
Abstract
The term multilocus imprinting disturbance (MLID) describes the aberrant methylation of multiple imprinted loci in the genome, and MLID occurs in patients suffering from imprinting disorder carrying methylation defects. First data indicate that functional variants in factors expressed from both the fetal as well as the maternal genome cause MLID. Molecular changes in such genes of the maternal genome are called maternal effect variants, they affect members of the subcortical maternal complex (SCMC) in the oocyte which plays an important role during early embryonic development. Whereas the contribution of variants in the SCMC genes NLRP2, NLRP5, NLRP7, and KHDC3L to the etiology of reproductive failure and aberrant imprinting is widely accepted, the involvement of PADI6 variants in the formation of MLID is in discussion. We now report on the identification of biallelic variants in a woman suffering from different miscarriages and giving birth to two children with MLID. Thereby the role of PADI6 in maintaining the proper imprinting status during early development is confirmed. Thus, PADI6 variants do not only cause (early) pregnancy losses, but maternal effect variants in this gene cause the same spectrum of pregnancy outcomes as variants in other SCMC encoding genes, including chromosomal aberrations and disturbed imprinting. The identification of maternal effect variants requires genetic and reproductive counseling as carriers of these variants are at high risks for reproductive failure.
Similar content being viewed by others

Introduction
Multilocus imprinting disturbance (MLID) is defined as the aberrant methylation of multiple imprinted loci in the genome. In some imprinting disorders like Beckwith–Wiedemann syndrome (BWS), Silver–Russell syndrome (SRS), and transient neonatal diabetes mellitus, MLID can be identified in up to 50% of carriers of methylation defects (epimutations) (for review: [1,2,3]). Whereas the vast majority of isolated epimutations (i.e., disturbed methylation at only one imprinted locus) occur sporadically, there is growing evidence that a significant ratio of MLID cases are familiar and caused by monogenetic variants (e.g., [2, 4]). These genomic variants comprise pathogenic alterations of genes expressed by the patients genome (e.g., ZFP57 [2]), as well as changes in maternal genes encoding members of the subcortical maternal complex (SCMC) in the oocyte [5]. The SCMC plays an important role during early embryonic development and consists of at least seven members (NLRP2, NLRP5, NLRP7, PADI6, KHDC3L, TLE6, and OOEP). These proteins are expressed exclusively from the maternal genome (so-called maternal effect genes) in the oocytes and the early embryo, and they are degraded when the embryonic genome becomes active (for review: [5]). As a result, functional variants in the SCMC genes are associated with female reproductive failure (e.g., [6, 7, 4, 8]). Whereas the contribution of maternal effect variants in the genes NLRP2, NLRP5, NLRP7, and KHDC3L to the etiology of hydatidiform moles, miscarriages, chromosomal aberrations and/or MLID in the progeny of their carries is meanwhile well established, the involvement of other SCMC factors is in discussion. With the increasing number of studies using whole exome sequencing (WES) approaches to identify monogenetic causes of reproductive failure, there is growing evidence that functional variants in the SCMC gene PADI6 also belong to the spectrum of maternal effect variants [9, 10, 4, 11,12,13].
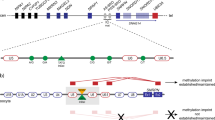
In mice, ablation of the maternal Padi6 allele causes disrupted localization of ribosomal components in the zygote, loss of stored mRNA, and arrest between the two-cell and four-cell stage [14, 15]. Accordingly, early embryonic arrest by the 4-cell stage appears to be associated with functionally relevant variants in the human PADI6 gene (Fig. 1). Thereby they potentially impair proper embryonic development [9, 10, 12, 13], and might result in infertility or early pregnancy loss. In addition, there is increasing evidence that PADI6 variants cause aberrant imprinting and MLID in children of maternal effect variant carriers [4].

With the identification of biallelic variants in PADI6 in a family with recurrent miscarriages, MLID, and triploidy, we now confirm that altered PADI6 is associated with aberrant imprinting marks, among them loss of methylation (LOM) of the imprinting center 2 (IC2, KCNQ1OT1:TSS-DMR) in 11p15.5 as a typical finding in BWS.
Case reports
A 38-year-old German woman was referred for genetic counseling after experiencing three failed pregnancies and the birth of two children with MLID (Fig. 2) from the same father. Her first child (II-1) was born at gestational week (gw) 33 + 3. The boy died immediately after birth; medical documentation was not available. The second pregnancy ended at gw 11, and a 69,XXY karyotype (II-2) was reported by an external lab, further data were not available. Karyotyping in a further miscarriage (II-4) showed a normal karyotype.

