
Abstract
The ever-growing research on lymphatic biology has clearly identified lymphatic vessels as key players that maintain human health through their functional roles in tissue fluid homeostasis, immunosurveillance, lipid metabolism and inflammation. It is therefore not surprising that the list of human diseases associated with lymphatic malfunctions has grown larger, including issues beyond lymphedema, a pathology traditionally associated with lymphatic drainage insufficiency. Thus, the discovery of factors and pathways that can promote optimal lymphatic functions may offer new therapeutic options. Accumulating evidence indicates that aside from biochemical factors, biomechanical signals also regulate lymphatic vessel expansion and functions postnatally. Here, we review how mechanical forces induced by fluid shear stress affect the behavior and functions of lymphatic vessels and the mechanisms lymphatic vessels employ to sense and transduce these mechanical cues into biological signals.
Similar content being viewed by others

Introduction
The advances in molecular, cellular, genetic, and imaging approaches in the past decade have led to the identification of specific lymphatic endothelial cell (LEC) markers, allowing the distinction between blood and lymphatic vessels and identification of the mechanisms controlling the development and stabilization of lymphatic vessels and their functions. Together, these discoveries have changed our perception of the lymphatic vasculature; namely, it is no longer believed to be a secondary vascular system but rather a system that is vital for human well-being and health. Indeed, the lymphatic system, with its extensive plexus of vessels and interconnected lymph nodes, plays essential roles in tissue homeostasis by clearing waste, in immunosurveillance by controlling immune cell trafficking and responses, and in organ morphogenesis by coordinating tissue regeneration [1, 2]. Moreover, the importance of the functional roles of lymphatic vessels in humans diseases is being increasingly recognized. Until recently, the traditional pathology associated with malfunctions in lymphatics was lymphedema. However, with increasing research on lymphatics, novel functional roles of the lymphatic vasculature in diseases have been discovered. As such, the number of human diseases or disorders associated with lymphatic defects has increased, and inflammatory bowel diseases, eye diseases, neurological disorders, obesity, and cardiovascular diseases are now included [2]. These new findings imply that restoring optimal lymphatic function may be a promising nonconventional strategy to managing these diseases. Thus, a deeper understanding of the mechanisms regulating lymphatic physiology and functions will be useful for the design of such an approach. In adult tissues, lymphatic vessel phenotype and behavior has been shown to be shaped by biochemical signals such as vascular endothelial growth factors (VEGFs). However, other cell-extrinsic factors likely impact lymphatic vessels. For example, cells commonly experience different mechanical forces triggered by changes in the magnitude or type of fluid shear stress, extracellular matrix composition, and stiffness. Likewise, emerging evidence indicates that lymphatics can also sense and transduce nearby mechanical forces into signals that regulate molecular pathway gene expression, controlling LEC cytoskeleton remodeling, proliferation, migration, the cell cycle, permeability and other functions [3,4,5]. The effect of changes in fluid osmolarity, hydraulic pressure, temperature, matrix composition and stiffness in the microenvironment has been reviewed [6, 7]. Therefore, in this review, we focus our attention on mechanical forces generated by interstitial fluid flow experienced by lymphatic vessels and describe the responses induced by this type of mechanical force in lymphatics, as well as the underlying sensing and mechanotransduction mechanisms, after summarizing the biological functions of lymphatic vessels in health and diseases.
Biological functions of lymphatic vessels and implications in diseases
Here, we provide a brief overview of conventional and newly discovered biological functions of lymphatic vessels and their implications in health and diseases, as this topic has been well described in recent reviews [1, 2].
