
Abstract
Various cellular stress conditions trigger mitochondrial DNA (mtDNA) release from mitochondria into the cytosol. The released mtDNA is sensed by the cGAS-MITA/STING pathway, resulting in the induced expression of type I interferon and other effector genes. These processes contribute to the innate immune response to viral infection and other stress factors. The deregulation of these processes causes autoimmune diseases, inflammatory metabolic disorders and cancer. Therefore, the cGAS-MITA/STING pathway is a potential target for intervention in infectious, inflammatory and autoimmune diseases as well as cancer. In this review, we focus on the mechanisms underlying the mtDNA-triggered activation of the cGAS-MITA/STING pathway, the effects of the pathway under various physiological and pathological conditions, and advances in the development of drugs that target cGAS and MITA/STING.
Similar content being viewed by others
Cytoplasmic nucleic acid-sensing pathways
The innate immune arm of the immune system forms the first line of host defense against pathogen infection, and it is initiated via the recognition of conserved microbial structures by cellular pattern recognition receptors (PRRs) [1,2,3,4,5,6]. After viral infection, invading viral nucleic acids, such as viral RNA (vRNA) or DNA (vDNA), are recognized by PRRs, which initiate signaling pathways that ultimately induce the expression of type I interferon (IFN), proinflammatory cytokine, and other antiviral effector genes [3,4,5,6,7]. These downstream effectors inhibit viral replication, induce apoptosis in infected cells, and promote activation of the adaptive immune response, leading to antiviral immune responses [1,2,3,4,5,6]. In contrast, viruses evolve multiple strategies to evade the host immune response to maintain their survival and persistent infection [7,8,9,10,11,12,13,14]. The relative power of these two opposing forces determine the eventual outcomes of the viral infection of a host.
After RNA virus infection of mammalian cells, vRNA invading the cytoplasm is sensed by RIG-I-like receptors (RLRs), which include RIG-I and MDA5 [1,2,3, 7]. After sensing vRNA, RLRs undergo conformational changes, oligomerization, and then translocation to mitochondria [15, 16], where they interact with Virus-Induced Signaling Adaptor (VISA, also called MAVS, Cardif, and IPS-1) [17,18,19,20]. VISA then recruits WDR5, TRAF and cIAP proteins, the kinases TBK1 and IKK, and the transcription factors IRF3 and NF-κB [21,22,23,24,25,26]. In these complexes, activated TBK1 and IKK phosphorylate and activate IRF3 and NF-κB, respectively, leading to their translocation into the nucleus and expression of downstream antiviral genes [1, 2]. The RLR-VISA axis activity is regulated by cofactors, distinct posttranslational modifications and regulators of posttranscriptional modifications to ensure efficient initiation of innate antiviral immunity and its timely termination in the late phase of infection [16, 23, 24, 27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43].
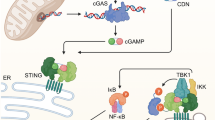
After DNA virus infection of mammalian cells, vDNA is sensed by a widely expressed enzyme called cyclic GMP–AMP synthase (cGAS) [4, 44,45,46]. Various studies have demonstrated that cGAS not only recognizes a wide range of microbial DNA but also senses mitochondrial DNA (mtDNA) and cellular nuclear DNA (nDNA) aberrantly localized to the cytosol after infection or under stress or pathological conditions [47,48,49,50,51,52,53,54]. After binding to cytosolic DNA, cGAS undergoes phase separation and forms “DNA-cGAS” liquid droplets to achieve optimal activation, after which it catalyzes the synthesis of cyclic GMP–AMP (cGAMP) from the substrates GTP and ATP [46, 55]. Newly synthesized cGAMP binds to Mediator of IRF3 Activation (MITA) (also known as STING and ERIS) in the endoplasmic reticulum (ER), which promotes MITA/STING oligomerization and trafficking from the ER to perinuclear punctate structures [56,57,58,59,60,61,62]. During the cellular trafficking processes, MITA/STING recruits TBK1 and IRF3, leading to IRF3 phosphorylation, dimerization and translocation into the nucleus to drive the transcription of type I IFN genes [56, 60, 63,64,65,66]. Additionally, NF-κB is activated by the MITA/STING-associated complex, leading to the transcription of inflammatory cytokine genes [67]. The cGAS-MITA/STING axis is extensively regulated by cofactors. For example, ZCCHC3, G3BP1 and PCBP1 promote cGAS binding to DNA [68,69,70]; ZDHHC1 and sulfated glycosaminoglycans (sGAGs) are important for cGAMP-triggered dimerization/oligomerization of MITA/STING [59, 61]; and iRhom2, SNX8, VPS34 and ARMH3 are critically involved in MITA/STING trafficking and activation [60, 62, 71]. The cGAS-MITA/STING axis is also extensively regulated by posttranslational modifications, such as cGAS acetylation [72, 73] and phosphorylation [74,75,76,77], MITA/STING phosphorylation [38, 78,79,80], and polyubiquitination [81,82,83,84,85,86,87,88,89,90]. These positive and negative regulating modification of cGAS-MITA/STING axis components ensures efficient and proper innate immune responses to DNA viruses. Because of the critical importance of the cGAS-MITA/STING pathway in the innate immune response to DNA viruses, it is extensively targeted by DNA viruses to enable viral immune evasion [13, 91,92,93,94,95,96,97,98,99,100,101,102,103]. Interestingly, in addition to induction of the innate immune response, activation of the cGAS-STING axis has also been reported to cause other effects, such as controlling autophagy [104], mRNA translation [105], IRF3-independent function [106], which contribute to several pathological processes, including clearance of invading pathogens, cell senescence and organ fibrosis, as well as the antitumor response.
It has been established that infection with RNA viruses is mainly sensed by RIG-I and MDA5, while infection with DNA viruses is mostly sensed by cGAS. It has been shown that Rig-i-/- mouse embryo fibroblasts (MEFs) do not produce type I IFNs in response to Sendai virus (SeV), vesicular stomatitis virus (VSV), influenza virus, paramyxovirus, Japanese encephalitis virus (JEV), and hepatitis C virus (HCV) [107,108,109], while Mda5-/- MEFs do not respond to picornaviruses such as encephalomyocarditis virus (EMCV) and Theiler’s virus [110]. Moreover, Rig-i-/- and Mda5-/- mice show increased susceptibility to infection with VSV and EMCV, respectively [111]. These studies suggest that different RNA viruses can be sensed by distinct RLR family members. Various studies indicated that cGas-/- and Mita-/- knockout mice produce much lower levels of type I IFNs and other cytokines after DNA virus infection and are highly susceptible to infection with DNA viruses, such as herpes simplex virus-1 (HSV-1) and vaccinia virus (VACV), suggesting that the cGAS-MITA/STING pathway is critically important for the innate immune response to DNA viruses [44, 68]. However, various studies have shown that deficiency in cGAS or MITA/STING did not completely abolish the expression of antiviral effector genes triggered by DNA viruses [44]. It has been shown that some AT-rich DNA viruses can be transcribed into vRNA by RNA polymerase III, which is sensed by RIG-I, initiating innate immune signaling [112,113,114,115]. On the other hand, various reports have demonstrated that the expression of type I IFNs and downstream interferon-stimulating genes (ISGs) following infection with certain RNA viruses is abrogated in cGas-/- and Mita-/- knockout cells, suggesting that the cGAS-MITA/STING pathway also participates in the innate immune response to certain RNA viruses [48, 116,117,118,119,120]. Clearly, the cytoplasmic RNA- and DNA-sensing pathways are common in host cells and antagonize RNA or DNA viruses (Fig. 1). Moreover, infection with certain RNA viruses causes mtDNA release from mitochondria into the cytosol, which triggers the innate immune response via the cGAS-MITA/STING axis [48, 121]. Various studies have demonstrated that mtDNA is released after cells undergo a specific type of stress, and this mtDNA is critically involved in inflammatory and autoimmune responses [122, 123]. These studies suggest that mtDNA release under various stress conditions may be a convergent and common mechanism underlying cellular defense.

