
Abstract
A high rate of consanguinity leads to a high prevalence of autosomal recessive disorders in inbred populations. One example of inbred populations is the Arab communities in Israel and the Palestinian Authority. In the Palestinian Authority in particular, due to limited access to specialized medical care, most patients do not receive a genetic diagnosis and can therefore neither receive genetic counseling nor possibly specific treatment. We used whole-exome sequencing as a first-line diagnostic tool in 83 Palestinian and Israeli Arab families with suspected neurogenetic disorders and were able to establish a probable genetic diagnosis in 51% of the families (42 families). Pathogenic, likely pathogenic or highly suggestive candidate variants were found in the following genes extending and refining the mutational and phenotypic spectrum of these rare disorders: ACO2, ADAT3, ALS2, AMPD2, APTX, B4GALNT1, CAPN1, CLCN1, CNTNAP1, DNAJC6, GAMT, GPT2, KCNQ2, KIF11, LCA5, MCOLN1, MECP2, MFN2, MTMR2, NT5C2, NTRK1, PEX1, POLR3A, PRICKLE1, PRKN, PRX, SCAPER, SEPSECS, SGCG, SLC25A15, SPG11, SYNJ1, TMCO1, and TSEN54. Further, this cohort has proven to be ideal for prioritization of new disease genes. Two separately published candidate genes (WWOX and PAX7) were identified in this study. Analyzing the runs of homozygosity (ROHs) derived from the Exome sequencing data as a marker for the rate of inbreeding, revealed significantly longer ROHs in the included families compared with a German control cohort. The total length of ROHs correlated with the detection rate of recessive disease-causing variants. Identification of the disease-causing gene led to new therapeutic options in four families.
Similar content being viewed by others


Introduction
Consanguinity is a deeply rooted cultural trait in Middle Eastern societies, especially in the Arab rural populations due to socio-cultural factors like maintenance of the family structure, property, or ease of marital arrangements [1]. Despite the fact that this type of marriages is discouraged by the major religions, recent studies estimated the prevalence of consanguineous marriages among the Palestinian Arab and Israeli Arab population to 44.3% and 25.9%, respectively, representing some of the highest rates in the world [2,3,4,5]. This high inbreeding rate leads to a high prevalence of autosomal recessive disorders. In first cousin relations, the risk of significant birth defects is increased up to 2.5 times as compared with the general population [3]. Especially in rural Palestinian areas these patients do not have access to advanced medical diagnostics and can often not be assessed by trained specialists. Therefore, most of the time a genetic diagnosis is not established.
In this study, we performed first-line whole-exome sequencing in 83 Arab families with suspected neurogenetic disorders due to at least two similar affected patients to identify the genetic cause of disease. Starting from only minimal clinical information, WES identified potentially disease-causing variants that were confirmed in a second step by targeted reverse phenotyping. Through this, 37 families received a definite genetic diagnosis and 5 families a likely diagnosis with novel candidate variants, leading to a high diagnostic yield of ~51%. Moreover, a specific therapy was made possible in four families due to WES.
Methods
Families were identified and enrolled in Israel or the Palestinian territories by cooperating physicians from 2012 to 2017. The inclusion criteria were defined as follows: (1) patients had to present with so far unexplained neurological symptoms and (2) at least two family members, including the index patient, had to suffer from similar symptoms. Initial phenotyping was often performed by medical staff who were not trained as neurologists and was thus mostly limited to broad categories such as “movement disorder,” “intellectual disability,” and “epilepsy.”
Written informed consent was obtained from the patients or the parents of the underage patients for diagnostic procedures and next-generation sequencing. The study has been approved by the local Institutional Review Board (vote 180/2010BO1).
Genetics
Patients were screened for exonic variants using a whole-exome enrichment approach (SureSelectXT Human All Exon V5 or SureSelectXT Human All Exon V6; Agilent, Santa Clara CA). Sequencing was performed on a HiSeq2500 (200 cycle chemistry) or NextSeq500 (300 cycle chemistry) platform (Illumina, San Diego, CA) in paired-end mode according to the manufacturers’ protocol.
