
Abstract
The centrosomal protein 55 kDa (CEP55 (OMIM 610000)) plays a fundamental role in cell cycle regulation and cytokinesis. However, the precise role of CEP55 in human embryonic growth and development is yet to be fully defined. Here we identified a novel homozygous founder frameshift variant in CEP55, present at low frequency in the Amish community, in two siblings presenting with a lethal foetal disorder. The features of the condition are reminiscent of a Meckel-like syndrome comprising of Potter sequence, hydranencephaly, and cystic dysplastic kidneys. These findings, considered alongside two recent studies of single families reporting loss of function candidate variants in CEP55, confirm disruption of CEP55 function as a cause of this clinical spectrum and enable us to delineate the cardinal clinical features of this disorder, providing important new insights into early human development.
Similar content being viewed by others


Introduction
The centrosomal protein 55 kDa (CEP55) is a centrosome- and midbody-associated protein that has been shown to play a central role in cell cycle regulation and is recognised as a key protein involved in the abscission process, the final stage of cytokinesis [1,2,3]. CEP55 facilitates abscission through the recruitment of two ESCRT (endosomal sorting complex required for transport)-I subunit associated proteins to the midbody: tumour susceptibility gene 101 (TSG101) and apoptosis-linked gene 2 interacting protein X (ALIX) [4]. The transcribed CEP55 centrosomal protein has three central coiled-coil domains and is expressed at the perinuclear membrane, cytoplasm, and nucleus [2, 5].
Recently candidate homozygous nonsense variants in CEP55 have been identified in five foetuses from two families in association with a lethal disorder, the features of which include dysplastic kidneys and complex brain malformations [6, 7]. Homozygous cep55l knockout mutant zebrafish display a significant reduction in the size of brain structures, and a decreased number of renal tubules, consistent with the human phenotype [6].
Here we describe a novel Amish homozygous founder frameshift CEP55 variant in two affected Amish foetuses presenting with hydranencephaly and Potter sequence secondary to cystic renal dysplasia and anhydramnios.
Materials and methods
Samples were taken with informed consent (University of Arizona protocol 10-0050-01 and Akron Children's Hospital project number986876-3) for DNA extraction. Single-nucleotide polymorphism (SNP) genotyping was performed using the HumanCytoSNP-12 v2.1 beadchip array (Illumina). Whole-exome sequencing (WES) analysis (NextSeq500: Illumina) involved: Agilent Sureselect Whole Exome v6 targeting, read alignment (BWA-MEM (v0.7.12), mate-pairs fixed and duplicates removed (Picard v1.129), InDel realignment/base quality recalibration (GATK v3.4-46), single-nucleotide variant (SNV)/InDel detection (GATK HaplotypeCaller), annotation (Alamut v1.4.4), and read depth (GATK DepthOfCoverage). Dideoxy sequencing was undertaken using standard techniques. The CEP55 variant (NM_018131.4: c.514dup; p.(Ile172Asnfs*17) and associated phenotype data were submitted to ClinVar (www.ncbi.nlm.nih.gov/clinvar, accession SCV000808984).
Results
Subjects
An Ohio Amish couple, distantly related through a fourth-generation common ancestor (Fig. 1b), presented in their second pregnancy with dichorionic diamniotic twins, one male (twin A) and one female (twin B). Antenatal ultrasound scanning (USS), undertaken at 21 + 2 weeks of gestation, revealed twin B to have hydranencephaly, a multicystic dysplastic right kidney (the left kidney was not visualised), shortened bowed long bones and anhydramnios. Twin A was found to have bilateral renal pelvis dilatation, but no further abnormalities were detected. A subsequent USS undertaken at 28 weeks of gestation to further examine twin B revealed intrauterine growth restriction (IUGR) and pericardial effusion, and the bladder and stomach were not identified. The twins were born following an uncomplicated spontaneous vaginal delivery at 42 weeks of gestation. Twin B survived for 90 min after birth; the clinical features of Potter Sequence with cutaneous syndactyly were documented (Fig. 1c). The couple had previously experienced a stillbirth of a male foetus at 42 weeks of gestation. The foetus presented with multiple foetal anomalies at 19 weeks of gestation. Anomalies included: IUGR, anhydramnios, hydranencephaly, described as ‘hydrocephalus seen throughout the cranium’, and bilateral hydronephrosis. The foetal birth weight was 3 lb 14 oz (1.76 kg) and features of Potter sequence, bilateral lower limb bowing, talipes, and syndactyly were identified. (Fig. 1c). Karyotyping of placental samples was normal and post-mortem (PM) examination was declined for both foetuses. Assuming that a founder variant was responsible for the condition, we used a combination of autozygosity mapping and WES to study this novel syndrome and identify the underlying molecular cause.

