
Abstract
Contour integration, a key visual function to deal with occlusion and discontinuity in natural scenes, is essential to human survival. However, individuals are not equally well equipped with this ability. In particular, contour integration deficiencies are commonly detected in patients with mental disorders, especially schizophrenia. To understand the underlying sources of these individual differences, the current study investigated the genetic basis of contour integration in humans. A total of 2619 normal participants were tested on their ability to detect continuous contours embedded in a cluttered background. Quantitative genomic analysis was performed, involving heritability estimation based on single nucleotide polymorphisms (SNPs) and association testing at SNP, gene, and pathway levels. Heritability estimation showed that common SNPs contributed 49.5% (standard error of the mean = 15.6%) of overall phenotypic variation, indicating moderate heritability of contour integration. Two-stage genome-wide association analysis (GWAS) detected four SNPs reaching genome-wide significance in the discovery test (N = 1931) but not passing the replication test (N = 688). Gene-level analysis further revealed a significant genome-wide association of a microRNA-encoding gene MIR1178 in both the discovery and replication cohorts. Another gene poly(A)-binding protein nuclear 1 like, cytoplasmic (PABPN1L) showed suggestive significance in the discovery cohort (p < 1 × 10−4) and was replicated in the replication cohort (p = 0.009). The pathway analysis did not detect any significant pathway. Taken together, this study identified significant gene associations with contour integration and provided support for a genetic transmission of the ability to perceive continuous contours in the environment.
Similar content being viewed by others

Introduction
The innate ability of contour integration is a key visual function for humans and animals. In a natural environment, due to occlusions, continuous contours and intact objects are often projected onto the retina as isolated elements. Failure to recognize them not only affects visual perception but also poses threats to individual survival [1]. For instance, delayed detection, due to poor contour integration ability, of a snake whose body is partly shielded by bushes might cost a person’s life. Indeed, people are not equally well equipped with the contour integration ability. A prominent finding is that some psychiatric disorders [2,3,4], especially schizophrenia, are accompanied with abnormal contour integration ability. So far, more than 50 studies have reported deficits of perceptual organization, including contour integration, in schizophrenia [5, 6]. Specifically, contour integration deficit has been found more frequently in schizophrenia patients than in healthy and psychiatric control groups [2, 7], and is more serious in patients with more severe disease [8]. Taken together, contour integration is an important visual function whose deficit affects normal life and is relevant to schizophrenia. However, the source of individual differences in this visual function and its molecular mechanisms are still unknown.
To address the questions above, the current study explored the genetic basis of contour integration ability in humans using a genome-wide association study (GWAS) technique in combination with psychophysics. The GWAS technique takes advantage of high-throughput gene chips to identify single-nucleotide polymorphisms (SNPs) and genes associated with a measurable phenotype. It has been used successfully to discover the genetic basis of both normal and abnormal physiological characteristics, as well as complex cognitive abilities including intelligence level [9,10,11,12], memory capacity [13,14,15,16,17,18], and social conformity tendency [19]. Psychophysics, a procedure that quantitatively investigates the relationship between physical stimuli and the perception they produce, gives a stable and precise measurement of human perception and has recently been applied to GWAS studies on human vision functions [20].
In the present study, we first performed linkage analysis on the common SNP heritability of contour integration using the GCTA program [21], and then conducted a GWAS on contour integration. The GWAS procedure consisted of a discovery stage and a replication stage, and was performed at multiple levels, including SNP, gene, and pathway levels. At the discovery stage, we performed a pilot GWAS on all tested SNPs in the discovery cohort (N = 1931) to discover SNPs showing a genome-wide association with contour integration. At the replication stage, we tested these discovered SNPs in another independent cohort (N = 688) to confirm their associations with the phenotype. Association analyses at gene and pathway levels were conducted based on SNP results and were performed with the same discovery and replication cohorts. This study provides information about the heritability of contour integration and identifies specific genes involved in this ability.
