
Abstract
Rab proteins coordinate inter-organellar vesicle-mediated transport, facilitating intracellular communication, protein recycling, and signaling processes. Dysfunction of Rab proteins or their direct interactors leads to a wide range of diseases with diverse manifestations. We describe seven individuals from four consanguineous Arab Muslim families with an infantile-lethal syndrome, including failure to thrive (FTT), chronic diarrhea, neonatal respiratory distress, variable pituitary dysfunction, and distal arthrogryposis. Exome sequencing analysis in the independent families, followed by an internal gene-matching process using a local exome database, identified a homozygous splice-site variant in MADD (c.2816 + 1 G > A) on a common haplotype. The variant segregated with the disease in all available family members. Determination of cDNA sequence verified single exon skipping, resulting in an out-of-frame deletion. MADD encodes a Rab guanine nucleotide exchange factor (GEF), which activates RAB3 and RAB27A/27B and is thus a crucial regulator of neuromuscular junctions and endocrine secretory granule release. Moreover, MADD protects cells from caspase-mediated TNF-α-induced apoptosis. The combined roles of MADD and its downstream effectors correlate with the phenotypic spectrum of disease, and call for additional studies to confirm the pathogenic mechanism and to investigate possible therapeutic avenues through modulation of TNF-α signaling.
Similar content being viewed by others


Introduction
Rab proteins are low molecular weight GTPases that serve as central organizers of vesicle-mediated transport between organelles [1]. Approximately 70 different Rabs in human cells facilitate intracellular communication, protein recycling, and signaling processes. Rabs function as molecular switches, alternating between active GTP-bound and inactive GDP-bound conformations that are temporally and spatially regulated by specific guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs) [2,3,4,5]. Dysfunction of Rabs or Rab proteins lead to a plethora of inherited genetic disorders and acquired diseases, consistent with their prominent role in membrane trafficking and cell homeostasis. These range from neurological disorders (Warburg Micro syndrome, Charcot-Marie-Tooth disease, Parkinson disease, Alzheimer disease) to immunodeficiency syndromes (Griscelli syndrome) and cancer [4].
The differentially expressed in normal and neoplastic cells (DENN) domain proteins constitute a subfamily of Rab GEFs with 17 recognized members, each of which specifically activates particular Rabs [6]. Among these, MADD (MAPK-activating death domain-containing protein) activates RAB3 and RAB27A/27B [3, 7] and is thus involved in regulation of secretory vesicle exocytosis [8]. RAB3 is essential for Ca2+-triggered synaptic vesicle exocytosis and for survival in mice, and its four murine homolog Rabs are functionally redundant despite differential distribution [9]. RAB27 regulates the transport and exocytosis of secretory granules and lysosomal-related organelles in various cell types, including cytotoxic T cells, platelets, and pancreatic beta cells [10]. RAB27A also has a role in proper melanosome distribution, thus providing pigmentation and photoprotection [11].
The murine homolog of MADD, Rab3 GEF, is a critical regulator of neurotransmitter release in synapses [12]. In addition to its role as a Rab GEF, MADD has a key regulatory role in neuroprotection, conferring resistance to TNF-α–induced, caspase-mediated apoptosis [13]. It also links type 1 tumor necrosis factor receptor (TNFR1) with MAP kinase activation [14]. Notably, different MADD isoforms, with varying tissue and temporal expression patterns, have been associated with divergent and even opposing functions on cell proliferation [13, 15].
We describe four families of consanguineous Arab Muslim descent with an infantile-lethal syndrome consisting of neonatal respiratory distress, severe failure to thrive (FTT), chronic diarrhea, metabolic acidosis, hypopituitarism, pancreatic exocrine insufficiency, normocytic anemia, variable dysmorphic features, congenital clubfoot or distal arthrogryposis, and genital abnormalities. In all families, a homozygous splice-site variant in MADD segregated with the disease phenotype. We present the clinical and molecular features of this syndrome in light of the current knowledge regarding the function of MADD in neuronal synapse formation, pituitary hormone expression, and TNF-α-induced apoptosis.
Materials and methods
Exome analysis
Following informed consent, exome analysis was pursued on DNA extracted from whole blood of the proband and her parents in Family A (trio exome) and on the proband only in Families B–D (Fig. 1A). Exonic sequences from DNA were enriched with the SureSelect Human All Exon 50 Mb V5 Kit (Agilent Technologies, Santa Clara, CA, USA). Sequences were generated on a HiSeq2500 or NovaSeq6000 sequencing system (Illumina, San Diego, CA, USA) as 125- or 150-bp paired-end runs. Read alignment and variant calling were performed with DNAnexus (Palo Alto, California, USA) using default parameters with the human genome assembly hg19 (GRCh37) as reference. Exome analysis of the probands yielded 60.43–80.2 million reads, with a mean coverage of 86–106X.

