
Abstract
Angelman syndrome (AS) is a rare neurogenetic imprinting disorder caused by the loss of function of UBE3A. In ~3–5% of AS patients, the disease is due to an imprinting defect (ID). These patients lack DNA methylation of the maternal SNRPN promotor so that a large SNRPN sense/UBE3A antisense transcript (SNHG14) is expressed, which silences UBE3A. In very rare cases, the ID is caused by a deletion of the AS imprinting centre (AS-IC). To search for sequence alterations, we sequenced this region in 168 patients without an AS-IC deletion, but did not detect any sequence alteration. However, the AS-IC harbours six common variants (five single nucleotide variants and one TATG insertion/deletion variant), which constitute five common haplotypes. To determine if any of these haplotypes is associated with an increased risk for an ID, we investigated 119 informative AS-ID trios with the transmission disequilibrium test, which is a family-based association test that measures the over-transmission of an allele or haplotype from heterozygous parents to affected offspring. By this we observed maternal over-transmission of haplotype H-AS3 (p = 0.0073). Interestingly, H-AS3 is the only haplotype that includes the TATG deletion allele. We conclude that this haplotype and possibly the TATG deletion, which removes a SOX2 binding site, increases the risk for a maternal ID and AS. Our data strengthen the notion that the AS-IC is important for establishing and/or maintaining DNA methylation at the SNRPN promotor and show that common genetic variation can affect genomic imprinting.
Similar content being viewed by others

Introduction
Angelman syndrome (AS, #105830) is a rare neurogenetic imprinting disorder characterised by severe intellectual disability with absence of speech, microcephaly, ataxia, seizures and a happy demeanour. It is caused by genetic and epigenetic alterations on chromosome 15q11q13 leading to loss of expression of the imprinted gene UBE3A, which in neurons is expressed from the maternal allele only [1] (Fig. 1a). In about 3–5% of AS patients, the disease is due to an imprinting defect (ID). These patients have biparentally inherited chromosomes 15, but the maternal allele carries a paternal epigenotype and is unmethylated. Lack of DNA methylation at the SNRPN promotor (SNURF:TSS-DMR [2]) leads to the expression of a large SNRPN sense/UBE3A antisense transcript (SNHG14) on the maternal allele, which silences UBE3A [1]. In very rare cases the ID is caused by a deletion of the AS imprinting centre (IC; previously termed AS-SRO for smallest region of deletion overlap [3, 4]) and associated with a recurrence risk of 50% if the mother is a non-mosaic carrier. In contrast, in families with non-IC-deletion AS-ID patients the recurrence risk does not seem to be elevated. The AS-IC has a size of about 880 bp and is located ~35 kb upstream of the SNRPN promotor. The AS-IC is responsible for the establishment of the maternal imprint in the female germ line [3]. Recently, oocyte-specific transcription start sites at SNRPN upstream exons located within the AS-IC were identified, demonstrating that the AS-IC serves as an oocyte-specific promotor [5]. The transcripts originating here or the process of transcription through the locus is believed to be necessary for the establishment of the maternal methylation imprint [5,6,7]. Similar observations have been made at four other imprinted loci: GNAS/Gnas [8], KCNQ1/Kcnq1 [9,10,11,12], Peg3 [13] and Zrsr1 [14]. Therefore, sequence alterations in the promoter or upstream exons located within the AS-IC might affect transcription in this region and thereby the establishment of the methylation imprint. To investigate this possibility, we performed Sanger sequencing of this region in a total of 168 AS-ID patients. Furthermore, following up on our previous study [15], we investigated the transmission of the five common haplotypes in a new, larger cohort of AS-ID trios and in a combined cohort, consisting of 48 AS-ID trios from the Zogel study and 72 new AS-ID trios. So far, common genetic variation had only been implicated in another imprinting disorder, the Beckwith–Wiedemann syndrome [16].