Cytoplasmic nucleic acid-sensing pathways. After RNA virus infection of mammalian cells, the invading vRNA is sensed by RLRs, which include RIG-I and MDA5, in the cytoplasm. After sensing vRNA, RLRs are translocated to mitochondria, where they interact with VISA. VISA then recruits WDR5 and TRAF proteins, the kinases TBK1 and IKK, and the transcription factors IRF3 and NF-κB. In these complexes, activated TBK1 and IKK phosphorylate and activate IRF3 and NF-κB, respectively, leading to their translocation into the nucleus and expression of downstream antiviral genes. After DNA virus infection of mammalian cells, vDNA is sensed by cGAS. After binding to cytosolic DNA, cGAS catalyzes the synthesis of cGAMP from the substrates GTP and ATP. cGAMP binds to MITA/STING at the endoplasmic reticulum (ER), which promotes MITA/STING oligomerization and trafficking from the ER to perinuclear punctate structures. During trafficking, MITA/STING recruits TBK1 and IRF3, leading to IRF3 phosphorylation, dimerization and translocation into the nucleus to drive the transcription of type I IFN genes. Additionally, NF-κB is activated by the MITA/STING-associated complex, leading to the transcription of inflammatory cytokine genes. The cGAS-MITA/STING axis is extensively regulated by cofactors, such as ZCCHC3, G3BP1, PCBP1, ZDHHC1, iRhom2 and ARMH3. Some AT-rich viral DNA sequences can be transcribed to vRNA by RNA polymerase III, and they are sensed by RIG-I to initiate innate immune signaling. Infection with certain RNA or DNA viruses causes mtDNA release from mitochondria into the cytosol, which triggers the innate immune response via the cGAS-MITA/STING axis. Cytoplasmic RNA- and DNA-sensing pathways are common used in host cells and antagonize either RNA or DNA viruses. RIG-I retinoic acid-inducible gene I; MDA5 melanoma differentiation-associated protein 5; VISA virus-induced signaling adaptor; WDR5 WD repeat-containing protein 5; TRAF TNF receptor-associated factor; TBK1 TANK-binding kinase 1; IKK inhibitor of nuclear factor kappa-B kinase; IRF3 interferon regulatory factor 3; cGAS cyclic GMP-AMP synthase; MITA mediator of IRF3 activation; STING stimulator of interferon gene; ZCCHC3 zinc finger CCHC domain-containing protein 3; G3BP1 GTPase activating protein (SH3 domain) binding protein 1; PCBP1 Poly(rC)-binding protein 1; ZDHHC1 zinc finger DHHC domain-containing protein 1; iRhom2 inactive rhomboid protein 2; ARMH3 Armadillo-like helical domain-containing protein 3
mtDNA is released from mitochondria into the cytosol under stress conditions
Mitochondria are important organelles in eukaryotic cells, as they synthesize ATP and metabolites [124]. Mitochondria can respond to external or endogenous stresses that trigger mitochondrial autophagy (mitophagy), mtDNA release, and apoptosis, and thus, they ultimately regulate of the survival or death of stressed cells [124,125,126]. Although the mechanisms underlying mitophagy and apoptosis have been extensively studied in recent decades, the mechanisms and effects of mtDNA release have only been studied in recent years. In cells under stress, mtDNA is released into the cytoplasm where it is sensed as a danger signal and thus activates a variety of signaling pathways in cells, including the cGAS-mediated innate immune response [48], absent in melanoma 2 (AIM2)- or NLR family pyrin domain-containing 3 (NLRP3)-mediated inflammation [127,128,129,130,131] and genomic DNA damage repair [132]. Recently, Z-DNA-binding protein 1 (ZBP1) has been reported to stabilize Z-form mtDNA and act as a cooperative partner for the cGAS response to mitochondrial genome instability [133]. In this article, we focus on the mechanisms of mtDNA release and its effects on the innate immune response under stress conditions or after viral infection.
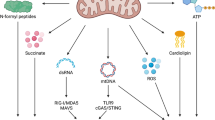
Circular mtDNA in vertebrates is maternally inherited and encodes eleven subunits of the mitochondrial electron transport chain and two subunits of ATP synthase. These proteins provide the wiring for the oxidation phosphorylation (OXPHOS) system [134, 135]. mtDNA also encodes 22 tRNAs and two rRNAs that are essential for mRNA translation in the mitochondrial matrix. The other mitochondrial proteins, including the various factors needed for mtDNA replication, repair and gene expression, are encoded by cellular DNA and are targeted to or imported into mitochondria [124]. In addition to acting as coding genes, mtDNA can be released into the cytosol under various cellular stress conditions (Fig. 2), leading to innate immune and inflammatory responses [48]. The release of mtDNA from mitochondria to the cytosol can be triggered by different factors, including radiation exposure, microbial infection, inflammatory conditions, toxic substances or drugs, and gene mutation or deletion [122, 123]. For example, radiation therapy can cause mitochondrial stress (mitostress) in tumor cells, which results in the release of mitochondrial contents, including cytochrome c and mtDNA, into the cytosol [136,137,138]. Viral infection can cause mitochondrial stress and release of mtDNA via Ca2+ uptake by the mitochondrial calcium uniporter (MCU) in a variety of cells [48, 118, 121]. The proinflammatory cytokines TNF and IL-1β have been reported to trigger mtDNA release in a variety of cells, such as myeloid cells, fibroblasts and epithelial cells, leading to activation of cGAS [139, 140]. A variety of foreign substances, including liposomes and crystalline silicon, have been reported to induce mtDNA release and thus activate innate immune and inflammatory responses [74, 141]. In addition, the release of mtDNA from mitochondria to the cytosol can be triggered by mutation or deletion of certain mitochondrion-related genes, which are involved in maintaining mitochondrial structure, mtDNA stabilization or mitophagy [48, 142,143,144]. These studies indicate that mtDNA release from mitochondria to the cytosol is triggered by divergent factors in different cell types, suggesting that mtDNA release is a basic and common cellular response to stress conditions.

Mechanisms of mtDNA release and its effects on innate immune response. The release of mtDNA from mitochondria into the cytosol is triggered by different factors, including radiation exposure, microbial infection, inflammatory conditions, toxic substances or drugs, and gene mutation or deletion. Under apoptotic conditions, mitochondrial contents, including oxidized mtDNA and cytochrome c, are released from the mitochondrial matrix into the cytosol, leading to the activation of the cGAS-MITA/STING pathway and caspases, respectively. Activated caspases cleave cGAS and IRF3 to suppress type I IFN expression. Under nonapoptotic conditions, only oxidized mtDNA is released to activate the cGAS-MITA/STING pathway. In this context, mtDNA undergoes fragmentation into 500–650 bp fragments by the DNA enzyme FEN1 before release into the cytosol, while OGG1 can mediate mtDNA glycosylation and thereby protect mtDNA from fragmentation. Under some circumstances (e.g., viral infection), MCU-mediated Ca2+ import into mitochondria is important for the initiation of mtDNA oxidation and MPTP opening. mtDNA-induced activation of the cGAS-MITA/STING pathway and subsequent type I IFN expression play important roles in antiviral host defense, antitumor response, and development of autoimmune, neurodegenerative and metabolic diseases. cGAS cyclic GMP-AMP synthase, MITA mediator of IRF3 activation, STING stimulator of interferon gene, FEN1 Flap endonuclease 1, OGG1 8-oxoguanine DNA glycosylase, VRK2 vaccinia related kinase 2, MCU mitochondrial calcium uniporter, MPTP mitochondrial permeability transition pore, OMM outer mitochondrial membrane, IMM inner mitochondrial membrane
Molecular mechanisms of mtDNA release
mtDNA is normally wrapped with Transcriptional Factor A Mitochondrial (TFAM) in the mitochondrial matrix [124]. Studies have shown that TFAM-coated mtDNA is stable and more resistant to ROS oxidation, while newly synthesized naked mtDNA is more susceptible to oxidation and leakage into the cytosol [128, 145]. Studies have shown that mtDNA undergoes oxidation and then fragmentation into 500–650 bp fragments by the DNA enzyme Flap Endonuclease 1 (FEN1) before its release into the cytosol, while 8-oxoguanine DNA glycosylase (OGG1) can mediate mtDNA glycosylation and thereby protect mtDNA from fragmentation [145] (Fig. 2). Studies have proven that oxidized mtDNA induces a greater immunostimulatory effect both in vitro and in vivo [146, 147]. However, the molecular mechanisms of the oxidation and fragmentation of mtDNA under different stress conditions need to be further elucidated.