Lymphatic fluid and lipid transport
Lymphatic vessels have long been recognized to function in the transport of interstitial fluid from the tissue to the draining lymph node (LN) and back to the venous system via the thoracic duct (Fig. 1). Thus, defective lymphatic drainage leads to fluid accumulation and subsequent tissue swelling. This process results in a disorder known as lymphedema, and it can be either congenital (primary lymphedema) or acquired (secondary lymphedema) [8]. Numerous genes associated with primary lymphedema have been identified, and they often overlap with genes implicated in the development of lymphatic vessels [2]. Secondary lymphedema occurs because of surgery, radiation therapy, infection or trauma that damages lymphatic vessels and compromises their draining function. Lymphedema is a progressive and chronic disease that lacks a cure [9, 10]. In addition to fluid transport, lymphatic vessels are also involved in lipid transport. Lacteals together with submucosal lymphatic vessels form the intestinal lymphatic vasculature and control the absorption of dietary fat. Briefly, dietary lipids are absorbed by enterocytes and transformed into large triglyceride-loaded lipoprotein particles, called chylomicrons. These lipoproteins are taken up by lacteals, blunt-ended initial lymphatics that transport them into the intestinal submucosal lymphatics and mesenteric collecting vessels. Intestinal then lymph drain to the mesenteric LNs and thoracic duct and further to the blood circulation [1]. Lacteals are surrounded by longitudinal villus SMCs, which facilitate efficient drainage of absorbed lipids by actively contracting and squeezing lacteals [11]. Because lacteals control dietary lipid absorption, they are also involved in regulating body weight [12,13,14,15]. Moreover, maintaining a healthy and functional lymphatic vasculature might be important for preventing metabolic diseases and obesity since a growing body of evidence indicates that lipid escape from leaky lymphatics can lead to fat accumulation [16,17,18,19]. Similarly, lymphatic vessels are involved in the transport of lipoproteins from the interstitial tissue to the blood [20,21,22]. The idea that lymphatic vessels function in the transport of lipoproteins from interstitial tissue to blood was proposed in the late 1980s [23], and this lymphatic function was revisited more recently by two separate groups. The study of Martel et al. [24] in Chy mice, a model of dermal lymphatic insufficiency, and our study [25] using wild-type mice in which lymphatic vessels were surgically excised provided the first clear, direct evidence that impaired lymphatic drainage significantly decreases the efficiency of cholesterol transport mediated by high-density lipoprotein (HDL) from the interstitium back to the blood circulation, a process known as reverse cholesterol transport (RCT). Excess cellular cholesterol can be highly toxic and eventually leads to cell death. RCT regulates the abundance of the intracellular cholesterol pool by removing excess cholesterol from peripheral tissues and transporting it to the liver via plasma for excretion [26]. We also further proposed that the transport of effluxed cholesterol from peripheral tissue to the circulation via lymphatics not only is passive as believed but also can be an active process dependent on HDL and scavenger receptor class BI (SR-BI) [25]. These findings support the concept that lymphatic vessels tightly regulate the clearance of cholesterol from the peripheral tissues through the transport of HDL-cholesterol [21]. Notably, this concept may also be true in humans based on a few case reports describing skin cholesterol deposition in primary lymphedema patients [27,28,29,30,31]. Because they can prevent cholesterol and other lipids from accumulating in tissues, lymphatic functions may play a role in cardiac- and metabolic diseases associated with lipid alterations, such as atherosclerosis and diabetes. Thus, strategies promoting efficient lymphatic drainage might be useful for these diseases [32,33,34,35,36].

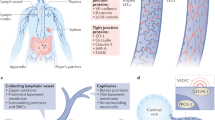
Structure and function of the lymphatic system. A The lymphatic vasculature (green) forms part of the circulatory system. Fluid that extravasates from the blood capillary bed into the tissue interstitium is absorbed into initial lymphatics vessels and flows through larger collecting lymphatic vessels that actively transport lymph fluid into draining lymph nodes before returning into the venous system via the thoracic duct. B Interstitial fluid, macromolecules and immune cells leave the tissue interstitium to enter discontinuous button-like initial lymphatic vessels that lack a continuous basement membrane. Collecting lymphatic vessels have a continuous basement membrane, smooth muscle cell coverage to provide contractile activity to assist blood flow and intraluminal valves to prevent lymph backflow. Collecting LECs are organized into tight continuous zipper-like junctions and do not absorb fluid from surrounding tissues. C Initial lymphatic vessels are composed of overlapping LECs that allow interstitial components to enter the vessels when interstitial pressure is high. The overlapping cells also act as valves, preventing fluid from leaking out. Anchoring filaments connect LECs to the surrounding extracellular matrix and facilitate fluid, macromolecule and cell entry into initial lymphatic vessels. D The collecting lymphatic vessels are composed of several lymphangions that propagate lymph flow. Coordinated contraction/expansion of each lymphangion and opening/closing of intraluminal valves ensure efficient lymph transport. LEC lymphatic endothelial cell, BM basement membrane, SMC smooth muscle cell. Created with BioRender.com
