
Abstract
Periventricular nodular heterotopia (PNH) is a brain malformation in which nodules of neurons are ectopically retained along the lateral ventricles. Genetic causes include FLNA abnormalities (classical X-linked PNH), rare variants in ARFGEF2, DCHS1, ERMARD, FAT4, INTS8, MAP1B, MCPH1, and NEDD4L, as well as several chromosomal abnormalities. We performed array-CGH in 106 patients with different malformations of cortical development (MCD) and looked for common pathways possibly involved in PNH. Forty-two patients, including two parent/proband couples, exhibited PNH associated or not with other brain abnormalities, 44 had polymicrogyria and 20 had rarer MCDs. We found an enrichment of either large rearrangements or cryptic copy number variants (CNVs) in PNH (15/42, 35.7%) vs polymicrogyria (4/44, 9.1%) (i.e., 5.6 times increased risk for PNH of carrying a pathogenic CNV). CNVs in seven genomic regions (2p11.2q12.1, 4p15, 14q11.2q12, 16p13.3, 19q13.33, 20q13.33, 22q11) represented novel, potentially causative, associations with PNH. Through in silico analysis of genes included in imbalances whose breakpoints were clearly detailed, we detected in 9/12 unrelated patients in our series and in 15/24 previously published patients, a significant (P < 0.05) overrepresentation of genes involved in vesicle-mediated transport. Rare genomic imbalances, either small CNVs or large rearrangements, are cumulatively a frequent cause of PNH. Dysregulation of specific cellular mechanisms might play a key pathogenic role in PNH but it remains to be determined whether this is exerted through single genes or the cumulative dosage effect of more genes. Array-CGH should be considered as a first-line diagnostic test in PNH, especially if sporadic and non-classical.
Similar content being viewed by others

Introduction
Periventricular nodular heterotopia (PNH) is a malformation of cortical development (MCD) in which nodules of heterotopic gray matter fail to migrate into the developing cortex and accumulate along the walls of the lateral ventricles. PNH is usually detected using brain magnetic resonance imaging (MRI) in patients with epilepsy, intellectual disability, or both, and is highly heterogeneous in severity and distribution, occurring as either an isolated abnormality or part of more complex developmental brain disorders. In the so-called “classical form”, bilateral clusters of confluent nodules extend along the walls of the lateral ventricles [1]. Less typical forms include less prominent bilateral or unilateral nodules of different size, variably positioned along the lateral ventricles [2]. Genetic etiology of PNH is also heterogeneous, with single-nucleotide variants (SNVs) or deletions of the FLNA gene being the most frequent cause of the classical X-linked form (Mendelian Inheritance in Man (MIM): #300049) [1, 3]. Rare variants in other genes account for a few reports (Supplementary Table 1A). In particular, biallelic variants in ARFGEF2 [4], INTS8 [5], MCPH1 [6], FAT4, and DCHS1 [7] cause recessive forms of PNH, with or without syndromic features, while heterozygous variants in NEDD4L [8] and ERMARD (C6orf70) [9, 10] have been associated to syndromic dominant forms of PNH. Heterozygous SNVs in MAP1B have recently been reported in sporadic and familial epilepsy-associated PNH [11]. Overall, 24 distinct genomic rearrangements have been described in 66 patients with PNH, exhibiting either syndromic or non-syndromic phenotypes (Table 1b, Supplementary Table 1B).
We present clinical and genetic data on 15 patients with PNH carrying pathogenic genomic imbalances, including two parent/proband couples. Eight such patients had been included in previous publications from our team [9, 12,13,14], while seven (five sporadic and two familial) had not been described before (Table 1a, b). We report seven genomic regions (2p11.2q12.1, 4p15, 14q11.2q12, 16p13.3, 19q13.33, 20q13.33, 22q11) which represent novel, potentially causative, associations with PNH.
This series points to an increased prevalence of both large rearrangements and cryptic copy number variants (CNVs) in PNH (15/42, 35.7%) with respect to polymicrogyria (4/44, 9.1%). In spite of clinical and genetic heterogeneity of PNH, it appears that genes coding for vesicle trafficking proteins are overrepresented in CNVs associated with this malformation.
