
Abstract
An intact and dynamic microtubule cytoskeleton is crucial for the development, differentiation, and maintenance of the mammalian cortex. Variants in a host of structural microtubulin-associated proteins have been identified to cause a wide spectrum of malformations of cortical development and alterations of microtubule dynamics have been recognized to cause or contribute to progressive neurodegenerative disorders. TBCD is one of the five tubulin-specific chaperones and is required for reversible assembly of the α-/β-tubulin heterodimer. Recently, variants in TBCD, and one other tubulin-specific chaperone, TBCE, have been identified in patients with distinct progressive encephalopathy with a seemingly broad clinical spectrum. Here, we report the clinical, neuroradiological, and neuropathological features in eight patients originating from the Faroe Islands, who presented with an early onset, progressive encephalopathy with features of primary neurodegeneration, and a homogenous clinical course. These patients were homozygous for a TBCD missense variant c.[3099C>G]; p.(Asn1033Lys), which we show has a high carrier frequency in the Faroese population (2.6%). The patients had similar age of onset as the previously reported patients (n = 24), but much shorter survival, which could be caused by either differences in supportive treatment, or alternatively, that shorter survival is intrinsic to the Faroese phenotype. We present a detailed description of the neuropathology and MR imaging characteristics of a subset of these patients, adding insight into the phenotype of TBCD-related encephalopathy. The finding of a Faroese founder variant will allow targeted genetic diagnostics in patients of Faroese descent as well as improved genetic counseling and testing of at-risk couples.
Similar content being viewed by others

Introduction
Variants that affect function in genes encoding tubulins and microtubule-associated proteins lead to neurodevelopmental disorders with malformations of cortical development as well as neurodegenerative disorders. Microtubules are assembled from heterodimers of different α- and β-tubulin isotypes, which display specific spatial and temporal expression patterns in post-mitotic neurons during cortical development. Microtubules participate in a range of essential cellular processes including neuronal migration, polarity, intracellular trafficking, and differentiation [1]. Microtubule dynamics are at least in part controlled by α-/β-tubulin heterodimer assembly and disassembly, which depends on several chaperone proteins including five tubulin-specific chaperones, termed cofactors A–E (TBCA-E), and a specific GTPase [2].
Variants that affect function in the tubulin-encoding genes TUBA1A, TUBB2A, TUBB2B, TUBB3, and TUBB5 result in partly convergent phenotypes, termed tubulinopathies, including malformations of cortical development with lissencephaly, pachygyria and polymicrogyria, basal ganglia malformation, microcephaly, and autism spectrum disorder without overt cerebral malformations, while other tubulinopathies result in more distinct phenotypes without neurological features [3, 4]. Interestingly, specific variants in TUBB4A, TUBA4A, and TBCE have been identified as disease-causing or contributing to neurodegeneration in hypomyelination with atrophy of basal ganglia and cerebellum (H-ABC) and related diseases, in amyotrophic lateral sclerosis, and in early-onset progressive encephalopathy, respectively [5,6,7]. It is thought that microtubule dynamics and function as well as cell-type-specific effects are involved in disease-specific pathogenesis [5, 7, 8]. More recently, variants in the tubulin-specific cofactor D (TBCD) have been identified in a series of patients with early-onset, progressive encephalopathy, extending the spectrum of microtubule-related neurodegenerative disease [9,10,11,12,13].
Here, we report a homozygous TBCD variant in eight patients from six families originating from the Faroe Islands, an archipelago of 18 islands with around 49,000 inhabitants, which is situated between Iceland and Norway in the North Atlantic Sea. The Faroese population originates from a small settlement and several genetic disorders have an increased incidence in the present population due to the founder effect, e.g., mitochondrial encephalomyopathy due to a SUCLA2 variant, MERTK-related retinitis pigmentosa, and Aicardi Goutières syndrome [14,15,16]. Patients with the TBCD variant had an apparently normal presentation at birth, and normal early development with onset of progressive encephalopathy within the first year, followed by rapid progression with median age at death of 24 months. We present a detailed description of the neuropathology and MR imaging characteristics of a subset of these patients, adding insight into the phenotype of TBCD-related encephalopathy. The finding of a Faroese founder variant will allow targeted genetic diagnostics in Faroese patients as well as improved genetic counseling and testing of at-risk couples.