Methods
Participants
All participants were college students recruited from the Chongqing Medical University in South China. The discovery cohort consisted of 1931 participants, with a mean age of 19 years (standard deviation (SD) = 1), 80% female, 90% Han; the replication cohort consisted of 688 participants, with a mean age of 20 years (standard deviation (SD) = 1), 86% female, 92% Han. No significant differences in ethnicity or place of origin were found between the discovery and replication cohorts by Pearson’s Chi-square tests. All participants were naïve to the experimental stimuli and procedure. Their psychiatric and medication history were screened by medical examination at the time of college entrance and by self-reported questionnaires just before the current study. None of the participants reported having neurological diseases or vision disorders. Visual acuity for each participant was measured with the software program FrACT (Version 3.8.2), which is based on the Freiburg Vision Test [22]. A Landolt-C, as an opto-type, was presented at the center of the screen. The viewing distance was 1.4 m. The participants made an eight-alternative forced choice judgment on the orientation of the Landolt-Cs. The program estimated the acuity threshold using a method called best parameter estimation by sequential testing. All participants had normal or corrected-to-normal visual acuity and gave written informed consent before experiments. The experimental procedures were approved by the human subject review committee in the School of Psychological and Cognitive Sciences at Peking University. The behavioral data were collected between September 2014 and June 2015.
Behavioral assay
Stimuli
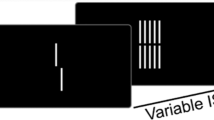
Contour integration stimuli consisted of an array of white line segments (luminance: 77 cd/m2) on a gray background (luminance: 31 cd/m2). To construct the stimuli, a circular area of 19.5° in radius was divided into 24 by 24 compartments, each containing a randomly oriented line segment (Fig. 1a) [23]. A straight contour with 4–8 adjacent collinear line segments was embedded in the array. The contour was 45° left or right tilted. The number of line segments of the contour (i.e., the length of the contour) determined the difficulty of the contour integration task (see below). Each line segment had the same length, which was 0.43° for the first two experimental runs and 0.325° for the last run.

Experimental stimuli and procedure. a Examples of two contour stimuli (within the circular area) used in the behavioral test. Visual contours were formed by collinear line segments embedded in a background of randomly oriented lines. Within the same background, perceptual saliency of contours varies with the number of collinear lines (e.g., four in the left and eight in the right figure). The stimuli were created based on Li and Gilbert (2002). b Participants judged the orientation (i.e. left or right) of the contour, which was tilted 45° to the left or right from the vertical direction
The stimuli were presented on an LCD monitor with a refresh rate of 60 Hz and a spatial resolution of 1920 × 1080. Luminance was measured with a MINOLTACS-100A Chroma Meter (Konica Minolta Sensing Americas, Inc., USA). The stimuli were generated and controlled using MATLAB and PsychToolbox [24].
Procedure
We adopted the classical psychophysical protocol [23, 25, 26]. In each trial, a contour integration stimulus was presented for 500 ms. Participants were asked to maintain fixation at the central dot on the screen and to judge the tilt direction of the contour (left or right) (Fig. 1b). No time limit was imposed. There were five difficulty levels corresponding to the five lengths of the contour (i.e., 4, 5, 6, 7, 8 line segments). Each difficulty level contained 120 trials, resulting in 600 trials in total. Trials with different difficulty levels were intermixed during test and all trials were split into three experimental runs of 200 trials. The mean accuracy of contour integration in the 600 trials was used as the behavioral phenotype of interest. To make sure that participants fully understood the task requirement, a practice session was carried out prior to the experiment. During practice, the same orientation discrimination procedure was adopted with the exception that the stimulus stayed on the computer screen until participants made a response. This manipulation decreased the task difficulty and ensured that all participants understood the task requirement. Participants kept practicing until they reached an accuracy over 75%. None of them failed.
Genotyping and quality control
Discovery cohort
DNA was extracted from peripheral blood of participants using the QuickGene whole-blood genome DNA extract system (Kurabo Industries Ltd., Japan), and was genotyped for 894,517 common SNPs using the HumanOmniZhongHua-8 Bead chip v1.2 (Illumina, Inc., San Diego, CA, USA). Common quality control parameters were applied to the genotype data which retained 830,937 SNPs. SNPs were included in the analysis if they met the following criteria: call rate ≥ 0.95, minor allele frequency (MAF) ≥ 0.01 and Hardy–Weinberg equilibrium test with p ≥ 10−4. Differences in allele frequencies between the discovery and replication cohorts were examined using paired sample t-test, and no significant deviation was observed. Individuals with genotyping call rate < 0.95 were excluded. Potential duplicates or close relatives were screened by calculating an identity-by-state (IBS) similarity matrix, and none were excluded based on the genome-wide IBS information (IBS distance for any pair of individuals > 0.9). Population stratification within this cohort was examined with EIGENSTRAT [27] via principal component analysis. Genetic outliers were detected and excluded automatically with the default mode (the maximum number of outlier removal iterations was five; an individual outside six SDs from the population mean of genetic principal components (PC) was removed as an outlier). A total of 1931 participants with both phenotypic and genotypic data available were included in the final analysis.
