
Abstract
The valorization of lignin to value-added basic chemicals is one of the most important technologies for efficient carbon recycling. While many catalytic systems have been developed for cleavage of monolignol linkages, especially for β-O-4 structures, the low solubility of lignin, which originates from its complicated polymeric structure, often makes it difficult to apply these catalytic process to degradation of real lignin-derived materials. Here, we investigated the degradation of poly(ethylene glycol)-modified lignin with transition metal complexes. Monolignols (4-methyl, 4-ethyl- and 4-propyl-guaiacol) were obtained as the degradation products. Although low solubility after detachment of the PEG moiety hampered efficient degradation, the addition of PEG for in situ protection of the hydroxy group was effective in maintaining the lignin solubility and improving the monolignol yields.
Similar content being viewed by others
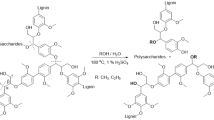

Lignin is one of the most abundant carbon resources, accounting for approximately 30% of woody biomass. In seeking efficient recycling of carbon resources, the valorization of lignin has been a topic of emerging interest [1,2,3]. Along with various methods developed to crack natural and modified lignin (Fig. 1a) [4], transition metal-mediated C–O and C–C bond cleavage in lignin-model compounds has been actively studied to improve the catalytic performance and to understand the mechanisms behind bond cleavage. In particular, C–O and C–C bonds in the so-called β-O-4 linkages are the main targets, and various catalytic systems utilizing Group 7 to Group 11 late transition metals have been studied [5,6,7]. We recently reported that iridium complexes bearing cyclopentadienone ligands degraded a lignin-model dimer to a monomer, guaiacol, in 74.9% yield (Fig. 1b) [8, 9]. However, the low solubility derived from the complicated polymeric structure often prevents the use of such model systems for the depolymerization of lignin polymers. Considerable effort has, therefore, been devoted to the solubilization of lignin [10, 11]. The degradation of soluble modified lignin with transition metal complexes is thus worth investigating to understand the connection of model systems and real lignin degradation and future development of an efficient homogeneous cracking process. In this study, we focused our attention on the poly(ethylene glycol)-modified lignin developed by Nge et al. [12], which has the potential to be a raw material precursor to biobased plastics, and hydrogenative degradations to monolignol mediated by late transition metal complexes [13, 14].