Subjects and methods
Patients
Eight patients with encephalopathy from six families originating from the Faroe Islands were collected from the Faroe Islands and Denmark over a period of 28 years. They had all tested negative for the Faroese SUCLA2 founder variant (c.534+1G>A) [16] and were without an etiological diagnosis despite standard molecular genetic and biochemical investigations. The follow-up study of the patients has been reviewed and permitted by the local ethical board. The patients’ parents had given informed consent for follow-up genetic testing and publication.
Published patients were identified from the literature by searching Pubmed with the search term “TBCD” on 28 December, 2017.
Genetic analyses
To search for the disease-causing variant, whole-exome sequencing (WES) was performed using DNA isolated from blood from patients 6 and 7 using the Ion AmpliSeq™ exome RDY kit (Thermo Fisher, Waltham, MA, USA). The IonChef was used to prepare libraries, which were sequenced using the IonProton system (Thermo Fisher). Base calling, pre-processing of the reads, short read alignment, and variant calling were performed using the Torrent Suite including the Torrent Variant Caller (Version 4.4–5.0) (Thermo Fisher). VarSeq (GoldenHelix, Bozeman, MT, USA) was used for annotation and filtration of the genomic variants.
Sanger sequencing was performed to confirm the TBCD variant discovered by WES. A TaqMan (Thermo Fisher) assay was developed and 306 DNA samples from Faroese control subjects, were screened to assess the carrier frequency, essentially as reported previously [16]. NM_005993.4 was used as reference sequence. The clinical and variant details have been submitted to the LOVD database (https://databases.lovd.nl/shared/individuals/00163916; 00164375; 00164376; 00164436; 00164437; 00164438; 00164439; 00164440).
Western blot analysis
Western blot analysis of protein from cultured fibroblasts was performed essentially as described previously [16]. An antibody against TBCD was obtained from Proteintech (Rosemont, IL, USA), whereas an antibody against actin (GeneTex, Irvine, CA, USA) was used as a loading control.
Neuropathology
Brains were fixed for 6 weeks in 25% saturated picrid acid/50% formaldehyde (37%) solution before macroscopic examination and routine sectioning. Tissue blocks were post fixed, transferred to 4% formalin, dehydrated, and embedded into paraffin wax. Sections were stained with H&E, van Gieson’s, and Klüver–Barrera stain. Immunohistochemistry included staining for glial fibrillary acid protein (GFAP Z0334, Dako, Santa Clara, CA, USA, diluted 1:6400), neurofilament (NF M0762, Dako, diluted 1:3200), microglial cells and macrophages (CD68 M0876, PGM-1, Dako, diluted 1:200), tau (Tau AB1020MN, Thermo Scientific, Waltham, MA, USA, diluted 1:500), alfa-synuclein (ASYN AB5038, Merck, Darmstadt, Germany, diluted 1:5000), and beta-amyloid (BETA-A M0872, Dako, diluted 1:100). Frozen sections from formalin-fixed brain were stained with Oil Red O and Sudan black. The tissue sections were then deparaffinized through xylene and ethanol to tap water. Monoclonal primary antibodies were detected with biotinylated rabbit anti-mouse IgG (E0354, Dako). Polyclonal primary antibodies were detected with biotinylated swine anti-rabbit IgG (E0353, Dako). Final detection was performed by ABComplex/HRP (K0355, Dako) and 3-amino-9-ethylcarbazole.
Results
Patients
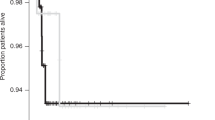
The clinical findings in all 8 patients are summarized in Table 1. A more detailed description of individual case histories and clinical progression is provided in Supplementary Data S1. Three of the patients were male and there were two sibling pairs (cases 2 and 3 as well as cases 6 and 7). The case histories revealed some invariable features, including apparently normal presentation at birth with normal birth measurements, normal Apgar scores if reported, and normal early development. The patients came to medical attention between the ages of 4 to 9 months (median 5.5 months) (Fig. 1a) and were referred due to developmental delay or stagnation of development, truncal hypotonia, reduced motor activity especially of legs, reduced attentiveness, beginning spasticity, or seizures (patient 3 only). In the further course, developmental regression, truncal hypotonia, spasticity, epilepsy, and the development of pronounced respiratory problems with stagnation of secretions, aspiration, and recurrent pneumonias was found in all. Bilateral hip luxation was found in four patients, and hip dysplasia described in one. All patients had severe, treatment-resistant epilepsy with onset between 6 and 19 months (median 9.5 months). Head circumference was documented at several time points for a few patients only, however, the available measurements indicated development of secondary microcephaly in some patients. Neurological progression and severe respiratory complications, and in one patient severe intractable seizures, led to premature death between the ages of 10 and 56 months (median age at death 24 months) (Fig. 1b).