Subjects and methods
Patient ascertainment and genetic investigations
We obtained written informed consent for genetic investigations and publication of the results for all patients participating in the study. The Ethics Committee of the Tuscany Region, Italy, approved this study within the framework of the EU project “DESIRE” (FP7, grant agreement 602531).
We reviewed clinical and MRI data of 106 individuals with MCD who were studied with array-comparative genomic hybridization (array-CGH) and grouped them in three categories according to the MCD subtype (Supplementary Table 2).
Group 1, including 42 patients with PNH, isolated or associated with other malformations, including polymicrogyria, corpus callosum abnormalities, enlarged or dysmorphic lateral ventricles; group 2, including 44 patients with polymicrogyria (PMG) only; group 3, including 20 patients with other MCDs (specified in Supplementary Table 2).
In all patients in group 1, we ruled out SNVs and deletions/duplications of the FLNA gene using Sanger sequencing and Multiplex Ligation-dependent Probe Amplification, as previously described [1]. In all the 106 patients we performed array-CGH analysis using the 180K Agilent platform (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s instructions and reporting the genomic coordinates according to the NCBI37/hg19 assembly. To confirm the deletions/duplications identified by array-CGH, we used quantitative SYBR Green I-based real-time PCR on a LightCycler Real-Time PCR (Roche Diagnostics, Basel, Switzerland). Primers and conditions are available upon request.
We classified CNVs as pathogenic or benign considering their frequency in the general population (http://dgv.tcag.ca/dgv/app/home) and their inheritance status. In patients 2, 3, and 5 and their parents, we performed karyotype and fluorescence in situ hybridization on peripheral blood lymphocytes by standard procedures to confirm or better characterize the copy number changes highlighted by array-CGH and investigate parental carrier status for balanced chromosome translocations. We evaluated the genomic context of each CNV using the University of California, Santa Cruz (UCSC) Genome Browser (https://genome-euro.ucsc.edu/). Relevant data were submitted to DECIPHER (DatabasE of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources; https://decipher.sanger.ac.uk/index).
Statistical analysis
To test the hypothesis that patients with PNH might be more likely to harbor pathogenic genomic imbalances with respect to those with the other most frequent MCD, we calculated and compared the frequency of such imbalances between group 1 (PNH) and group 2 (PMG). We could not identify pathogenic imbalances in group 3. We calculated the odds ratio (OR) using the EpiInfo software v6 (http://www.cdc.gov/EpiInfo/) and considered a P value < 0.05 as statistically significant.
To explore whether pathogenic imbalances carried by patients with PNH were enriched for specific classes of genes, we used the statistical overrepresentation test tool available at the PANTHER (Protein ANnotation THrough Evolutionary Relationships) database website (http://www.pantherdb.org/) [15]. We performed the overrepresentation test in PNH-associated CNVs in our database (15 patients, Table 1a, b) and repeated the analysis for those CNVs whose genomic boundaries were previously published in detail by other groups (24 patients, Supplementary Table 1B).
Patient 7, carrying a complex rearrangement probably originating from a chromothripsis, was excluded from the analysis. From genes included in the imbalances, we selected those to be analyzed based on their expression profile inferred using the Mouse Genome Informatics (MGI) Gene Expression Data tool (http://www.informatics.jax.org/gxd). We performed the statistical overrepresentation test using the Homo sapiens database, the default settings suggested by PANTHER, and the Bonferroni correction for multiple testing. We evaluated gene overrepresentation in the “GO biological process complete” and the “GO cellular component complete” PANTHER datasets. To reduce spurious observations, we manually reviewed the overrepresented classes of genes, excluding from further analysis those unrelated to specific developmental/functional processes (e.g., cognition, rhythmic processes) and considered as potentially associated with PNH pathogenesis only those exhibiting the highest enrichment values.
Results
Clinical features
Clinical features of patients carrying pathogenic CNVs are summarized in Table 1a, b. More details on newly reported patients are available in the Supplementary Clinical Notes and in Supplementary Figure 1. Eight patients, including a mother and her son, both exhibiting bilateral PNH, had already been described in previous publications from our team [9, 12,13,14]. These individuals were included in this study only for the purposes of assessing the prevalence of CNVs in PNH versus other MCDs and for gene enrichment analysis.