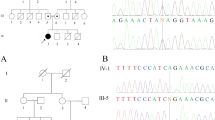
A Pedigrees of four families with affected individuals indicated in solid fill. Probands are indicated by arrows. Genotypes for the MADD c.2816 + 1 G > A variant, as determined by Sanger sequencing, are shown where available. In family C, individuals II-2 and II-3 had Stargardt disease. B Sanger sequencing of homozygous wild-type (upper panel), heterozygous (middle panel), and homozygous variant (lower panel). C cDNA Sanger sequencing of RT-PCR product from wild-type (upper panel) and heterozygous (middle panel) individuals. Lower panel represents lower molecular weight band of homozygous individual extracted from agarose gel (Fig. S3), and shows skipping of exon 17. Exon 16 is an alternative exon and was not identified in cDNA from peripheral blood.
Segregation analysis
An amplicon containing the MADD variant was amplified by conventional PCR of genomic DNA from probands and all available parents and siblings, and analyzed by Sanger dideoxy nucleotide sequencing.
RNA/cDNA analysis
RNA was isolated from fresh lymphocytes of the patient, parents and control by TRIzol reagent extraction and cDNA was prepared from 1 µg RNA using qScript cDNA Synthesis Kit (Quantabio). The region encompassing exons 14–19 of MADD was amplified by PCR reaction using DreamTaq Green PCR Master Mix (Thermo Scientific) with forward primer 5′-AACTCCACCGTCTCCAACAC-3′ and reverse primer 5′-GAGGAGGTCTAACATTCCCTTG-3′. The resultant fragments were separated by 3% (w/v) agarose gel electrophoresis and their sequence determined by Sanger sequencing.
Results
Clinical reports
Family A
The proband in Family A was a female, the sixth child born to first cousins of Arab Muslim descent. She was initially evaluated at 5 weeks of age for FTT and dysmorphic features. Pregnancy was remarkable for fetal hypokinesia, prompting induction of labor at week 36 + 6. The infant was born via Cesarean section (C-section) due to fetal distress. Birth weight was 2030 grams (third percentile) (Table 1). She had transient tachypnea of the newborn (TTN), and required oxygen for 2 weeks. She was discharged from the hospital at 25 days, and presented to the emergency department several days later with FTT and chronic diarrhea. Physical exam revealed dysmorphic features including a prominent occiput, vaulted palate, relatively long philtrum, and micrognathia. She had prominent heels and lateral eversion of the left foot. Work-up included head US and brain MRI, both within normal limits, abdominal ultrasound which was normal, and an echocardiogram which showed multiple small ventricular septal defects (VSD). Ammonia and lactate were normal, as were pituitary hormones (cortisol, follicle-stimulating hormone (FSH), luteinizing hormone (LH), thyroid-stimulating hormone (TSH), and thyroxine (T4)). Lymphocyte count including CD4 levels was normal. Follow-up over the next several months showed anemia (Hb levels 6.5 gr%) requiring blood transfusions. Fecal elastase was undetectable, indicating pancreatic exocrine dysfunction, and the infant was maintained on pancreatic enzyme replacement therapy. Growth parameters at 4 months of age were weight 3.58 kg (Z score −3.74) and length 49.5 cm (Z score −4.85).
Family history was remarkable for a second-born child of the parents who died at 2.5 months of age. This male sibling had been born at 36 weeks and had TTN after birth requiring oxygen for 12 days. He had FTT, chronic diarrhea, cholestasis (albeit following total parenteral nutrition), undescended testes, micropenis, and bilateral inguinal hernias. Clinical course was significant for recurrent hypoglycemia. The latter, together with micropenis, prompted an evaluation of pituitary hormones, which revealed hypopituitarism with low prolactin, FSH, LH, TSH, and T4 levels. Growth hormone was borderline low. Cortisol levels were normal. He had normocytic anemia requiring blood transfusions. Head ultrasound showed slightly dilated ventricles, and head computed tomography (CT) had an absent pituitary signal. Echocardiography revealed an atrial septal defect (ASD)/ patent foramen ovale (PFO). Chromosome analysis showed a normal male karyotype, 46,XY. Additional evaluations included ammonia, lactate, very long chain fatty acids (VLCFA), isoelectric focusing of transferrin, plasma amino acids, and urine organic acids––all of which resulted normal. Immunological work-up included immunoglobulin levels and immune response to mitogens which was normal. Lymphocyte count showed low CD4 levels.