Lymphatic control of immune cell trafficking
In addition to fluid and lipid transport, lymphatic vessels transport soluble antigens and immune cells from peripheral tissues into regional LNs via afferent lymph (Fig. 1B). Classic cannulation studies carried out in humans and small and large animals have revealed that the major migrating cells in normal afferent lymph are T lymphocytes (80–90%), followed by antigen-presenting dendritic cells (DCs) and very small numbers of B lymphocytes, which together account for most of the remaining 10–15% of cells. The number of CD4-expressing T lymphocytes is much higher than that of CD8+ T cells [37,38,39]. Only low numbers of naive T cells are usually present in afferent lymph, and most of the T cells are antigen- experienced memory cells [40, 41], although a significant proportion of CD4+ T cells are T regulatory cells, as recently demonstrated using photoconvertible Kaede mice [42]. Advances in in vivo and in vitro methods to assess cell migration, such as time-lapse imaging in tissue explants and intravital microscopy [43, 44], have greatly contributed to elucidating the cellular and molecular mechanisms controlling lymphatic migration of DCs and T cells (as reviewed [43, 45, 46]). The chemokine receptor CCR7 expressed on the surface of DCs and its ligand CCL21 produced by lymphatic endothelial cells (LECs) [47,48,49] are the main molecules involved in DC migration via afferent lymphatics under steady-state and inflammatory conditions; CCR7 and CCL21 guide DCs through the interstitium toward afferent lymphatics and within the initial lymphatics toward collecting vessels [50,51,52]. Although CCR7 deficiency or blockade of CCL21 significantly impairs DC migration to draining LNs, other chemokines have been found to be involved in this process [43, 53, 54], including CXCL12 and CX3CL1, which bind to DC-expressed CXCR4 and CX3CR1, respectively [53, 54]. In contrast to leukocyte extravasation from blood vessels, which is a highly integrin-dependent process, DC migration into and through afferent lymphatics in steady state conditions is independent of DC-expressed integrins [55] and only becomes integrin-dependent when LECs upregulate ICAM-1 and VCAM-1 in response to inflammation [56, 57]. Similar to DCs, the CCR7-CCL21 pathway mediates T-cell migration from peripheral tissue via afferent lymphatics in steady state and in inflammation [58].
Lymphatic vessels in the LNs also play an important role in the trafficking and positioning of immune cells within the LNs, which are critical for proper immune responses [59]. In the LN, fibroblastic reticular cells are the major source of CCL21, and LECs lining the lymphatic sinus “ceiling” express high levels of a decoy receptor for CCL21, called CCRL1. This organization creates a CCL21 gradient supporting the migration of DCs into the LN medulla [60]. Subcapsular sinus LSCs have also been shown to control the entry of lymphocytes and soluble antigens into the LN parenchyma through the expression of plasmalemma vesicle-associated protein (PLVAP), which is normally restricted to fenestrated blood endothelial cells [61]. Finally, LN LECs control the egress of activated T cells such as effector T cells from LNs by releasing the signaling phospholipid sphingosine-1-phosphate (S1P) into the efferent lymph, which attracts T cells through its binding to the S1P receptor S1P1 [62, 63]. Notably, S1P is also produced by LECs in peripheral tissues and affects DC migration into draining LNs [63,64,65].
Lymphatic regulation of T-cell responses
In addition to contributing to immunity through the transport of soluble antigens and migration of immune cells, lymphatic vessels have emerged as key regulators of T cells responses in the past decade (as reviewed [66]). Several studies in mice have reported that steady-state LN LECs contribute to peripheral T-cell tolerance by presenting endogenously expressed tissue-restricted antigens [67, 68] through MHC class I (MHCI) molecules and eliminating autoreactive CD8+ T cells [69,70,71]. LN LECs can also control the elimination of antigen-specific CD8+ T cells by cross-presenting exogenous antigens in MHCI molecules, which in turn leads to T-cell apoptosis [72]. Although several studies indicate that LN LECs can also impact peripheral CD4+ T-cell responses, further studies in different immunological settings are necessary to confirm this role [66]. Indeed, the study by Rouhani et al. [73] showed that LECs were incapable of presenting antigenic peptides via MHC class II (MHCII) since they do not express H2-M at steady state. In contrast, Dubrot et al. reported that in addition to inducing antigen-specific CD4+ T-cell tolerance by presenting peptide-MHCII complexes acquired from DCs, LECs endogenously express MHCII molecules through the IFN-γ-inducible promoter IV (pIV) of the MHC class II (MHCII) transactivator CIITA [74]. Recently, the same group provided evidence that the absence of MHCII expression in LN LECs in aging mice impairs peripheral CD4+ T-cell tolerance by promoting defective regulatory T cells and increasing effector T cells, which results in peripheral organ T-cell infiltration and autoantibody production [75], further supporting the potential role of LECs in peripheral CD4+ T-cell responses.