Data analysis was performed using the megSAP [6] pipeline. The pipeline uses BWA-MEM [7] for alignment, freebayes [8] for variant calling, and Ensembl VEP [9] for variant annotation. Variants were first checked for pathogenic variants known to be associated with neurological disorders using the HGMD [10] database. If no known disease-causing variant was identified, variants were next filtered for rare variants (gnomAD [11] minor allele frequency <0.1%), considering both recessive and dominant inheritance, and prioritized according to gene function, conservation (pyhloP [12], GERP++ [13]) and in silico prediction scores (CADD [12], SIFT [14], PolyPhen2 [15]). If no clear candidate variant could be identified, a second exome was sequenced from another affected family member to reduce the number of potential variants. A total of 102 exomes were sequenced. In addition, copy number variants were determined from WES data using CnvHunter [16], a tool which compares the depth of coverage for each exon to a collective of reference samples to determine outlier exons. Runs of homozygosity (ROHs) were determined using RohHunter [16], which detects homozygous regions that are too long to occur by chance based on detected SNP genotypes and the allele frequency of the SNPs in public databases. Sanger sequencing was used to confirm the identified variants and test the segregation in all available family members. For nonsegregating variants, the possibility of two independent genetic disorders was taken into account. Variants have been classified as pathogenic or likely pathogenic if they fulfilled the respective ACMG criteria [17]. The respective conditions under which the variants are causing disease (dominant, recessive, X-linked) are specified for each pathogenic/likely pathogenic variant in Table 1 and Table S1. Patients presenting with cerebellar ataxia were additionally screened for repeat expansions in SCA 1, 2, 3, 6, 7, 17 and Friedreich’s ataxia. Patients with HSP phenotypes underwent MLPA for SPG4. All WES data from unsolved cases were reannotated and reanalyzed shortly before submission of the paper.
Statistical analysis
Statistical analysis was performed using JMP 14.2.0. The nonparametric comparison in Fig. 1 was done using the Wilcoxon method.

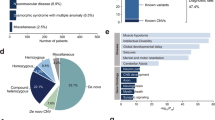
a Runs of homozygosity (ROHs) in a German control cohort, patients with no reported consanguinity and patients with reported consanguinity. Every dot represents the ROHs in one exome. The ROHs are significantly longer in patients with reported consanguinity vs patients without reported consanguinity. Interestingly they are also significantly longer in patients without reported consanguinity compared with German controls indicating inbreeding in the Arab communities (p = 0.0053). b ROHs in patients with “unsolved” exomes compared with patients with solved/likely solved exomes. ROHs were significantly longer in solved/likely solved cases suggesting that the higher ROHs result in a better chance to find the causative variant. c total number and percentage of families with pathogenic/likely pathogenic variants, candidate variants, and unsolved families.
Results
Eighty-three Arab families with at least two affected patients with similar neurological disorders were included in this study. Sixty families reported consanguineous marriages. Twenty-three families were not aware of any consanguinity in their family history. In total 102 individuals received exome sequencing. On average 116,282,290 sequence reads of 101-bp length were generated per sample. The mean sequencing depth was 114× while 92% of the target sequence was covered at least 20 times. Our standardized exome filtering approach revealed pathogenic/likely pathogenic variants in well-established disease genes in 35 families. Probably disease-causing candidate variants that could not be classified as pathogenic or likely pathogenic due to ACMG standards were identified in five families. In addition, two new disease genes (published separately) were identified in the context of this study [18, 19]. In total, 51% of the families (42 families) received either a definitive diagnosis (37 families, Table 1) or a likely diagnosis with a novel candidate variant (5 families, Table 2). Two independent genetic disorders were identified in one family (GPT2 and likely autosomal recessive deafness) as discussed elsewhere [20]. Based on exome sequencing (ROH) were determined for every sequenced patient. The total length of ROHs was used to estimate the degree of inbreeding. patients from families with reported consanguinity had a median ROHs length of 243 Mb. ROHs in these families were significantly longer compared with patients without reported consanguinity of the parents (median ROHs length 48 Mb; p < 0.0001) (Fig. 1). Compared with a German control cohort (median ROHs length 35 Mb) the ROHs were significantly longer not only in the Arab patient group with reported consanguinity but also in the Arab patient group without reported consanguinity. A considerably higher percentage of families with reported consanguinity received a genetic diagnosis and the ROHs were also significantly longer in patients with identified pathogenic/likely pathogenic variants compared with patients that did not receive a genetic diagnosis (median ROH in solved patients: 233 Mb, median ROH in patients without genetic diagnosis: 101 Mb; p < 0.0001, Fig. 1).