The maternal family (not shown) was empty in respect to histories of recurrent reproductive failure.
The first living daughter (II-3) was born by Caesarean section at gw 29 + 2 induced because of pathological cardiotocography and decreased fetal movements (APGAR 6/7/9). Birth parameters were in the normal range (weight 1240 g (0.00 z), length 39 cm (0.15 z), head circumference 28 cm (0.34 z)). An umbilical hernia was noted. Further clinical data were not available, at the age of 6 years the mother described her development as normal.
The second living child (II-5) was born small for gestational age (gw 34 + 6, weight: 1980 g (−1.11 SDS), length: 44 cm (−1.06 SDS), head circumference: 30.5 cm (−1.23 SDS)). The patient showed a prominent forehead. However, BWS was diagnosed as the patient exhibited placental mesenchymal dysplasia and macroglossia. These features are regarded as key features (two points each) of BWS according to the recently consented diagnostic BWS guidelines [16]. Three additional signs (one point each) suggestive for BWS were present comprising transient hypoglycemia, ear pits, and a small omphalocele which could be reponed. In summary, the patient showed the clinical picture of BWS with a clinical score of >4.
Molecular studies and results
Blood samples of the two girls were referred for molecular diagnostics of BWS and testing by methylation-specific multiplex ligation-dependent probe amplification was carried out (MS MLPA; assays ME030, ME032, ME034; MRC Holland, Amsterdam, The Netherlands), addressing 11 imprinted loci on 7 chromosomes (PLAGL1:alt-TSS-DMR (6q24), GRB10:alt-TSS-DMR (7p12), MEST:alt-TSS-DMR (7q32), H19/IGF2:IG-DMR (11p15), KCNQ1OT1:TSS-DMR (11p15), MEG3:TSS-DMR (14q32), SNURF:TSS-DMR (15q11), PEG3:TSS-DMR(19q13.43), GNAS-NESP:TSS-DMR (20q13), GNAS-AS1: TSS-DMR (20q13), and GNAS-XL: Ex1-DMR (20q13). MS MLPA revealed LOM at several imprinted loci on different chromosomes, thus indicating a MLID in both children (Fig. 3). In fact, the MS MLPA hybridization patterns indicated a mosaic occurrence of LOM at some loci. In both girls, LOM affected the KCNQ1OT1:TSS-DMR in 11p15.5, which is typically affected in BWS.

Whereas testing of the maternal DNA sample revealed normal hybridization patterns (I-2), (mosaic) LOM of the two 11p15.5 ICs could be detected in patients II-3 and of the IC2 in patient II-5 by the assay ME030-C2 (MRC Holland, Amsterdam, The Netherlands). Analyses of the multilocus MS MLPA ME034-A1 showed aberrant methylation at further loci in both patients. (The MS MLPA copy number runs are not shown as they gave normal results). The data were analyzed with the Coffalyser.Net software (MRC Holland, v.140721.1958).
In patient II-3, LOM, additionally, of the following differentially methylated regions (DMRs) was observed: PLAGL1:alt-TSS-DMR(6q24), GRB10:alt-TSS-DMR (7p12), MEST:alt-TSS-DMR (7q32), H19/IGF2:IG-DMR (11p15), GNAS-AS1:TSS-DMR, and GNAS-XL:Ex1-DMR. For GNAS-NESP:TSS-DMR (20q13) gain of methylation was observed.
MLID testing in patient II-5 revealed additional LOM at the loci GRB10:alt-TSS-DMR (7p12), MEG3:TSS-DMR (14q32), and SNURF: TSS-DMR (15q11).
Methylation analysis in the mother (I-2) was negative
For WES of the mother (I.2), the IDT xGen Exome Research Panel (v2.0) was used. The enriched libraries were sequenced on a NextSeq500 Sequencer with 2 × 75 cycles on a high-output flow cell. FastQ-files were generated using bcl2fastq2 (Illumina, San Diego, CA, USA). The automated SeqMule pipeline (v1.2.6) 7 was used for alignment and variant calling (GATKLite, 2.3-99. Average depth of coverage was 153x with 97.1% >20x for the target region. Annotation and bioinformatic prioritization of variants was performed using KGGSeq (v1.1, 07/Feb./2019). Variants with a minor allele frequency higher than 0.75% in public databases (i.e., gnomAD (http://gnomad.broadinstitute.org/), EXAC, 1000 GP, ESP) and synonymous variants were excluded. In total, all OMIM (https://www.ncbi.nlm.nih.gov/omim) annotated genes were analyzed for variants with putative functional relevance, with a specific focus on genes encoding members of the SCMC (NLRP2, NLRP5, NLRP7, PADI6, KHDC3L, TLE6, and OOEP).