Family history was further positive for several second cousins of the proband who died within the first year of life; no further clinical or molecular reports were available. In addition, the first pregnancy of the parents was of twins, one of which had hydrocephalus and was terminated; the other twin underwent intrauterine fetal death (IUFD).
Family B
The proband was a female infant, the second of five children born to first and second cousins of Arab Muslim origin, who died at 10.5 months due to chronic diarrhea, FTT, and secondary infections. She was born at 36 weeks after an uneventful pregnancy, with a birth weight of 1950 grams (fifth percentile). Apgar scores were 6 at 1 min and 8 at 5 min. Neonatal complications included respiratory distress requiring continuous positive airway pressure (CPAP) ventilation for 3 days and oxygen supplementation for 10 days, hyperbilirubinemia requiring phototherapy, and watery diarrhea since 1 week of age. She was readmitted to the hospital at 2.5 months of age for chronic diarrhea and severe FTT. Growth parameters on admission were weight 2.23 kg (Z score −4.49), length 46 cm (Z score −5.3), and head circumference 33.5 cm (Z score −5.82). On physical exam, she had dysmorphic features including long eyelashes, gum hyperplasia, high arched palate, micrognathia, and short neck. Multiple formulas were tried, as well as pancreatic enzyme replacement, steroids and total parenteral nutrition, yet with minimal response. The infant developed a wide anion gap metabolic acidosis attributed to hypernatremic dehydration. Polyuria with high serum osmolarity prompted work-up and diagnosis of possible nephrogenic diabetes insipidus. Upper GI endoscopy showed mild gastritis with relative duodenal atrophy, and biopsy showed relative shortening of the villi and chronic inflammation. Echocardiogram revealed patent foramen ovale (PFO). No episodes of hypoglycemia were documented. Evaluations included brain ultrasound, EEG, immunological work-up including immunoglobulin levels and T cell subtypes, metabolic studies, thyroid function tests, liver function tests, sweat chloride test, common CFTR variants, and chromosome analysis (Table 1), all of which resulted normal.
The fifth child of these parents was a male infant who died at 14 months due to chronic diarrhea, FTT, and secondary infections. He was born at 36 weeks after an uneventful pregnancy, with birth weight 2700 grams (48th percentile). Apgar scores were 2 at 1 min and 4 at 5 min. Neonatal complications included respiratory distress since birth requiring oxygen supplementation (possible TTN) and watery diarrhea since 5 days of life. Different formulas and pancreatic enzyme replacement therapy were to no avail. At 1 month of age, weight was 2.57 g (Z score −2.77), length 46 cm (Z score −3.71), and head circumference 35 cm (Z score −1.67). Additional medical issues included E. coli sepsis and meningitis, and normocytic anemia requiring blood transfusions. Work-up included metabolic evaluations, immunoglobulin levels, liver function tests, thyroid function tests, brain ultrasound, transferrin isoelectric focusing––all of which resulted normal. Exome sequencing was pursued, and was interpreted under an expected autosomal recessive mode of inheritance due to parental consanguinity and affected siblings.
Family C
The proband was a male, the only child of Arab Muslim parents who were double first cousins. The mother had Stargardt disease, which was molecularly confirmed (ABCA4: p.(Asn965Ser)) and the father was heterozygous for the ABCA4 variant. The parents had opted for prenatal genetic diagnosis (PGD) due to a 50% recurrence risk of Stargardt disease, yet PGD had failed and they conceived spontaneously. Fetal sonography demonstrated clubfoot and was otherwise within normal limits. Delivery was at 40 + 4 weeks by C-section due to non-reassuring fetal monitoring, at a birth weight of 2728 grams (Z score −2.16). Physical examination showed hyperterlorism, broad nasal bridge, microretrognathia, incomplete single palmar crease on right, bilateral clubfoot, micropenis, and unilateral cryptorchidism. At 7 weeks, he was evaluated for chronic diarrhea, FTT, metabolic acidosis, and central diabetes insipidus. Growth parameters were as follows: weight 2884 g (Z score −3.10), length 46.5 cm (Z score −4.93), and head circumference 36.5 cm (Z score −1.65). Pituitary hormone work-up revealed partial growth hormone deficiency, low gonadotropins and prolactin levels, TSH deficiency, and normal cortisol. Additional medical issues included macrocytic anemia, ASD and small patent ductus arteriosus (PDA), mild unilateral hydronephrosis. Head ultrasound was normal, as was brain MRI including the sella turcica. Initial genetic evaluation included chromosome analysis (46,XY) and fluorescent in situ hybridization (FISH) for 22q11 which resulted normal. The child died at 6 months of age from respiratory failure.