Mitochondria are bilayer membranous organelles that consist of the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM) [124]. mtDNA needs to cross the IMM and OMM for export from the mitochondrial matrix into the cytosol. Recent studies indicate that major IMM pores include the mitochondrial permeability transition pore (MPTP), which is a nonspecific mitochondrial channel that is activated by Ca2+ influx [49, 145, 148], and inner mitochondrial membrane (IMM) herniation, which is triggered by oligomerization of Bcl-2-associated X protein (BAX) and Bcl-2 homologous antagonist/killer (BAK) [50]. Under oxidative stress, voltage-dependent anion channel 1 (VDAC1) located at the OMM can oligomerize and mediate mtDNA release. In the presence of mtDNA fragments, VDAC1 oligomerization is accelerated [149]. VDAC1 was initially considered a component of the MPTP; however, an analysis of VDAC1-deficient mice showed that VDAC1 deficiency exerted no marked effects on MPTP formation, which indicated that VDAC1 is dispensable for MPTP formation or a compensatory system consisting of other VDAC family members is involved [150]. How MPTP opening and VDAC1 oligomerization synergistically mediate mtDNA release is still unknown. Of course, VDAC2/3 might compensate for VDAC1 deficiency. During apoptosis, the formation of BAX/BAK macropores causes OMM permeabilization and mtDNA extrusion [50]. Under certain stress conditions, mtDNA may cross the IMM and OMM via one or more of these pores and eventually leak into the cytosol. The detailed mechanisms explaining how these pores are formed and regulated need further investigation. Recently, vaccinia-related kinase 2 (VRK2) was identified as a regulator of VDAC1 oligomerization [121]. Virus-induced mtDNA release into the cytosol is markedly impaired in VRK2-deficient cells [121]. It has also been suggested that rapid Ca2+ uptake via the mitochondrial calcium uniporter (MCU) precedes MPTP opening and subsequent VDAC1 oligomerization [121, 145]. Additionally, Prohibitin 1 (PHB1) has been reported to regulate mtDNA release by inhibiting mPTP opening [151]. Whether other events, such as mitochondrial ROS accumulation, trigger MPTP opening and how stress conditions signal these events are unanswered questions. Furthermore, even though BAX/BAK oligomerization can lead to mtDNA release during apoptosis, the release of mtDNA mediated by VDAC but not BAX/BAK oligomerization has been shown to be involved in most diseases (see below).
Physiological and pathological effects of mtDNA release
mtDNA released into the cytosol is sensed by the widely expressed DNA sensor cGAS in a DNA sequence-independent manner. The cGAS response results in the synthesis of cGAMP and subsequent MITA/STING-dependent signaling events, leading to the expression of type I IFNs and various inflammatory cytokines, including TNF superfamily members, interleukins and chemokines [45, 63]. Notably, most of the physiological and pathological effects of mtDNA release are mediated by these cytokines (Fig. 2).
mtDNA release contributes to host defense against viral infection
Various studies have demonstrated that infection with either DNA or RNA viruses triggers mtDNA release from mitochondria into the cytosol, where it activates the cGAS-MITA/STING axis, leading to the activation of downstream antiviral effectors [48, 116, 121, 152, 153]. Depletion of mtDNA by ethidium bromide (EB) or dideoxycytidine (ddC) downregulates the transcription of IFNB1 and other antiviral genes after HSV-1 infection [48, 121]. Deficiency of VRK2, which is a regulator of VDAC1 oligomerization and mtDNA release, renders mice more susceptible to infection with both the DNA virus HSV-1 and the RNA virus EMCV [121]. Although rapid induction of type I IFNs limits virus propagation, a sustained increase in the levels of type I IFNs in the late phase of infection is associated with aberrant inflammation and poor clinical outcomes. It has been suggested that in patients with COVID-19, the cGAS-MITA/STING pathway is critical to the type I IFN immunopathology of extrapulmonary complications in lung endothelial cells after mtDNA release [11, 154, 155]. There is accumulating evidence showing that mtDNA sensing by the cGAS-MITA/STING pathway plays a critical role in antiviral host defense, but how viral infection triggers Ca2+ import into mitochondria via MCU to induce mitostress and how MPTP-VDAC oligomer-mediated mtDNA release is regulated after viral infection are not fully understood. Furthermore, whether specific viral mechanisms suppress mtDNA release to enable immune evasion is also an unanswered question.
Radiation-induced mtDNA release benefits antitumor therapy
Previous studies have demonstrated that ionizing radiation-mediated tumor regression is dependent on the activation of the cGAS-MITA/STING axis in dendritic cells (DCs) [138]. Accordingly, activation of MITA/STING by cGAMP enhances radiation-triggered antitumor immunity [138]. Recently, studies have also demonstrated that mtDNA released into the cytosol of irradiated tumor cells failed to activate the cGAS-MITA/STING pathway due to caspase 3/9-mediated suppression of this signaling pathway [136, 156]. In this context, caspases have been reported to cleave cGAS and IRF3 to suppress type I IFN activation [157]. The combination of radiation, a caspase inhibitor and an anti-PD-L1 antibody promoted antitumor therapy [136]. Additionally, it has been reported that mtDNA drives abscopal responses to radiation exposure that are inhibited by autophagy [137]. Autophagy-deficient cells secrete increased amounts of type I IFNs, suggesting that autophagy inhibitors may serve as potential drugs for increasing the efficacy of radiation therapy in cancer patients [137].
mtDNA release causes autoimmune, neurodegenerative and metabolic diseases
Activation of IRF3 and induction of type I IFNs protect the host against infection and cancer, but excessive IFN responses triggered by self-DNA (including mtDNA) under sterile conditions cause autoinflammatory conditions such as Aicardi–Goutières syndrome (AGS), STING-associated vasculopathy of infancy (SAVI) and systemic lupus erythematosus (SLE) [158, 159]. Neutrophil extracellular traps (NETs) have been implicated in autoimmunity, and NETs enriched with oxidized mtDNA are interferongenic and contribute to lupus-like disease [160]. Another study also indicated that mtDNA fragments released following VDAC oligomerization promoted lupus-like disease [149]. The VDAC inhibitor VBIT-4 reduced mtDNA release, the IFN response and disease severity in a mouse model of SLE [149].
Recently, studies have demonstrated that deregulation of the cGAS-MITA/STING axis is involved in multiple sterile inflammatory diseases, such as myocardial infarction, heart failure, cardiac hypertrophy, aortic aneurysm and dissection, obesity, and nonalcoholic fatty liver diseases [161,162,163,164,165,166,167,168,169]. This is because of the large loads of damage-associated molecular patterns, including mtDNA and/or DNA in extracellular vesicles liberated during recurrent injury to metabolic cellular organelles and tissues, which are sensed by the cGAS-MITA/STING pathway [170, 171]. Furthermore, although the cGAS-MITA/STING pathway-mediated immune response is often neuroprotective, excessive or sustained activation of this pathway in the brain causes neuroinflammation and neurodegeneration [172, 173]. Therefore, targeting the cGAS-MITA/STING pathway can have potential therapeutic benefits in patients with one of several neurodegenerative disorders, including Alzheimer’s disease [174], Parkinson’s disease [175], and amyotrophic lateral sclerosis (ALS) [49].