Furthermore, LECs have been shown to influence naive T-cell survival through the release of S1P, which stimulates mitochondrial function [76]. LN LECs are also capable of dampening T-cell activation and proliferation during inflammation either directly through the production of nitric oxide induced in response to T-cell cytokines [77] or indirectly by controlling the maturation of DCs [78, 79]. Thus, lymphatic vessels participate in important processes in immunosurveillance and immunomodulation by regulating immune cells, soluble antigen transport and T-cell responses. Lymphatic vessels have been shown to play active roles in infectious and inflammatory diseases, tissue transplants and cancer, which are often associated with the expansion of lymphatic vessels, through these immune functions [2].
In cancer, lymphatic vessels have been found to play a role in the spreading of solid tumor cells to distant organs by serving as channels connecting the primary tumors to LNs. During cancer progression, tumor cells and cells from the tumor environment produce growth factors that promote lymphatic cell proliferation, sprouting and enlargement in and around solid tumors, a process known as lymphangiogenesis. Tumor lymphangiogenesis correlates with metastasis and poor patient prognosis in several cancer types (as discussed [80]). For example, in melanoma patients, intratumoral lymphatics are associated with distant metastasis [81] and poor disease- free survival [82], and lymphatic vessel area is associated with poor overall survival [83]. Similarly, in colorectal, breast and lung cancers high lymphatic vessel density is an indicator of lymph node metastasis and associated with reduced overall survival [84,85,86,87,88,89]. In mice, lymphangiogenesis has also been shown to promote the dissemination of metastases to distal organs [90, 91]. Therefore, therapies aimed at blocking tumor lymphangiogenesis are currently being developed as promising approaches for the treatment of malignancies such as melanoma and colorectal cancer [92]. However, LECs embedded in the tumor environment may also modulate tumor immunity [93] and influence anticancer therapy responses. Notably, recent studies suggest that tumor-associated lymphatic vessels may exhibit positive and/or negative effects on tumor immunity depending on the tumor stage and the immunological environments of the tumor (which differ in cases of immune evasion/immune subversion or with immunotherapy) [66]. Tumor- associated lymphatic vessels are necessary for the initiation of antitumoral responses because they support DC trafficking [94, 95], and tumor-associated lymphangiogenesis in melanoma can potentiate immunotherapy by promoting T-cell infiltration [96]. However, LECs in the tumor environment can exhibit immunosuppressive functions that will conversely reduce antitumor immunity. During tumor progression, LECs have been shown to upregulate PD-L1 expression in response to IFNy secreted by tumor-specific CD8+ T cells and subsequently suppress T-cell accumulation in tumors [97, 98]. Although further investigations are needed to decipher the specific contributions of lymphatics to antitumor immunity, these findings reveal the potential utility of targeting lymphatic functions during tumor development. Moreover, it is unlikely that the regulatory roles of lymphatics will be limited to T cells given that LECs can potentially interact with a variety of immune cells depending on the tissue/organ in which they reside and shape their responses through the expression of multiple growth factors, cytokines and chemokines.
The finding that LECs in LNs affect medullary macrophages through production of the survival factor colony-stimulating factor-1 supports this idea [99]. Thus, the list of immune cells regulated by LECs is expected to grow. Furthermore, given that lymphatic vessels orchestrate immune responses and disposal of tissue fluid, proteins, and lipids, it is reasonable to postulate that lymphatic functions are important in many diseases associated with the accumulation of such biological waste and immune dysfunction, including cardiovascular diseases (e.g., myocardial infarction, atherosclerosis), ocular diseases (e.g., glaucoma), Crohn’s disease and neurological diseases (Alzheimer’s disease, Parkinson’s disease, stroke) [1, 100,101,102].