As expected most identified variants were homozygous variants in recessive disease genes (Table 1), all located within a region of homozygosity. Autosomal dominant pathogenic variants were identified in two families in the KCNQ2 gene and the KIF11 gene, respectively. One family had a hemizygous pathogenic variant in MECP2. Compound heterozygous pathogenic variants, as well as likely pathogenic copy number variants, were not identified. Of the 37 variants that were regarded as disease causing (in 42 families), 20 variants had been previously associated with disease, 2 variants were established in new disease genes, while 16 were novel variants in known disease genes (6 missense variants, 10 loss of function variants), thus expanding the genetic spectrum of these disorders (Table 1). Of the novel missense variants, only one missense variant in NTRK1 could be classified as likely pathogenic, due to the pathognomonic phenotype with insensitivity to pain and anhidrosis. The other five missense variants are thus listed below as novel candidate variants.
Novel candidate variants in established disease genes
Based on the ACMG criteria most novel missense variants cannot be classified as likely pathogenic or pathogenic, if they are found only in one family and no functional readout is available, even if other missense variants in patients with similar phenotypic features have been established in these disease genes. We still consider the following candidate variants as probably pathogenic:
-
(1)
MCOLN1 (homozygous c.230C>T: p.(Thr77Met)): this variant segregated in a large consanguineous family with 6 affected and 11 healthy family members was very rare in gnomAD (MAF 2 × 10−5, not observed homozygous) and had high in silico prediction scores (CADD 18.8). The affected patients suffered from mild intellectual disability, slowly progressive cerebellar ataxia, and variable affection of upper motor neuron, epilepsy, and pes cavus. Ophthalmological examination was not performed and corneal clouding was not obvious. Although not being the typical presentation of mucolipidosis type 4 (intellectual disability and ophthalmological abnormalities), the clinical features are compatible with this diagnosis.
-
(2)
MFN2 (homozygous c.1963A>G: p.(Lys655Glu)): this MFN2 variant segregated in a family with two affected and three healthy family members. Both patients suffered from peripheral neuropathy with distal muscle weakness, suggestive for Charcot–Marie–Tooth Disease. We thus consider CMT2A2B to be the likely diagnosis.
-
(3)
SEPSECS (homozygous c.181A>G: p.(Met61Val)): this variant was identified in a family with two siblings suffering from pontocerebellar hypoplasia and severe global developmental delay. The variant was very rare in gnomAD (MAF 4 × 10−6, not observed homozygous) and received a high in silico prediction score (CADD: 17). We concluded that pontocerebellar hypoplasia type 2D is the likely diagnosis.
-
(4)
SYNJ1 (homozygous c.1274G>T: p.(Cys425Phe)): this variant was present in a family with two affected children suffering from a severe early-onset epileptic encephalopathy with myoclonic epilepsy. The variant was not present in gnomAD, received high in silico prediction scores (CADD: 27.6), and segregated in the family with the parents and one unaffected sibling. We consider this variant to be another cause of autosomal recessive SYNJ1-associated epileptic encephalopathy. So far less than ten patients have been described [21,22,23].
-
(5)
TSEN54 (homozygous c.341C>T: p.(Pro114Leu)): two siblings with early-onset epilepsy, global developmental delay, and microcephaly shared this variant. The variant was absent from gnomAD, received high in silico prediction scores (CADD: 24.3), and segregated in this family with three healthy family members. Although a brain MRI was not available the clinical features are suspicious for TSEN54 associated pontocerebellar ataxia.