Comparison of the clinical course of patients with the Faroese founder variant (n = 8) and patients with other TBCD variants (n = 24), and characterization of the c.[3099C>G]; p.(Asn1033Lys) substitution. a Box plot showing similar age at onset of disease in the two groups of patients with a larger age range in the non-Faroese group. b Kaplan–Meier plot showing shorter survival in patients with the Faroese founder variant. c Conservation of TBCD p.Asn1033 in different species. d Western blot analysis showing a severely decreased amount of TBCD protein in fibroblasts from patients 4 and 7, compared to controls. An antibody against actin was used as a loading control
To our knowledge, only 24 patients with TBCD variants, originating from 15 families, have been described at present [9,10,11,12,13]. These patients had a similar age at onset (median 5 months), but considerably longer survival (Fig. 1a, b), compared to the patients with the Faroese founder variant.
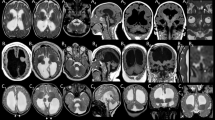
Cerebral MRI was performed in four Faroese patients between the ages of 8 months and 21 months, with two serial cerebral MRIs in patient 8. The MRIs showed cortical and global cerebral atrophy, which was most severe in the frontal region and most pronounced in older patients. In case 8, a progressively reduced volume of the white matter and corpus callosum was found, without signs of primary involvement of myelin. Infratentorial structures were involved to a much lesser degree (for details see Fig. 2).

Magnetic resonance imaging (MRI) of patient 4 (a–d) at 8 months, patient 7 (e–h) at 8 months, patient 5 (i–l) at 20 months, and patient 8 at 6 (m–o) and 21 (p–r) months of age including coronal T1- (a, e, i), axial T2- (b, f, j, m, p), sagittal T1- (c, g, k, n, k), and axial T2-weighted (d, h, l, o, r) imaging. MRI of patient 4 demonstrates cortical and global cerebral atrophy with frontal overweight, reduced volume of white matter, and myelination adequate for age (a, b). In sagittal midline, a severely reduced volume of corpus callosum and reduced volume of the brain stem is visible (c). Cerebellar peduncles and cerebellar white matter show mild atrophy (d). Similarly, MRI of patient 7 shows cortical and central atrophy with severely reduced volume of cerebral white matter, especially in the frontal region, with normal signal from myelin (e, f). Corpus callosum is thin and mesencephalon slightly atrophic (g). Cerebellum appears normal (h). MRI of patient 5 reveals profound cortical and central cerebral atrophy, most pronounced in the frontal regions. White matter is severely reduced, but T1 and T2 signal suggest normal myelination. The basal ganglia appear without pathology (i, j). Sagittal imaging (k) suggests absence of corpus callosum, however a normal septum pellucidum indicates severe corpus callosum atrophy. Brain-stem structures are only moderately reduced in volume. Cerebellum is atrophic with reduced white matter and slim cerebellar peduncles (l). MRI in patient 8 was performed at age 6 and 21 months and recapitulated the neuroradiological abnormalities described above (m–r). During the observation period of 15 months, a severe progression of the predominantly frontal cortical and central cerebral atrophy was seen, resulting in nearly complete absence of white matter (m, p). Corpus callosum was already severely atrophic at 6 months, which had progressed further on follow-up MRI. Cerebellum and brain-stem atrophy as well as atrophy of the cerebellar peduncles was more pronounced at 21 months (n, q, o, r)
Neuropathology
Neuropathological examination of brain had been performed in three patients, and histology slides were available for re-evaluation in patients 6 and 7 (Fig. 3). In patient 1, the macroscopic neuropathological examination reported symmetrical severe atrophy of the central cerebral hemispheres with increased space between sulci and ectatic ventricular system. Cerebellum was described with normal structure and volume. On microscopy, the cerebral cortex showed intact architecture, whereas the white matter was extremely atrophic with reactive gliosis. Mesencephalon, thalami, hippocampi, and pons were structurally normal. In medulla spinalis, a severe loss of anterior horn neurons was observed, starting from the level of medulla oblongata. The remaining anterior horn neurons showed signs of