Intervention of diseases by targeting the cGAS-MITA/STING pathway
Since deregulation of the mtDNA-cGAS-MITA/STING pathway causes aberrant innate immune and inflammatory responses and pathological effects, great efforts have been made to identify strategies for selective modulation of the cGAS-MITA/STING axis in various diseases, and these strategies include identification of agonists of the cGAS-MITA/STING axis to use as vaccine adjuvants or as anticancer and antiviral immunostimulatory agents, as well as identification of selective inhibitors of the axis to use as potential drugs for treating inflammatory and autoimmune diseases. Specific agents targeting cGAS and MITA/STING are summarized in Table 1.
MITA/STING agonists
Activating the cGAS-MITA/STING pathway can enhance the antitumor immune response [176]. In addition, MITA/STING agonists can be used as adjuvants to develop vaccines against certain infectious diseases [177]. To date, most MITA/STING activators have been synthetic cyclic dinucleotides (CDNs), such as ADU-S100 [178], BMS-986301 (Clinical Trials.gov ID: NCT03956680) and MK-1454 [179]. Intratumoral injection of these CDNs induces intense antitumor T-cell immune responses and generates immune memory, leading to complete tumor regression as well as prevention of distal metastasis of lung cancers [180, 181].
Considering the high polarity and proteolytic tendency of the abovementioned cyclic dinucleotide agonists, their clinical application potential is limited. In recent years, nonnucleotide derivatives have gained prominence due to their high specificity and effectiveness. For example, amide compounds, such as amide benzimidazole (ABZI) and its derivatives, Compounds 16 g, 24b, and 24e, N-(methylcarbamoyl)-2-[5] phenylacetamide (C11), 6-bromo-n-(naphthalen-1-yl)-benzo (d), and dioxole-5-carboxamide (BNBC), have been identified as human MITA/STING agonists [182,183,184]. These compounds, when given intravenously, exerted potent antitumor effects in mice bearing subcutaneous tumors. Furthermore, other compounds, such as MSA-2 [185] and SR-717 [186], have also been identified as MITA/STING nonnucleotide agonists. These compounds can induce IFN-β production in tumors and a long-lasting antitumor immune response and can also exert synergistic effect when administered with an anti-PD-1 antibody therapy [185, 186]. MSA-2 and SR-717 show the potential for clinical application because of their oral availability characteristics and simplified administration mode. Additionally, flavonoid compounds have been reported to be MITA/STING agonists; they include α-mangostin, G10, and dispiro diketopiperazine (DSDP) [187,188,189]. The effects of these agonists on antitumor activities need to be investigated.
MITA/STING inhibitors
Two main approaches have been utilized to identify MITA/STING inhibitors. The first approach involves the design of molecules that target the CDN-binding site, thereby functioning as competitive inhibitors of MITA/STING activation. This class of MITA/STING inhibitors mainly includes tetrahydroisoquinolines [190] and astin C [191]. The second approach is to identify molecules that bind to either the Cys88 or the Cys91 residue of the human MITA/STING protein, each of which is a target for palmitoylation [192]. Tetrahydroisoquinolines bind to MITA/STING dimers in 2:2 ratio and thus inhibit cGAMP-induced IFN-β secretion from THP-1 cells [190]. Astin C is a natural product that binds competitively to the CDN site and blocks the recruitment of IRF3 to the MITA/STING signalosome. Furthermore, astin C inhibits the expression of type I IFNs in Trex1-/- BMDMs and mice [191]. To date, several different chemicals targeting MITA/STING palmitoylation residues have been reported, including nitrofurans (C-176 and C-178) [193], indole urea (H-151) [193] and nitro fatty acids [194]. Mice administered C-176 exhibited markedly reduced production of serum type I IFNs induced by MITA/STING agonists. Additionally, pretreatment of Trex1-/- mice with C-176 led to a significant reduction in type I IFN levels and the number of inflammatory signatures in the heart [193]. Intraperitoneal administration of H-151 inhibited the systemic cytokine response triggered by a MITA/STING agonist [193]. Additionally, certain nitro fatty acids reduced type I IFNs in response to DNA viral infection in both THP-1 cells and BMDMs [194].
cGAS inhibitors
Two classes of cGAS inhibitors have been developed for the treatment of inflammatory and autoimmune diseases. The first class of cGAS antagonists bind to the enzymatic active site, resulting in competition with the ATP or GTP substrate or the product cGAMP. The second class of cGAS antagonists block DNA binding to cGAS, thereby inhibiting the initial step in cGAS activation. The catalytic site inhibitors of cGAS include PF-06928125 [195], RU.521 [196] and G150 [197], and the action of most has been validated only in vitro. For example, RU.521 has been shown to be a selective inhibitor of cGAS that reduced the Ifnb1 mRNA level in bone marrow-derived macrophages (BMDMs) from Trex1-/- mice [196]. Inhibitors that disrupt the DNA-binding activity of cGAS are mainly antimalarial drugs. It has been reported that antimalarial drugs, including hydroxychloroquine and quinacrine, can be used for SLE treatment since these drugs suppress IFN-β expression by blocking the cGAS–dsDNA interaction [198, 199]. It has also been reported that suramin can bind to cGAS and disrupt the formation of the cGAS–dsDNA complex [200]. In vivo studies on the functions of these selective cGAS inhibitors are needed to validate their roles in the intervention of inflammatory and autoimmune diseases in the future.
Concluding remarks
Studies in recent years have established critical roles for mtDNA release in cellular defense against various stress conditions, including stress caused by infection with various types of viruses, deregulation of autophagy and introduction of DNA aberrations. Studies have identified components involved in mtDNA release from mitochondria into the cytosol after viral infection or under other stress conditions. However, more studies are needed to identify the common and distinct mechanisms of mtDNA release under different stress conditions. Released mtDNA is sensed by cGAS, which triggers MITA/STING-mediated innate immune responses. However, deregulation of the mtDNA-cGAS-MITA/STING axis activity leads to inflammatory and autoimmune diseases as well as cancer. In recent years, small molecules targeting MITA/STING or cGAS have been developed for potential application in cancer immunotherapy or the treatment of inflammatory and autoimmune diseases. Further studies on the molecular mechanisms underlying the mtDNA-cGAS-MITA/STING axis will certainly provide a more comprehensive understanding of cellular defense against viral infection and other stress conditions and help in the development of novel strategies for the intervention of serious human diseases, including inflammatory/autoimmune diseases and cancer.
References
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20.
Hu MM, Shu HB. Cytoplasmic Mechanisms of Recognition and Defense of Microbial Nucleic Acids. Annu Rev Cell Dev Biol. 2018;34:357–79.
Hu MM, Shu HB. Innate immune response to cytoplasmic DNA: mechanisms and diseases. Annu Rev Immunol. 2020;38:79–98.
Yang Q, Shu HB. Deciphering the pathways to antiviral innate immunity and inflammation. Adv Immunol. 2020;145:1–36.
Hu MM, Shu HB. Multifaceted roles of TRIM38 in innate immune and inflammatory responses. Cell Mol Immunol. 2017;14:331–8.
Onomoto K, Onoguchi K, Yoneyama M. Regulation of RIG-I-like receptor-mediated signaling: interaction between host and viral factors. Cell Mol Immunol. 2021;18:539–55.
Ma Z, Damania B. The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe. 2016;19:150–8.
Jiang L, Chen H, Li C. Advances in deciphering the interactions between viral proteins of influenza A virus and host cellular proteins. Cell Insight. 2023;2:100079.
Leung DW, Basler CF, Amarasinghe GK. Molecular mechanisms of viral inhibitors of RIG-I-like receptors. Trends Microbiol. 2012;20:139–46.
Chen D, Zhao YG, Zhang H. Endomembrane remodeling in SARS-CoV-2 infection. Cell Insight. 2022;1:100031.
Lockhart A, Mucida D, Parsa R. Immunity to enteric viruses. Immunity. 2022;55:800–18.
Fu YZ, Su S, Gao YQ, Wang PP, Huang ZF, Hu MM, et al. Human Cytomegalovirus Tegument Protein UL82 Inhibits STING-Mediated Signaling to Evade Antiviral Immunity. Cell Host Microbe. 2017;21:231–43.