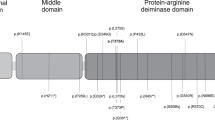
a The figure gives an overview of the genes within the imprinted cluster on chromosome 15q11q13. Genes depicted in blue are paternally expressed, the UBE3A gene is depicted in red for maternal only expression (situation in neurons). The biallelically expressed genes are shown in black. The bipartite structure of the IC is shown with the light grey ovals indicating the AS- and the PWS-IC. The AS-IC region is shown expanded below including the oocyte-specific transcription start sites at the upstream exons U5, U6 and U6.5 (rounded rectangles) and the six common variants (green circles for single nucleotide substitutions, a green triangle for the 4 bp InDel). b The figure shows part of the SNRPN gene with some of its upstream exons (rounded rectangles). Upstream exons U5 and U6 are located within the AS-IC (grey region) and U6.5 is located directly 3′. All three upstream exons have been shown to be oocyte-specific transcription start sites indicated by their red colour [5]. The transcripts originating at these exons all splice onto exon 2 of the SNRPN gene (blue rectangle), indicated above. The 4 bp InDel variant (green triangle), that is only present in haplotype H-AS3, affects a SOX2 binding site (orange triangle; [18]). This variant might affect SOX2 binding and - possibly in conjunction with other factors - could have an effect that hinders establishment of the methylation imprint at the AS-IC. Not drawn to scale. cen centromeric, tel telomeric, IC imprinting centre, mat maternally methylated.
Material and methods
Material
All patients have a molecular diagnosis of AS due to an ID. Older cases had been identified using methylation-specific (MS)-PCR [15], newer cases by MS-MLPA (methylation-specific multiplex ligation-dependent probe amplification, kit ME028, different versions, MRC Holland, Amsterdam, Netherlands). IC-deletions were excluded either by MS-MLPA or in older cases by quantitative real-time PCR. Uniparental disomy of chromosomes 15 was excluded in all cases by microsatellite analyses.
To the best of our knowledge, none of the AS-ID patients in this study had affected siblings.
In our former study, 48 AS-ID trios were described [15]. Informed consent was obtained for the so far unpublished participants. The study was approved by the ethics committee of the University Duisburg-Essen (AZ 08–3858).
The data has been submitted to the LOVD database (https://urldefense.proofpoint.com/v2/url?u=https-3A__databases.lovd.nl_shared_references_DOI-3A10.1038_s41431-2D020-2D0595-2Dy&d=DwIDaQ&c=vh6FgFnduejNhPPD0fl_yRaSfZy8CWbWnIf4XJhSqx8&r=FZZECvVW6LToxnfMxEanqbIvvLKGJ1qJ06OvmG6rJQw&m=wa5MA9FPSo0fZqkeit2NK63qqY5WxBmglJ02Z6IuDPw&s=TkrBfjHMyvmyTHm6y9e-WMEQsb5fIIdQScoJEFEZFxY&e=).
DNA was extracted from peripheral blood using the Flexigene Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol or was obtained as extracted DNA.
The AS-IC region was amplified and sequenced using standard methods. PCR products were sequenced on an ABI3130XL capillary sequencer (Applied Biosystems). Data analysis was performed using the Sequencing Analysis (Applied Biosystems) and the Geneious software (Biomatters, Auckland, New Zealand). For details see Supplementary.
Statistical analyses
Statistical analyses were performed as described before, i.e. the biallelic TDT [15] (see Supplementary). A goodness-of-fit-test for Hardy–Weinberg equilibrium showed no deviation (chi square ≤ 3.841).
Results and discussion
First, we investigated whether ID in AS-ID patients without IC-deletions could be due to sequence alterations within the AS-IC and especially within the newly described oocyte-specific transcription start sites at the SNRPN upstream exons U5, U6 and U6.5 [5]. For this, we performed Sanger sequencing of this region in 168 AS-ID patients but did not detect any sequence alterations beside the known common variants. This suggests that nucleotide substitutions in this region are either extremely rare, not causative for AS-ID or are not tolerated.