Of the remaining seven patients, five were females (including a mother and daughter, #6b and #6a), with a mean age at the time of the study of 15 ± 1.3 years (range: 1–47 years).
Epilepsy was present in four patients, with seizure onset in the first month of life in two and at 8 and 18 years of age in the remaining two. Seizures were variable in type, including clonic, myoclonic, tonic, epileptic spasms and focal, and were drug resistant in three patients (#1, #5, #6b).
All seven patients had intellectual disability, ranging from mild (#4, #6b) to moderate (#1, #6a) and severe (#2, #3 and #5). In four patients PNH was part of a syndromic disorder also featuring dysmorphic facial features (#1–3, #5).
MRI features
Brain MRI in the seven unreported patients (Table 1a, b, Fig. 1) revealed classical bilateral PNH in one (#6b), and other subtypes of PNH in the remaining six, often associated with additional brain abnormalities including dysmorphic lateral ventricles (five patients), thin or hypoplastic corpus callosum (four patients), and white matter hyperintensities (two patients). Patient 5 exhibited a complex MCD featuring PNH and bilateral parieto-temporal polymicrogyria.

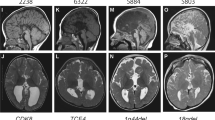
Brain magnetic resonance imaging (MRI) scan of the reported patients. Patient 1 (16p13.3p13.2 deletion): T2–weighted (W) axial (a) and T1W coronal (b) sections showing a single heterotopic nodule (arrows) on the right occipital horn, enlarged lateral ventricles, and dilated subarachnoid spaces. Patient 2 (19q13.33q13.43 duplication and 20q12.33 deletion): T1W axial (c) and T2W coronal (d) sections showing few, small, and non-contiguous nodules of gray matter (white arrows) lining the walls of the lateral ventricles, mostly on the left, simplified gyral pattern, enlarged dysmorphic lateral ventricles, and subarachnoid spaces. Patient 3 (7q33q34 duplication and 7q34qter mosaic duplication): T2W axial section (e) showing a nodule (white arrow) located along the body of left lateral ventricle, asymmetric lateral ventricles, and diffuse periventricular white matter hyperintensities; T1W sagittal section (f) revealing four non-contiguous nodules of heterotopic gray matter (white arrows) protruding into the lumen of left lateral ventricle, thin corpus callosum, white matter hyperintensities, and enlarged subarachnoid spaces, mainly in the frontal region. Patient 4 (2p11.2q12.1 mosaic amplification and 2q12.2 duplication): T2W axial (g) and T2W coronal (h) sections showing multiple, small, non-contiguous, asymmetrical nodules of gray matter (white arrows) lining the wall of the right lateral ventricle and the left frontal horn. Patient 5 (14q11.2q12 duplication and 15q11.1q14 deletion): T1W axial sections (i, j) demonstrating few scattered, bilateral, and asymmetric nodules of heterotopic gray matter (black arrows) along the walls of lateral ventricles, hypomyelination, and bilateral parietal-temporal polymicrogyria (black asterisk); Patient 6a (22q11.22q11.23 duplication): T2W axial (k) and T1W coronal (l) sections showing multiple, bilateral, asymmetrical, scattered nodules of heterotopic gray matter in the lateral ventricles and enlargement of the left lateral ventricle (arrows). Patient 6b (22q11.22q11.23 duplication): T1W coronal (m, n) sections showing bilateral, diffuse subependymal PNH along both lateral ventricles. Patient 7 (4p15 deletion in a complex rearrangement): T1W axial sections (o, p) showing multiple bilateral nodules of gray matter (black arrows) located along the walls of lateral ventricles, most evident prominent on lateral and superior walls of the right side. For this patient, MRI images were not previously published
Genetic features and CNV enrichment analysis
None of the patients with PNH harbored SNVs/CNVs involving the FLNA gene. We observed pathogenic CNVs, either de novo or inherited from an affected parent, in 15 patients from group 1 (PNH: 15/42, 35.7%), and in four patients from group 2 (PMG: 4/44: 9.1%) (Tables 1a, b, and 2 and Supplementary Table 2). We identified no pathogenic imbalances in group 3.