Yuan Y, Fang A, Wang Z, Tian B, Zhang Y, Sui B, et al. Trim25 restricts rabies virus replication by destabilizing phosphoprotein. Cell Insight. 2022;1:100057.
Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–20.
Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, Finberg RW, et al. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity. 2013;38:437–49.
Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–40.
Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–82.
Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–8.
Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–72.
Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–61.
Liu S, Chen J, Cai X, Wu J, Chen X, Wu YT, et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife. 2013;2:e00785.
Lei CQ, Zhong B, Zhang Y, Zhang J, Wang S, Shu HB. Glycogen synthase kinase 3beta regulates IRF3 transcription factor-mediated antiviral response via activation of the kinase TBK1. Immunity. 2010;33:878–89.
Mao AP, Li S, Zhong B, Li Y, Yan J, Li Q, et al. Virus-triggered ubiquitination of TRAF3/6 by cIAP1/2 is essential for induction of interferon-beta (IFN-beta) and cellular antiviral response. J Biol Chem. 2010;285:9470–6.
Wang YY, Liu LJ, Zhong B, Liu TT, Li Y, Yang Y, et al. WDR5 is essential for assembly of the VISA-associated signaling complex and virus-triggered IRF3 and NF-kappaB activation. Proc Natl Acad Sci USA 2010;107:815–20.
Li S, Zheng H, Mao AP, Zhong B, Li Y, Liu Y, et al. Regulation of virus-triggered signaling by OTUB1- and OTUB2-mediated deubiquitination of TRAF3 and TRAF6. J Biol Chem. 2009;285:4291–7.
Zhang J, Hu MM, Shu HB, Li S. Death-associated protein kinase 1 is an IRF3/7-interacting protein that is involved in the cellular antiviral immune response. Cell Mol Immunol. 2014;11:245–52.
Lian H, Zang R, Wei J, Ye W, Hu MM, Chen YD, et al. The Zinc-Finger Protein ZCCHC3 Binds RNA and Facilitates Viral RNA Sensing and Activation of the RIG-I-like Receptors. Immunity. 2018;49:438–48.e435.
Yan BR, Zhou L, Hu MM, Li M, Lin H, Yang Y, et al. PKACs attenuate innate antiviral response by phosphorylating VISA and priming it for MARCH5-mediated degradation. PLoS Pathog. 2017;13:e1006648.
Lei CQ, Zhang Y, Xia T, Jiang LQ, Zhong B, Shu HB. FoxO1 negatively regulates cellular antiviral response by promoting degradation of IRF3. J Biol Chem. 2013;288:12596–604.
Chen LT, Hu MM, Xu ZS, Liu Y, Shu HB. MSX1 Modulates RLR-Mediated Innate Antiviral Signaling by Facilitating Assembly of TBK1-Associated Complexes. J Immunol. 2016;197:199–207.
Lei CQ, Zhang Y, Li M, Jiang LQ, Zhong B, Kim YH, et al. ECSIT bridges RIG-I-like receptors to VISA in signaling events of innate antiviral responses. J Innate Immun. 2015;7:153–64.
Ran Y, Liu TT, Zhou Q, Li S, Mao AP, Li Y, et al. SENP2 negatively regulates cellular antiviral response by deSUMOylating IRF3 and conditioning it for ubiquitination and degradation. J Mol Cell Biol. 2011;3:283–92.
Chen H, Li Y, Zhang J, Ran Y, Wei J, Yang Y, et al. RAVER1 is a coactivator of MDA5-mediated cellular antiviral response. J Mol Cell Biol. 2013;5:111–9.
Yan J, Li Q, Mao AP, Hu MM, Shu HB. TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J Mol Cell Biol. 2014;6:154–63.
Luo WW, Li S, Li C, Zheng ZQ, Cao P, Tong Z, et al. iRhom2 is essential for innate immunity to RNA virus by antagonizing ER- and mitochondria-associated degradation of VISA. PLoS Pathog. 2017;13:e1006693.
Guo W, Wei J, Zhong X, Zang R, Lian H, Hu MM, et al. SNX8 modulates the innate immune response to RNA viruses by regulating the aggregation of VISA. Cell Mol Immunol. 2020;17:1126–35.
Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347:aaa2630.
Li S, Zhu M, Pan R, Fang T, Cao YY, Chen S, et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat Immunol. 2016;17:241–9.
Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat Immunol. 2016;18:214–24.
Hu MM, He WR, Gao P, Yang Q, He K, Cao LB, et al. Virus-induced accumulation of intracellular bile acids activates the TGR5-beta-arrestin-SRC axis to enable innate antiviral immunity. Cell Res. 2019;29:193–205.
Hu MM, Liao CY, Yang Q, Xie XQ, Shu HB. Innate immunity to RNA virus is regulated by temporal and reversible sumoylation of RIG-I and MDA5. J Exp Med. 2017;214:973–89.
Guo W, Wei J, Shu HB, Yang Q. SNX8 modulates innate immune response to RNA virus by regulating the aggregation of VISA. Cell Mol Immunol. 2019;17:1126–35.
Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341:1390–4.
Xiao TS, Fitzgerald KA. The cGAS-STING pathway for DNA sensing. Mol Cell. 2013;51:135–9.
Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–91.
White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–62.
West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–7.
Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell. 2020;183:636–49.e618.
McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018;359:eaao6047.
Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–6.
Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461–5.
Bakhoum SF, Cantley LC. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell. 2018;174:1347–60.
Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553:467–72.
Du M, Chen ZJ. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science. 2018;361:704–9.
Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–50.
Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–92.
Sun W, Li Y, Chen L, Chen H, You F, Zhou X, et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci USA 2009;106:8653–8.
Zhou Q, Lin H, Wang S, Wang S, Ran Y, Liu Y, et al. The ER-associated protein ZDHHC1 is a positive regulator of DNA virus-triggered, MITA/STING-dependent innate immune signaling. Cell Host Microbe. 2014;16:450–61.
Luo WW, Li S, Li C, Lian H, Yang Q, Zhong B, et al. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat Immunol. 2016;17:1057–66.
Fang R, Jiang Q, Guan Y, Gao P, Zhang R, Zhao Z, et al. Golgi apparatus-synthesized sulfated glycosaminoglycans mediate polymerization and activation of the cGAMP sensor STING. Immunity. 2021;54:962–75.e968.
Fang R, Jiang Q, Jia X, Jiang Z. ARMH3-mediated recruitment of PI4KB directs Golgi-to-endosome trafficking and activation of the antiviral effector STING. Immunity. 2023;56:500–15.e506.
Zhang ZD, Zhong B. Regulation and function of the cGAS-MITA/STING axis in health and disease. Cell Insight. 2022;1:100001.
Liu Y, Zhou Q, Zhong L, Lin H, Hu MM, Zhou Y, et al. ZDHHC11 modulates innate immune response to DNA virus by mediating MITA-IRF3 association. Cell Mol Immunol. 2018;15:907–16.
Luo WW, Shu HB. Emerging roles of rhomboid-like pseudoproteases in inflammatory and innate immune responses. FEBS Lett. 2017;591:3182–9.
Luo WW, Shu HB. Delicate regulation of the cGAS-MITA-mediated innate immune response. Cell Mol Immunol. 2018;15:666–75.
Yum S, Li M, Fang Y, Chen ZJ. TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci USA 2021;118:e2100225118.
Lian H, Wei J, Zang R, Ye W, Yang Q, Zhang XN, et al. ZCCHC3 is a cosensor of cGAS for dsDNA recognition in innate immune response. Nat Commun. 2018;9:3349.
Liao CY, Lei CQ, Shu HB. PCBP1 modulates the innate immune response by facilitating the binding of cGAS to DNA. Cell Mol Immunol. 2021;18:2334–43.
Liu ZS, Cai H, Xue W, Wang M, Xia T, Li WJ, et al. G3BP1 promotes DNA binding and activation of cGAS. Nat Immunol. 2019;20:18–28.