Family D
The proband was a male, the second of three children born to first cousin parents of Arab Muslim ethnicity who had multiple miscarriages. Pregnancy was complicated by oligohydramnios, asymmetric IUGR), left talipes deformity, and bilateral clenched hands. Parents opted not to do amniocentesis. He was born at 34 + 4 weeks of gestation, at a birth weight of 1680 grams (fourth percentile), length 43 cm (16th percentile), and head circumference 31 cm (35th percentile). Apgar scores were 1 at 1 min and 5 at 5 min with positive pressure ventilation. Neonatal complications included respiratory distress syndrome (RDS), PDA, complete bilateral retinal vascularization, and hyperbilirubinemia requiring phototherapy. Physical examination revealed dysmorphic features including dolichocephaly, low-set undeveloped ears, micrognathia, umbilical hernia, right inguinal hernia, small penis with hypoplastic scrotum, and bilateral undescended testes. In addition, he had distal arthrogryposis with limited movements of almost all upper and lower limb joints, bilateral clenched hands with single palmar creases, right rocker-bottom foot, and club foot. He had recurrent admissions to the hospital due to respiratory distress and transient hypoalbuminemia and lower limb edema, and was suspected to have protein losing enteropathy. Brain CT demonstrated prominent occipital horns of the lateral ventricles and mega cisterna magna. Abdominal ultrasound was unremarkable. Chromosome analysis was normal. The infant died at 7 months of respiratory issues.
A younger brother of the proband died at 1 month of age due to respiratory difficulties. Antenatal ultrasound showed bilateral clenched hands and bilateral talipes deformity. He was born at 35 + 1 weeks gestation, at a birth weight of 1860 grams (sixth percentile), length 44 cm (19th percentile), and head circumference 33 cm (74th percentile). Apgar scores were 2 at 1 min and 7 at 5 min with positive pressure ventilation. Dysmorphic features included a flat forehead, scoliosis and abnormal vertebrae, as well as bilateral clenched hands and bilateral clubfeet. Neonatal complications included RDS, bowel perforation requiring bowel resection and anastomosis, neonatal sepsis, and pancytopenia. Echocardiogram demonstrated a VSD. He had hypoalbuminemia and suspected protein losing enteropathy. Brain US and renal US were normal. The infant died at 1 moth of age due to the above mentioned problems. Genetic evaluation included chromosome analysis which resulted normal (46,XY), and exome sequencing was pursued.
Family history was significant for several other affected family members (four siblings of the father); as well as common cousins of the parents with similar symptoms by report (Fig. 1).
Consistent infantile-lethal phenotype with multi-organ involvement
Overall, the clinical presentation of the seven affected individuals was relatively consistent and included neurological, pulmonary, gastrointestinal, endocrine, cardiac, hematological, genital, and skeletal manifestations (Table 1, Fig. 2). Notably, none presented with abnormal skin or hair color. Six of seven infants were born preterm (at 34–36 weeks) at relatively low birth weights (Z scores ranged from −2.16 to −0.06). Neurological evaluation was difficult to assess properly due to early infantile death or recurrent hospitalizations. Fetal akinesia or hypokinesia was reported in two individuals, and congenital distal arthrogryposis was observed in five. Brain imaging was unremarkable in most, aside from absent pituitary signal (Family A, Individual II-2) and slightly dilated ventricles. Respiratory distress (TTN/RDS) was reported in six of seven neonates. All individuals had severe FTT and chronic diarrhea, with pancreatic exocrine insufficiency or response to pancreatic enzyme replacement therapy documented in two families. Cholestasis was also reported. Endocrine abnormalities included anterior pituitary dysfunction in two families and possible posterior pituitary dysfunction in one family. Echocardiogram revealed ASD/PFO, VSD, or PDA in all individuals, and all individuals had anemia. Immune deficiency was suspected in several cases due to recurrent secondary infections, yet immunological work-up was within normal limits. Genital abnormalities such as inguinal hernias, cryptorchidism and micropenis were apparent in three of five males. Skeletal abnormalities included the aforementioned distal arthrogryposis, and scoliosis and abnormal vertebrae in one individual. Initial genetic evaluation (i.e., chromosome analysis) was negative, and therefore exome sequencing was pursued in all probands.

Bar graph indicating number of individuals affected by specific endophenotype. Abbreviations: ASD atrial septal defect, FTT failure to thrive, PDA patent ductus arteriosus, PFO patent foramen ovale, RDS respiratory distress syndrome, TTN transient tachypnea of newborn, VSD ventral septal defect.