Extensions of the phenotypic spectrum and novel disease genes
Two distally related families were included in this study with two sibs in each branch suffering from early-onset generalized muscle weakness. Spinal muscular atrophy was suspected due to severe muscle weakness, atrophy, and fasciculations of the tongue in all patients. Additional diagnostics like electrophysiology were not available to these patients from the Palestinian Authority. WES was carried out in one affected individual of both families and revealed a novel homozygous MTMR2 frameshift variant c.766_767delAA segregating with the disease in both families (Fig. 2). We reexamined all patients clinically to confirm the diagnosis of severe Charcot–Marie–Tooth type 4b1, caused by recessive pathogenic variants in MTMR2. The four patients were between 7 and 23 years old. All shared distally pronounced symmetric flaccid weakness, more severe in the older patients. Reflexes were reduced (in the 7-year-old girl) or absent (in all other patients). While the 7-year-old girl was still able to walk independently, the 15-year-old patient was using a walker, and the two oldest patients were wheelchair bound since the age of 14 and 15 years, respectively. All had severe respiratory problems with stridor and breathing restricted to the diaphragm in the two oldest patients. Three of the four patients presented with hoarseness. Facial weakness, chewing, and swallowing difficulties were present in all four patients. Only some minor distal sensory deficits were reported by the patients, and there were only minor deficits in position sense at the toes. Taken together the core features of early-onset disease with respiratory distress, distal symmetric weakness and atrophy, and vocal cord involvement were in agreement with the genetic diagnosis of CMT type 4B1. The early involvement of the vocal cord and stridor is increasingly recognized in patients with CMT type 4B1 [24]. This example shows the importance of reverse phenotyping in these cases, once a genetic diagnosis was suspected.

The mother in AQ18 is the sister of the father in AQ19 while the mother in AQ19 is the sister of the father in AQ18. In addition, both couples are first cousins. The MTMR2 variant (NM_016156:exon8:c.766_767del) was homozygous in all affected patients and heterozygous in the parents. mt pathogenic variant; wt wild type.
In fact the a priori clinical diagnosis of the referring local physicians differed several times from the diagnosis achieved after genetically guided reverse phenotyping. Another example for this was a family in which we identified a known pathogenic homozygous SPG11 variant via WES (AQ54, Table 1), whereas the a prior diagnosis of this family was muscular dystrophy. The more detailed genetically guided phenotyping finally confirmed a complicated form of hereditary spastic paraplegia (cHSP) compatible with the homozygous pathogenic variant in the SPG11 gene.
Another interesting finding was a homozygous frameshift variant (c.446delG: p.(Ser149Thrfs*45)) in SCL25A15 in two sisters presenting with spastic paraparesis, mild cerebellar ataxia, and polyneuropathy, best summarized as complicated form of HSP. Although pyramidal and cerebellar affections are well-described in patients with recessive pathogenic SLC25A15 variants [25, 26], hyperornithinemia–hyperammonemia–homocitrullinemia (HHH) syndrome is most likely not on the list when seeing patients clinically presenting with cHSP. An obvious learning disability was not present in the patient’s medical history or clinical impression.
Besides new variants in established disease genes, the WES-first approach enabled us to identify new disease genes for ultra-rare disorders. In families without likely disease-causing variants in the index patient we performed additional WES of a second or third affected family member. This helped to reduce the number of candidate genes and helped (i) to identify two novel disease genes (WWOX and PAX7, both published elsewhere [18, 19]) and (ii) to establish substantial expansions of the gene-associated phenotypic spectrum like in GPT2, CNTNAP1, and POLR3A [20, 27, 28]. (iii) We identified additional families with pathogenic or likely pathogenic variants in genes that had been described only in single families. This helped to confirm the pathogenic role of these variants and to establish its causal relation for the disease like the homozygous splice variant c.2023-2A>G in SCAPER that has recently been associated with retinitis pigmentosa and intellectual disability [29]. Interestingly, all three patients with this pathogenic SCAPER variant in our series had nuclear cataracts in addition to retinitis pigmentosa and intellectual disability.
WES enables specific therapy
In 4 of 83 families we identified genetic diseases that offer causal treatment options most likely with beneficial effects on the course of disease. The above mentioned molecular genetic diagnosis of HHH syndrome, caused by an defect of the urea cycle, enabled a dietary treatment with supplementation of ornithine, and restriction of protein [25].