Adult lymphatic vessel anatomy and plasticity
The lymphatic system is composed of an extensive network of vessels and interconnected lymph nodes. This unidirectional vascular network includes blind-ended capillaries or initial lymphatic vessels and larger lymphatic vessels, known as the collecting lymphatic vessels, on which the initial lymphatics converge (Fig. 1). Both lymphatic types are lined with LECs that express platelet endothelial cell adhesion molecule (PECAM-1 or CD31), a marker shared with blood endothelial cells (BECs), the homeobox transcription factor prospero-related homeobox 1 (PROX1) and the receptor tyrosine kinase vascular endothelial growth factor (VEGF) receptor-3 (VEGFR-3) [103, 104]. Initial lymphatic vessels can be identified by the expression of podoplanin and lymphatic vessel hyaluronan receptor 1 (LYVE-1) [105] and the absence of mural cells (e.g. pericyte or smooth muscle cells). They are characterized by a discontinuous basal membrane and specialized button-like cell junctions that allow interstitial fluid, lipids, soluble antigens and immune cells to have easy access to the lumen of the initial lymphatics [106] (Fig. 1B). Moreover, fluid enters through the opening of overlapping flaps localized between the button junctions of adjacent LECs that are operated by anchoring filaments that tether the initial lymphatics to surrounding extracellular matrix and sense changes in interstitial pressure [107, 108] (Fig. 1C). Under conditions of increased interstitial pressure, the anchoring filaments pull the LECs and open the overlapping flaps, which allows the uptake of lymph tissue fluid [109, 110]. Once the vessel is filled, the increasing luminal pressure closes the overlaps, thus preventing leakage of lymph out of the vessels [111]. The overlapping flaps also serve as lymphatic valves to prevent backflow of fluid from the lymphatic lumen (Fig. 1C). To our knowledge, few molecules regulating the connections of LECs with the anchoring filaments and extracellular matrix have been identified thus far. Among those that have been identified, the elastic microfibril-associated protein Emilin1 is highly expressed in LECs and is a component of the anchoring filaments in lymphatic vessels [112]. Deficiency in murine Emilin1 leads to structural defects in lymphatic vasculature, including reduction in anchoring filaments, lymphatic hyperplasia and disorganization, which impair lymphatic drainage and lead to lymph leakage. Collecting vessels equipped with zipper-like junctions also express podoplanin but downregulate LYVE-1 expression. They are covered with a basement membrane and circumferential smooth muscle cells and have luminal valves that propel and maintain unidirectional lymph flow to draining lymph nodes and eventually into the venous circulation via the thoracic duct (Fig. 1B, D) [113, 114]. Unlike the blood system, the lymphatic system does not possess a central pump; instead, lymph is propelled against an overall hydraulic pressure gradient from interstitial spaces to central veins thanks to two pumping mechanisms, which rely on extrinsic forces (contraction of surrounding skeletal muscles and arterial pulsations) or the intrinsic rhythmic contractility of lymphatic muscle cells. Coordinated contraction of muscle cells facilitates transport of lymph back to the blood circulation [115]. Notably, lymphatic contraction malfunctions, barrier dysfunction and valve defects in collecting vessels have been associated with pathologies either directly involving the lymphatic system, such as primary and secondary lymphedema, or indirectly involving the lymphatic system, such as inflammation, obesity, metabolic syndrome, and inflammatory bowel disease [116].
Recent progress in lymphatic biology research has unveiled the plasticity of adult lymphatic vessels, which can actively sense and adapt to changes within their tissue environment [117]. Notably, inflammation triggered by pathogen and nonpathogen (damaged cells, irritants) stimuli has been shown to alter the structure and function of lymphatic vessels in adult mammals [118]. Postnatal lymphangiogenesis, the formation of new lymphatic vessels from pre-existing ones, has been shown to occur in experimental and clinical inflammatory conditions including renal [119, 120] and corneal transplant rejection [121,122,123], inflammatory bowel disease [124,125,126,127], rheumatoid arthritis [128,129,130], chronic airway inflammation [131, 132], atopic dermatitis and psoriasis [133,134,135]. To date, several factors have been reported to promote lymphatic vessel expansion in vitro and in animal models including VEGF-A, VEGFC, VEGF-D, FGF-2, PDGF, IGF-1, IGF-2, angiopoietin-1, and HGF [107, 136], and the list continues to rapidly expand. VEGF-C and its receptor VEGFR-3 are the main drivers of both developmental and inflammatory lymphangiogenesis (as reported [117]) and are believed to be the most promising therapeutic targets for the treatment of human lymphedema [136]. In addition, cytokines and chemokines can also regulate lymphatic expansion directly or indirectly by inducing VEGF-C [137, 138].