Identification of a homozygous variant in MADD
Exome sequencing was undertaken in all four families in search of an underlying genetic diagnosis. Following alignment to the reference genome [hg19] and variant calling, variants were filtered out if the total read depth was less than 8X and if they were off-target (>8 bp from splice junction), synonymous, or had minor allele frequency (MAF) > 0.005 in the gnomAD database. Exome data of each family was analyzed independently; homozygous variants of interest that survived filtering are provided in Table S1. In all four probands, the top candidate was a splice-site variant in MADD (chr11:g.47311045 G > A [hg19]; NC_000011.9(NM_003682.4): c.2816 + 1 G > A; r.2714_2816del) located in intron 17 of 35 introns, affecting all protein-encoding transcripts (Fig. S1). This is predicted to cause mis-splicing of exon 17 (exons numbered according to NG_029462.1) due to alteration of the wild-type donor site. Segregation studies in all available family members by Sanger sequencing (Fig. 1A-B) revealed that parents were heterozygous carriers, affected individuals were homozygous for the variant, and unaffected siblings were either heterozygous or homozygous WT (LOD score (θ = 0) was Z = 3.913). Paternal grandparents in Family D were both heterozygous, and indeed had four children who died from similar symptoms in infancy. Although the families denied being related, the MADD variant occurred on a shared haplotype of 15.643MB (chr11:43,911,365-59,554,495 [hg19]), suggesting that it is a founder variant in the local Arab Muslim population. Expression data indicate that MADD is highly expressed in pituitary tissue, brain cortex, and cerebellum (GTEx Portal [16], Fig. S2).
The MADD variant causes exon skipping at the RNA level
In order to determine whether the MADD variant affects splicing, we analyzed the cDNA sequence of individual III-12 in Family A, both parents, and a wild-type control. RT-PCR revealed the existence of a lower molecular weight band in individual III-12 of Family A and in the parents, in addition to the wild-type band. The lower molecular band was non-existent in the wild-type control (Fig. S3), indicating a leaky splice site variant. Sequencing of this band after extraction from the agarose gel revealed skipping of exon 17 (Fig. 1C). Notably, exon 16 is an alternative exon (Fig. S1) and was not seen in the transcript amplified from peripheral blood. As predicted, the skipping of exon 17 leads to an out-of-frame deletion of 103 bp.
Discussion
In this study, we report seven affected individuals in four independent nuclear families within a clinical spectrum of FTT, chronic diarrhea, pancreatic exocrine insufficiency, pituitary dysfunction, dysmorphic facies, and congenital clubfoot or arthrogryposis (Table 1, Fig. S4). Exome sequencing in each of the families, followed by internal gene-matching in the local exome database, revealed a shared homozygous splice-site variant in MADD: c.2816 + 1 G > A on a founder haplotype of ~15.6MB shared absence of heterozygosity (AOH). The variant was shown to lead to mis-splicing of exon 17, leading to an out-of-frame deletion [13].
Recently, biallelic variants in MADD were described in families with macrocephaly and cognitive delay [17], and in families with global developmental delay, FTT and autism [18]. Monoallelic de novo variants in MADD have been described in individuals with autism [19] and muscular dystrophy; [20] future studies will help determine whether these are truly causal and whether specific monoallelic variants (and not only biallelic variants) can cause disease manifestations. MADD variants have also been associated with fasting hyperglycemia and diabetes due to impaired glucose-induced insulin release [21, 22]. In addition, reduced endogenous MADD expression and consequent enhanced apoptosis correlate with neuronal cell death in hippocampal neurons and Alzheimer disease [23,24,25]. As compared to clinical features previously published in association with MADD variants [17, 18], the phenotype described herein is strikingly more severe and pleiotropic. While this manuscript was under review, Schneeberger et al. (2020) published a series of 23 patients with 21 different MADD variants; these included a group of 14 individuals with multi-systemic manifestations similar to those described herein, and a second group of nine individuals with a predominant neurological phenotype. The authors stated that they could not establish a genotype–phenotype correlation [26], and this will be interesting to revisit as additional cases are identified.