Similarly, in three apparently independent families we found a well-established pathogenic GAMT variant causing cerebral creatine deficiency syndrome 2 (CCDS2). In CCDS2 standardized treatment recommendations, including creatine supplementation to reduce cerebral creatinine deficiency are available and are likely to improve or stabilize symptoms. Unfortunately, the treatment response could only be monitored in two patients with CCDS2. These two patients both showed improvement of aggressive behavior and autistic features, as well as a reduction of seizures frequency comparable to the previously reported positive effects of creatine supplementation [30]. In summary, WES enabled specific treatment in nine patients from four families.
Discussion
We have shown that first-line exome diagnostic in neurological patients from consanguineous Arab communities in Israel and the Palestinian Authority reaches a high diagnostic yield. In this study 42 of 83 (51%) families received a definite genetic diagnosis or at least a very likely candidate variant. This is comparable to the diagnostic yield from similar studies using next-generation sequencing in consanguineous populations (55–60%) [31,32,33].
ROHs were significantly longer in the included Arab patients compared with a German control cohort, even in the subgroup of Arab patients without reported consanguinity. This confirms the basic assumption that there is generally a higher inbreeding rate in the Palestinian and Israeli Arab communities than in other populations even if consanguinity is not documented in the family, and demonstrates the potential of ROHs calculations based on NGS data as a marker for inbreeding. Furthermore, a correlation between longer ROHs and the likelihood of finding a homozygous disease-causing variant was shown.
In our exome-first approach we could establish a genetic diagnosis even with only basic clinical information and without extensive additional diagnostics like electrophysiology, laboratory screening, or brain imaging. For patients from a consanguineous background with limited access to medical diagnostics, first-line WES in combination with careful reverse clinical phenotyping might be the fastest and the most cost-efficient way to establish a genetic diagnosis.
Data availability
Human variants and phenotypes have been reported to ClinVar (submission name “TLP001,” accession numbers for all variants are found in Table S1; www.ncbi.nlm.nih.gov/clinvar).
Change history
28 May 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41431-021-00909-7
References
Zlotogora J, Shalev S, Habiballah H, Barjes S. Genetic disorders among Palestinian Arabs: 3. autosomal recessive disorders in a single village. Am J Med Genet. 2000;92:343–5.
Sharkia R, Zaid M, Athamna A, Cohen D, Azem A, Zalan A. The changing pattern of consanguinity in a selected region of the Israeli Arab community. Am J Hum Biol. 2008;20:72–7.
Tadmouri GO, Nair P, Obeid T, Al Ali MT, Al Khaja N, Hamamy HA. Consanguinity and reproductive health among Arabs. Reprod Health. 2009;6:17.
Zlotogora J. Genetic disorders among Palestinian Arabs: 1. Effects of consanguinity. Am J Med Genet. 1997;68:472–5.
Al-Gazali L, Hamamy H, Al-Arrayad S. Genetic disorders in the Arab world. BMJ. 2006;333:831–4.
Sturm, M. megSAP—a medical genetics sequence analysis pipeline. https://github.com/imgag/megSAP. Accessed 5 May 2019.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95.
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv. https://arxiv.org/abs/1207.3907. 2012.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The Ensembl variant effect predictor. Genome Biol. 2016;17:122.
Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human gene mutation database (HGMD): 2003 update. Hum Mutat. 2003;21:577–81.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. biorxiv. https://www.biorxiv.org/content/10.1101/531210v2. 2019.
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–21.
Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol. 2010;6:e1001025.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Sturm, M. ngs-bits—short-read sequencing tools. https://github.com/imgag/ngs-bits. Accessed 5 May 2019.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Mallaret M, Synofzik M, Lee J, Sagum CA, Mahajnah M, Sharkia R, et al. The tumour suppressor gene WWOX is mutated in autosomal recessive cerebellar ataxia with epilepsy and mental retardation. Brain. 2014;137:411–9.
Feichtinger RG, Mucha BE, Hengel H, Orfi Z, Makowski C, Dort J, et al. Biallelic variants in the transcription factor PAX7 are a new genetic cause of myopathy. Genet Med. 2019;21:2521–31.
Hengel H, Keimer R, Deigendesch W, Riess A, Marzouqa H, Zaidan J, et al. GPT2 mutations cause developmental encephalopathy with microcephaly and features of complicated hereditary spastic paraplegia. Clin Genet. 2018;94:356–61.
