
Abstract
Sudan, a northeastern African country, is characterized by high levels of cultural, linguistic, and genetic diversity, which is believed to be affected by continuous migration from neighboring countries. Consistent with such demographic effect, genome-wide SNP data revealed a shared ancestral component among Sudanese Afro-Asiatic speaking groups and non-African populations, mainly from West Asia. Although this component is shared among all Afro-Asiatic speaking groups, the extent of this sharing in Semitic groups, such as Sudanese Arab, is still unknown. Using genotypes of six polymorphic human leukocyte antigen (HLA) genes (i.e., HLA-A, -C, -B, -DRB1, -DQB1, and -DPB1), we examined the genetic structure of eight East African ethnic groups with origins in Sudan, South Sudan, and Ethiopia. We identified informative HLA alleles using principal component analysis, which revealed that the two Semitic groups (Gaalien and Shokrya) constituted a distinct cluster from the other Afro-Asiatic speaking groups in this study. The HLA alleles that distinguished Semitic Arabs co-exist in the same extended HLA haplotype, and those alleles are in strong linkage disequilibrium. Interestingly, we find the four-locus haplotype “C*12:02-B*52:01-DRB1*15:02-DQB1*06:01” exclusively in non-African populations and it is widely spread across Asia. The identification of this haplotype suggests a gene flow from Asia, and likely these haplotypes were brought to Africa through back migration from the Near East. These findings will be of interest to biomedical and anthropological studies that examine the demographic history of northeast Africa.
Similar content being viewed by others

Introduction
The considerable level of cultural, linguistic, and genetic diversity of populations inhabiting East Africa and the Nile Valley reflects the complex demographic history of this part of the world. Such complexity demonstrates the large effective population size [1] and the great regional substructure [2] that characterize North-Eastern Africa. Furthermore, this substantial diversity has been shaped, not only by a contribution of native populations, as several studies have shown the role of demographic events as evidenced by the bi-directional genetic exchange between Northeastern Africans and neighboring populations from West Asia [3,4,5,6,7]. Studies of the genetic diversity of Africans indicate that both geography and linguistic affiliation contribute in shaping the genetics of African populations [2]. Linguistic families of African populations are categorized into four macro-families, three of which are present in the Sudanic region of East Africa (an area encompasses Sudan and South Sudan): Afro-Asiatic, Nilo-Saharan, and Niger-Congo. The Afro-Asiatic family is mainly found in the northern and eastern parts of Africa (includes Arabic, Bedawiyet; spoken by Beja, and the Cushitic languages). The Nilo-Saharan family covers most of the Sudanese languages, including those spoken by Nubians in the north, Darfurians in the west, and Nilotes of South Sudan. Furthermore, the Niger-Congo/Kordofanian geographically spans most of Sub-Saharan Africa [8, 9]; however, it is restricted in the Sudanic region to few groups from Nuba Mountains and Fulani (Fig. 1).

The Inset indicates the location of Sudan, South Sudan, and Ethiopia within Africa.
The genetic diversity of Sudanese populations (Sudan and South Sudan) has been explored using different types of autosomal [2, 10], Y chromosome, and mitochondrial DNA markers [11], which collectively showed the various genetic contributions from African and non-African ancestries. More recently, the genetic diversity of East African populations has been studied using genome-wide SNP data [5, 7]. These studies have identified a northeast/south-west cline that defines two main population substructures, in addition to a North-African ancestral component identified in Afro-Asiatic speaking groups. Although these studies, including the genome-wide approach, have unraveled the influences and genetic relationships between populations of East Africa and Western Asia, exploration of the demographic history that shaped the current peopling of the region is an ongoing quest.
The human major histocompatibility complex (MHC) is a genomic region spanning 3.6 Mb of the human chromosome 6. Based on their genomic location and function, genes of the MHC region have been categorized into three classes (class I, class II, and class III), of which classical genes in class I and II are highly polymorphic. Currently, more than 26,000 human leukocyte antigen (HLA) alleles have been registered in the IMGT/HLA public database [12]. Such excessive variability is thought to be maintained through interaction with the environment, particularly selective pressure from infectious pathogens [13]. The significant variations in the distribution of HLA alleles and haplotypes between human populations provide valuable markers for comparative analysis of human populations [14]. These variations have been thoroughly studied benefiting from the established donor registries of the transplantation programs in many developed countries [15]. However, in low-income countries, such as those in Africa, including Sudan, large-scale studies of HLA diversity are hampered by the high cost of HLA typing [16]. In Sudan, few HLA studies have been conducted to understand the genetic diversity of HLA genes (Supplementary Table S1). These studies have limitations in terms of HLA-typing resolution and a wide representation of ethnically defined populations [17, 18]. Hence, a study with broader geographic coverage, more ethno-linguistically defined populations, and high-resolution HLA-typing allow a better assimilation of the genetic diversity of populations living in this region.