Despite the undisputable role of the above biochemical signals in controlling lymphatic expansion and behavior during physiological and pathophysiological conditions, it is conceivable that lymphatic vessels also respond to mechanical signals produced by changes in microenvironment surrounding lymphatic vessels, which in turn impacts lymphatic vasculature and functions.
Biomechanical control of lymphatic physiology and functions
Lymphatics are constantly exposed to shear stress produced by fluid flow at their luminal and abluminal surface. During lymph fluid uptake by the initial lymphatics, LECs may encounter interstitial fluid flow with in basal-to-luminal (or transmural) direction at their intercellular junctions. The lymph fluid then flows into the downstream collecting lymphatics, inflicting laminar shear stress on the luminal surface of capillary LECs. The collecting lymphatics transport lymph to the draining lymph node and back to the venous circulation. LECs lining the collecting vessels may more frequently face oscillatory flows. Moreover, lymphatic vessels are surrounded by a wide range of supportive structural cells, such as SMCs, in collecting vessels and diverse extracellular matrix components forming the basement membrane of the vessels and filling the interstitial space, which can exert other physical forces, such as stretching and stiffness forces. Thus, it is proposed that lymphatic vessels are exposed to several physical forces, and they are capable of translating this mechanical information into biological responses such as vessel remodeling, expansion, and stabilization, which are important for maintaining their physiological functions [4,5,6, 139]. Alterations in mechanical forces are often observed during disease and may result in abnormal lymphatic structure and function and thus contribute to pathology.
Therefore, it is critical to understand which aspects of lymphatic vessel biology are influenced by mechanical forces and to characterize the mechanisms by which lymphatics sense and respond to physical forces. In this section of the review, we first focus our attention on describing in vitro and in vivo studies supporting the responses to fluid-induced mechanical forces in lymphatics followed by a description of mechanotransduction and sensing mechanisms employed by lymphatics in response to fluid shear stress (FSS) (Fig. 2).

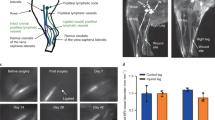
Fluid shear stress induced changes in lymphatics. A Effects of fluid shear stress on lymphatic vasculature and function. B Mechanosensory and mechanotransduction pathways in lymphatic endothelial cells. Lymphatic endothelial cells sense shear stress produced by interstitial fluid flow via 2 possible mechanisms. The mechanosensory complex, including PECAM, VE-cadherin, VEGFR2 and VEGFR3, senses fluid shear stress and activates the PI3K/Akt pathway, induces cytoskeleton reorganization and regulates YAP/TAZ signaling. In addition to effects on this complex, flow sensing leads to the activation of VEGFR3 phosphorylation via its ligand VEGF-C or interaction with β1 integrin and induces the downstream pathway activation mentioned above and the regulation of key genes, including the master transcription factors PROX-1, FOXC2, GATA2 and others depicted in this schematic diagram. The second mechanosensor in LECs, PIEZO1, activates the membrane-bound calcium channel ORAI1, leading to intracellular calcium entry. The major intracellular calcium sensor calmodulin forms a protein complex with KLF2, which subsequently drives lymphatics-related downstream gene upregulation or downregulation. Abbreviations: PIEZO1 Piezo type mechanosensitive ion channel component 1, ORAI1 calcium release-activated calcium channel protein 1, PECAM platelet and endothelial cell adhesion molecule, VE-cadherin vascular endothelial cadherin, VEGFR2 vascular endothelial growth factor receptor 2, VEGFR3 vascular endothelial growth factor receptor 2, YAP/TAZ yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), PI3K phosphoinositide 3-kinases, AKT serine/threonine-protein kinase, GATA2 GATA binding protein 2, KLF2 Krüppel-like factor 2, PROX-1 prospero homeobox protein 1, FOXC2 Forkhead box protein C2, NFATc1 calcineurin/NFAT, FOXP2 Forkhead box protein P2, Cx37 connexin37, Itga9 integrin alpha- 9/beta-1, Fat4 FAT tumor suppressor homolog 4, Vegfc vascular endothelial growth factor C, Vegfa vascular endothelial growth factor A, Fgfr3 fibroblast growth factor receptor 3, Dtx1 deltex E3 ubiquitin ligase 1, Dtx3 L deltex E3 ubiquitin ligase 3L. A letter P in the yellow circles indicates phosphorylation. Created with BioRender.com
Responses to flow-induced mechanical forces in lymphatics
LECs are subjected to fluid shear stress (FSS). Unlike blood vessels, which are controlled by a central pump and subjected to high FSS, initial lymphatics are exposed to interstitial fluid flow and thus lower FSS, approximately 10-fold less than blood vessels [140]. The hydrostatic and osmotic pressure between lymphatic vessels, blood vessels and the interstitial space guide the uptake of interstitial fluid and resulting forces (known as Starling forces) [141, 142]. Because collecting lymphatic vessels have an intrinsic pumping capacity and smooth muscle cell coverage (Fig. 1A), they experience greater FSS than the initial lymphatics, which ensures efficient transport of the lymph back to the venous circulation [116].