Al Zaabi N, Al Menhali N, Al-Jasmi F. SYNJ1 gene associated with neonatal onset of neurodegenerative disorder and intractable seizure. Mol Genet Genom Med. 2018;6:109–13.
Dyment DA, Smith AC, Humphreys P, Schwartzentruber J, Beaulieu CL, Consortium FC, et al. Homozygous nonsense mutation in SYNJ1 associated with intractable epilepsy and tau pathology. Neurobiol Aging. 2015;36:1222. e1221–1225.
Hardies K, Cai Y, Jardel C, Jansen AC, Cao M, May P, et al. Loss of SYNJ1 dual phosphatase activity leads to early onset refractory seizures and progressive neurological decline. Brain. 2016;139:2420–30.
Zambon AA, Natali Sora MG, Cantarella G, Cerri F, Quattrini A, Comi G, et al. Vocal cord paralysis in Charcot-Marie-Tooth type 4b1 disease associated with a novel mutation in the myotubularin-related protein 2 gene: a case report and review of the literature. Neuromuscul Disord. 2017;27:487–91.
Martinelli D, Diodato D, Ponzi E, Monne M, Boenzi S, Bertini E, et al. The hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Orphanet J Rare Dis. 2015;10:29.
Salvi S, Santorelli FM, Bertini E, Boldrini R, Meli C, Donati A, et al. Clinical and molecular findings in hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Neurology. 2001;57:911–4.
Hengel H, Magee A, Mahanjah M, Vallat JM, Ouvrier R, Abu-Rashid M, et al. CNTNAP1 mutations cause CNS hypomyelination and neuropathy with or without arthrogryposis. Neurol Genet. 2017;3:e144.
Minnerop M, Kurzwelly D, Wagner H, Soehn AS, Reichbauer J, Tao F, et al. Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia. Brain. 2017;140:1561–78.
Tatour Y, Sanchez-Navarro I, Chervinsky E, Hakonarson H, Gawi H, Tahsin-Swafiri S, et al. Mutations in SCAPER cause autosomal recessive retinitis pigmentosa with intellectual disability. J Med Genet. 2017;54:698–704.
Stockler-Ipsiroglu S, van Karnebeek C, Longo N, Korenke GC, Mercimek-Mahmutoglu S, Marquart I, et al. Guanidinoacetate methyltransferase (GAMT) deficiency: outcomes in 48 individuals and recommendations for diagnosis, treatment and monitoring. Mol Genet Metab. 2014;111:16–25.
Jalkh N, Corbani S, Haidar Z, Hamdan N, Farah E, Abou Ghoch J, et al. The added value of WES reanalysis in the field of genetic diagnosis: lessons learned from 200 exomes in the Lebanese population. BMC Med Genom. 2019;12:11.
Yavarna T, Al-Dewik N, Al-Mureikhi M, Ali R, Al-Mesaifri F, Mahmoud L, et al. High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum Genet. 2015;134:967–80.
Reuter MS, Tawamie H, Buchert R, Hosny Gebril O, Froukh T, Thiel C, et al. Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiatry. 2017;74:293–9.
Sharkia R, Wierenga KJ, Kessel A, Azem A, Bertini E, Carrozzo R, et al. Clinical, radiological, and genetic characteristics of 16 patients with ACO2 gene defects: Delineation of an emerging neurometabolic syndrome. J Inherit Metab Dis. 2019;42:264–75.
Spiegel R, Pines O, Ta-Shma A, Burak E, Shaag A, Halvardson J, et al. Infantile cerebellar-retinal degeneration associated with a mutation in mitochondrial aconitase, ACO2. Am J Hum Genet. 2012;90:518–23.
Alazami AM, Hijazi H, Al-Dosari MS, Shaheen R, Hashem A, Aldahmesh MA, et al. Mutation in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes intellectual disability and strabismus. J Med Genet. 2013;50:425–30.
Accogli A, Iacomino M, Pinto F, Orsini A, Vari MS, Selmi R, et al. Novel AMPD2 mutation in pontocerebellar hypoplasia, dysmorphisms, and teeth abnormalities. Neurol Genet. 2017;3:e179.
Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet. 2001;29:189–93.
Gan-Or Z, Bouslam N, Birouk N, Lissouba A, Chambers DB, Veriepe J, et al. Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia. Am J Hum Genet. 2016;98:1038–46.