a Depolymerization of lignin. Representative structures from the guaiacyl unit are shown. b Degradation of the β-O-4 linkage in the lignin model compound by the iridium complex and c iridium-mediated depolymerization of PEG-modified lignin (this work)
The lignin modified with poly(ethylene glycol) (PEG-modified lignin) was first treated with 1 atmosphere of dihydrogen in the presence of various transition metal complexes to afford monolignols 1, 2 and 3 (Table 1). The starting material was derived from Japanese cedar biomass and mainly consisted of β-O-4 linkages with PEG modification at the α-OH group, as reported in the literature (The lignin sample used in this work has 75% β-O-4 linkage decollated with PEG at the α-hydroxy group (GL200M in the original literature [12])). A spectroscopic analysis of the PEG-modified lignin by 1H-NMR in dmso-d6 showed partial introduction of PEG200 to the hydroxy groups, with the remaining -OH providing a signal at approximately 9.0 ppm. The average molecular mass for a disubtituted aromatic ring was calculated as 413 by quantitative 13C NMR spectroscopy with 1,1,2,2-tetrachloroethane as an internal standard, which closely matched the guaiacyl monolignol unit bearing one PEG200 group (see the supporting information for 1H and 13C NMR spectra). All of the iridium and ruthenium complexes examined in this study showed activity in degrading PEG-modified lignin to generate 4-methyl-, 4-ethyl-, and 4-propylguaiacol as the main monolignol products (1, 2 and 3 in Table 1). Among the complexes examined, the iridium complexes, in general, were superior to ruthenium (see the supporting information for a complete set of entries). The best total yield was achieved with a very simple cyclooctadiene iridium complex bearing one triphenylphosphine ligand (E), which afforded a 2.4 μmol (2.0%) total yield of the three guaiacol derivatives (entry 5). Longer reaction times up to 96 h gave only a slight improvement in the total yield to 3.3 μmol (2.7%).
Although the complexes provided degradation of the linkages between the monolignols, the yields were not satisfactory. The total yields remained up to 3.3 μmol (2.7%), which was smaller than that of the loaded iridium complex. In these investigations, we realized that there was considerable insoluble material in the reaction vessel after 24 h, which was insoluble even in dimethyl sulfoxide (DMSO), dimethyl formamide (DMF) or any other common organic solvent. It should be noted that the starting PEG-modified lignin was soluble in many organic solvents, such as DMSO, DMF and THF. The amount of insoluble material was 30 mg in entry 5, which corresponded to approximately 60 wt% of the starting lignin. To investigate the structural changes that caused the insolubility of the lignin, the insoluble material was analyzed by 13C CP-MAS-NMR spectroscopy. The spectra of the original PEG-modified lignin and the insoluble material produced during the reaction are shown in Fig. 2. The signal at approximately 73 ppm (highlighted with red) was assigned to the ethylene carbon in the PEG moiety. In comparison to the PEG-modified lignin before the reaction, the spectrum after the hydrogen treatment showed a much smaller signal at approximately 73 ppm, which indicated the loss of PEG due to the harsh hydrogenolysis conditions. The α- and β-methine carbons appeared at approximately 80–90 ppm (highlighted in blue). The significant decreases seen in the signals for the α- and β-methine carbons implied that cleavage of either the Cα–O and/or Cβ–O bonds produced the insoluble material or that the lignin became insoluble after C–O bond cleavage so that further fragmentation to monolignol was retarded. Partial removal of oxygen functionality was also supported by the emergence of an alkyl carbon signal at approximately 30 ppm (highlighted in green). The degradation of PEG was also supported by an independent reaction with PEG-300 under the same reaction conditions, which showed the emergence of ethoxy and methoxy termini, suggesting C–O and C–C bond cleavage (Scheme 1 and Supplementary Fig. S3 in the supporting information). This insolubility problem was not solved by the use of ortho-dichlorobenzene (ODCB), a good solvent with a high boiling point (Table 2, entry 2). The dilution provided even a negative effect in terms of the monolignol yield (0.83% yield in total with 100 μL of ODCB, entry 3, Table 2). A significant improvement in the monolignol yield was achieved when PEG-300 was used as the solvent (entry 4, Table 2). Monolignols 1, 2 and 3 were obtained in 3.1% yield (3.7 μmol). It should be noted that in the PEG-300 solvent, the lignin was kept soluble throughout the reaction and generated only a trace amount of insoluble material. The better yield observed in the PEG solvent was most likely the result of the high solubility of the substrate lignin in PEG-300. Furthermore, the suppression of insolubility enabled us to analyze the changes in the molecular weight through degradation. Figure 3 shows the GPC data for the starting PEG-modified lignin (pink) and the reaction mixture (black). Contrary to our expectation, the number-average molecular weight of the reaction mixture (Mn = 1700) was not decreased but was even higher than that of the starting PEG-modified lignin (Mn = 1500). The emergence of the peak at 19 min (Mn ~ 200) was consistent with the formation of monolignols. Since the lignin polymer remained dissolved and the increase in the molecular weight was small (ca. 200 Da), the origin of the increased molecular weight was not a cross-linking of multiple polymer molecules but most likely additional PEG functionalization of the remaining phenolic- or γ-hydroxy groups in situ. These analytical data (13C NMR and GPC analysis) suggested that although the PEG moiety, which was introduced to provide higher solubility, was cleaved under harsh reaction conditions, in situ functionalization of the hydroxy group by the excess PEG could be key to keeping the lignin polymer in solution.