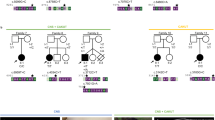
CEP55 variants, family pedigree, genotype, and images of affected individuals. a Schematic representation indicating the position of the disease-associated CEP55 gene variants in relation to genomic organisation. The CEP55 c.514dup; p.(Ile172Asnfs*17) variant identified in the current study is highlighted by red text (exons; coloured boxes, introns; black lines). The correlation between coding regions and CEP55 polypeptide functional domains is also indicated; tumour suppressor gene 101 (TSG101) and apoptosis-linked gene 2 interacting protein X (ALIX) binding domains (green), and terminal region of the protein involved in localisation during cytokinesis (yellow). b Simplified pedigree of the Amish family investigated, with electropherograms showing the DNA sequence at the position of the CEP55 variant (c.514dup) confirmed as homozygous in affected twin B and the deceased male sibling, and heterozygous in both parents and unaffected twin A. CEP55 genotype is shown in red under electropherograms in generations V and VI (+, c.514dup; –, WT). c–h Clinical features of individuals homozygous for the CEP55 variant (c.514dup). c–e VI:1 showing features of Potter sequence (also known as oligohydramnios sequence and used to describe a combination of distinctive facial and other associated phenotypic features that are a result of too little amniotic fluid. Features include epicanthic folds, retrognathia, a flattened nose, low-set ears, pulmonary hypoplasia, and limb contractures including talipes) [15]. Additional features observed included bilateral 2–5 toe syndactyly with a widened first web space and a bulbous nasal tip. f–h VI:3 (twin B) showing features of Potter sequence. The features of both affected foetuses that can be attributed as secondary to oligohydramnios include redundant skin folds, short neck, flattened face, short pinched nose, retrognathia, small palpebral fissures and low-set ears, brachydactyly, tapering fingers, and short fifth fingers with clinodactyly
Genetic studies
Whole-genome SNP genotyping of both twins and the affected brother identified a number of homozygous genomic regions particular to the affected individuals, the largest of which was a 18.3 Mb region of 10q22.3-q24.1 (rs1769756-rs7081796, chr10:g.79,164,647-97,444,769 [hg38]). Subsequent WES of DNA from the affected twin excluded previously described Amish founder variants associated with cystic kidney disease, including NPHP3 [8]. Rare variants predicted to have a functional consequence were cross-referenced with SNP genotyping data, identifying a single candidate homozygous variant of relevance to the phenotype. This variant (NM_018131.4: c.514dup; p.(Ile172Asnfs*17), chr10:g.93507042dup, [hg38]) in CEP55, predicted to result in a premature stop, is within the chromosome 10 locus. It is present in a single heterozygote in gnomAD and was not listed in ClinVar, NCBI (National Center for Biotechnology Information), or HGMDpro (Human Gene Mutation Database Professional) databases. Dideoxy sequencing confirmed the presence and cosegregation of the variant (Fig. 1b). A total of 179 samples from healthy Amish adults were analysed and 7 heterozygous carriers were identified, corresponding to an estimated allele frequency of 0.02 in this population.
Discussion
This is the third reported family with likely homozygous loss of function variants in CEP55 identified in association with a lethal foetal disorder (comparison of cases; Table 1). Frosk et al. [6] reported a family with Dutch–German Mennonite ancestry and three affected foetuses homozygous for a CEP55 nonsense variant (NM_018131.4) c.1274C>A; p.Ser425* presenting with dysplastic kidneys hydraencephaly, cerebellar hypoplasia, and multinucleated neurones at PM. The authors termed this disorder MARCH syndrome (multinucleated neurons, anhydramnios, renal dysplasia, cerebellar hypoplasia and hydranencephaly syndrome (Online Mendelian Inheritance in Man (OMIM) 236500)) and highlighted an additional nine cases in the literature with phenotypic overlap including features of hydranencephaly, renal dysplasia, and syndactyly [9,10,11,12,13]. However, as far as we are aware, CEP55 genetic analysis has not been undertaken in these individuals. Interestingly, in two cases neuropathological PM findings identified multinucleated neurons [9, 10]. Bondeson et al. [7] reported a Swedish couple with two affected foetuses homozygous for CEP55 c.256C>T; p.Arg86* (NM_018131.4) with features in one including: hydranencephaly, enlarged cystic kidneys, oligohydramnios, and cystic hygroma [7]. The second foetus had a slightly different phenotype comprising occipital encephalocele, cerebral cyst, and cystic hygroma, and the kidneys were severely degraded due to foetal autolysis. PM examination was only possible for one foetus, and although neither were identified to have polydactyly or liver abnormalities, the authors classified the combination of clinical features as a Meckel-like syndrome.