Here, to investigate the distribution of HLA alleles and haplotypes in East Africa, and to have an insight on the peopling and demographic history of the region, we examined the genetic diversity of eight east African populations from Sudan, South Sudan, and Ethiopia by using allele and haplotype frequencies of six HLA loci (HLA-A, -C, -B, -DRB1, -DQB1, and -DPB1) determined by phase-defined analysis [19]. Integration of HLA data from these ethnic groups with data from populations in Sub-Saharan Africa, Middle East, and Asia allowed a wider context for understanding the genetic history of the East African populations.
Materials and methods
Sample information
Saliva samples were collected from 329 individuals between July and September 2010 from various ethnic groups in Sudan, South Sudan, and Ethiopia. The Ethiopian samples were collected from Ethiopians people living in Sudan. The ethnic groups are categorized based on the linguistic classification of African populations [8]. The samples represent three major linguistic families: Afro-Asiatic (Gaalien, Shokrya, Beja, and Ethiopians), Nilo-Saharan (Darfurians, Nubians, and Nilotes), and Nuba who belongs to, both, Nilo-Saharan and Niger-Congo families. Geographic locations and linguistic affiliations of the study populations are shown in Supplementary Tables S2 and S3 and Fig. 1. Informed consent was obtained from all participants and the study protocol was approved by the ethics committee of the Sudan Medical and Scientific Research Institute, Sudan (SUM 2010/7), and of the National Institute of Genetics, Mishima, Japan (nig1508, 2015.11.30). Sample collection and DNA isolation were done using the Oragene™ collection kit (OG-500) (DNA Genotek, Ontario, Canada) following the recommended protocol. To compare the populations in this study and other African and Middle Eastern populations, a total of 41 datasets were used, including 30,518 samples from the Allele Frequency Net Database (AFND) [20]. The datasets include populations from North Africa, Sub-Saharan Africa (West, Central, and South Africa), the Middle East, Europe, and South and East Asia, Supplementary Table S4. In order to compare these datasets to our study populations we downloaded only datasets with four-digit typing resolution. Furthermore, as many of these datasets do not include class-II genes, we used only class-I genes for this comparison. We further filtered these by requiring the total allele frequencies in each gene to be at least 0.95 (Supplementary data). Of the 41 datasets, we found that 19 met the allele frequency criterion and were used for the principal component analysis (PCA) analysis.
NGS library preparation and analysis
Genotyping of HLA genes was done using a next-generation sequencing (NGS) method. The full protocol of the NGS library preparation and sequencing were described by Ahmadloo et al. [21]. The DNA libraries (328 samples pooled in four 96-well plates) were sequenced using Illumina MiSeq® platform. The sequencing method was paired-end type (forward read 350 bp and reverse one 250 bp) with an average expected insert size of 600 bp. The details of the analysis pipeline are described in Supplementary File S1.
Sanger sequencing of new HLA alleles
Mismatched positions in the samples that were candidates for new HLA alleles were confirmed by Sanger sequencing. For each position, we designed specific primers flanking the variant and amplified the position using, mostly, allele-specific PCR. Furthermore, we performed Sanger sequencing using the amplified products and BigDye® Terminator V3.1 Cycle Sequencing kit (Life Technologies). The sequencing was done using, both, forward and reverse primers on an ABI 3130xl Genetic Analyzer (Applied Biosystems).
Population genetics analysis
The genetic diversity indicators, such as the number of alleles in each locus and the degree of heterozygosity, were calculated using Python for Population Genomics [22] (PyPop v.0.7.0). The expected heterozygosity (Ĥ), assuming Hardy–Weinberg equilibrium (HWE), was estimated using the following formula: \(\widehat {\rm{H}} = \frac{n}{{n - 1}}\left( {1 - \mathop {\sum}\nolimits_{i = 1}^k {P_i^2} } \right)\) [23], where Pi is the frequency of the ith allele and n is the number of samples for k number of alleles. We tested whether the identified genotypes follow or deviate from HWE using Arlequin [23], which uses Guo and Thompson’s procedure [24]. To understand the genetic relatedness among the study populations using HLA genes, we computed the pairwise FST statistic between all pairs of populations using Arlequin. The statistical significance (P value <0.05) is determined after permuting haplotypes (10,000 permutations) under the assumption of no difference between populations. The pairwise genetic distances were used to build a neighbor-joining phylogenetic tree [25] using MEGA v.6 [26].
To test whether natural selection is operating on any HLA loci in the study populations, we performed Ewens–Watterson homozygosity test of neutrality [27] implemented in PyPop, v.0.7.0 [22]. The homozygosity F statistic is given by: \(F = \mathop {\sum}\nolimits_{i = 1}^k {P_i^2}\), where Pi is the frequency of the ith for k number of unique alleles. The normalized deviate of homozygosity (Fnd) is calculated as the difference between observed and expected homozygosity, divided by the square root of the variance of the expected homozygosity [28]. The reported P values in this test are the probability of obtaining a homozygosity F statistic under neutrality assumptions that is less than or equal to the observed one. The implementation of the test is based on the exact test written by Slatkin [29], which uses a Markov-Chain Monte-Carlo method to obtain the null distribution of homozygosity. A negative significant Fnd value indicates that the observed homozygosity is deviated in the direction of balancing selection, while a significant positive value indicates directional selection. To calculate multi-locus haplotype frequencies, we used the expectation maximization (EM) method, which uses an iterative maximum likelihood estimation approach [30], as implemented in Arlequin.