Replication cohort
The replication cohort used an independent cohort of participants who were genotyped on the HumanOmniZhongHua-8 Bead chip v1.1. The same quality control protocol as that at the discovery stage was applied, resulting in a final data set of 830,937 SNPs genotyped in a homogenous sample of 688 individuals. Array-based data has been deposited at the European Genome–phenome Archive (EGA), which is hosted by the EBI and the CRG, under accession number EGAS00001003639.
Statistical genetic analyses
Heritability estimation
Estimation of the phenotypic variance explained by all common SNPs (heritability) was carried out by GCTA version 1.24 [21]. This method measures the variance in the trait that is due to linkage disequilibrium (LD) between genotyped SNPs and unknown causal variants. The discovery and replication cohorts were combined for analysis. The genetic relationship matrix (GRM) was estimated on each pair of participants using all autosomal markers that were genotyped and checked for quality control as described above (altogether 830,937). One individual in each pair was excluded if the estimated genetic relatedness of that pair was larger than 0.025 [28]. Thirty-five individuals were excluded and 2584 individuals remained. The proportion of the total phenotypic variance explained by all common SNPs was estimated by the restricted maximum likelihood (REML) algorithm, with the first 20 eigenvectors from GCTA-PCA included as covariates. We used the online GCTA-GREML Power Calculator [29] to calculate the statistical power.
Imputation
Genotypes were pre-phased into haplotypes with SHAPEIT [30, 31]. Imputation was then performed using IMPUTE v2.3.1 [32] based on 1000 Genomes haplotype data (Phase I integrated variant set release (SHAPEIT2) in NCBI build 37/UCSC hg19) with 36,820,992 SNPs, 1,384,273 short bi-allelic indels and 14,017 structural variations. Quality control retained those variants with missing data proportion <1 × 10−6, INFO > 0.5, and MAF > 0.01 for subsequent association analyses.
GWAS
The mean accuracy of the contour orientation judgment was taken as the quantitative trait. Effects of demographic factors including gender and ethnicity were also tested using linear regression with phenotypes. None of those factors showed significant associations with the phenotype. Since subjects were all college freshmen and sophomore within a very narrow age range, age was not considered as a covariate in the analysis. Thus, no covariate was included in subsequent association tests. Based on the Score Method in the Frequentist Test framework, association tests of imputed SNPs were performed with SNPTEST v2.5 [33]. assuming an additive model. To generate Manhattan plots the full set of p-values that emerged from association analyses was loaded and visualized in Haploview v4.2 [34]. Basic statistical analyses were performed, the genomic inflation factor λ was calculated, and quantile–quantile (Q–Q) plots for observed vs. expected p-values were generated (Fig. 2), all by R v3.2.1 (https://www.R-project.org). The genome-wide significance threshold was set at 5 × 10−8. For variants that had passed quality control, regional association plots were drawn for two mega-bases around top candidate SNPs from the discovery GWAS by the web-based LocusZoom (http://locuszoom.sph.umich.edu/locuszoom) program. Quanto v1.2 was used for power calculation [35]. The power of this study to discover those genome-wide significant SNPs exceeded 0.85.

Quantile–quantile (Q–Q) plots of GWAS results at the discovery stage. Observed p-values relative to expected p-values are plotted based on p-values calculated using linear regression and with significant principal components included as covariates. The red line indicates the null hypothesis of no association
Candidate SNP selection and replication
Genome-wide significant SNPs at the discovery stage were selected as candidates for follow-up replication. Genotypes of the selected SNPs were obtained from the replication cohort and were tested by SNPTEST v2.5 under a general linear regression model. All genomic positions reported in the main text were based on the hg19 protocol unless otherwise specified.