degeneration. The longitudinal tracts were intact. Neuropathological examination in cases 6 and 7 (Fig. 3a–e, g, case 7; Fig. 3f, case 6) revealed low brain weight (645 and 628 g at age 10 and 11 months, respectively). The external brain examination showed focal hyperconvolution of gyri with an otherwise normal gyration pattern. The focal hyperconvolution of gyri was interpreted as secondary to loss of white matter. In coronal sections, the cerebral white matter volume was substantially reduced with an extremely thin corpus callosum and marked ventriculomegaly. The gray matter had a normal appearance (Fig. 3a). Histology of the cortex showed a normal six-layered architecture with substantial vacuolization with loss of neurons, reactive gliosis and areas with a variable degree of laminar necrosis (Fig. 3b, d). Reactive gliosis and vacuolization was also found in the basal ganglia and thalamus (not shown). The cerebral white matter had profound gliosis in case 7 while gliosis was slightly more discrete in case 6 (Fig. 3b, e). Remarkably, myelination of the white matter was normal despite severe volume reduction (Fig. 3c). In the hippocampus, loss of neurons in the cornu ammonis area 1 and 2 was seen and was more pronounced in case 6 (Fig. 3g). Histology of the cerebellum showed mild loss of purkinje cells and neurons in the dentate nucleus (Fig. 3f). In the brain stem, vacuolization and gliosis was less severe than in the cerebrum and the loss of neurons was mild to moderate. Axonal swelling, inclusions or immunoreactivity with alfa-synuclein, tau or beta-amyloid were not found. Muscle histology (available from patients 1, 2, and 6) showed muscle fibers that were too small for age, and fiber type grouping, typical of denervated muscle (for details see Supplementary data S1).

Macroscopic (a) and histopathological (b–g) examination of brains of case 6 at 10 months of age (f) and case 7 at 11 months of age (a–e, g). Staining is hematoxylin & eosin (b; d–g) and Klüver–Barrera (c). White matter volume is severely reduced leading to ventriculomegaly of lateral ventricles and extreme thinning of corpus callosum (a). The yellow discoloration of the brain is due to fixation in picrid acid. Histologically, cortex is showing marked vacuolization and severe neuronal loss (b, d), substantially reduced volume of the white matter (b), but normal myelin content in Klüver–Barrera stain (c), exemplified in the frontal lobe. A profound gliosis in the white matter of the frontal lobe is seen (e). These findings were verified in the other cortical areas of both patients (not shown). The cerebellum displayed mild loss of purkinje cells in patient 6 (f) and in hippocampus severe neuronal loss in cornu ammonis area 1 and area 2 in patient 7 (g)
Molecular genetics
Filtering of the WES data revealed a homozygous variant in TBCD (NM_005993.4), c.[3099C>G]; p.(Asn1033Lys), which was considered because of the clinical link to the description of TBCD variants as causative for neurodegenerative encephalopathy [9, 10]. A summary of the WES data output is provided in Supplementary table S2. Sanger sequencing confirmed the homozygous presence of the variant in all eight patients, and, when available, segregation analysis confirmed the carrier status of the parents (patients 6 and 7). Asparagine at position 1033 is moderately conserved (Fig. 1c) and the substitution is predicted tolerated by SIFT (http://sift.jcvi.org/), possibly damaging (score 0.928) by Polyphen2 (genetics.bwh.harvard.edu/pph2/), disease-causing by MutationTaster (http://www.mutationtaster.org/), and moderately damaging in CADD (score 25.2) (http://cadd.gs.washington.edu/home). Western blot analysis of fibroblast protein from patients 4 and 7 showed a severely decreased amount of TBCD protein (Fig. 1d).
The c.[3099C>G] variant is recorded in gnomAD with an allele frequency in non-Finnish Europeans of 3/126,686 (0.00002368), but absent from other populations (global MAF 3/277,166). Analysis of carrier frequency showed that 8 of 306 Faroese controls were heterozygous for the variant, equivalent to a carrier frequency of 1 in 38 (2.6%). This corresponds to a calculated incidence of the disorder of 1 in 1444 in the Faroese population. The eight patients were identified in the period from 1993–2016, which would equal an incidence of 1 in 2875 (estimated number of Faroese born per year ~1000).