Detection of informative alleles
To determine the informative alleles, we used PCA. We performed PCA on a covariance matrix of normalized allele frequencies in all groups using the function (prcomp) from the STATS package in R version 3.2.1 [31], which uses a singular value decomposition method. We used a matrix of n rows and m columns, where n is the number of alleles and m is the number of populations. For each allele, we calculated the normalized allele frequency (Pno) which is given by: \(P_{\rm{no}} = \frac{{P_i - \mu }}{\sigma }\), where Pi is the frequency of the ith allele, µ is the mean allele frequency in all groups, and σ is the standard deviation. We assumed that the informative alleles are the ones that are associated with the clustering patterns in the PC plots. Therefore, to determine the alleles, we selected those with PC scores greater than one standard deviation. All graphical presentations in this study were done using R version 3.2.1 [31].
Estimation of linkage disequilibrium
We used PyPop [22] (v.0.7.0) program to calculate pairwise linkage disequilibrium (LD) between the different HLA loci (i.e., C, B, DRB1, and DQB1). We calculated the pairwise LD between alleles in the same multi-locus haplotypes using the normalized D measure (Lewontin’s D’) defined as: \(D_{ij}^\prime = \frac{{D_{ij}}}{{D_{\rm{max}}}}\).
Results
Number of HLA alleles and heterozygosity
The number of observed HLA alleles and heterozygosity for each locus in the study populations are presented in Table 1. In all loci, most groups showed remarkable variation in the number of alleles. This is clearly seen in HLA-A as Nubians has 16 alleles while Gaalien has 27 alleles. Such allelic diversity, particularly in class-I genes, is common and has been reported in African populations [32]. We also found that most populations show no deviation from HWE proportions (Table 1 and Supplementary Table S5). However, in three Sudanese populations (Gaalien, Nubians, and Nuba), we find that three of class-II HLA genes (HLA-DRB1, HLA-DQB1, and HLA-DPB1) show lower than expected heterozygosity under HWE. On the other hand, in Nilotes of South Sudan, we see HLA-C and HLA-B with deviated heterozygosity values.
Identification of new HLA alleles
In two populations (Nubians and Darfurians) several sequences did not fully match any known HLA alleles in the IMGT database (i.e., has at least one mismatch). In those samples, we confirmed the variant positions by Sanger sequencing. In total, we identified four new HLA alleles (Supplementary Table S6 and Supplementary Fig. S1), one of which is HLA-C allele, and the others are HLA-B alleles. The novel HLA-C allele differs by two nonsynonymous variants (L10I and L11V) from the closest C*14:02:01 allele. The second allele, which also found in Nubians, has one synonymous variant (P291P) in a background of B*51:01:01:01 allele. The two remaining novel HLA-B alleles were both in the Darfurian group, a synonymous variant (L12L), and nonsynonymous variant (S27C). The closest HLA-B alleles to these novel ones are B*35:01:01:01 and B*39:10:01, respectively. All of the new HLA sequences were submitted to the Genbank database and accession numbers are in Supplementary Table S6.
Population differentiation and test of natural selection
Pairwise comparisons between the study populations showed differences in FST indices (Table 2). In general, the genetic affinities between populations of the Afro-Asiatic, and Nilo-Saharan and Niger-Congo families were larger than those within the same linguistic family. The only exception was Nubians, which showed close distance to the groups from Afro-Asiatic family despite being from the Nilo-Saharan family: all pairwise comparisons between Nubians and Afro-Asiatic groups have low FST values. In almost all pairwise comparisons, populations were significantly differentiated from each other. However, in two comparisons, the pairwise distances were relatively close with low FST values (Beja–Ethiopians and Ethiopians–Nubians pairs). Furthermore, phylogenetic analysis based on the calculated genetic distances showed that the most distant populations were Nilotes (Nilo-Saharan) on the one side, and the two Arab groups (Gaalien and Shokrya) on the other (Supplementary Fig. S2). Furthermore, to test whether any of the HLA loci is under selection, we performed Ewens–Watterson test of selective neutrality. Results show that, in four populations (Beja, Nubians, Nuba, and Ethiopians), several loci showed deviated homozygosity (Supplementary Table S7). With the exception of HLA-DPB1, the remaining loci show deviated homozygosity values in at least one population, which is common for HLA loci [33]. All deviated loci have significant negative Fnd values (normalized deviate of homozygosity, see Methods), suggesting that balancing selection is acting on these loci.