Gene-based and pathway-based analyses
Gene-based association tests and pathway analyses were carried out by VEGAS2 [36]. 20,919 genes were tested. Mapping of SNPs onto genes was based on the NCBI37 (hg19) protocol. VEGAS2 used SNPs within genes or within LD (r2) > 0.8 of a SNP in genes, from Southern Han Chinese in the 1000 Genomes-ASIAN database. All autosomal chromosomes were taken into consideration, while no allele frequency difference between males and females was assumed during LD calculation. Pathway databases included GO, KEGG, REACTOME, BIOCARTA, and PANTHER (6211 pathways in total). The genome-wide significance threshold was set at family-wise error rate (FWER) of 0.05, which corresponds to p = 0.05/20,919 = 2.4 × 10−6 for gene-based analysis and 0.05/6211 = 8 × 10−6 for pathway analysis. The significance thresholds for replication were 0.05/n, where n = total number of candidate SNPs.
Results
Behavioral summary
The mean accuracy across the 2619 participants in the discovery and replication cohorts was 73.93% (SD = 9.6%). We estimated reliability of the measurement across the three experimental runs. We observed high correlations between each pair of runs (Spearman’s rank correlation coefficient r1,2 = 0.83, r2,3 = 0.84, and r1,3 = 0.78), confirming a high test–retest reliability of the behavioral result.
Heritability estimation
The GREML [21] method was used on 2584 individuals after quality control. This model estimated the narrow heritability of the contour integration ability through quantifying the proportion of the phenotypic variance explained by all genotyped autosomal SNPs (830,937 SNPs). We found that 49.5% (SEM = 15.6%, p = 8.0 × 10−4) of the phenotypic variance could be explained by the genotyped SNPs. This SNP-based analysis reveals a moderate heritability for contour integration, providing evidence for the heritability of basic visual functions in humans.
Genome-wide study of single-marker association
A total of 1931 participants were retained in the discovery cohort. Quantitative GWAS was performed on the imputed markers after quality control. The genomic inflation factor λ was 1.016. All association results across the whole genome are shown in the ‘Manhattan’ plots in Fig. 3. This analysis identified four SNPs meeting the standard of genome-wide significance (p < 5 × 10−8) (Table 1).

Manhattan plots of GWAS results at the discovery stage. GWAS results [−log10(p-value)] are shown in chromosomal order for individually genotyped SNPs that were tested for linear regression with contour integration performance. Results were based on an additive genetic model. Each dot represents one SNP. The red dotted line indicates the genome-wide significance level (5 × 10−8). Genome-wide significant SNPs are shown as enlarged red dots. Chromosomes are shown in different colors for clarity
These four SNPs were then selected and tested for associations in the replication cohort of 688 participants (Table 1). However, none of the four SNPs reached significance in the replication test. Consequently, no confirmed association was found between single SNPs and contour integration ability in the current cohort.
Gene-level and pathway-level association analyses
The gene-based analysis tests the joint association of phenotype with SNP markers belonging to the same gene. It reduces the number of tests and makes it possible to detect effects consisting of multiple weak associations. One gene, microRNA 1178 (MIR1178) located on chromosome 12, reached Bonferroni-corrected genome-wide significance (p = 2.4 × 10−6) at the discovery stage with a p-value of 2 × 10−6. This gene also reached genome-wide significance (p = 1 × 10−6) at the replication stage, suggesting an important role in contour integration (Table 2). Three other genes showing potential associations (2.4 × 10−6 < p < 1 × 10−4) at the discovery stage were also submitted to the replication test. One gene, poly(A)-binding protein nuclear 1 like, cytoplasmic (PABPN1L) showed significance in the replication cohort (p = 0.009 < 0.05/4).
We further carried out a pathway analysis to explore potential biological pathways involved in contour integration. None of the pathways reached significance after Bonferroni correction (threshold = 8 × 10−6). Table 3 lists all 16 pathways with p-values lower than 0.05 in both the discovery and replication cohorts.
Discussion
This study is the first systematic whole-genome analysis on contour integration in humans. By using a GREML method, we showed that common SNPs alone contribute 49.5% of overall phenotypic variations, suggesting at least moderate heritability of contour integration in humans. Gene-based analysis revealed genome-wide significant associations of the gene MIR1178 in both the discovery and replication cohorts. Another gene, PABPN1L, showed suggestive association with contour integration. To sum up, the current study begins to reveal the genetic basis of contour integration in humans.