Wei J, Lian H, Guo W, Chen YD, Zhang XN, Zang R, et al. SNX8 modulates innate immune response to DNA virus by mediating trafficking and activation of MITA. PLoS Pathog. 2018;14:e1007336.
Song ZM, Lin H, Yi XM, Guo W, Hu MM, Shu HB. KAT5 acetylates cGAS to promote innate immune response to DNA virus. Proc Natl Acad Sci USA 2020;117:21568–75.
Dai J, Huang YJ, He X, Zhao M, Wang X, Liu ZS, et al. Acetylation Blocks cGAS Activity and Inhibits Self-DNA-Induced Autoimmunity. Cell. 2019;176:1447–60.e1414.
Yang YL, Cao LB, He WR, Zhong L, Guo Y, Yang Q, et al. Endocytosis triggers V-ATPase-SYK-mediated priming of cGAS activation and innate immune response. Proc Natl Acad Sci USA 2022;119:e2207280119.
Zhong L, Hu MM, Bian LJ, Liu Y, Chen Q, Shu HB. Phosphorylation of cGAS by CDK1 impairs self-DNA sensing in mitosis. Cell Discov. 2020;6:26.
Seo GJ, Yang A, Tan B, Kim S, Liang Q, Choi Y, et al. Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Rep. 2015;13:440–9.
Li M, Shu HB. Dephosphorylation of cGAS by PPP6C impairs its substrate binding activity and innate antiviral response. Protein Cell. 2020;11:584–99.
Gao P, Hu MM, Shu HB. CSK promotes innate immune response to DNA virus by phosphorylating MITA. Biochem Biophys Res Commun. 2020;526:199–205.
Xia T, Yi XM, Wu X, Shang J, Shu HB. PTPN1/2-mediated dephosphorylation of MITA/STING promotes its 20S proteasomal degradation and attenuates innate antiviral response. Proc Natl Acad Sci USA 2019;116:20063–9.
Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155:688–98.
Sun H, Zhang Q, Jing YY, Zhang M, Wang HY, Cai Z, et al. USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat Commun. 2017;8:15534.
Zhang M, Zhang MX, Zhang Q, Zhu GF, Yuan L, Zhang DE, et al. USP18 recruits USP20 to promote innate antiviral response through deubiquitinating STING/MITA. Cell Res. 2016;26:1302–19.
Qin Y, Zhou MT, Hu MM, Hu YH, Zhang J, Guo L, et al. RNF26 temporally regulates virus-triggered type I interferon induction by two distinct mechanisms. PLoS Pathog. 2014;10:e1004358.
Zhang J, Hu MM, Wang YY, Shu HB. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J Biol Chem. 2012;287:28646–55.
Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity. 2009;30:397–407.
Hu MM, Yang Q, Xie XQ, Liao CY, Lin H, Liu TT, et al. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity. 2016;45:555–69.
Guo Y, Jiang F, Kong L, Wu H, Zhang H, Chen X, et al. OTUD5 promotes innate antiviral and antitumor immunity through deubiquitinating and stabilizing STING. Cell Mol Immunol. 2021;18:1945–55.
Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng MS, et al. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun. 2017;8:945.
Wang Y, Lian Q, Yang B, Yan S, Zhou H, He L, et al. TRIM30alpha Is a Negative-Feedback Regulator of the Intracellular DNA and DNA Virus-Triggered Response by Targeting STING. PLoS Pathog. 2015;11:e1005012.
Zhang HY, Liao BW, Xu ZS, Ran Y, Wang DP, Yang Y, et al. USP44 positively regulates innate immune response to DNA viruses through deubiquitinating MITA. PLoS Pathog. 2020;16:e1008178.
Nukui M, Roche KL, Jia J, Fox PL, Murphy EA. Protein S-Nitrosylation of Human Cytomegalovirus pp71 Inhibits Its Ability To Limit STING Antiviral Responses. J Virol. 2020;94:e00033–20.
Ren Y, Wang A, Wu D, Wang C, Huang M, Xiong X, et al. Dual inhibition of innate immunity and apoptosis by human cytomegalovirus protein UL37x1 enables efficient virus replication. Nat Microbiol. 2022;7:1041–53.
Albright ER, Mickelson CK, Kalejta RF. Human Cytomegalovirus UL138 Protein Inhibits the STING Pathway and Reduces Interferon Beta mRNA Accumulation during Lytic and Latent Infections. mBio. 2021;12:e0226721.
Zou HM, Huang ZF, Yang Y, Luo WW, Wang SY, Luo MH, et al. Human Cytomegalovirus Protein UL94 Targets MITA to Evade the Antiviral Immune Response. J Virol. 2020;94:e00022–20.
Huang ZF, Zou HM, Liao BW, Zhang HY, Yang Y, Fu YZ, et al. Human Cytomegalovirus Protein UL31 Inhibits DNA Sensing of cGAS to Mediate Immune Evasion. Cell Host Microbe. 2018;24:69–80.e64.
Choi HJ, Park A, Kang S, Lee E, Lee TA, Ra EA, et al. Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type I interferon immune responses. Nat Commun. 2018;9:125.
Biolatti M, Dell’Oste V, Pautasso S, Gugliesi F, von Einem J, Krapp C, et al. Human Cytomegalovirus Tegument Protein pp65 (pUL83) Dampens Type I Interferon Production by Inactivating the DNA Sensor cGAS without Affecting STING. J Virol. 2018;92;e01774–17.
Paijo J, Doring M, Spanier J, Grabski E, Nooruzzaman M, Schmidt T, et al. cGAS Senses Human Cytomegalovirus and Induces Type I Interferon Responses in Human Monocyte-Derived Cells. PLoS Pathog. 2016;12:e1005546.
Lou M, Huang D, Zhou Z, Shi X, Wu M, Rui Y, et al. DNA virus oncoprotein HPV18 E7 selectively antagonizes cGAS-STING-triggered innate immune activation. J Med Virol. 2023;95:e28310.
Yu K, Tian H, Deng H. PPM1G restricts innate immune signaling mediated by STING and MAVS and is hijacked by KSHV for immune evasion. Sci Adv. 2020;6:eabd0276.
Zhang G, Chan B, Samarina N, Abere B, Weidner-Glunde M, Buch A, et al. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc Natl Acad Sci USA 2016;113:E1034–43.
Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe. 2015;18:333–44.
Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, et al. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci USA 2015;112:E4306–15.
Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;567:262–6.
Zhang D, Liu Y, Zhu Y, Zhang Q, Guan H, Liu S, et al. A noncanonical cGAS-STING-PERK pathway facilitates the translational program critical for senescence and organ fibrosis. Nat Cell Biol. 2022;24:766–82.
Wu J, Dobbs N, Yang K, Yan N. Interferon-Independent Activities of Mammalian STING Mediate Antiviral Response and Tumor Immune Evasion. Immunity. 2020;53:115–26.e115.
Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28.
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–5.
Sumpter R Jr., Loo YM, Foy E, Li K, Yoneyama M, Fujita T, et al. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689–99.
Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA 2006;103:8459–64.
Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–45.
Naesens L, Haerynck F, Gack MU. The RNA polymerase III-RIG-I axis in antiviral immunity and inflammation. Trends Immunol. 2023;44:435–49.
Ran Y, Li D, Xiong MG, Liu HN, Feng T, Shi ZW, et al. African swine fever virus I267L acts as an important virulence factor by inhibiting RNA polymerase III-RIG-I-mediated innate immunity. PLoS Pathog. 2022;18:e1010270.
Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. 2009;10:1065–72.
Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–91.
Sun B, Sundstrom KB, Chew JJ, Bist P, Gan ES, Tan HC, et al. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci Rep. 2017;7:3594.
Moriyama M, Koshiba T, Ichinohe T. Influenza A virus M2 protein triggers mitochondrial DNA-mediated antiviral immune responses. Nat Commun. 2019;10:4624.
Liu H, Zhu Z, Xue Q, Yang F, Li Z, Xue Z, et al. Innate sensing of picornavirus infection involves cGAS-STING-mediated antiviral responses triggered by mitochondrial DNA release. PLoS Pathog. 2023;19:e1011132.