In the mother (I-2), compound heterozygosity for two basepair substitutions in the coding sequence of PADI6 could be identified (NM_207421.3:c.[1114A>G];[2069G>])(Fig. 2). The variant NM_207421.3:c.1114A>G is a missense variant (p.(Thr372Ala)) and has already been listed in public databases with a low frequency (dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/) (151): rs374615037; ESP6500SIV2: Eur. Am.: G = 0.01%—Afr. Am.: G = 0.00%). It was classified as rather benign by bioinformatic prediction tools (CADD = 10.6; Alamut Visual v.2.15.0). The missense variant c.1114G>A is localized in the protein-arginine deiminase domain in which the majority of the currently known functional variants are localized (Fig. 1; for review: [13]).
The second variant NM_207421.3:c.2069G>A leads to a premature stop codon (p.(Trp690*)) and showed a higher CADD score (18.03) than the variant NM_207421.3:c.1114A>G (p.(Thr372Ala)). It has not yet been reported but it has been submitted to LOVD (https://databases.lovd.nl/shared) (Genomic variant #0000665756). It affects the protein-arginine deiminase domain as well and is localized close to other already published variants (Fig. 1).
By Sanger sequencing, the inheritance of the variant NM_207421.3:c.2069G>A (p.(Trp690*)) could be confirmed in the first daughter (patient II-3), whereas, the second daughter (patient II-5) was heterozygous for the variant NM_207421.3:c.1114A>G (p.(Thr372Ala)).
Discussion
In contrast to the widely accepted association between maternal effect variants in NLRP genes, reproductive failure, and disturbed imprinting/MLID, the pathologic effects of PADI6 variants are still in discussion. Several functional studies and case reports indicated that embryonic development arrest is linked to pathogenic PADI6 variants [9,10,11,12,13], but there is only one study suggesting their putative impact on disturbed imprinting and MLID: Begemann et al. [4] described PADI6 variants in four mothers of MLID carriers, but the pathogenicity of these variants was difficult to estimate as the variants either occurred in heterozygote state (probands 11, 12), or compound heterozygosity could not be proven (proband 9), or due the ambiguous bioinformatic classification (proband 10). The problem to finally confirm the pathogenicity of maternal effect variants is due to the difficulty to conduct functional assays for PADI6 variants in a suitable cell system and model organism, and to the incomplete reliability of bioinformatic prediction tools. In particular, genomic variant databases do not record information on fertility or the reproductive outcome, and maternal effect variants are often listed in variant databases with higher frequencies than expected for pathogenic alterations because they appear apathogenic in men and in women without reproductive history. Therefore, database searches are only of limited value to evaluate maternal effect variants. The latter becomes obvious for the variant NM_207421.3:c.1114A>G (p.(Thr372Ala)) in our family which is classified as rather benign by bioinformatics tools, and is reported in public databases with a very low frequency. However, due to its localization in the gene and the biallelic occurrence of a second potentially damaging PADI6 variant, as well as to the reproductive history and occurrence of MLID in two children, the pathogenic nature of the variant NM_207421.3:c.1114A>G (p.(Thr372Ala)) is highly probable.
The identification of biallelic variants in a woman suffering from different miscarriages and giving birth to two children with MLID now strengthens the suggestion that PADI6 variants do not only cause (early) pregnancy losses, but that maternal effect variants in this gene cause the same spectrum of pregnancy outcomes as variants in NLRP genes, including chromosomal aberrations and disturbed imprinting. The broad spectrum of reproductive failure and pregnancy outcome of women with maternal effect is not yet fully understood. In fact, PADI6 variants are rather associated with early embryonic arrest, but as reports on variants in other genes encoding other SCMC members show that the functional consequences can be broad and are influenced by the type of variant, its localization, and zygosity [6].