MADD is one of seven isoforms alternatively spliced from a single transcript [15]. The full length IG20 (insulinoma–glucagonoma, clone 20) gene is locate on chromosome 11p11.2 and has 36 exons. Alternative splicing of exons 13 L, 16, 21, 26, and 34 results in splice variants with different expression patterns and protein functions, including IG20, MADD/DENN, DENN-SV, and KIAA0358. The IG20 isoforms regulate cell proliferation, survival, and death through alternative mRNA splicing. The different splice variants differentially affect TNF-α-induced apoptosis mediated by caspase-8 and caspase-3 [13]. The MADD isoform is an Akt substrate and confers resistance to TRAIL (TNF-related apoptosis-inducing ligand)- and TNF-α–induced apoptosis, thus playing an important role in cancer cell survival [27, 28]. Abrogation of MADD expression renders cells highly susceptible to TNF-α–induced apoptosis, and re-expression of MADD in the absence of endogenous IG20 expression rescues cells from TNF-α–induced apoptosis. The requirement for MADD is highly specific for TNF-α–induced activation of mitogen-activated protein kinase (MAPK) but not for related JNK and p38 kinases [29]. Schneeberger et al. (2020) showed that patient-derived fibroblasts from individuals with biallelic MADD variants had impaired TNF- α signaling and increased apoptosis as compared to control cells [26], and much of the clinical phenotype of the patients may result from increased apoptosis.
Additionally, MADD functions as a Rab GEF activating RAB3 and RAB27A/27B [3, 13, 22]. In Drosophila, the MADD homolog rab3-GEF controls Rab3 localization and regulates the active zone development at the neuromuscular junction, potentially as a Rab3 effector protein to dock vesicles at the synapse [30]. Schneeberger et al. (2020) showed a defect in endocytosis of epidermal growth factor (EGF) in patient cells as compared to control cells, and suggest a possible link between the vesicle trafficking defect and impaired exocytosis [26]. Additional studies are required to determine whether the fetal hypokinesia and distal arthrogryposis in the affected individuals result from aberrant neuromuscular junction formation. Notably, Rab3 knockout (KO) mice exhibited shallow and irregular breathing patterns [9], reminiscent of the respiratory difficulties in affected infants. Moreover, both Rab3 and Rab27 have been implicated in trafficking of the CFTR chloride channel and the epithelial sodium channel (ENaC), possibly providing a link between MADD deficiency and the pulmonary and intestinal manifestations of cystic fibrosis [31]. This requires further investigation before reaching definitive conclusions.
RAB27A has a critical role in intracellular transport of secretory lysosomes, such as melanosomes in melanocytes, lytic granules in cytotoxic T lymphocytes, and platelet-dense granules [32]. Accordingly, biallelic RAB27A variants cause Griscelli syndrome type 2 [MIM 607624], with symptoms reflecting dysfunction of these organelles such as pigmentary dilution, immunodeficiency and increased bleeding time [33, 34]. It is of interest that the patients described herein did not present with abnormal skin or hair color, possibly indicating genetic redundancy upstream of RAB27A, yet do have a clinical suspicion of immunodeficiency, perhaps due to disrupted RAB27-regulated granule secretion [10]. Members of the RAB27 subfamily also regulate exocytosis of dense core granules containing peptide hormones in endocrine cells, and overexpression of Rab27b mutants significantly inhibits ACTH secretion in vitro. The lack of endocrine abnormalities in Griscelli syndrome has been attributed to functional compensation for RAB27A by RAB27B [35]. Indeed, double KO mice of Rab27a/Rab27b demonstrated markedly impaired distribution of secretory granules in the pituitary of mice [36]. Thus, the anterior pituitary dysfunction in individuals with biallelic MADD loss-of-function variants may be attributed to downstream RAB27 dysregulation. In this series of patients, only two had documented pituitary dysfunction, although not all individuals underwent thorough hormonal work-up (Table 1). Nonetheless, pituitary dysfunction was recently described in eight individuals with biallelic MADD variants [26], and hormonal work-up is thus critical in the context of suspected MADD deficiency.
Regarding the intestinal phenotype, Rab-27 has been shown to be expressed in intestinal cells in C. elegans [7]. In mice, Rab27b is widely expressed in canonical secretory cells and additionally in neurons and cells involved in surface protection and mechanical extension [36]. Notably, both the Rab3 isoform Rab3D and Rab27 are expressed in pancreatic acinar cells and may be involved in regulated exocytosis [37,38,39,40,41], possibly accounting for the exocrine pancreatic deficiency encountered in Family A. Moreover, the intestinal manifestations may result from increased TNF-α-induced apoptosis, consistent with the central role of TNF-α in intestinal inflammation and inflammatory bowel disease [42].
In conclusion, the multiple roles of MADD in TNF-α-induced apoptosis and as a Rab GEF controlling RAB27A/B and RAB3 in the pituitary and neuronal synapses, as well as other tissues, seem consistent with the clinical phenotype encountered herein (Fig. S4), and are supported by the recent publication of a series of individuals with MADD variants. Further functional studies are called for in order to understand the pathogenic mechanism and to determine whether pharmacologic alteration of the TNF-α pathway may be beneficial in this infantile lethal disorder. Identification of additional families with MADD-associated disorders will aid in substantiating the phenotypic spectrum and in establishing genotype-phenotype correlations.