All seven cases described with biallelic CEP55 variants display phenotypical similarity, with renal dysplasia or cysts resulting in oligohydramnios and Potter sequence, and central nervous system (CNS) abnormalities comprising hydranencephaly or cerebral cysts, as the cardinal clinical features (Table 1). Interestingly, as noted by Bondeson et al. [7], the presence of these two congenital anomalies fulfils two of the characteristic Meckel syndrome (MKS) clinical triad [7]. However, in the absence of polydactyly, none of the seven cases described to date fulfils the triad. Although PM examination was undertaken in only three cases, hepatic ductal plate malformation, a frequent finding in classical MKS, was not identified. Occipital encephalocele, the most frequent CNS abnormality in MKS (83.8% of cases) [14], was documented in one foetus [7]. A spectrum of other CNS abnormalities have been described in MKS cases, including cerebral cysts and hydrocephalus, of which hydranencephaly can be considered a severe form. Several other features reported in MKS were identified in this patient cohort, including syndactyly, hydronephrosis, and short bowed limbs. In view of the wide variability seen in MKS, the lack of diagnostic criteria, and the phenotypic overlap with CEP55 patients, this disorder should be considered a MKS-like condition [7].
The Amish siblings described here enable us to more precisely delineate the clinical consequences of CEP55 loss of function, with hydranencephaly and cystic renal dysplasia as the predominant features. The identification of seven additional Amish control samples heterozygous for the CEP55 frameshift variant highlights the importance of testing when an Amish affected foetus presents with Potter sequence or an MKS-like phenotype. Antenatal USS and PM investigations following stillbirth or neonatal death are infrequently undertaken by the Amish and, despite no previous reports, the allele frequency suggests that this condition is underrecognised.
Unfortunately, while no tissue was available for cerebral histological analysis from the Amish and Bondeson cases [7], all three Frosk cases [6] displayed multinucleated neurons in cerebral tissue, and in one case multinucleated hepatocytes. Disruption of the CEP55 binding site for TSG101 and ALIX likely results from both the p.Arg86* and p.(Ile172Asnfs*17) variants (Fig. 1a), which may prevent the cytokinesis abscission process resulting in incomplete cell division, providing a plausible explanation for the multinucleated neurons. The c.256C>T; p.Arg86* variant, located in exon 3, would be predicted to result in nonsense mediated mRNA decay; however, studies on heterozygous parents identified equal levels of wild-type and truncated transcript [7]. The p.Ser425* variant is predicted to delete the terminal 40 amino acids critical for localisation during cytokinesis. Consistent with this, subcellular localisation studies showed the variant disrupts localisation to the midbody during cell division [6].
The lack of development of cerebral structures suggests that loss of CEP55 function may also play a role in cell migration during embryogenesis and development. Impaired cytokinesis may contribute to impaired neuronal migration in cells with aberrant cellular division, potentially explaining the MKS-like phenotype as both impaired cilia function and cell division processes may cause abnormal neuronal migration. However, further studies of CEP55 function are required to fully determine the precise pathomolecular basis of this lethal multisystem congenital anomaly disorder. Taken together, our findings consolidate CEP55 as a molecule fundamental to normal human development, with homozygous loss of function associated with a Meckel-like lethal foetal disorder profoundly affecting brain and kidney development.
References
Zhao WM, Seki A, Fang G. Cep55, a microtubule-bundling protein, associates with centralspindlin to control the midbody integrity and cell abscission during cytokinesis. Mol Biol Cell. 2006;17:3881–96.
Fabbro M, Zhou BB, Takahashi M, Sarcevic B, Lal P, Graham ME, et al. Cdk1/Erk2- and Plk1-dependent phosphorylation of a centrosome protein, Cep55, is required for its recruitment to midbody and cytokinesis. Dev Cell. 2005;9:477–88.
Xu ZY, Ma XS, Qi ST, Wang ZB, Guo L, Schatten H, et al. Cep55 regulates spindle organization and cell cycle progression in meiotic oocyte. Sci Rep. 2015;5:16978.