Comparing how frequently pathogenic imbalances occurred in patients with PNH (group 1) with respect to those with PMG only (group 2), we found a 5.6 times increased risk of carrying a causative chromosomal rearrangement in the PNH group (OR 5.6, 95% confidence interval: 1.66 to 18.55, P = 0.0053, Table 2).
All pathogenic genomic imbalances identified in patients with PNH were de novo, except in patients 6a and 13a, who had inherited pathogenic CNV from their affected mothers (#6b and #13b). In four patients (#2–5), the genomic lesion consisted in a cytogenetically visible large rearrangement, which in patient 5 had resulted from the unbalanced segregation of a balanced translocation carried by a healthy parent.
In patient 7, molecular cytogenetic investigations had demonstrated a de novo complex rearrangement with at least seven breakpoints, consisting of two non-consecutive 2p regions translocated at 4q24 and 4q28, and a 17.6 Mb interstitial 4p15 deletion [16]. This rearrangement could have resulted from chromotripsis [17], although we could not test this hypothesis because no further DNA was available. We excluded this patient from the enrichment analysis due to the complexity of the rearrangement, which was only partially elucidated by molecular cytogenetics. With the exception of patient 7, all previously reported patients included in this study (#8–13) carried simple deletions in four distinct genomic regions: 5q14.3q15, 6p25, 6q27, 17p11.2 [9, 12,13,14]. Among the seven previously unreported patients, only three carried a single cryptic CNV, namely the 16p13.3p13.2 deletion (#1) and the 22q11.22q11.23 duplication (#6a-b).
All pathogenic CNVs identified in group 2 (PMG, n = 4) were de novo, consisting of simple deletions/duplications (Supplementary Table 2).
Overrepresented classes of genes
All mouse orthologs of the known causative genes for PNH whose expression data are available in the MGI database (ARFGEF2, FAT4, FLNA, MCPH1, and NEDD4L) are expressed in the cerebral cortex (Supplementary Figure 2). In order to delineate shared dysfunctional cellular processes possibly leading to PNH in patients with CNVs in this series, we explored whether the genes mapping in the different CNVs belonged to specific subclasses.
Using the MGI Gene Expression Data tool, we found that out of 1094 known genes mapping in the different CNVs in group 1, 157 were expressed in the cerebral cortex (Supplementary Table 3). Among these 157 genes, the statistical overrepresentation test revealed 25 different classes with significant overrepresentation (P < 0.05) in both the “GO biological process complete” and the “GO cellular component complete” PANTHER datasets (Supplementary Tables 4 and 5). The two gene classes with the highest enrichment were those involved in vesicle-mediated transport regulation (4.31-fold increase in Gene Ontology (GO) biological process complete dataset, P = 1.8E–02, Supplementary Tables 4 and 6) and those encoding plasma membrane receptor complexes (7.79-fold increase in GO cellular component complete dataset, P = 3.2E–04, Supplementary Tables 5 and 6). Most of the genes in the latter class also belonged to ion channel complexes, the second most overrepresented class in the GO cellular component complete dataset (5.72 fold increase, P = 8.20E–04, Supplementary Tables 5 and 6). CNVs found in nine unrelated patients (#2, #3–6, #9, #10, #12, and #13) harbored at least one of the genes included in the three above overrepresented classes (Supplementary Table 7).
The same analysis, performed for previously published PNH-related CNVs (Supplementary Table 1B), revealed an overrepresentation of genes encoding for vesicle components (2.56 fold increase in GO cellular component complete dataset, P = 4.28E–02, Supplementary Table 8) but did not confirm a statistically significant overrepresentation of genes encoding for plasma membrane receptor complexes or ion channel complexes.