Identification of informative alleles
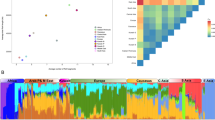
To understand the genetic structure of the study populations, we performed PCA on a covariance matrix of HLA allele frequencies. The analysis revealed that the first two principal components (PCs) explained 41.5% of the variance (Fig. 2a and Supplementary Fig. S3). PC1 splits Nilo-Saharan and Niger-Congo populations from the Afro-Asiatic family members, consistent with the pairwise distance analysis based on FST statistics. Furthermore, PC1 also shows that the Nilo-Saharan (Nubians) is found in the same direction as Ethiopians and Beja and separate from the other Nilo-Saharan and Niger-Congo family members (Darfurians, Nilotes, and Nuba). In PC2, the Afro-Asiatic Arabs (Gaalien and Shokrya) clustered away from the other members of the same linguistic family (Beja and Ethiopians).

a Plot of the first (25.7%) and second (15.8%) PCs showing clustering patterns according to linguistic affiliations. b Plot of the first and second PCs of individual HLA alleles (see Methods). Dotted vertical and horizontal lines indicate ±1 standard deviation from the first and second PC means, respectively. HLA alleles that deviate by more than 1 standard deviation are considered informative alleles. Some of these alleles are highlighted as cluster-1, cluster-2, and cluster-3.
To identify alleles informative for discrimination between populations, we plotted the principal component scores (PCS, also known as individual plot), which are the projections of each allele in relation to the PC (Fig. 2b). Alleles, whose projections are markedly deviate from the PCs, make greater contributions to explain the differences between populations. We found that the deviated alleles (±1 standard deviation) follow patterns of allele frequencies that can be categorized into three clusters. Alleles of the first cluster have a high frequency in Nilo-Saharan and Niger-Congo members (Fig. 3; cluster-1). In the second cluster, most alleles show high frequencies in the Afro-Asiatic members, in addition to Nubians from the Nilo-Saharan (Fig. 3; cluster-2). Finally, the last cluster is almost unique to the Arabs (Gaalien and Shokrya), and its alleles are characterized by high frequencies among them (Fig. 3; cluster-3). This distinction between Arabs and non-Arabs in the Afro-Asiatic family was particularly interesting because it has not been detected in the genome-wide SNP studies [5, 7].

HLA alleles are arranged in rows, where each corresponds to alleles found in a cluster in Fig. 2b. Alleles in the same cluster show similar patterns of frequency distribution.
Common shared multi-locus haplotypes
To investigate whether the alleles identified in the PCA co-occur in the same haplotype, we estimated multi-locus haplotype frequencies using EM algorithm. Table 3 shows the most common (>4%) four-locus HLA haplotypes among the study populations. Apart from “H7” and “H11” haplotypes, all haplotypes are not shared by more than three populations and mostly the haplotypes are not shared by groups in the same linguistic family. LD analysis of the gene pairs (C-B and DRB1-DQB1) in Table 3 haplotypes revealed that most pairs are not strongly linked, with the exception of H7 in Gaalien and, both, H12 and H13 in Gaalien and Shokrya (Supplementary Table S8). Expectedly, several alleles in the same haplotype are also found in the same cluster in PCS plots (Fig. 2b). The haplotypes “H1,” “H11,” and “H14” have at least two alleles in cluster-1 (Fig. 2b). These haplotypes are common in the Nilo-Saharan and Niger-Congo, except for Nubians. Other haplotypes that are shared among Afro-Asiatic members composed of alleles that reside in cluster-2 (H4, H7, and H15). Likewise, some of the alleles that were found in cluster-3 compose haplotypes that are mostly found in the two Arab groups (e.g., H12: C*12:02-B*52:01-DRB1*15:02-DQB1*06:01 and H13: C*15:02-B*51:01-DRB1*03:01-DQB1*02:01). Noticeably, we find that Gaalien and Shokrya share more extended haplotypes than any other populations. Interestingly, the haplotype “H12,” which is common among these two Arab groups, has all its alleles in cluster-3. This suggested tight LD between alleles of H12 haplotype. We estimated LD (D’) between allele pairs in the two haplotypes (H12 and H13) (Fig. 4). In “H12” haplotype, the extent of LD was high in all pairwise comparisons; in fact, there is complete LD between allelic components of “H12” haplotype in the Shokrya group. On the other hand, in “H13” haplotype, the LD was strong only between the pairs HLA-C-HLA-B and HLA-DRB1-HLA-DQB1.

Pairwise LD (D’) values of HLA alleles composing H12 haplotype “C*12:02” (left panel) and H13 haplotype “C*15:02” (right panel).