Twins and linkage studies have previously demonstrated a heritable basis for multiple visual functions including binocular rivalry, facial expression recognition, and biological motion. In twin studies, stronger phenotype correlations have been found between monozygotic twins than between dizygotic twins for these functions. Genetic modeling further revealed moderate heritability of binocular rivalries [37, 38], facial expression detection [39], and biological motion [40]. Using genetic linkage studies, our recent work identified weak but significant heritability of perceptual rivalries [20]; Robinson et al. found that the degree of heritability of facial expression detection was around 35% [41], although it was not replicated by Coleman et al. [42]. Using linkage studies, in this analysis we found moderate heritability of visual contour integration. Together, these studies provide support for the idea that genetic factors contribute to basic visual functions. Future studies could focus on other more basic visual functions, such as detection and discrimination of visual contrast, orientation, motion direction, and so on, whose genetic bases are unexplored.
We did not use a detection threshold (e.g. the number of line segments at 75% detection accuracy) as a measure of contour integration ability. This is because some participants showed either a ceiling effect or a floor effect in the task, which renders the threshold estimation impossible. To verify that the detection accuracy method and the detection threshold method could quantify participants’ contour integration ability equally well, we calculated Pearson’s correlations between the accuracy and the threshold across the remaining participants. We observed very high correlation for each test run (run 1: −0.94; run 2: −0.92; run 3: −0.94) and for the three runs overall (−0.97). Furthermore, we also calculated the common SNP heritability in participants (N = 2442) whose threshold could be measured. The heritability based on the detection threshold was 0.440 (SEM = 16.7%), not very different from that based on the detection accuracy (0.495, SEM = 15.6%).
Our study is the first one to establish heritability of contour integration. The moderate heritability in the current study was estimated using the GREML method, which is based on an assumption different from the heritability analysis in twin studies [21]. Notably, the heritability reflects the genetic variance explained by the SNPs submitted to the analysis. That is, the SNP-based method only includes the common SNPs. Therefore, the actual heritability (which is contributed by both common and uncommon SNPs) of contour integration could be larger than the estimate in the current study. The finding that at least 49.5% of the phenotypic variance in contour integration can be accounted for by genetics alone indicates that it is worthwhile to further screen SNPs underlying contour integration ability.
GWAS on the imputed SNPs discovered four SNPs with genome-wide significance in the discovery cohort, yet none of them was replicated at the replication stage. Due to the small sample size, we only reported SNPs with MAF higher than 0.01. When we lowered the MAF criterion from 0.01 to 0.001, 414 SNPs were found to be genome-wide significant in the discovery cohort. For the replication test, three of these SNPs, namely rs142151205 (NC_000012.11: g.68270009G>A), rs73324772 (NC_000010.10: g.86568755T>C), and rs12257701 (NC_000010.10: g.86561743C>A), displayed significant association after multiple test corrections (p < 0.05/414 = 1.21 × 10−4); and one SNP, rs181147991 (NC_000004.11: g.187411841C>W) showed a false discovery rate below 0.05 (Table 4). SNP rs142151205 is located in gene LOC107984526 and alters a transcription binding motif, GKLF_01 (by 3DSNP: http://cbportal.org/3dsnp/). SNPs rs73324772 and rs12257701 are close to each other and the nearest gene to them is LOC105378400. SNP rs181147991 is located in the intron of a gene encoding for an anti-sense RNA for F11, and also 42 kb downstream from the gene MTNR1A which, encodes the melatonin receptor. Further replication with larger cohorts would be helpful to confirm these results.
The most significant finding of the current study is the identification of genes MIR1178 and PABPN1L in contour integration. A previous study has found that gene expression of MIR1178 contributes to pancreatic cancer cell proliferation, G1/S transition, migration, and invasion [43]. The microRNA encoded by MIR1178 is hsa-mir-1178, which is predicted to bind SLC39A6 and RGS9BP with the highest affinity (ranked in the top 10 by two miRNA target prediction programs, TargetScan and miRDB). SLC39A6 is a zinc transporter, while RGS9BP is specifically expressed in the retina and functions as a regulator of G protein-coupled receptor signaling in photo-transduction (as per GenBank). In such cases, MIR1178 might indirectly contribute to visual functions. Notably, MIR1178 has in a recent GWAS study also been reported to be associated with individuals’ ability to voluntarily modulate the switching rate of perceptual rivalry [20]. Although the specific role of MIR1178 in perceptual rivalry and contour integration is unclear, it may contribute to some general biological mechanisms in visual functions. PABPN1L encodes a protein that binds the poly(A) tail of mRNA. It is overexpressed in brain cerebellum (from GTEx) and has been discovered in a GWAS study on Parkinson’s disease [44]. So far, none of the previous studies have investigated the relationship between these two genes and visual cognition. Our study suggests future research potential for these two genes.