In vivo, LECs are exposed to a broad range of shear stresses depending on the tissue location of the lymphatic vessels and the tissue conditions (normal versus lymphedema). For example, LECs experience shear stress ranging from 0 to 12 dynes/cm2 in rat mesenteric prenodal lymphatics [140] and up to 40 dynes/cm2 in models of lymphedema [143]. Shear stress can be steady laminar (where fluid moves in one direction at a steady magnitude), disturbed laminar (where flow separates, recirculates and subsequently reattaches), oscillatory (where laminar flow fluid moves in a bidirectional manner) or turbulent (where flow is chaotic and moves in all directions) [5]. Both oscillatory and turbulent flows are often classified as disturbed.
Early studies in the 2000s unveiled that lymphatic expansion and remodeling can be triggered by FSS, which acts as a critical lymphangiogenic mediator by controlling LEC migration, VEGF- C expression, and initial lymphatic formation (Fig. 2A) [144]. An in vitro study using 3D collagen gel cultures showed that interstitial flow is a morphogenetic mediator of LEC organization and stabilization [145] (Fig. 2A). More recent studies have confirmed and further extended the effects of FSS on LECs to include effects on LEC shape, alignment, migration, and cell cycle and, importantly, revealed that these effects depend on the direction, magnitude, velocity and strength of pump pulses [146]. For example, under 4 dynes/cm2 laminar shear stress (LSS), LECs elongate and form stress fibers aligned with the flow direction, whereas LECs adopt a more cuboidal cell shape under 4 dynes/cm2 ¼ Hz oscillatory shear stress (OSS), and the amount of short perinuclear F- actin stress fibers increases; furthermore, their alignment is less dependent on flow direction [147]. Notably, the formation of cuboidal cells upon exposure of LECs to OSS resembles that of lymphatic valve cells in vivo, whereas the elongated and aligned LECs that form under LSS tend to show lymphangion cell morphology [147]. In an in vivo model of loss or gain of interstitial flow, Planas- paz et al. [139] showed that increased fluid can affect LEC stretching and proliferation (Fig. 2A).
Interestingly, the dynamic changes in LEC morphology observed in vitro in response to LSS have also been reported to alter lymphatic barrier function through a mechanism dependent upon Rac1-mediated actin dynamics [148]. In line with the effect of FSS on lymphatic physiological responses, sensitivity of LECs to shear stress has been postulated to be essential for adapting lymphatic function to meet tissue drainage needs and thus maintain body fluid homeostasis. Baeyens et al. [149] proposed the concept of the FSS set point, which assumes the existence of a preferred range of shear stress that determines vascular remodeling.
Consistent with the low and high levels of shear stress experienced by lymphatic and blood vasculature, respectively, the set point of BECs ranged between 10 and 20 dyne/cm2, as determined in vitro based on cell alignment and NF-kB translocation, which is a well-studied response associated with vessel stabilization and suppression of inflammatory pathways [150]. On In contrast, LECs are markedly more sensitive, with a set point of 4–10 dyne/cm2, which is in the range of shear stress values observed in the rat mesenteric lymphatic collecting vessels [140]. The high sensitivity of LECs to low levels of shear stress is determined by the increased expression of VEGFR3 in LECs compared to BECs. Reducing VEGFR3 expression in LECs increases the FSS setpoint, whereas increasing VEGFR3 expression in BEC decreases the FSS setpoint [149].