Searching for informative haplotypes in the Arab groups
The two Arab groups share a genetic component that distinguishes them from other Afro-Asiatic speaking groups. In particular, the strongly linked and most common H12 haplotype seem to be informative for tracing past demographic events in these groups. We searched the literature (Pubmed) and AFND [20] for “H12” haplotype frequency data and found no evidence of any Sub-Saharan African population having “H12” haplotype (Table 4 and Supplementary Table S9). Although African-American (AA) population has low frequency of H12 haplotype, given the reported admixed ancestries in some AA communities [34], acquiring the haplotype through admixture with other populations cannot be ruled out. Then we extended the H12 haplotype by finding the linked HLA-A alleles in the two Arab groups (Supplementary Table S10). We found two strongly linked (LD > 0.70) HLA-A alleles (i.e., A*11:01 and A*03:02) in the Shokrya and Gaalien groups, respectively (Supplementary Table S11). None of these haplotypes is carried by an African population in the AFND database (Supplementary Table S12). Interestingly, the highest frequency of the extended “H12” haplotype (containing A*11:01 allele) is seen in an Arab population from the Middle East (UAE, 2.8%), while the one with A*03:02 allele is found in the Turkish minority dataset.
To understand the genetic structure of our study groups in the context of other populations, we integrated 19 datasets from other populations obtained from the AFND: datasets from Middle Eastern, Asian, and Sub-Saharan African populations (Supplementary Table S13). PCA of the combined datasets was consistent with the previous PCA of East African samples (Supplementary Figs. S4 and S5). Overall, both Sudanese and Ethiopians were located between non-African and Sub-Saharan African populations. PC1 (11.9%) separates non-African (Middle Eastern and Asian populations) from the Sudanese and Sub-Saharan African populations. Also, PC1 shows that Gaalien and Shokrya, among other Sudanese, were the closest to all non-African populations. On the other hand, the Nilo-Saharan and Niger-Congo speaking groups (Darfurians, Nilotes, and Nuba) were close to the other Sub-Saharan Africans. PC2 (9.1%) separated all Sudanese and Ethiopians from other populations. PC2 mainly differentiated between Afro-Asiatic speaking groups and Nubians in the one hand and Sub-Saharan Africans on the other, with Sudanese Nilo-Saharan/Niger-Congo speaking groups in the middle. Extending the analysis by including PC3 (Supplementary Fig. S6) showed that the maximum variance in this PC is between populations from Cameroon and other populations, with the Nilo-Saharan from Kenya (Luo) being at the end of the distribution.
Discussion
As one of the candidate places where modern humans potentially originated and evolved [35], East Africa is characterized by high levels of linguistic, cultural, and genetic diversity [2]. Here, using allelic distribution of six polymorphic HLA genes, we studied the genetic diversity of eight ethno-linguistic groups from Sudan, South Sudan, and Ethiopia. Our several analyses revealed the complexity of the HLA region in these populations and shedded light on the migratory history of the region, hinting to the possibility of gene flow in the Semitic groups of Sudan (Gaalien and Shokrya).
The apparent diversity of the study populations is demonstrated by the large number of observed alleles per locus (Table 1), the high heterozygosity values (Table 1), and the clear population differentiation as measured by the FST statistic (Table 2). The few exceptions to this were the low heterozygosity and deviation from HWE observed in several loci (i.e., DPB1 in Gaalien and Nubians, DQB1 in Nuba, DRB1 in Nubians and Nuba, and both C and B in Nilotes). Furthermore, the interpretation of such deviation is not simple as several factors could be involved, including genotyping errors, sample ascertainment, admixture, and non-random mating as well as natural selection. In terms of allele count across all loci, we find the East African groups have comparable numbers to other Sub-Saharan African populations [33]. The increased diversity, as indicated by allele counts, and the level of heterozygosity point to balancing selection effects on these loci. Furthermore, Ewens–Watterson homozygosity test showed that, in all populations with significant Fnd values, homozygosity is deviated from expectations under neutrality assumption, signifying that balancing selection could be acting on these loci. The observation of deviated homozygosity is consistent with findings from several other populations [36]. The FST statistic showed that the study populations are differentiated at the superfamily level (i.e., between Afro-Asiatic and Nilo-Saharan/Niger-Congo), suggesting limited gene flow between these different families, and culture, particularly languages, have a role in shaping the genetic structure of East African populations. Furthermore, this culture/language role was previously explored in African populations that showed several such examples, including the notable one of lactase persistence trait among nomadic pastoralists along the Sahel [37].
In this study, we found four alleles in HLA-B and HLA-C that were previously not described in the IMGT/HLA database (Supplementary Table S6). The variants in these new alleles are exonic variants, and three of the five variants are nonsynonymous that may have functional implications at the protein level. The populations in which the new alleles are identified belong to the Nilo-Saharan family (Nubians and Darfurians), suggesting more HLA alleles are yet to be discovered in this linguistic family.