To sum up, the current study presents the first whole-genomic association analyses on contour integration. Through combining the GWAS technique with the psychophysical protocol, our study reveals moderate heritability of contour integration and identifies two genes associated with it. Additional GWAS studies involving larger samples are necessary to identify additional genetic factors in this visual function.
References
Field DJ, Hayes A, Hess RF. Contour integration by the human visual-system—evidence for a local association field. Vis Res. 1993;33:173–93.
Chandna A, Pennefather PM, Kovacs I, Norcia AM. Contour integration deficits in anisometropic amblyopia. Invest Ophth Vis Sci. 2001;42:875–8.
Phillips WA, Silverstein SM. Convergence of biological and psychological perspectives on cognitive coordination in schizophrenia. Behav Brain Sci. 2003;26:65–82.
Silverstein SM, Keane BP. Perceptual organization impairment in schizophrenia and associated brain mechanisms: review of research from 2005 to 2010. Schizophr Bull. 2011;37:690–9.
Feigenson KA, Gara MA, Roche MW, Silverstein SM. Is disorganization a feature of schizophrenia or a modifying influence: evidence of covariation of perceptual and cognitive organization in a non-patient sample. Psychiat Res. 2014;217:1–8.
Feigenson KA, Keane BP, Roche MW, Silverstein SM. Contour integration impairment in schizophrenia and first episode psychosis: state or trait? Schizophr Res. 2014;159:515–20.
Kovacs I. Human development of perceptual organization. Vis Res. 2000;40:1301–10.
Silverstein SM, Hatashita-Wong M, Schenkel LS, Wilkniss S, Kovacs I, Feher A, et al. Reduced top-down influences in contour detection in schizophrenia. Cogn Neuropsychiatry. 2006;11:112–32.
Davies G, Tenesa A, Payton A, Yang J, Harris SE, Liewald D, et al. Genome-wide association studies establish that human intelligence is highly heritable and polygenic. Mol Psychiatr. 2011;16:996–1005.
Piffer D. A review of intelligence GWAS hits: Their relationship to country IQ and the issue of spatial autocorrelation. Intelligence. 2015;53:43–50.
Sniekers S, Stringer S, Watanabe K, Jansen PR, Coleman JRI, Krapohl E, et al. Genome-wide association meta-analysis of 78,308 individuals identifies new loci and genes influencing human intelligence (vol 49, pg 1107, 2017). Nat Genet. 2017;49:1558.
Zabaneh D, Krapohl E, Gaspar HA, Curtis C, Lee SH, Patel H, et al. A genome-wide association study for extremely high intelligence. Mol Psychiatr. 2018;23:1226–32.
Debette S, Verbaas CAI, Bressler J, Schuur M, Smith A, Bis JC, et al. Genome-wide studies of verbal declarative memory in nondemented older people: the cohorts for heart and aging research in genomic epidemiology consortium. Biol Psychiat. 2015;77:749–63.
Milnik A, Heck A, Vogler C, Heinze HJ, de Quervain DJF, Papassotiropoulos A. Association of KIBRA with episodic and working memory: a meta-analysis. Am J Med Genet B. 2012;159b:958–69.
Papassotiropoulos A, Henke K, Stefanova E, Aerni A, Muller A, Demougin P, et al. A genome-wide survey of human short-term memory. Mol Psychiatr. 2011;16:184–92.
Papassotiropoulos A, Stefanova E, Vogler C, Gschwind L, Ackermann S, Spalek K, et al. A genome-wide survey and functional brain imaging study identify CTNNBL1 as a memory-related gene. Mol Psychiatr. 2013;18:255–63.
Vogler C, Gschwind L, Coynel D, Freytag V, Milnik A, Egli T, et al. Substantial SNP-based heritability estimates for working memory performance. Transl Psychiatry. 2014;4:e438.