Aguirre S, Fernandez-Sesma A: Collateral Damage during Dengue Virus Infection: Making Sense of DNA by cGAS. J Virol. 2017;91:e01081–16.
Liu XN, Li LW, Gao F, Jiang YF, Yuan WZ, Li GX, et al. cGAS Restricts PRRSV Replication by Sensing the mtDNA to Increase the cGAMP Activity. Front Immunol. 2022;13:887054.
He WR, Cao LB, Yang YL, Hua D, Hu MM, Shu HB. VRK2 is involved in the innate antiviral response by promoting mitostress-induced mtDNA release. Cell Mol Immunol. 2021;18:1186–96.
Newman LE, Shadel GS. Mitochondrial DNA Release in Innate Immune Signaling. Annu Rev Biochem. 2023;92:299–332.
West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017;17:363–75.
Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–59.
Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40:e104705.
Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21:85–100.
Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477–89.
Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560:198–203.
Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–8.
Dombrowski Y, Peric M, Koglin S, Kaymakanov N, Schmezer V, Reinholz M, et al. Honey bee (Apis mellifera) venom induces AIM2 inflammasome activation in human keratinocytes. Allergy. 2012;67:1400–7.
Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell. 2017;171:1057–71.e1011.
Wu Z, Oeck S, West AP, Mangalhara KC, Sainz AG, Newman LE, et al. Mitochondrial DNA Stress Signaling Protects the Nuclear Genome. Nat Metab. 2019;1:1209–18.
Lei Y, VanPortfliet JJ, Chen YF, Bryant JD, Li Y, Fails D, et al. Cooperative sensing of mitochondrial DNA by ZBP1 and cGAS promotes cardiotoxicity. Cell. 2023;186:3013–32.e3022.
Wallace DC. Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu Rev Biochem. 2007;76:781–821.
Bayona-Bafaluy MP, Fernandez-Silva P, Enriquez JA. The thankless task of playing genetics with mammalian mitochondrial DNA: a 30-year review. Mitochondrion. 2002;2:3–25.
Han C, Liu Z, Zhang Y, Shen A, Dong C, Zhang A, et al. Tumor cells suppress radiation-induced immunity by hijacking caspase 9 signaling. Nat Immunol. 2020;21:546–54.
Yamazaki T, Kirchmair A, Sato A, Buque A, Rybstein M, Petroni G, et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol. 2020;21:1160–71.
Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41:843–52.
Willemsen J, Neuhoff MT, Hoyler T, Noir E, Tessier C, Sarret S, et al. TNF leads to mtDNA release and cGAS/STING-dependent interferon responses that support inflammatory arthritis. Cell Rep. 2021;37:109977.
Aarreberg LD, Esser-Nobis K, Driscoll C, Shuvarikov A, Roby JA, Gale M Jr. Interleukin-1beta Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol Cell. 2019;74:801–15.e806.
Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–56.
Lepelley A, Della Mina E, Van Nieuwenhove E, Waumans L, Fraitag S, Rice GI, et al. Enhanced cGAS-STING-dependent interferon signaling associated with mutations in ATAD3A. J Exp Med. 2021;218:e20201560.
Chen L, Dong J, Liao S, Wang S, Wu Z, Zuo M, et al. Loss of Sam50 in hepatocytes induces cardiolipin-dependent mitochondrial membrane remodeling to trigger mtDNA release and liver injury. Hepatology. 2022;76:1389–408.
Cobo I, Tanaka TN, Chandra Mangalhara K, Lana A, Yeang C, Han C, et al. DNA methyltransferase 3 alpha and TET methylcytosine dioxygenase 2 restrain mitochondrial DNA-mediated interferon signaling in macrophages. Immunity. 2022;55:1386–401.e1310.
Xian H, Watari K, Sanchez-Lopez E, Offenberger J, Onyuru J, Sampath H, et al. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity. 2022;55:1370–85.e1378.
Alam K, Moinuddin, Jabeen S. Immunogenicity of mitochondrial DNA modified by hydroxyl radical. Cell Immunol. 2007;247:12–7.
Pazmandi K, Agod Z, Kumar BV, Szabo A, Fekete T, Sogor V, et al. Oxidative modification enhances the immunostimulatory effects of extracellular mitochondrial DNA on plasmacytoid dendritic cells. Free Radic Biol Med. 2014;77:281–90.
Bernardi P, Rasola A, Forte M, Lippe G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol Rev. 2015;95:1111–55.
Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. 2019;366:1531–6.
Krauskopf A, Eriksson O, Craigen WJ, Forte MA, Bernardi P. Properties of the permeability transition in VDAC1(-/-) mitochondria. Biochim Biophys Acta. 2006;1757:590–5.
Liu H, Fan H, He P, Zhuang H, Liu X, Chen M, et al. Prohibitin 1 regulates mtDNA release and downstream inflammatory responses. EMBO J. 2022;41:e111173.
Xu X, Cai J, Wang X, Lu Y, Guo B, Lai M, et al. Human cytomegalovirus infection activates NLRP3 inflammasome by releasing mtDNA into the cytosol in human THP-1 cells. Microbiol Immunol. 2023;67:303–13.
Combs JA, Norton EB, Saifudeen ZR, Bentrup KHZ, Katakam PV, Morris CA, et al. Human Cytomegalovirus Alters Host Cell Mitochondrial Function during Acute Infection. J Virol. 2020;94:e01183–19
Domizio JD, Gulen MF, Saidoune F, Thacker VV, Yatim A, Sharma K, et al. The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature. 2022;603:145–51.
Li G, Tang Z, Fan W, Wang X, Huang L, Jia Y, et al. Atlas of interactions between SARS-CoV-2 macromolecules and host proteins. Cell Insight. 2023;2:100068.
Buque A, Rodriguez-Ruiz ME, Fucikova J, Galluzzi L. Apoptotic caspases cut down the immunogenicity of radiation. Oncoimmunology. 2019;8:e1655364.
Ning X, Wang Y, Jing M, Sha M, Lv M, Gao P, et al. Apoptotic Caspases Suppress Type I Interferon Production via the Cleavage of cGAS, MAVS, and IRF3. Mol Cell. 2019;74:19–31.e17.
Crowl JT, Gray EE, Pestal K, Volkman HE, Stetson DB. Intracellular Nucleic Acid Detection in Autoimmunity. Annu Rev Immunol. 2017;35:313–36.
Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N. Engl J Med. 2014;371:507–18.
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22:146–53.
Cao DJ, Schiattarella GG, Villalobos E, Jiang N, May HI, Li T, et al. Cytosolic DNA Sensing Promotes Macrophage Transformation and Governs Myocardial Ischemic Injury. Circulation. 2018;137:2613–34.
Lai L, Zhang A, Yang B, Charles EJ, Kron IL, Yang Z. Plasmacytoid Dendritic Cells Mediate Myocardial Ischemia/Reperfusion Injury by Secreting Type I Interferons. J Am Heart Assoc. 2021;10:e020754.
Rech L, Abdellatif M, Pottler M, Stangl V, Mabotuwana N, Hardy S, et al. Small molecule STING inhibition improves myocardial infarction remodeling. Life Sci. 2022;291:120263.
Han J, Dai S, Zhong L, Shi X, Fan X, Zhong X, et al. GSDMD (Gasdermin D) Mediates Pathological Cardiac Hypertrophy and Generates a Feed-Forward Amplification Cascade via Mitochondria-STING (Stimulator of Interferon Genes) Axis. Hypertension. 2022;79:2505–18.
Yang LL, Xiao WC, Li H, Hao ZY, Liu GZ, Zhang DH, et al. E3 ubiquitin ligase RNF5 attenuates pathological cardiac hypertrophy through STING. Cell Death Dis. 2022;13:889.
Cho CS, Park HW, Ho A, Semple IA, Kim B, Jang I, et al. Lipotoxicity induces hepatic protein inclusions through TANK binding kinase 1-mediated p62/sequestosome 1 phosphorylation. Hepatology. 2018;68:1331–46.
