakov O. Li T. Rootletin, a novel coiled-coil protein, is a structural component of the ciliary rootlet. J Cell Biol. 2002;159:431–40.
Yang J, Gao J, Adamian M, Wen X-H, Pawlyk B, Zhang L, et al. The ciliary rootlet maintains long-term stability of sensory cilia. Mol Cell Biol. 2005;25:4129–37.
Mohan S, Timbers TA, Kennedy J, Blacque OE, Leroux MR. Striated rootlet and nonfilamentous forms of rootletin maintain ciliary function. Curr Biol. 2013;23:2016–22.
Berto S, Usui N, Konopka G, Fogel BL. ELAVL2-regulated transcriptional and splicing networks in human neurons link neurodevelopment and autism. Hum Mol Genet. 2016;25:110.
Akamatsu W, Okano HJ, Osumi N, Inoue T, Nakamura S, Sakakibara SI, et al. Mammalian ELAV-like neuronal RNA-binding proteins HuB and HuC promote neuronal development in both the central and the peripheral nervous systems. Proc Natl Acad Sci U.S.A. 1999;96:9885–90.
Pacal M, Bremner R. Mapping differentiation kinetics in the mouse retina reveals an extensive period of cell cycle protein expression in post-mitotic newborn neurons. Dev Dyn. 2012;241:1525–44.
Amato MA, Boy S, Arnault E, Girard M, Della Puppa A, Sharif A, et al. Comparison of the expression patterns of five neural RNA binding proteins in the Xenopus retina. J Comp Neurol. 2005;481:331–9.
Kim JH, Mukherjee A, Madhavan SM, Konieczkowski M, Sedor JR. WT1-interacting protein (Wtip) regulates podocyte phenotype by cell-cell and cell-matrix contact reorganization. Am J Physiol - Ren Physiol. 2012;302:103–15.
Bubenshchikova E, Ichimura K, Fukuyo Y, Powell R, Hsu C, Morrical SO, et al. Wtip and Vangl2 are required for mitotic spindle orientation and cloaca morphogenesis. Biol Open. 2012;1:588–96.
Osawa T, Noda NN. Atg2: a novel phospholipid transfer protein that mediates de novo autophagosome biogenesis. Protein Sci. 2019;28:1005–12.
Osawa T, Kotani T, Kawaoka T, Hirata E, Suzuki K, Nakatogawa H, et al. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat Struct Mol Biol. 2019;26:281–8.
Kortvely E, Ueffing M. Common mechanisms for separate maculopathies?. In: LaVail M, Ash J, Anderson R, Hollyfield J, Grimm C, editors. Retinal degenerative diseases. advances in experimental medicine and biology. Boston, MA: Springer; 2012. vol 723. https://doi.org/10.1007/978-1-4614-0631-0_9.
Acknowledgements
This study was funded by Retina Foundation Canada (RFC) (LD), University of Alberta Hospital Foundation (Olive Young Fund for Macular Degeneration) (IMM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Doucette, L.P., Noel, N.C.L., Zhai, Y. et al. Whole exome sequencing reveals putatively novel associations in retinopathies and drusen formation. Eur J Hum Genet 29, 1171–1185 (2021). https://doi.org/10.1038/s41431-021-00872-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00872-3
This article is cited by
-
A new impact factor for European Journal of Human Genetics
European Journal of Human Genetics (2021)