Zhu Z, Chen B, Yan H, Fang W, Zhou Q, Zhou S, et al. Multi-level genomic analyses suggest new genetic variants involved in human memory. Eur J Hum Genet. 2018;26:1668–78.
Chen BQ, Zhu ZJ, Wang YY, Ding XH, Guo XB, He MG, et al. Nature vs. nurture in human sociality: multi-level genomic analyses of social conformity. J Hum Genet. 2018;63:605–19.
Chen BQ, Zhu ZJ, Na R, Fang W, Zhang WX, Zhou Q, et al. Genomic analyses of visual cognition: perceptual rivalry and top-down control. J Neurosci. 2018;38:9668–78.
Yang JA, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Bach M. The Freiburg visual acuity test—automatic measurement of visual acuity. Optom Vis Sci. 1996;73:49–53.
Li W, Piech V, Gilbert CD. Learning to link visual contours. Neuron. 2008;57:442–51.
Brainard DH. The psychophysics toolbox. Spat Vis. 1997;10:433–6.
Li W, Gilbert CD. Global contour saliency and local colinear interactions. J Neurophysiol. 2002;88:2846–56.
Li W, Piech V, Gilbert CD. Contour saliency in primary visual cortex. Neuron. 2006;50:951–62.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904.
Yang JA, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–U131.
Visscher PM, Hemani G, Vinkhuyzen AAE, Chen GB, Lee SH, Wray NR, et al. Statistical power to detect genetic (Co)variance of complex traits using SNP data in unrelated samples. PLoS Genet. 2014;10:e1004269.
Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2012;9:179–81.
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–9.
Howie B, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529.
Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–13.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5.
Gauderman WJ, Morrison JM. QUANTO 1.1: a computer program for power and sample size calculations for genetic-epidemiology studies. http://hydrauscedu/gxe. 2006.
Liu JZ, Mcrae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, et al. A versatile gene-based test for genome-wide association studies. Am J Hum Genet. 2010;87:139–45.
Miller SM, Hansell NK, Ngo TT, Liu GB, Pettigrew JD, Martin NG, et al. Genetic contribution to individual variation in binocular rivalry rate. Proc Natl Acad Sci USA. 2010;107:2664–8.
Shannon RW, Patrick CJ, Jiang Y, Bernat E, He S. Genes contribute to the switching dynamics of bistable perception. J Vis. 2011;11:1–7.
Lau JYF, Burt M, Leibenluft E, Pine DS, Rijsdijk F, Shiffrin N, et al. Individual differences in children’s facial expression recognition ability: the role of nature and nurture. Dev Neuropsychol. 2009;34:37–51.
Wang Y, Wang L, Xu Q, Liu D, Chen LH, Troje NF, et al. Heritable aspects of biological motion perception and its covariation with autistic traits. Proc Natl Acad Sci USA. 2018;115:1937–42.
Robinson EB, Kirby A, Ruparel K, Yang J, McGrath L, Anttila V, et al. The genetic architecture of pediatric cognitive abilities in the Philadelphia Neurodevelopmental Cohort. Mol Psychiatr. 2015;20:454–8.
Coleman JRI, Lester KJ, Keers R, Munafo MR, Breen G, Eley TC. Genome-wide association study of facial emotion recognition in children and association with polygenic risk for mental health disorders. Am J Med Genet B. 2017;174:701–11.
Cao Z, Xu J, Huang H, Shen P, You L, Zhou L, et al. MiR-1178 promotes the proliferation, G1/S transition, migration and invasion of pancreatic cancer cells by targeting CHIP. PLoS ONE. 2015;10:e0116934.
Beecham GW, Dickson DW, Scott WK, Martin ER, Schellenberg G, Nuytemans K, et al. PARK10 is a major locus for sporadic neuropathologically confirmed Parkinson disease. Neurology. 2015;84:972–80.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Projects 31421003 and 31671168). We are grateful to Zhangyan Guan and Huizhen Yang for help with DNA preparation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhu, Z., Chen, B., Na, R. et al. Heritability of human visual contour integration—an integrated genomic study. Eur J Hum Genet 27, 1867–1875 (2019). https://doi.org/10.1038/s41431-019-0478-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0478-2
This article is cited by
-
A genome-wide association study reveals a substantial genetic basis underlying the Ebbinghaus illusion
Journal of Human Genetics (2021)