13C-CP-MAS NMR spectrum of PEG-modified lignin (bottom) and the insoluble material obtained in entry 5, Table 1 (top)

Degradation of PEG-300 by iridium complex E under dihydrogen

GPC data for the lignin polymer before (pink) and after (black) the reaction. (THF solutions, and the molecular weight was calibrated with polystyrene standards)
In conclusion, we investigated the reductive degradation of PEG-modified lignin with dihydrogen and late-transition metal complexes to overcome the low solubility of the lignin material. Iridium and ruthenium complexes were found to degrade PEG-modified lignin to the monolignols 4-methyl-, 4-ethyl-, and 4-propylguaiacol. Dissociation of the PEG at high temperatures was confirmed by solid-state 13C CP-MAS NMR. The coexistence of excess poly(ethylene glycol) kept the lignin soluble via in situ modification of the hydroxy groups in lignin to afford an improved yield of the monolignols. Although the reaction efficiency was not yet satisfactory, the reversible PEG modification could be the key to overcoming the low reactivity of insoluble polymeric lignin materials.
References
Li C, Zhao X, Wang A, Huber GW, Zhang T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem Rev 2015;115:11559–624.
Zhang C, Wang F. Scientific questions for lignin conversion and a brief summary of methods for lignin depolymerization. In: Lignin Conversion Catalysis: Transformation to Aromatic Chemicals : Wiley‐VCH GmbH; 2022. p. 79–130.
Chio C, Sain M, Qin W. Lignin utilization: a review of lignin depolymerization from various aspects. Renew Sustain Energy Rev. 2019;107:232–49.
Sun Z, Fridrich B, de Santi A, Elangovan S, Barta K. Bright side of lignin depolymerization: toward new platform chemicals. Chem Rev 2018;118:614–78.
Deuss PJ, Barta K. From models to lignin: transition metal catalysis for selective bond cleavage reactions. Coord Chem Rev. 2016;306:510–32.
Das A, König B. Transition metal- and photoredox-catalyzed valorization of lignin subunits. Green Chem. 2018;20:4844–52.
Zhang C, Shen X, Jin Y, Cheng J, Cai C, Wang F. Catalytic strategies and mechanism analysis orbiting the center of critical intermediates in lignin depolymerization. Chem Rev 2023;123:4510–601.
Kusumoto S, Kishino M, Nozaki K. Cleavage of C–C and C–O bonds in β-O-4 linkage of lignin model compound by cyclopentadienone group 8 and 9 metal complexes. Chem Lett. 2020;49:477–80.
Kusumoto S, Nozaki K. Direct and selective hydrogenolysis of arenols and aryl methyl ethers. Nat Commun. 2015;6:6296.
Baruah J, Nath BK, Sharma R, Kumar S, Deka RC, Baruah DC, et al. Recent trends in the pretreatment of lignocellulosic biomass for value-added products. Front Energy Res. 2018;6:141.
Soria AJ, McDonald AG, Shook SR. Wood solubilization and depolymerization using supercritical methanol. Part 1: Process optimization and analysis of methanol insoluble components (bio-char). Holzforschung. 2008;62:402–8.
Nge TT, Tobimatsu Y, Takahashi S, Takata E, Yamamura M, Miyagawa Y, et al. Isolation and characterization of polyethylene glycol (PEG)-modified glycol lignin via PEG solvolysis of softwood biomass in a large-scale batch reactor. ACS Sustain Chem Eng. 2018;6:7841–8.
Nge TT, Tobimatsu Y, Takahashi S, Umezawa T, Yamada T. Hydrogen peroxide treatment of softwood-derived poly(ethylene glycol)-modified glycol lignin at room temperature. Molecules. 2023;28:1542.
Nge TT, Tobimatsu Y, Yamamura M, Takahashi S, Takata E, Umezawa T, et al. Effect of heat treatment on the chemical structure and thermal properties of softwood-derived glycol lignin. Molecules. 2020;25:1167.
Acknowledgements
This research was performed by the Environment Research and Technology Development Fund (JPMEERF20223R03) of the Environmental Restoration and Conservation Agency provided by the Ministry of the Environment of Japan, Inoue Foundation for Science and Noguchi Institute Research Fund. This work was supported by JSPS KAKENHI Grant-in-Aid for Scientific Research (S) and Grant Number JP18H05259.
Funding
Open access funding provided by The University of Tokyo.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kusumoto, S., Higashi, T., Matsumoto, Y. et al. Hydrogenative degradation of PEG-functionalized lignin. Polym J 56, 353–357 (2024). https://doi.org/10.1038/s41428-023-00867-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-023-00867-5