Discussion
Data gathered over the last 20 years indicate that PNH is not a single disorder, but rather comprises several subtypes with considerable clinical, anatomic, and genetic heterogeneity [18]. Combining all previously published patients with PNH and CNVs (n = 66) and the seven novel reported in this study, we identified 73 individuals carrying 30 different genomic rearrangements (Table 1a, b and Supplementary Table 1B), 12 of which (40%) involving more than one chromosome, or consisting of a mosaic supernumerary marker chromosome. This observation suggests that PNH, at least in a subset of individuals, might result from the cumulative dosage effect of more genes.
The diagnostic yield of molecular cytogenetics analysis, in our PNH cohort, was 35.7%. Comparing the frequency of pathogenic imbalances observed in patients with PNH with that observed in those with polymicrogyria only (9.1%), we estimated that patients with PNH have a 5.6 times increased risk of carrying a causative de novo chromosomal rearrangement.
Clinical and brain MRI findings in the seven novel patients reported here revealed PNH with varying distribution and size. In patients 6a and 6b, the same genomic rearrangement was associated with intra-familial phenotypic variability, leading to classical PNH in the mother (#6b) and bilateral scattered nodules in her daughter (#6a). Periventricular nodules were accompanied in all patients by additional, albeit most often minor, brain abnormalities including dysmorphic lateral ventricles, areas of white matter hyperintensity, and thin or hypoplastic corpus callosum (Table 1a, Fig. 1). One patient (#5) had associated bilateral parieto-temporal polymicrogyria, confirming that these MCDs can co-occur [9, 13, 18, 19]. Patients 1 and 3 had one and three periventricular nodules respectively. Although isolated heterotopic nodules are a common, rather unspecific finding, which can also appear at times in otherwise healthy individuals, their co-occurring with additional phenotypic anomalies in the above patients allows defining their phenotype as a syndromic disorder with isolated heterotopic nodules. Intellectual disability, though variable in severity, was a constant feature in our cohort (7/7). Epilepsy was present in 4/7 patients (57%) at last follow-up. In addition, 4/7 patients (57%) exhibited dysmorphic facial features (Table 1a and Supplementary Clinical Notes).
Overrepresented classes of genes
In the CNVs we submitted to the overrepresentation test (12 unrelated patients), we found genes involved in the regulation of vesicle-mediated transport or encoding plasma membrane receptor complexes to exhibit the highest enrichment in the “GO biological process complete” (P < 0.05) and the “GO cellular component complete” (P < 0.05) PANTHER datasets.
FLNA, ARFGEF2, and MAP1B are implicated in vesicle trafficking [11, 20] and ERMARD is a putative vesicle-associated protein [9]. The overrepresentation of genes involved in vesicle-related trafficking in the CNVs observed in 9/12 unrelated patients in our series (4.31-fold, P < 0.05) and in 15/24 already published by other authors (Supplementary Table 1B) (2.56 fold, P < 0.05) further suggests an association between altered vesicle trafficking and PNH. Absence of genes belonging to this class in the CNVs of patients 1, 8, and 11 might be due to either incomplete data in public databases on expression profile and function for some of the genes mapping within the CNVs analyzed or to other classes of genes contributing to PNH pathogenesis in these three patients.
Although we could not confirm a statistically significant overrepresentation of genes encoding for components of plasma receptor and ion channel complexes in PNH-associated CNVs published by other authors, genes belonging to these classes might still play a role in PNH pathogenesis. Loss of the C-terminal dimerization domain and repeats required for FLNA interaction with membrane receptors and cell signaling molecules has indeed been proposed as a possible mechanism for PNH pathogenesis [21, 22]. In addition, knockdown of KCNK2, KCNK9, or KCNK10 (potassium two pore domain channel subfamily K members 2, 9 and 10) impair the migration of late-born cortical excitatory neurons destined to become layer II/III neurons [23], while SCN1B (voltage-gated sodium channel beta subunit 1) participates as cell adhesion molecule to cell–cell communication, neuronal migration, and neurite outgrowth [24, 25]. These observations have been corroborated by the recent report of patients carrying SCN1A variants who, in addition to Dravet syndrome, exhibited different MCDs including PNH [26].