Thus, PADI6 represents the fourth SCMC member in which maternal effect variants have a severe impact on the proper genomic imprinting of the reproductive outcome. PADI6 does not only share the localization in the oocyte with NLRP2, NLPR5, and NLRP7 and mediate the proper cleavage in early embryogenesis by altering the spindle microtubule assembly [17], but PADI6 is probably also involved in methylation at imprinted loci. Pathogenic PADI6 maternal effect variants can therefore result in altered imprinting marks. However, it is currently unclear how members of the SCMC contribute to the proper methylation of imprinted loci. It has been suggested that NLRP7 and KHDC3L are rather involved in the establishment of maternal imprinting marks, whereas NLRP2 and NLRP5 might account for maintenance mechanisms (for review: [18]). The finding that the aberrant (mosaic) methylation in our family with PADI6 variants affects both paternally and maternally methylated DMRs indicates that PADI6 also has an impact on imprinting maintenance. Nevertheless, it currently remains unclear whether variants in SCMC factors indirectly affect the methylation at imprinted loci by disturbing the integrity of the SCMC [19] as a spatial prerequisite for the methylation machinery, or directly by impaired recruitment of factors which are essential for imprinting maintenance.
As the methylation results in the two children in our family show, the pattern of altered methylation are arbitrary, but it appears that the KCNQ1OT1:TSS-DMR in 11p15.5 is particularly prone as it is hypomethylated in both children exhibiting BWS features. This assumption corresponds to the findings from other MLID reports showing that BWS patients with IC2 LOM present the major group of MLID carriers: This locus is affected in a significant proportion of patients suffering from imprinting disorders, whereas a common aberrant methylation pattern at other imprinted loci is not obvious despite the same clinical features.
Corresponding to these common molecular observations, the reproductive outcomes associated with PADI6 variants and those of NLRP variant carriers are comparable. With the proof of MLID in our PADI6 family, the whole clinical spectrum associated with NLRP variants is also present in PADI6 associated families. Furthermore, it is conceivable that a correlation exists between the severity of the reproductive failure and its genetic basis, i.e., homozygosity/compound heterozygosity and heterozygosity for NLRP7 variants [6], or modifier genes with an impact on the phenotypic outcome [8]. In fact, the phenotype of children with MLID born to mothers with maternal effect variants is difficult to predict as the pattern of aberrant imprinting in the same family can vary considerably and cannot be foreseen [6]. Probably due to the mode of ascertainment, the majority of reported MLID carriers appear to exhibit symptoms specific for single imprinting disorders (e.g., BWS, SRS), but as the children from our family show this phenotype might be less specific or overlapping with features of different imprinting disorders (patient II-5: clinical signs of BWS, but SGA and protruding forehead as features of SRS [2]).
The family reported here confirms that functional variants in genes encoding SCMC members cause recurrent and severe complications in human reproduction. It is, therefore, necessary to identify women carrying maternal effect variants to avoid a long history of reproductive failures and diagnostic odysseys. Thus, physicians, gynecologists, and genetic counselors should be aware of the molecular link between infertility, miscarriages, aberrant chromosomal constitutions and imprinting disorders. A careful documentation of the reproductive and family history as well as molecular and (histo)pathological studies of the reproductive outcomes of these patients are required and might help to identify the molecular bases.
In case pathogenic maternal effect variants are identified, genetic and reproductive counseling should be offered as carriers of these variants are at high risks for further reproductive wastages. Furthermore, the clinical picture of the progeny of maternal effect carriers can hardly be predicted, due to the mosaic distribution and heterogeneity of imprinting disturbances (e.g., [6]). To circumvent these imponderability, oocyte donation should be discussed and has already been proven as a feasible option (for review: [20]).
References
Sanchez-Delgado M, Riccio A, Eggermann T, Maher ER, Lapunzina P, Mackay D, et al. Causes and consequences of multi-locus imprinting disturbances in humans. Trends Genet. 2016;32:444–55.
Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40:949–51.
Azzi S, Rossignol S, Steunou V, Sas T, Thibaud N, Danton F, et al. Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell Silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum Mol Genet. 2009;18:4724–33.
Begemann M, Rezwan FI, Beygo J, Docherty LE, Kolarova J, Schroeder C, et al. Maternal variants in NLRP and other maternal effect proteins are associated with multilocus imprinting disturbance in offspring. J Med Genet. 2018;55:497–504.
Monk D, Sanchez-Delgado M, Fisher R. NLRPs, the subcortical maternal complex and genomic imprinting. Reproduction. 2017;154:R161–R70.
Soellner L, Begemann M, Degenhardt F, Geipel A, Eggermann T, Mangold E. Maternal heterozygous NLRP7 variant results in recurrent reproductive failure and imprinting disturbances in the offspring. Eur J Hum Genet. 2017;25:924–9.