Lee HH, Elia N, Ghirlando R, Lippincott-Schwartz J, Hurley JH. Midbody targeting of the ESCRT machinery by a noncanonical coiled coil in CEP55. Science. 2008;322:576–80.
Chen CH, Lu PJ, Chen YC, Chen CY, Su LJ, Lee YC et al. FLJ10540-elicited cell transformation is through the activation of PI3-kinase/AKT pathway. Oncogene. 2007;26:4272–83.
Frosk P, Arts HH, Philippe J, Gunn CS, Brown EL, Chodirker B, et al. A truncating mutation in CEP55 is the likely cause of MARCH, a novel syndrome affecting neuronal mitosis. J Med Genet. 2017;54:490–501.
Bondeson ML, Ericson K, Gudmundsson S, Ameur A, Pontén F, Wesström J et al. A nonsense mutation in CEP55 defines a new locus for a Meckel-like syndrome, an autosomal recessive lethal fetal ciliopathy. Clin Genet. 2017;92:510–6.
Simpson MA, Cross HE, Cross L, Helmuth M, Crosby AH. Lethal cystic kidney disease in Amish neonates associated with homozygous nonsense mutation of NPHP3. Am J Kidney Dis. 2009;53:790–5.
Bendon RW, Siddiqi T, de Courten-Myers G, Dignan P. Recurrent developmental anomalies: 1. Syndrome of hydranencephaly with renal aplastic dysplasia; 2. Polyvalvular developmental heart defect. Am J Med Genet Suppl. 1987;3:357–65.
Gschwendtner A, Mairinger T, Soelder E, lge A, Kreczy A. Hydranencephaly with renal dysgenesis: a coincidental finding? Case report with review of the literature. Gynecol Obstet Invest. 1997;44:206–10.
Hamby WB, Krauss RF, Beswick WF. Hydranencephaly; clinical diagnosis; presentation of 7 cases. Pediatrics. 1950;6:371–83.
Strauss S, Bouzouki M, Goldfarb H, Uppal V, Costales F. Antenatal ultrasound diagnosis of an unusual case of hydranencephaly. J Clin Ultrasound. 1984;12:420–2.
Chu G, Miller W, Norton M, Kinney H, Genest D, Folkerth R, et al. Hydranencephaly with binucleate neurons-renal dysplasia-syndactyly syndrome in three siblings. J Neuropathol Exp Neurol. 1998;54:483.
Barisic I, Boban L, Loane M, Garne E, Wellesley D, Calzolari E, et al. Meckel-Gruber Syndrome: a population-based study on prevalence, prenatal diagnosis, clinical features, and survival in Europe. Eur J Hum Genet. 2015;23:746–52.
Curry CJ, Jensen K, Holland J, et al. The Potter sequence: a clinical analysis of 80 cases. Am J Med Genet. 1984;19:679–702.
Acknowledgements
We are grateful to the parents of the affected children for taking part in this study and to the Amish community and colleagues at the New Leaf Centre: Clinic for Special Children, Ohio, for their continuing support of our work. We would like to acknowledge Katie Guegan for technical assistance with DNA extraction from foetal tissue and Anna-Marie Bussell and Jessica Settle for their assistance with the whole-exome sequencing which was undertaken at the Royal Devon & Exeter NHS Foundation Trust, UK. The work was supported by MRC Clinical Research Training fellowship G1001931 (to ELB), MRC grant G1002279 (to AHC), MRC Proximity to Discover and Confidence in Concept (UEMSto ELB and AHC), the Newlife Foundation for disabled children (to LER, ELB, and AHC).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Rawlins, L.E., Jones, H., Wenger, O. et al. An Amish founder variant consolidates disruption of CEP55 as a cause of hydranencephaly and renal dysplasia. Eur J Hum Genet 27, 657–662 (2019). https://doi.org/10.1038/s41431-018-0306-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0306-0
This article is cited by
-
Evidence that the Ser192Tyr/Arg402Gln in cis Tyrosinase gene haplotype is a disease-causing allele in oculocutaneous albinism type 1B (OCA1B)
npj Genomic Medicine (2022)
-
MicroRNA-449a Suppresses Mouse Spermatogonia Proliferation via Inhibition of CEP55
Reproductive Sciences (2021)
-
Cytokinetic Abscission Regulation in Neural Stem Cells and Tissue Development
Current Stem Cell Reports (2021)
-
Cep55 overexpression promotes genomic instability and tumorigenesis in mice
Communications Biology (2020)
-
Cep55 promotes cytokinesis of neural progenitors but is dispensable for most mammalian cell divisions
Nature Communications (2020)