Discussion
Here, we report 8 patients affected with an early-onset progressive encephalopathy due to a a novel variant in TBCD. The likely effect of this variant on TBCD function is supported by the following points: 1. The phenotype of the eight patients is in concordance with that found in other patients with TBCD variants; 2. The patients had a severely decreased amount of TBCD protein in fibroblasts; 3. The affected amino acid is moderately conserved and the amino acid substitution is predicted to affect function in three out of four in silico prediction programs including the comprehensive CADD score. Asn1033 is predicted to be located within a loop between antiparallel α helices, and the substitution of asparagine with lysine may influence protein conformation, but would also change a neutral to a positive amino acid on the protein surface, which could impair formation of the tubulin chaperone multiprotein complex.
Because the amount of TBCD protein is severely reduced in patient fibroblasts (Fig. 1d; patient 4 and 7) it is possible that TBCD p.(Asn1033Lys) is subject to misfolding and proteasomal degradation. TBCD protein level in patient skin fibroblasts and immortalized lymphocytes has previously been examined in 6 individuals homozygous for TBCD missense variants [9, 11, 13]. Although not directly quantifiable, the results indicate relatively higher residual mutant TBCD protein levels than detected in this study in all except one individual (F118_346; [9]).
Altogether, 21 different TBCD variants have been reported, including three splice variants, one nonsense variant, and 17 missense variants [9,10,11,12,13]. The missense variants affect amino acids located in different parts of TBCD, and are generally predicted to interfere with folding of α solenoid repeats, loop structures, and/or interactions with other molecules, thus affecting protein conformation and function.
The clinical findings in the Faroese patients are in accordance with what has been reported in published patients [9,10,11,12,13]: an early onset (most often before age 12 months), progressive encephalopathy with intellectual disability, motor impairment, secondary microcephaly, and epilepsy. Many patients were reported with hypotonia and/or spasticity, which were found in all Faroese patients. Optic atrophy was also a frequent feature in published patients (described in 14 of 20 patients for whom information regarding optic nerve pathology was provided), but results of ophthalmologic investigations were available for only one of the Faroese patients showing pale optic discs. Despite a similar age of onset of disease in Faroese and non-Faroese patients, there was a clear difference in survival between the two groups, shown in the Kaplan–Meier plot (Fig. 1b). Thus, the phenotype might progress faster in the Faroese patients, although it cannot be ruled out that ascertainment bias in favor of longer surviving patients or differences in supportive treatment explain the differences in survival. On the other hand, length of survival has not changed significantly over the 28 years, in which patients were collected for this study, possibly arguing that shorter survival is intrinsic to the Faroese phenotype. A more detailed understanding of TBCD function, genetic interactions, and the pathogenetic consequences of perturbed TBCD function might render further clarification of this observation possible in the future. Very low or absent protein level of p.(Asn1033Lys) TBCD in the Faroese patient population could be part of the explanation.
Cerebral MRI (Fig. 2) confirms absence of cortical malformation in the Faroese TBCD patients (patients 4, 5, 7, 8) as observed in the previously reported cases [9,10,11,12,13]. Serial cerebral MRI of patient 8 (Fig. 2m–r) illustrates very severe progression of cortical and central atrophy with marked volume loss of cerebral white matter over an observation period of 15 months (6 and 21 months of age). In patient 5, a cerebral MRI at 20 months of age seems to indicate a similar development of atrophy. Neuroradiological documentation of fast progressing cerebral atrophy confirms the clinical impression that the neurodegenerative course of disease is uniform and rapid in this patient population. Progressive cerebral atrophy in TBCD patients has previously been well documented [10], accompanied by seemingly more pronounced cerebellar atrophy. Others found comparingly subtle signs of cortical atrophy and corpus callosum volume loss at higher ages [9]. One patient with onset of symptoms at birth had severe volume loss and global ventriculomegaly already at first cerebral MRI aged 8 months [10], but long survival (individual 2, deceased at age 19 years). As opposed to previous descriptions [9, 11], we observed age-adequate myelination in atrophic white matter in all patients examined by cerebral MRI. This was consistent with normal cerebral myelin staining on histopathological examination (exemplified in frontal lobe, Fig. 3e, patient 7) which otherwise revealed a picture of diffuse neuronal loss with vacuolization, areas with laminar necrosis and diffuse gliosis in the cortex, as well as in basal ganglia, cerebellum, and brain stem (Fig. 3). Miyake et al. [10] described very similar neuropathology in one patient who was asphyctic at birth and our findings confirm that the picture of diffuse neuronal loss likely was attributable to TBCD deficiency and not asphyxia in this case. Severe white matter gliosis and atrophy were interpreted as secondary phenomena, driven by primary neuronal pathology and cell death.