Candidate genes
Since searching for single candidate genes for PNH within large rearrangements would be impractical, we limited our search in relation to two novel, simple cryptic CNVs, namely the 16p13.3p13.2 deletion (#1), and the 22q11.22q11.23 duplication (#6a and #6b).
Patient 1 carried a de novo 16p13 deletion of about 3.1 Mb, containing eight protein-coding genes, five of which are expressed in the cerebral cortex (Supplementary Table 7) but none belongs to the three overrepresented classes. Two of these genes, namely PMM2 and ABAT, are associated to recessive disorders (MIM: #212065 and #613163) whose clinical features are inconsistent with those observed in patient 1. Among the remaining genes, USP7, encoding an ubiquitin-specific protease, is essential for WASH (Wiskott–Aldrich syndrome protein and SCAR homolog)-mediated endosomal actin assembly and protein recycling [27]. This gene has been implicated in neurodevelopmental disorders in seven individuals with haploinsufficiency [27]. The CNV detected in patient 1 DNA largely overlaps those reported (Supplementary Figure 3). Clinical features shared by our patient and those with USP7 haploinsufficiency [27] are developmental delay, seizures, dyspraxia, hypotonia, strabismus, and cryptorchidism, whereas our patient exhibited neither autism nor aggressive behavior.
RBFOX1, also included in the deleted interval in patient 1, encodes a neuron-specific splicing factor previously implicated in autism, intellectual disability, and epilepsy which regulates tissue-specific alternative splicing of a network of neuronal genes involved in cell differentiation and proliferation, including FLNA [28]. Knockdown of Rbfox1-iso1 by in utero electroporation causes abnormal neuronal positioning during corticogenesis in mice, which has been attributed to impaired radial migration [29]. Both USP7 and RBFOX1 are therefore interesting potential candidates.
Patients 6a and 6b, a daughter and mother, share an approximately 2 Mb 22q11 duplication containing more than 30 protein-coding genes. According to the MGI database, four of them, including the two morbid genes BCR and SMARCB1, are expressed in the cerebral cortex (Supplementary Table 7).
BCR, which belongs to the overrepresented class of vesicle-mediated trafficking genes, has been extensively studied in the context of chronic myeloid leukemia, as the BCR-ABL fusion protein, resulting from a recurrent t(9;22) translocation, is the most frequent cause of this condition [30, 31]. This gene plays a role in neuronal development and migration [32, 33], thus representing a good candidate for the brain phenotype we observed in these two patients.
Numerous loss-of-function variants, either germline or somatic, have been described in SMARCB1, which encodes a member of the SWI/SNF chromatin-remodeling complex, in association to rhabdoid tumors (MIM: #609322) and schwannomatosis (MIM: #162091). In contrast, a gain-of-function or dominant-negative effect has been proposed for the missense variants identified in patients with Coffin–Siris syndrome (CSS, MIM: #614608) [34]. Although PNH has never been reported in CSS, we cannot exclude a contribution of this gene to the brain phenotype of our two patients.
Overall, the above data suggest that a complex series of dynamic and interacting processes regulating neuronal migration in the developing brain can lead to PNH, as a consequence of either variants affecting the function of single genes or the cumulative dosage effect of more genes.
Considering all PNH-associated CNVs, deletions represent 73.3% (22/30) of imbalances, thus suggesting haploinsufficiency as the most likely pathogenic mechanism. However, we are aware that establishing pathogenicity of structural variations only based on their effect on gene dosage may be quite simplistic, as the impact of CNVs on the overall genomic architecture remains to be determined. The expression of genes located far away from the breakpoint of CNVs or of translocations may be influenced by the removal, duplication, or mis-localization of regulatory elements, whose role in human diseases is increasingly recognized [35]. Alterations involving the same genomic region may result in different phenotypes, possibly depending on the final configuration of topologically associated domains [35,36,37].
Several factors may explain why not all individuals sharing CNVs at the same locus exhibit PNH on brain MRI, as observed for example with 6q26-q27 and 5q14.3 deletions [9, 13]. A main reason might reside in the heterotopia being too small to be picked up by MRI. A second possibility is that isolated or scattered nodules may be missed in the presence of concomitant malformations that greatly widen the ventricular surface such as, for example, severe colpocephaly, or be overlooked or misdiagnosed as a ventricular dimple caused by the depth of a sulcus. Finally, it is possible that in some conditions, small areas of PNH might just form as a mere secondary physical consequence of impaired proliferation or migration—therefore becoming an inconstant feature—and not as a direct and constant consequence of the primary genetically induced molecular pathology.