Docherty LE, Rezwan FI, Poole RL, Turner CL, Kivuva E, Maher ER, et al. Mutations in NLRP5 are associated with reproductive wastage and multilocus imprinting disorders in humans. Nat Commun. 2015;6:8086.
Sparago A, Verma A, Patricelli MG, Pignata L, Russo S, Calzari L, et al. The phenotypic variations of multi-locus imprinting disturbances associated with maternal-effect variants of NLRP5 range from overt imprinting disorder to apparently healthy phenotype. Clin Epigenetics. 2019;11:190.
Xu Y, Shi Y, Fu J, Yu M, Feng R, Sang Q, et al. Mutations in PADI6 cause female infertility characterized by early embryonic arrest. Am J Hum Genet. 2016;99:744–52.
Wang X, Song D, Mykytenko D, Kuang Y, Lv Q, Li B, et al. Novel mutations in genes encoding subcortical maternal complex proteins may cause human embryonic developmental arrest. Reprod Biomed Online. 2018;36:698–704.
Robbins SM, Thimm MA, Valle D, Jelin AC. Genetic diagnosis in first or second trimester pregnancy loss using exome sequencing: a systematic review of human essential genes. J Assist Reprod Genet. 2019;36:1539–48.
Zheng W, Chen L, Dai J, Dai C, Guo J, Lu C, et al. New biallelic mutations in PADI6 cause recurrent preimplantation embryonic arrest characterized by direct cleavage. J Assist Reprod Genet. 2020;37:205–12.
Qian J, Nguyen NMP, Rezaei M, Huang B, Tao Y, Zhang X, et al. Biallelic PADI6 variants linking infertility, miscarriages, and hydatidiform moles. Eur J Hum Genet. 2018;26:1007–13.
Yurttas P, Vitale AM, Fitzhenry RJ, Cohen-Gould L, Wu W, Gossen JA, et al. Role for PADI6 and the cytoplasmic lattices in ribosomal storage in oocytes and translational control in the early mouse embryo. Development. 2008;135:2627–36.
Liu X, Morency E, Li T, Qin H, Zhang X, Zhang X, et al. Role for PADI6 in securing the mRNA-MSY2 complex to the oocyte cytoplasmic lattices. Cell Cycle. 2017;16:360–6.
Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. 2018;14:229–49.
Kan R, Yurttas P, Kim B, Jin M, Wo L, Lee B, et al. Regulation of mouse oocyte microtubule and organelle dynamics by PADI6 and the cytoplasmic lattices. Dev Biol. 2011;350:311–22.
Monk D, Mackay DJG, Eggermann T, Maher ER, Riccio A. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat Rev Genet. 2019;20:235–48.
Demond H, Anvar Z, Jahromi BN, Sparago A, Verma A, Davari M, et al. A KHDC3L mutation resulting in recurrent hydatidiform mole causes genome-wide DNA methylation loss in oocytes and persistent imprinting defects post-fertilisation. Genome Med. 2019;11:84.
Elbracht M, Mackay D, Begemann M, Kagan KO, Eggermann T. Disturbed genomic imprinting and its relevance for human reproduction: causes and clinical consequences. Hum Reprod Update. 2020;26:197–213.
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, EG110/15–1; 948/32–1 FUGG). This work was supported by the Genomics Facility, a core facility of the Interdisciplinary Center for Clinical Research (IZKF) Aachen within the Faculty of Medicine at RWTH Aachen University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Eggermann, T., Kadgien, G., Begemann, M. et al. Biallelic PADI6 variants cause multilocus imprinting disturbances and miscarriages in the same family. Eur J Hum Genet 29, 575–580 (2021). https://doi.org/10.1038/s41431-020-00762-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-00762-0
This article is cited by
-
Molecular characterisation of 36 multilocus imprinting disturbance (MLID) patients: a comprehensive approach
Clinical Epigenetics (2023)
-
Loss of the Maternal Effect Gene Nlrp2 Alters the Transcriptome of Ovulated Mouse Oocytes and Impacts Expression of Histone Demethylase KDM1B
Reproductive Sciences (2023)
-
Trans-acting genetic variants causing multilocus imprinting disturbance (MLID): common mechanisms and consequences
Clinical Epigenetics (2022)
-
Germline variants in genes of the subcortical maternal complex and Multilocus Imprinting Disturbance are associated with miscarriage/infertility or Beckwith–Wiedemann progeny
Clinical Epigenetics (2022)
-
Novel biallelic mutations in PADI6 in patients with early embryonic arrest
Journal of Human Genetics (2022)