In conclusion, the present study confirms TBCD encephalopathy as a progressive, neurodegenerative disorder with early onset and, for some patients, early death. In the Faroese population, we have identified a TBCD founder variant, which is associated with a severe phenotype. The disorder is likely underdiagnosed, and identification of additional Faroese cases may elucidate if the founder variant is indeed associated with a severe phenotype.
References
Kapitein LC, Hoogenraad CC. Building the neuronal microtubule cytoskeleton. Neuron. 2015;87:492–506.
Francis JW, Goswami D, Novick SJ, et al. Nucleotide binding to ARL2 in the TBCD ∙ ARL2 ∙ β-tubulin complex drives conformational changes in β-tubulin. J Mol Biol. 2017;429:3696–716.
Tischfield MA, Cederquist GY, Gupta ML, Engle EC. Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr Opin Genet Dev. 2011;21:286–94.
Breuss MW, Leca I, Gstrein T, Hansen AH, Keays DA. Tubulins and brain development—the origins of functional specification. Mol Cell Neurosci. 2017;84:58–67.
Curiel J, Bey GR, Takanohashi A, et al. TUBB4A mutations result in specific neuronal and oligodendrocytic defects that closely match clinically distinct phenotypes. Hum Mol Genet. 2017;26:4506–18.
Smith BN, Ticozzi N, Fallini C, et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron. 2014;84:324–31.
Sferra A, Baillat G, Rizza T, et al. TBCE mutations cause early-onset progressive encephalopathy with distal spinal muscular atrophy. Am J Hum Genet. 2016;99:974–83.
Dubey J, Ratnakaran N, Koushika SP. Neurodegeneration and microtubule dynamics: death by a thousand cuts. Front Cell Neurosci. 2015;9. https://doi.org/10.3389/fncel.2015.00343.
Flex E, Niceta M, Cecchetti S, et al. Biallelic mutations in TBCD, encoding the tubulin folding cofactor D, perturb microtubule dynamics and cause early-onset encephalopathy. Am J Hum Genet. 2016;99:962–73.
Miyake N, Fukai R, Ohba C, et al. Biallelic TBCD mutations cause early-onset neurodegenerative encephalopathy. Am J Hum Genet. 2016;99:950–61.
Pode-Shakked B, Barash H, Ziv L, et al. Microcephaly, intractable seizures and developmental delay caused by biallelic variants in TBCD: further delineation of a new chaperone-mediated tubulinopathy. Clin Genet. 2017;91:725–38.
Ikeda T, Nakahara A, Nagano R, et al. TBCD may be a causal gene in progressive neurodegenerative encephalopathy with atypical infantile spinal muscular atrophy. J Hum Genet. 2017;62:473–80.
Edvardson S, Tian G, Cullen H, et al. Infantile neurodegenerative disorder associated with mutations in TBCD, an essential gene in the tubulin heterodimer assembly pathway. Hum Mol Genet. 2016;25:4635–8.
Ostergaard E, Joensen F, Sundberg K, et al. A novel RNASEH2B splice site mutation responsible for Aicardi-Goutieres syndrome in the Faroe Islands. Acta Paediatr Int J Paediatr 2012;101. https://doi.org/10.1111/j.1651-2227.2012.02807.x.
Ostergaard E, Duno M, Batbayli M, Vilhelmsen K, Rosenberg T. A novel MERTK deletion is a common founder mutation in the Faroe Islands and is responsible for a high proportion of retinitis pigmentosa cases. Mol Vis. 2011;17:1485–92.
Ostergaard E, Hansen FJ, Sorensen N, et al. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain. 2007;130:853–61.
Acknowledgements
We thank the families for their participation and Rikke Kammersgaard Nysted and Helle Nørreskov Jensen for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Grønborg, S., Risom, L., Ek, J. et al. A Faroese founder variant in TBCD causes early onset, progressive encephalopathy with a homogenous clinical course. Eur J Hum Genet 26, 1512–1520 (2018). https://doi.org/10.1038/s41431-018-0204-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0204-5
This article is cited by
-
Biallelic pathogenic variants in TBCD-related neurodevelopment disease with mild clinical features
Neurological Sciences (2019)