Data availability
The ClinVar accession number for the DNA variant data is SCV001364046.1.
References
Segev N. Ypt and Rab GTPases: insight into functions through novel interactions. Curr Opin Cell Biol. 2001;13:500–11.
Pfeffer S, Aivazian D. Targeting Rab GTPases to distinct membrane compartments. Nat Rev Mol Cell Biol. 2004;5:886–96.
Yoshimura S, Gerondopoulos A, Linford A, Rigden DJ, Barr FA. Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors. J Cell Biol. 2010;191:367–81.
Banworth MJ, Li G. Consequences of Rab GTPase dysfunction in genetic or acquired human diseases. Small GTPases. 2018;9:158–81.
Langemeyer L, Fröhlich F, Ungermann C. Rab GTPase function in endosome and lysosome biogenesis. Trends Cell Biol. 2018;28:957–70.
Wada M, Nakanishi H, Satoh A, Hirano H, Obaishi H, Matsuura Y, et al. Isolation and characterization of a GDP/GTP exchange protein specific for the Rab3 subfamily small G proteins. J Biol Chem. 1997;272:3875–8.
Mahoney TR, Liu Q, Itoh T, Luo S, Hadwiger G, Vincent R, et al. Regulation of synaptic transmission by RAB-3 and RAB-27 in Caenorhabditis elegans. Mol Biol Cell. 2006;17:2617–25.
Handley MT, Haynes LP, Burgoyne RD. Differential dynamics of Rab3A and Rab27A on secretory granules. J Cell Sci. 2007;120:973–84.
Schlüter OM, Schmitz F, Jahn R, Rosenmund C, Südhof TC. A complete genetic analysis of neuronal Rab3 function. J Neurosci. 2004;24:6629–37.
Fukuda M. Rab27 effectors, pleiotropic regulators in secretory pathways. Traffic. 2013;14:949–63.
Wu X, Hammer JA. Melanosome transfer: it is best to give and receive. Curr Opin Cell Biol. 2014;29:1–7.
Tanaka M, Miyoshi J, Ishizaki H, Togawa A, Ohnishi K, Endo K, et al. Role of Rab3 GDP/GTP exchange protein in synaptic vesicle trafficking at the mouse neuromuscular junction. Mol Biol Cell. 2001;12:1421–30.
Al-Zoubi AM, Efimova EV, Kaithamana S, Martinez O. El-Idrissi Mel-A, Dogan RE, et al.: Contrasting effects of IG20 and its splice isoforms, MADD and DENN-SV, on tumor necrosis factor alpha-induced apoptosis and activation of caspase-8 and -3. J Biol Chem. 2001;276:47202–11.
Schievella AR, Chen JH, Graham JR, Lin LL. MADD, a novel death domain protein that interacts with the type 1 tumor necrosis factor receptor and activates mitogen-activated protein kinase. J Biol Chem. 1997;272:12069–75.
Efimova EV, Al-Zoubi AM, Martinez O, Kaithamana S, Lu S, Arima T, et al. IG20, in contrast to DENN-SV, (MADD splice variants) suppresses tumor cell survival, and enhances their susceptibility to apoptosis and cancer drugs. Oncogene. 2004;23:1076–87.
Consortium G. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–5.
Hu H, Kahrizi K, Musante L, Fattahi Z, Herwig R, Hoseeini M, et al. Genetics of intellectual disability in consanguineous families. Mol Psychiatry. 2019;24:1027–39.
Anazi S, Maddirevula S, Salpietro V, Asi YT, Alsahli S, Alhashem A, et al. Expanding the genetic heterogeneity of intellectual disability. Hum Genet. 2017;136:1419–29.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21.
Farwell Hagman KD, Shinde DN, Mroske C, Smith E, Radtke K, Shahmirzadi L, et al. Candidate-gene criteria for clinical reporting: diagnostic exome sequencing identifies altered candidate genes among 8% of patients with undiagnosed diseases. Genet Med. 2017;19:224–35.
Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–16.
Li LC, Wang Y, Carr R, Haddad CS, Li Z, Qian L, et al. IG20/MADD plays a critical role in glucose-induced insulin secretion. Diabetes. 2014;63:1612–23.
Miyoshi J, Takai Y. Dual role of DENN/MADD (Rab3GEP) in neurotransmission and neuroprotection. Trends Mol Med. 2004;10:476–80.