Ulzi G, Lecchi M, Sansone V, Redaelli E, Corti E, Saccomanno D, et al. Myotonia congenita: novel mutations in CLCN1 gene and functional characterizations in Italian patients. J Neurol Sci. 2012;318:65–71.
Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim YI, Zenvirt S, et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE. 2012;7:e36458.
Item CB, Mercimek-Mahmutoglu S, Battini R, Edlinger-Horvat C, Stromberger C, Bodamer O, et al. Characterization of seven novel mutations in seven patients with GAMT deficiency. Hum Mutat. 2004;23:524.
Beryozkin A, Zelinger L, Bandah-Rozenfeld D, Shevach E, Harel A, Storm T, et al. Identification of mutations causing inherited retinal degenerations in the Israeli and palestinian populations using homozygosity mapping. Investig Ophthalmol Vis Sci. 2014;55:1149–60.
Bassuk AG, Wallace RH, Buhr A, Buller AR, Afawi Z, Shimojo M, et al. A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am J Hum Genet. 2008;83:572–81.
Nisipeanu P, Inzelberg R, Abo Mouch S, Carasso RL, Blumen SC, Zhang J, et al. Parkin gene causing benign autosomal recessive juvenile parkinsonism. Neurology. 2001;56:1573–5.
McNally EM, Duggan D, Gorospe JR, Bonnemann CG, Fanin M, Pegoraro E, et al. Mutations that disrupt the carboxyl-terminus of gamma-sarcoglycan cause muscular dystrophy. Hum Mol Genet. 1996;5:1841–7.
Sharkia R, Mahajnah M, Zalan A, Sourlis C, Bauer P, Schols L. Sanfilippo type A: new clinical manifestations and neuro-imaging findings in patients from the same family in Israel: a case report. J Med Case Rep. 2014;8:78.
Weber B, Guo XH, Wraith JE, Cooper A, Kleijer WJ, Bunge S, et al. Novel mutations in Sanfilippo A syndrome: implications for enzyme function. Hum Mol Genet. 1997;6:1573–9.
Stevanin G, Azzedine H, Denora P, Boukhris A, Tazir M, Lossos A, et al. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain. 2008;131:772–84.
Schonewolf-Greulich B, Tejada MI, Stephens K, Hadzsiev K, Gauthier J, Brondum-Nielsen K, et al. The MECP2 variant c.925C>T (p.Arg309Trp) causes intellectual disability in both males and females without classic features of Rett syndrome. Clin Genet. 2016;89:733–8.
Acknowledgements
We are grateful to the patients and family members who participated in this study. HH and LS are members of the European Reference Network for Rare Neurological Diseases - Project ID No 739510.
Funding
This work was supported by grants from the German Research Foundation (Reference number SCHO 754/5-1 and -2). HH was supported by the intramural fortüne program (#2554-0-0). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the paper. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
HH, RB, MS, TBH, YS, MM, RS, AA, GB, SA, RK, WD, JZ, and HM report no disclosures. PB received speaker honoraria from Actelion and is a paid consultant for Centogene AG. LS received grants from EU FP7 and the BMBF during conduct of this study outside the submitted work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hengel, H., Buchert, R., Sturm, M. et al. First-line exome sequencing in Palestinian and Israeli Arabs with neurological disorders is efficient and facilitates disease gene discovery. Eur J Hum Genet 28, 1034–1043 (2020). https://doi.org/10.1038/s41431-020-0609-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-0609-9
This article is cited by
-
Identification of founder and novel mutations that cause congenital insensitivity to pain (CIP) in palestinian patients
BMC Medical Genomics (2023)
-
Exome sequencing for structurally normal fetuses—yields and ethical issues
European Journal of Human Genetics (2023)
-
Structural basis for sequence-independent substrate selection by eukaryotic wobble base tRNA deaminase ADAT2/3
Nature Communications (2022)
-
Analysis of the Clinical Features and Imaging Findings of Pontocerebellar Hypoplasia Type 2D Caused by Mutations in SEPSECS Gene
The Cerebellum (2022)
-
Increasing involvement of CAPN1 variants in spastic ataxias and phenotype-genotype correlations
neurogenetics (2021)