The genetic structure of Sudanese populations was studied by Dobon et al. [5] using genome-wide SNP data. Their study demonstrated two main population substructures that correlated with linguistic classification of the studied populations (Afro-Asiatic on one side and Nilo-Saharan/Niger-Congo on the other). In this study, we used a set of the same samples that were genotyped by Dobon et al. [5]. We aimed to expand upon the previous data by studying the diversity of HLA genes to have an insight on the demographic history of East Africa. We employed PCA to portray the genetic affinities of the study populations, which revealed that the clustering pattern in our HLA data is in harmony with the clustering based on genome-wide SNP data by Dobon et al. [5], with few exceptions (Fig. 2a). This concordance implies the maintenance of population structure, despite HLA loci being under various selective forces; therefore, it is possible to infer the genetic history of these populations using HLA data. The split of the study populations in PC1 correlates with geography (Nile Valley) and linguistic affiliation at the superfamily level. It has been shown in East African populations that both geography and language are associated with genetic diversity [2]. The deviated alleles in PCS plot (Fig. 2b) revealed an HLA profile that is distinct between the Afro-Asiatic groups and those of the Nilo-Saharan and Niger-Congo families. Although these populations are geographically close, the PCA distinction implies a restricted gene flow between these populations and in consistency with previous Y-chromosome data [11]. The three groups (Darfurians, Nuba, and Nilotes) are geographically and linguistically separate at the subfamily level; however, their clustering as one group in the PCS plot (Fig. 2b) indicates a similar HLA profile and supports their past genetic affinity, since the plot shows that some of the defining alleles in cluster-1 are common among Sub-Saharan African populations [33] (i.e., A*30:04, B*42:04, and B*47:03) (Fig. 3). Furthermore, in East Africans, the association between genetic diversity and linguistic affiliation is not complete, as observed by Tishkoff et al. [2]. In the Nilo-Saharan family, geography has better correlation with genetic diversity than language. The PCA shows the Nilo-Saharan Nubians (PC1) falls in the direction of the Afro-Asiatic speaking groups (Fig. 2a), suggesting gene flow from these neighboring populations, as Nubians were influenced by Arabs as a direct result of the migration of large numbers of Arabs into the Nile Valley over the long period of time following arrival of Islam around 651 AD [38, 39]. The deviation of Nubians from their linguistic family has been previously reported, as other studies have shown a similar clustering pattern [3, 5].
Four groups in this study belong to the Afro-Asiatic family, two of which (Gaalien and Shokrya) speak the Arabic language, and the other two groups (Beja and Ethiopians) have their own indigenous languages. The PCA analysis reveals the close genetic affinity between Beja and Ethiopians, which is consistent with previous findings showing the two groups share several Y-chromosome markers including the J1 haplogroup [7]. Interestingly, the haplogroup J1 is known to be of Eurasian origin [40], suggesting both Beja and Ethiopians had experienced gene flow from West Asia. Although the Afro-Asiatic speaking groups in PC1 are placed in the same direction, the distinction between Arabs (Gaalien and Shokrya) and non-Arabs (Beja and Ethiopians) is revealed in PC2 (Fig. 2a). Contrary to this “Arabs/Non-Arabs” division, previous analyses using genome-wide SNP data [5] showed Afro-Asiatic speaking groups as a single group. The defining alleles in cluster-3 (Fig. 3) are almost unique to the Arab groups so we thought these alleles are informative for exploring the demographic history of these Semitic groups. A possible explanation of why only Arabs, among Afro-Asiatic speaking groups, have the specific alleles of cluster-3 is that both language and geography are probably important in shaping the genetic structure in these populations. Furthermore, provided the defining alleles in cluster-3 are not shared by all individuals in those groups, the split between Arab and non-Arab in PC2 is not associated with a substructure at the genomic level, although the frequency of cluster-3 alleles are relatively high among the Arabs (Table 3).
Identification of multi-locus haplotypes depends on the strength of association between the different loci. Apart from Arab groups, our data show few sharing of multi-locus haplotypes among the study populations, which is consistent with the fact that Africans have increased heterogeneity and lower LD compared to other populations [35]. In contrast, several studies have shown the high levels of LD within the HLA complex, which sometimes extends to more than 1 Mb [41]. The strong LD in the HLA region, combined with the excessive allelic diversity among human populations, permits the identification of demographic signatures by using population-specific extended haplotypes [32]. Our re-construction of the multi-locus HLA haplotypes showed that many of these are composed of alleles that reside in the same cluster (Table 3). Although most of these extended haplotypes show evidence of LD decay, the strength of LD in the H12 haplotype among Semitic (Arab) groups (Table 3) suggests that LD is maintained between the four HLA loci. Moreover, our data show that H12 haplotype is exclusively found in non-African populations and it has a high frequency in South and Southeast Asia [20]. Previously, Abi-Rached et al. suggested that the C*12:02-bearing haplotype (H12) evolved in South-East Asia before the out-of-Africa migration and it is acquired by admixture with Denisovans [42]; however, their last conclusion was later challenged by Yasukochi et al. [43]. Given the documented demographic movements [39] between West Asia, Middle East, and East Africa and the possible origin of H12 haplotype, we hypothesize that the identification of H12 haplotype in the Semitic groups of Sudan indicates that a gene flow event brought the haplotype back to Africa through back-migration. Although it is possible to explain the identification of H12 in Gaalien and Shokrya in terms of common ancestry between the Sudanese Arabs and non-African populations, the gene-flow hypothesis is more supported because: (1) Dobon et al. [5] studied the same samples using SNP-array markers and their data did not show Arab as a separate cluster from other Afro-Asiatic groups, (2) the findings from Y-chromosome that show several non-African haplogroups (e.g., the European haplogroup “J-12f2”) in populations inhabiting north Sudan [3], which demonstrates the role of demography in shaping the genetic structure of the Sudanese populations, (3) the HLA-A alleles that are linked to H12 haplotype in Shokrya and Gaalien coincide with those found in populations with documented migration history to Sudan (i.e., Middle Eastern Arabs and Turkish) [39].