Mo Y, Williams C, Miller CA. DENN/MADD/IG20 alternative splicing changes and cell death in Alzheimer’s disease. J Mol Neurosci. 2012;48:97–110.
Del Villar K, Miller CA. Down-regulation of DENN/MADD, a TNF receptor binding protein, correlates with neuronal cell death in Alzheimer’s disease brain and hippocampal neurons. Proc Natl Acad Sci USA. 2004;101:4210–5.
Schneeberger PE, Kortüm F, Korenke GC, Alawi M, Santer R, Woidy M, et al. Biallelic MADD variants cause a phenotypic spectrum ranging from developmental delay to a multisystem disorder. Brain. 2020;143:2437–53.
Mulherkar N, Ramaswamy M, Mordi DC, Prabhakar BS. MADD/DENN splice variant of the IG20 gene is necessary and sufficient for cancer cell survival. Oncogene. 2006;25:6252–61.
Li LC, Jayarama S, Pilli T, Qian L, Pacini F, Prabhakar BS. Down-modulation of expression, or dephosphorylation, of IG20/MADD in tumor necrosis factor-related apoptosis-inducing ligand-resistant thyroid cancer cells makes them susceptible to treatment with this ligand. Thyroid. 2013;23:70–78.
Kurada BR, Li LC, Mulherkar N, Subramanian M, Prasad KV, Prabhakar BS. MADD, a splice variant of IG20, is indispensable for MAPK activation and protection against apoptosis upon tumor necrosis factor-alpha treatment. J Biol Chem. 2009;284:13533–41.
Bae H, Chen S, Roche JP, Ai M, Wu C, Diantonio A, et al: Rab3-GEF controls active zone development at the drosophila neuromuscular junction. eNeuro. e0031-16.2016:1–19.
Farinha CM, Matos P. Rab GTPases regulate the trafficking of channels and transporters––a focus on cystic fibrosis. Small GTPases. 2018;9:136–44.
Deacon SW, Gelfand VI. Of yeast, mice, and men. Rab proteins and organelle transport. J Cell Biol. 2001;152:F21–24.
Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupois S, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25:173–6.
Wilson SM, Yip R, Swing DA, O’Sullivan TN, Zhang Y, Novak EK, et al. A mutation in Rab27a causes the vesicle transport defects observed in ashen mice. Proc Natl Acad Sci USA. 2000;97:7933–8.
Zhao S, Torii S, Yokota-Hashimoto H, Takeuchi T, Izumi T. Involvement of Rab27b in the regulated secretion of pituitary hormones. Endocrinology. 2002;143:1817–24.
Gomi H, Mori K, Itohara S, Izumi T. Rab27b is expressed in a wide range of exocytic cells and involved in the delivery of secretory granules near the plasma membrane. Mol Biol Cell. 2007;18:4377–86.
Chen X, Edwards JA, Logsdon CD, Ernst SA, Williams JA. Dominant negative Rab3D inhibits amylase release from mouse pancreatic acini. J Biol Chem. 2002;277:18002–9.
Ohnishi H, Samuelson LC, Yule DI, Ernst SA, Williams JA. Overexpression of Rab3D enhances regulated amylase secretion from pancreatic acini of transgenic mice. J Clin Invest. 1997;100:3044–52.
Piiper A, Leser J, Lutz MP, Beil M, Zeuzem S. Subcellular distribution and function of Rab3A-D in pancreatic acinar AR42J cells. Biochem Biophys Res Commun. 2001;287:746–51.
Chen X, Li C, Izumi T, Ernst SA, Andrews PC, Williams JA. Rab27b localizes to zymogen granules and regulates pancreatic acinar exocytosis. Biochem Biophys Res Commun. 2004;323:1157–62.
Williams JA, Chen X, Sabbatini ME. Small G proteins as key regulators of pancreatic digestive enzyme secretion. Am J Physiol Endocrinol Metab. 2009;296:E405–14.
Ruder B, Atreya R, Becker C: Tumour necrosis factor alpha in intestinal homeostasis and gut related diseases. Int J Mol Sci. 2019;20:1887.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Abu-Libdeh, B., Mor-Shaked, H., Atawna, A.A. et al. Homozygous variant in MADD, encoding a Rab guanine nucleotide exchange factor, results in pleiotropic effects and a multisystemic disorder. Eur J Hum Genet 29, 977–987 (2021). https://doi.org/10.1038/s41431-021-00844-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00844-7
This article is cited by
-
New insights into the clinical and molecular spectrum of the MADD-related neurodevelopmental disorder
Journal of Human Genetics (2024)
-
Clinical genomics—but faster
European Journal of Human Genetics (2021)