Qiao JT, Cui C, Qing L, Wang LS, He TY, Yan F, et al. Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism. 2018;81:13–24.
Luo W, Wang Y, Zhang L, Ren P, Zhang C, Li Y, et al. Critical Role of Cytosolic DNA and Its Sensing Adaptor STING in Aortic Degeneration, Dissection, and Rupture. Circulation. 2020;141:42–66.
Mao Y, Luo W, Zhang L, Wu W, Yuan L, Xu H, et al. STING-IRF3 Triggers Endothelial Inflammation in Response to Free Fatty Acid-Induced Mitochondrial Damage in Diet-Induced Obesity. Arterioscler Thromb Vasc Biol. 2017;37:920–9.
King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP Jr., et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. 2017;23:1481–7.
Oduro PK, Zheng X, Wei J, Yang Y, Wang Y, Zhang H, et al. The cGAS-STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy. Acta Pharm Sin B. 2022;12:50–75.
Paul BD, Snyder SH, Bohr VA. Signaling by cGAS-STING in Neurodegeneration, Neuroinflammation, and Aging. Trends Neurosci. 2021;44:83–96.
Cao Y, Wang Y, Yang J. NAD(+)-dependent mechanism of pathological axon degeneration. Cell Insight. 2022;1:100019.
Hou Y, Wei Y, Lautrup S, Yang B, Wang Y, Cordonnier S, et al. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc Natl Acad Sci. 2021;118:e2011226118.
Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–62.
Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. 2019;20:657–74.
Van Herck S, Feng B, Tang L. Delivery of STING agonists for adjuvanting subunit vaccines. Adv Drug Deliv Rev. 2021;179:114020.
Meric-Bernstam F, Sweis RF, Kasper S, Hamid O, Bhatia S, Dummer R, et al. Combination of the STING Agonist MIW815 (ADU-S100) and PD-1 Inhibitor Spartalizumab in Advanced/Metastatic Solid Tumors or Lymphomas: An Open-Label, Multicenter, Phase Ib Study. Clin Cancer Res. 2023;29:110–21.
Chang W, Altman MD, Lesburg CA, Perera SA, Piesvaux JA, Schroeder GK, et al. Discovery of MK-1454: A Potent Cyclic Dinucleotide Stimulator of Interferon Genes Agonist for the Treatment of Cancer. J Med Chem. 2022;65:5675–89.
Corrales L, Gajewski TF. Molecular Pathways: Targeting the Stimulator of Interferon Genes (STING) in the Immunotherapy of Cancer. Clin Cancer Res. 2015;21:4774–9.
Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11:1018–30.
Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018;564:439–43.
Gall B, Pryke K, Abraham J, Mizuno N, Botto S, Sali TM, et al. Emerging Alphaviruses Are Sensitive to Cellular States Induced by a Novel Small-Molecule Agonist of the STING Pathway. J Virol. 2018:92;e01913–17.
Zhang X, Liu B, Tang L, Su Q, Hwang N, Sehgal M, et al. Discovery and Mechanistic Study of a Novel Human-Stimulator-of-Interferon-Genes Agonist. ACS Infect Dis. 2019;5:1139–49.
Pan BS, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, et al. An orally available nonnucleotide STING agonist with antitumor activity. Science. 2020;369:eaba6098.
Chin EN, Yu C, Vartabedian VF, Jia Y, Kumar M, Gamo AM, et al. Antitumor activity of a systemic STING-activating nonnucleotide cGAMP mimetic. Science. 2020;369:993–9.
Zhang Y, Sun Z, Pei J, Luo Q, Zeng X, Li Q, et al. Identification of alpha-Mangostin as an Agonist of Human STING. ChemMedChem. 2018;13:2057–64.
Banerjee M, Middya S, Shrivastava R, Basu S, Ghosh R, Pryde DC, et al. G10 is a direct activator of human STING. PLoS One. 2020;15:e0237743.
Liu B, Tang L, Zhang X, Ma J, Sehgal M, Cheng J, et al. A cell-based high throughput screening assay for the discovery of cGAS-STING pathway agonists. Antivir Res. 2017;147:37–46.
Siu T, Altman MD, Baltus GA, Childers M, Ellis JM, Gunaydin H, et al. Discovery of a Novel cGAMP Competitive Ligand of the Inactive Form of STING. ACS Med Chem Lett. 2019;10:92–7.
Li S, Hong Z, Wang Z, Li F, Mei J, Huang L, et al. The Cyclopeptide Astin C Specifically, Inhibits the Innate Immune CDN Sensor STING. Cell Rep. 2018;25:3405–21.e3407.
Hansen AL, Mukai K, Schopfer FJ, Taguchi T, Holm CK. STING palmitoylation as a therapeutic target. Cell Mol Immunol. 2019;16:236–41.
Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. Targeting STING with covalent small-molecule inhibitors. Nature. 2018;559:269–73.
Hansen AL, Buchan GJ, Ruhl M, Mukai K, Salvatore SR, Ogawa E, et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc Natl Acad Sci USA 2018;115:E7768–75.
Hall J, Brault A, Vincent F, Weng S, Wang H, Dumlao D, et al. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS One. 2017;12:e0184843.
Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun. 2017;8:750.
Lama L, Adura C, Xie W, Tomita D, Kamei T, Kuryavyi V, et al. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat Commun. 2019;10:2261.
An J, Woodward JJ, Sasaki T, Minie M, Elkon KB. Cutting edge: Antimalarial drugs inhibit IFN-beta production through blockade of cyclic GMP-AMP synthase-DNA interaction. J Immunol. 2015;194:4089–93.
An J, Minie M, Sasaki T, Woodward JJ, Elkon KB. Antimalarial Drugs as Immune Modulators: New Mechanisms for Old Drugs. Annu Rev Med. 2017;68:317–30.
Wang M, Sooreshjani MA, Mikek C, Opoku-Temeng C, Sintim HO. Suramin potently inhibits cGAMP synthase, cGAS, in THP1 cells to modulate IFN-beta levels. Future Med Chem. 2018;10:1301–17.
Dubensky TW Jr., Kanne DB, Leong ML. Rationale, progress and development of vaccines utilizing STING-activating cyclic dinucleotide adjuvants. Ther Adv Vaccines. 2013;1:131–43.
Ding C, Song Z, Shen A, Chen T, Zhang A. Small molecules targeting the innate immune cGAS‒STING‒TBK1 signaling pathway. Acta Pharm Sin B. 2020;10:2272–98.
Xi Q, Wang M, Jia W, Yang M, Hu J, Jin J, et al. Design, Synthesis, and Biological Evaluation of Amidobenzimidazole Derivatives as Stimulator of Interferon Genes (STING) Receptor Agonists. J Med Chem. 2020;63:260–82.
Shi J, Liu CL, Zhang B, Guo WJ, Zhu J, Chang CY, et al. Genome mining and biosynthesis of kitacinnamycins as a STING activator. Chem Sci. 2019;10:4839–46.
Padilla-Salinas R, Sun L, Anderson R, Yang X, Zhang S, Chen ZJ, et al. Discovery of Small-Molecule Cyclic GMP-AMP Synthase Inhibitors. J Org Chem. 2020;85:1579–600.
Acknowledgements
The work in the authors’ laboratory was supported by grants from the State Key R&D Program of China (2022YFA1304900), the Fundamental Research Funds for the Central Universities (2042022dx0003), the National Natural Science Foundation of China (32188101, 31830024), the CAMS Innovation Fund for Medical Sciences (2019-I2M-5-071), and the Translational Medicine and Interdisciplinary Research Joint Fund of Zhongnan Hospital of Wuhan University (Grant No. ZNJC202205).
Author information
Authors and Affiliations
Contributions
H-BS conceived the framework of this review; M-MH and H-BS wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, MM., Shu, HB. Mitochondrial DNA-triggered innate immune response: mechanisms and diseases. Cell Mol Immunol 20, 1403–1412 (2023). https://doi.org/10.1038/s41423-023-01086-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-023-01086-x