These findings, in addition to the usefulness of utilizing the HLA system to study population history, have biomedical implications as HLA has been associated with numerous diseases. Although our conclusion is supported by the presented data, the relatively small sample size does not warrant extending the finding of “H12” haplotype to other Semitic groups in Sudan. Further studies with more samples and inclusion of other Arab groups would strengthen the evidence presented in this study. Moreover, the fact that several HLA genes are known for being under balancing selection, there is still a possibility that H12 haplotype evolved in the Arab groups in response to selective pressure from local pathogens.
References
Elhassan N, Gebremeskel EI, Elnour MA, Isabirye D, Okello J, Hussien A, et al. The episode of genetic drift defining the migration of humans out of Africa as derived from a large East African population size. PLoS One. 2014;9:e97674.
Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, et al. The genetic structure and history of Africans and African Americans. Science. 2009;324:1035–44.
Hassan HY, Underhill PA, Cavalli‐Sforza LL, Ibrahim ME. Y‐chromosome variation among Sudanese: restricted gene flow, concordance with language, geography, and history. Am J Phys Anthropol. 2008;137:316–23.
Hodgson JA, Mulligan CJ, Al-Meeri A, Raaum RL. Early Back-to-Africa migration into the Horn of Africa. PLoS Genet. 2014;10:e1004393.
Dobon B, Hassan HY, Laayouni H, Luisi P, Ricano-Ponce I, Zhernakova A, et al. The genetics of East African populations: a Nilo-Saharan component in the African genetic landscape. Sci Rep. 2015;5:9996.
Rodriguez-Flores JL, Fakhro K, Agosto-Perez F, Ramstetter MD, Arbiza L, Vincent TL, et al. Indigenous Arabs are descendants of the earliest split from ancient Eurasian populations. Genome Res. 2016;26:151–62.
Hollfelder N, Schlebusch CM, Günther T, Babiker H, Hassan HY, Jakobsson M. Northeast African genomic variation shaped by the continuity of indigenous groups and Eurasian migrations. PLoS Genet. 2017;13:e1006976.
Greenberg JH. The languages of Africa. 1st ed. Bloomington, Indiana, US: Indiana University; 1963.
Eberhard DM, Simons GF, Fennig CD. Ethnologue: languages of Africa and Europe. 22nd ed. Summer Institute of Linguistics, Academic Publications; 2019.
Babiker HM, Schlebusch CM, Hassan HY, Jakobsson M. Genetic variation and population structure of Sudanese populations as indicated by 15 Identifiler sequence-tagged repeat (STR) loci. Investigative Genet. 2011;2:12–12.
Hassan, HY. Genetic patterns of Y-chromosome and mitochondrial DNA variation, with implications to the peopling of the Sudan. PhD thesis, University of Khartoum; 2009.
Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SGE. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 2015;43:D423–31.
Radwan J, Babik W, Kaufman J, Lenz TL, Winternitz J. Advances in the evolutionary understanding of MHC polymorphism. Trends Genet. 2020;36:298–311.
Sanchez-Mazas A, Meyer D. The relevance of HLA sequencing in population genetics studies. J Immunol Res. 2014;2014:1–12.
Hernández-Frederick CJ, Cereb N, Giani AS, Ruppel J, Maraszek A, Pingel J, et al. Detection of 549 new HLA alleles in potential stem cell donors from the United States, Poland and Germany: detection of 549 new HLA alleles. HLA. 2016;87:31–5.
Rizvi SAH, Naqvi SA, Hussain Z, Hashmi A, Akhtar F, Hussain M, et al. Renal transplantation in developing countries. Kidney Int. 2003;63:S96–100.
Magzoub MM, Stephens HA, Sachs JA, Biro PA, Cutbush S, Wu Z, et al. HLA-DP polymorphism in Sudanese controls and patients with insulin-dependent diabetes mellitus. Tissue Antigens. 1992;40:64–8.
Dafalla AM, McCloskey DJ, Alemam AA, Ibrahim AA, Babikir AM, Gasmelseed N, et al. HLA polymorphism in Sudanese renal donors. Saudi J kidney Dis Transplant. 2011;22:834–40.