In conclusion, our data demonstrate that genomic imbalances, either small CNVs or large rearrangements, are frequently associated with PNH. Array-CGH should be considered as a first-line diagnostic genetic test in patients with sporadic, non-classical PNH. In this series, 11/15 patients with PNH and CNVs exhibited this malformation subtype, corresponding to a 28.9% overall (11/38) diagnostic yield, while frequency of variants in FLNA, the major PNH gene, is around 5% in sporadic non-classical PNH [1]. Gene expression tools suggest that dysregulation of vesicle-mediated trafficking might play a key role in the pathogenesis of PNH. To which extent this role is exerted via single gene mechanisms or the cumulative dosage effect of more genes remains to be determined.
References
Parrini E, Ramazzotti A, Dobyns WB, et al. Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin A mutations. Brain. 2006;129:1892–906.
van Kogelenberg M, Ghedia S, McGillivray G, et al. Periventricular heterotopia in common microdeletion syndromes. Mol Syndromol. 2010;1:35–41.
Fox JW, Lamperti ED, Eksioglu YZ, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315–25.
Sheen VL, Ganesh VS, Topcu M, et al. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet. 2004;36:69–76.
Oegema R, Baillat D, Schot R, et al. Human mutations in integrator complex subunits link transcriptome integrity to brain development. PLoS Genet. 2017;13:e1006809.
Trimborn M, Bell SM, Felix C, et al. Mutations in microcephalin cause aberrant regulation of chromosome condensation. Am J Hum Genet. 2004;75:261–6.
Cappello S, Gray MJ, Badouel C, et al. Mutations in genes encoding the cadherin receptor-ligand pair DCHS1 and FAT4 disrupt cerebral cortical development. Nat Genet. 2013;45:1300–8.
Broix L, Jagline H, Ivanova E, et al. Mutations in the HECT domain of NEDD4L lead to AKT-mTOR pathway deregulation and cause periventricular nodular heterotopia. Nat Genet. 2016;48:1349–58.
Conti V, Carabalona A, Pallesi-Pocachard E, et al. Periventricular heterotopia in 6q terminal deletion syndrome: role of the C6orf70 gene. Brain. 2013;136:3378–94.
Peddibhotla S, Nagamani SC, Erez A, et al. Delineation of candidate genes responsible for structural brain abnormalities in patients with terminal deletions of chromosome 6q27. Eur J Hum Genet. 2015;23:54–60.
Heinzen EL, O’Neill AC, Zhu X, et al. De novo and inherited private variants in MAP1B in periventricular nodular heterotopia. PLoS Genet. 2018;14:e1007281.
Capra V, Biancheri R, Morana G, et al. Periventricular nodular heterotopia in Smith-Magenis syndrome. Am J Med Genet A. 2014;164A:3142–7.
Cardoso C, Boys A, Parrini E, et al. Periventricular heterotopia, mental retardation, and epilepsy associated with 5q14.3-q15 deletion. Neurology. 2009;72:784–92.
Cellini E, Disciglio V, Novara F, et al. Periventricular heterotopia with white matter abnormalities associated with 6p25 deletion. Am J Med Genet A. 2012;158A:1793–7.
Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–66.
Ciccone R, Giorda R, Gregato G, et al. Reciprocal translocations: a trap for cytogenetists? Hum Genet. 2005;117:571–82.
Kloosterman WP, Guryev V, van Roosmalen M, et al. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum Mol Genet. 2011;20:1916–24.
Guerrini R, Dobyns WB. Malformations of cortical development: clinical features and genetic causes. Lancet Neurol. 2014;13:710–26.
Wieck G, Leventer RJ, Squier WM, et al. Periventricular nodular heterotopia with overlying polymicrogyria. Brain. 2005;128:2811–21.