The GnomAD database (https://gnomad.broadinstitute.org/; accessed August 2019) shows only variants with very low frequencies (MAF between 0.017 and 0.000031; average 0.0009046) within the AS-IC region with the exception of six common variants (five single nucleotide variants and one TATG insertion/deletion (InDel) variant), which constitute five common haplotypes [15, 17]. None of these common variants is located within a SNRPN upstream exon. We analysed if any of these haplotypes might be associated with an increased risk for an ID using the transmission disequilibrium test (TDT). The TDT is a family-based association test that measures the over-transmission of an allele or haplotype from heterozygous parents to affected offspring. Of note, test results are not affected by population structure. This is important, because the patients were recruited from many different countries. In 48 parent-child trios tested previously, we had detected a trend for maternal over-transmission of the haplotype H-AS3 (p = 0.058; Table 1a [15]). We have now analysed an additional cohort of 72 AS-ID trios and found the same trend for maternal over-transmission of the haplotype H-AS3 (p = 0.058; Table 1b) supporting the trend seen previously in the smaller cohort. By combining the data from both studies, we could extend the cohort to 119 AS-ID trios. Again, we observed maternal over-transmission of haplotype H-AS3 (p = 0.0073; Table 1c; for haplotype data see Supplementary Table 1). No other haplotype showed over-transmission from either parent.
Interestingly, haplotype H-AS3 is the only haplotype that includes the TATG deletion allele. It has been shown that this deletion affects a SOX2 binding site [18]. SOX2 is a widely expressed transcription factor, which plays a crucial role in development [19, 20]. It is possible that the 4 bp deletion affects SOX2 binding and reduces transcription initiation at the AS-IC in the oocyte or early embryo. This may prevent imprint establishment or maintenance in some cells. (Fig. 1b). Since AS-IDs are extremely rare and we failed to see an effect of the TATG deletion in reporter gene assays in HEK293 cells (data not shown), additional cis- and trans-acting factors appear to be necessary for maternal imprinting in 15q11q13.
We conclude that DNA sequence alterations within the AS-IC do not seem to be a cause of AS-ID. This may explain why we have not seen AS-ID siblings in families without a maternal AS-IC deletion. However, the haplotype H-AS3 and possibly the TATG deletion, which removes a SOX2 binding site, seems to increase the risk for a maternal ID and AS.
In view of the fact that the population frequency of the H-AS3 haplotype is ~0.1 (see Supplementary) and the birth prevalence of a child with AS-ID is <1:1,000,000, the risk associated with this haplotype is too weak to warrant prenatal diagnosis.
It is also possible that the H-AS3 haplotype affects oocyte fitness. Due to sparse data we cannot investigate this at present. In the GnomAD database 66 women homozygous for the TATG deletion are listed but without further information. However, in our cohort, three mothers are homozygous for H-AS3 and all three have at least one unaffected child so that an effect, if present, cannot be severe.
In summary, this finding demonstrates that common genetic variation can affect genomic imprints and strengthens the notion that the AS-IC is important for establishing and/or maintaining DNA methylation at the SNRPN promotor.
References
Buiting K, Williams C, Horsthemke B. Angelman syndrome–insights into a rare neurogenetic disorder. Nat Rev Neurol. 2016;12:584–93.
Monk D, Morales J, den Dunnen JT, Russo S, Court F, Prawitt D, et al. Recommendations for a nomenclature system for reporting methylation aberrations in imprinted domains. Epigenetics. 2018;13:117–21.
Buiting K, Saitoh S, Gross S, Dittrich B, Schwartz S, Nicholls RD, et al. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet. 1995;9:395–400.
Buiting K, Lich C, Cottrell S, Barnicoat A, Horsthemke B. A 5-kb imprinting center deletion in a family with Angelman syndrome reduces the shortest region of deletion overlap to 880 bp. Hum Genet. 1999;105:665–6.
Lewis MW, Brant JO, Kramer JM, Moss JI, Yang TP, Hansen PJ, et al. Angelman syndrome imprinting center encodes a transcriptional promoter. Proc Natl Acad Sci USA. 2015;112:6871–5.
Lewis MW, Vargas-Franco D, Morse DA, Resnick JL. A mouse model of Angelman syndrome imprinting defects. Hum Mol Genet. 2019;28:220–9.