Hosomichi K, Jinam TA, Mitsunaga S, Nakaoka H, Inoue I. Phase-defined complete sequencing of the HLA genes by next-generation sequencing. BMC Genomics. 2013;14:355.
González-Galarza FF, Takeshita LY, Santos EJ, Kempson F, Maia MHT, Silva ALSda, et al. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 2015;43:D784–8.
Ahmadloo S, Nakaoka H, Hayano T, Hosomichi K, You H, Utsuno E, et al. Rapid and cost-effective high-throughput sequencing for identification of germline mutations of BRCA1 and BRCA2. J Hum Genet. 2017;62:561–7.
Lancaster AK, Single RM, Solberg OD, Nelson MP, Thomson G. PyPop update—a software pipeline for large-scale multilocus population genomics. Tissue Antigens. 2007;69:192–7.
Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–7.
Guo SW, Thompson EA. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 1992;48:361–72.
Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evolution. 1987;4:406–25.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evolution. 2013;30:2725–9.
Ewens WJ. The sampling theory of selectively neutral alleles. Theor Popul Biol. 1972;3:87–112.
Salamon H, Klitz W, Easteal S, Gao X, Erlich HA, Fernandez-Vina M, et al. Evolution of HLA class II molecules: allelic and amino acid site variability across populations. Genetics. 1999;152:393–400.
Slatkin M. An exact test for neutrality based on the Ewens sampling distribution. Genetical Res. 1994;64:71–4.
Excoffier L, Slatkin M. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol. 1995;12:921–7.
R Core Team. R: a language and environment for statistical computing. 2013. Available from: http://www.R-project.org.
Vina MAF, Hollenbach JA, Lyke KE, Sztein MB, Maiers M, Klitz W, et al. Tracking human migrations by the analysis of the distribution of HLA alleles, lineages and haplotypes in closed and open populations. Philos Trans Biol Sci. 2012;367:820–9.
Solberg OD, Mack SJ, Lancaster AK, Single RM, Tsai Y. Balancing selection and heterogeneity across the classical human leukocyte antigen loci: a meta-analytic review of 497 population studies. Hum Immunol. 2008;69:443–64.
Bryc K, Velez C, Karafet T, Moreno-Estrada A, Reynolds A, Auton A, et al. Genome-wide patterns of population structure and admixture among Hispanic/Latino populations. Proc Natl Acad Sci USA. 2010;107(Supplement_2):8954–61.
Tishkoff SA, Williams SM. Genetic analysis of African populations: human evolution and complex disease. Nat Rev Genet. 2002;3:611–21.
Buhler S, Sanchez-Mazas A. HLA DNA sequence variation among human populations: molecular signatures of demographic and selective events. PLoS One. 2011;6:e14643.
Černý V, Kulichová I, Poloni ES, Nunes JM, Pereira L, Mayor A, et al. Genetic history of the African Sahelian populations. HLA. 2018;91:153–66.
MacMichael HA.Tribes North Cent Kordofan. 1914;46:378.
Metz HC. Library of Congress Federal Research Division, Thomas Leiper Kane Collection. Sudan: a country study. 4th ed. Washington, DC: Federal Research Division, Library of Congress; 1992 (Area handbook series).
Di Giacomo F, Luca F, Popa LO, Akar N, Anagnou N, Banyko J, et al. Y chromosomal haplogroup J as a signature of the post-neolithic colonization of Europe. Hum Genet. 2004;115:357–71.
Sanchez-Mazas A, Djoulah S, Busson M, Le Monnier de Gouville I, Poirier JC, Dehay C, et al. A linkage disequilibrium map of the MHC region based on the analysis of 14 loci haplotypes in 50 French families. Eur J Hum Genet. 2000;8:33–41.
Abi-Rached L, Jobin MJ, Kulkarni S, McWhinnie A, Dalva K, Gragert L, et al. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science. 2011;334:89–94.
Yasukochi Y, Yasukochi Y, Ohashi J, Ohashi J. Elucidating the origin of HLA-B73 allelic lineage: did modern humans benefit by archaic introgression? Immunogenetics. 2017;69:63–7.
Acknowledgements
The authors would like to thank all participants who generously donated their DNA samples. MGN was supported by a Spinoza grant of the Netherlands Organization for scientific research.
Author information
Authors and Affiliations
Contributions
II, NH, and AW conceived and designed the study. II and NMG provided materials and supervision. AW performed the experiments with contribution from HHY, and JM. HHY collected participants data with contribution from TH. AW, HN, performed HLA data analysis and interpretation with contribution from HHY, HK, and AMH. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Aamer, W., Hassan, H.Y., Nakaoka, H. et al. Analysis of HLA gene polymorphisms in East Africans reveals evidence of gene flow in two Semitic populations from Sudan. Eur J Hum Genet 29, 1259–1271 (2021). https://doi.org/10.1038/s41431-021-00845-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00845-6