Sheen VL. Filamin A and Big2: a shared endocytic pathway. Bioarchitecture. 2014;4:53–57.
Sheen VL, Dixon PH, Fox JW, et al. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum Mol Genet. 2001;10:1775–83.
Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol. 2004;6:1034–8.
Bando Y, Hirano T, Tagawa Y. Dysfunction of KCNK potassium channels impairs neuronal migration in the developing mouse cerebral cortex. Cereb Cortex. 2014;24:1017–29.
Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10:19–26.
Brackenbury WJ, Isom LL. Na channel beta subunits: overachievers of the ion channel family. Front Pharmacol. 2011;2:53.
Barba C, Parrini E, Coras R, et al. Co-occurring malformations of cortical development and SCN1A gene mutations. Epilepsia. 2014;55:1009–19.
Hao YH, Fountain MD Jr, Fon Tacer K, et al. USP7 acts as a molecular rheostat to promote WASH-dependent endosomal protein recycling and is mutated in a human neurodevelopmental disorder. Mol Cell. 2015;59:956–69.
Fogel BL, Wexler E, Wahnich A, et al. RBFOX1 regulates both splicing and transcriptional networks in human neuronal development. Hum Mol Genet. 2012;21:4171–86.
Hamada N, Ito H, Nishijo T, et al. Essential role of the nuclear isoform of RBFOX1, a candidate gene for autism spectrum disorders, in the brain development. Sci Rep. 2016;6:30805.
Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36:93–99.
Heisterkamp N, Stam K, Groffen J, de Klein A, Grosveld G. Structural organization of the bcr gene and its role in the Ph’ translocation. Nature. 1985;315:758–61.
Park AR, Oh D, Lim SH, et al. Regulation of dendritic arborization by BCR Rac1 GTPase-activating protein, a substrate of PTPRT. J Cell Sci. 2012;125:4518–31.
Narayanan AS, Reyes SB, Um K, McCarty JH, Tolias KF. The Rac-GAP Bcr is a novel regulator of the Par complex that controls cell polarity. Mol Biol Cell. 2013;24:3857–68.
Tsurusaki Y, Okamoto N, Ohashi H, et al. Coffin-Siris syndrome is a SWI/SNF complex disorder. Clin Genet. 2014;85:548–54.
Lupianez DG, Kraft K, Heinrich V, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–25.
Vetro A, Dehghani MR, Kraoua L, et al. Testis development in the absence of SRY: chromosomal rearrangements at SOX9 and SOX3. Eur J Hum Genet. 2015;23:1025–32.
Kurth I, Klopocki E, Stricker S, et al. Duplications of noncoding elements 5’ of SOX9 are associated with brachydactyly-anonychia. Nat Genet. 2009;41:862–3.
den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 Update. Hum Mutat. 2016;37:564–9.
Acknowledgements
This work was supported by funding from the EU Seventh Framework Programme (FP7) under the project DESIRE grant agreement 602531 (to RG). We are grateful to Dr. Maria Clara Bonaglia (Cytogenetics Laboratory, Scientific Institute, IRCCS Eugenio Medea, Bosisio Parini, Lecco, Italy) and Professor Johan T. Den Dunnen (Human Genetics and Clinical Genetics, Leiden University Medical Center, Leiden, The Netherlands; curator of the HGVS nomenclature web page) for their precious help in revising the nomenclature of complex rearrangements.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Cellini, E., Vetro, A., Conti, V. et al. Multiple genomic copy number variants associated with periventricular nodular heterotopia indicate extreme genetic heterogeneity. Eur J Hum Genet 27, 909–918 (2019). https://doi.org/10.1038/s41431-019-0335-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0335-3
This article is cited by
-
Heterozygous truncating variant of TAOK1 in a boy with periventricular nodular heterotopia: a case report and literature review of TAOK1-related neurodevelopmental disorders
BMC Medical Genomics (2024)
-
Compound heterozygous variants in LAMC3 in association with posterior periventricular nodular heterotopia
BMC Medical Genomics (2021)
-
International consensus recommendations on the diagnostic work-up for malformations of cortical development
Nature Reviews Neurology (2020)