Smith EY, Futtner CR, Chamberlain SJ, Johnstone KA, Resnick JL. Transcription is required to establish maternal imprinting at the Prader-Willi syndrome and Angelman syndrome locus. PLoS Genet. 2011;7:e1002422.
Chotalia M, Smallwood SA, Ruf N, Dawson C, Lucifero D, Frontera M, et al. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 2009;23:105–17.
Beygo J, Joksic I, Strom TM, Ludecke HJ, Kolarova J, Siebert R, et al. A maternal deletion upstream of the imprint control region 2 in 11p15 causes loss of methylation and familial Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2016;24:1280–6.
Beygo J, Burger J, Strom TM, Kaya S, Buiting K. Disruption of KCNQ1 prevents methylation of the ICR2 and supports the hypothesis that its transcription is necessary for imprint establishment. Eur J Hum Genet. 2019;27:903–8.
Singh VB, Sribenja S, Wilson KE, Attwood KM, Hillman JC, Pathak S, et al. Blocked transcription through KvDMR1 results in absence of methylation and gene silencing resembling Beckwith-Wiedemann syndrome. Development. 2017;144:1820–30.
Valente FM, Sparago A, Freschi A, Hill-Harfe K, Maas SM, Frints SGM, et al. Transcription alterations of KCNQ1 associated with imprinted methylation defects in the Beckwith-Wiedemann locus. Genet Med. 2019;21:1808–20.
Bretz CL, Kim J. Transcription-driven DNA methylation setting on the mouse Peg3 locus. Epigenetics. 2017;12:945–52.
Joh K, Matsuhisa F, Kitajima S, Nishioka K, Higashimoto K, Yatsuki H, et al. Growing oocyte-specific transcription-dependent de novo DNA methylation at the imprinted Zrsr1-DMR. Epigenetics Chromatin. 2018;11:28.
Zogel C, Bohringer S, Gross S, Varon R, Buiting K, Horsthemke B. Identification of cis- and trans-acting factors possibly modifying the risk of epimutations on chromosome 15. Eur J Hum Genet. 2006;14:752–8.
Murrell A, Heeson S, Cooper WN, Douglas E, Apostolidou S, Moore GE, et al. An association between variants in the IGF2 gene and Beckwith-Wiedemann syndrome: interaction between genotype and epigenotype. Hum Mol Genet. 2004;13:247–55.
Buiting K, Gross S, Lich C, Gillessen-Kaesbach G, el-Maarri O, Horsthemke B. Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet. 2003;72:571–7.
Kaufman Y, Heled M, Perk J, Razin A, Shemer R. Protein-binding elements establish in the oocyte the primary imprint of the Prader-Willi/Angelman syndromes domain. Proc Natl Acad Sci USA. 2009;106:10242–7.
Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12:15–30.
Tang WW, Kobayashi T, Irie N, Dietmann S, Surani MA. Specification and epigenetic programming of the human germ line. Nat Rev Genet. 2016;17:585–600.
Acknowledgements
We thank the patients, their families and their doctors for their participation in the study and Stephanie Zimmer for expert technical assistance. This work was funded by the Bundesministerium für Bildung und Forschung (BMBF; Imprinting diseases, grant no. 01GM1513A). Open Access Funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beygo, J., Grosser, C., Kaya, S. et al. Common genetic variation in the Angelman syndrome imprinting centre affects the imprinting of chromosome 15. Eur J Hum Genet 28, 835–839 (2020). https://doi.org/10.1038/s41431-020-0595-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-0595-y
This article is cited by
-
Social attention and social-emotional modulation of attention in Angelman syndrome: an eye-tracking study
Scientific Reports (2023)
-
Identification of differentially methylated regions in rare diseases from a single-patient perspective
Clinical Epigenetics (2022)
-
Long Noncoding RNA SNHG14 Promotes Ischemic Brain Injury via Regulating miR-199b/AQP4 Axis
Neurochemical Research (2021)
-
Imitation in Angelman syndrome: the role of social engagement
Scientific Reports (2020)