Further supporting the involvement of flow-induced mechanical forces in maintaining the physiological functions of lymphatics, a study using in vitro and in vivo models revealed that transmural flow enhances lymphatic permeability, transport of macromolecules and DC migration into lymphatics [151] (Fig. 2A). This DC migration was found to be accompanied by increased lymphatic expression of the chemokine CCL21, which is essential for the homing and docking of CCR7 expressing DCs to initial lymphatics as well as increased expression of adhesion molecules such intercellular adhesion molecule-1 and E-selectin. Moreover, the authors used an experimental model of lymphedema in which lymphatic drainage is markedly decreased, and in the model, lymphatic endothelial expression of CCL21 was nearly absent. This study suggests that FSS can act as an early signal for initial lymphatics to regulate immune cell trafficking and soluble antigen transport from the interstitial tissue to the draining lymph nodes. This regulatory mechanism may thus impact immunity and the progression of inflammatory diseases.
The effects of FFS on lymphatic function are not limited to initial lymphatics and also apply to collecting lymphatic vessels (Fig. 2A). The contractility of collecting vessel SMCs contributes to the pumping function of the vessels and can rapidly adapt to changes in the microenvironment resulting, for example, from flow-induced wall shear stress. Low nitric oxide (NO) concentrations produced by endothelial NO synthase (NOS), such as those associated with pulsatile flow from spontaneous contractions, have been shown to increase contraction amplitude [152] whereas higher NO concentrations produced through the activation of inducible NOS and inflammatory cytokines [153, 154] inhibit both contraction frequency and amplitude [116]. Interestingly, a study revealed FSS as a modulator of NO release and lymphatic pump function in contractile lymphatics [155]. A more recent study by Kornuta et al. [156] reported that the rate of change in wall shear stress encountered by collecting lymphatic vessels significantly affects the contractile response of lymphatic vessels to flow and that fluid shear stress has the capacity to regulate the coordination of lymphatic pumping activity between lymphangions. In vitro, even low, 0.5 or 1.0 dyn/cm2 shear stress has been shown to trigger the release of ATP from LECs, which results in an increase in eNOS via activation of the purinergic P2X/2Y receptor and enhanced Ca2+ signaling [157]. Thus, shear stress-dependent NO production by eNOS in LECs may support the coordination between shear stress and lymphatic pumping by controlling the contractility of SMCs, which is essential for removing excess interstitial fluid load and preventing oedemic conditions.
Potential synergy between fluid flow-induced shear stress and other biological signals
Although the response to FSS itself in LECs can be sufficient to maintain the complex structure of the vessel necessary for lymphatic functions, LECs may also integrate biochemical and fluid shear stress to do so (Fig. 2A). There is evidence supporting that synergy between FSS and traditional biological factors such as VEGF-C promotes the remodeling of lymphatic vasculature [4]. For example, slow interstitial flow, with an average velocity of 4.2 m/s, synergizes in vitro with molecular factors promoting lymphatic vessel sprouting by enhancing the availability of matrix-bound growth factors to the cells [158]. Likewise, it was demonstrated more recently in a 3D in vitro model that interstitial flow with 1 m/s average velocity augments the effects of prolymphangiogenic molecular factors and determines the direction of lymphatic sprouting [159]. Intriguingly, FSS can also affect LECs by cooperating with S1P. S1P and one of its receptors, S1PR1, are commonly involved in individual and collective cell migration. As described above, the concentration of S1P is nearly zero in interstitial fluid, while it is high in blood and lymph, and LECs are the major source of lymph S1P. S1P can promote LEC sprouting in vitro in an S1PR1-dependent manner [160]. The study by Surya et al. [161] proposed a model in which S1P and FSS act synergistically during the development and remodeling of the lymphatic system. Previously, using an impinging flow chamber, the authors discovered that some LECs migrate upstream against flow direction [162]. Subsequently, they provided evidence that this upstream migratory phenotype requires both S1P and its receptor, suggesting that S1P may act as a biochemical cue in response to FSS. However, the mechanism by which FSS stimulates S1PR1 remains to be elucidated. Interestingly, another study reporting that LSS enhances VEGF-C signaling and LEC sprouting, further supporting LSS as a lymphangiogenic mediator, revealed that this process is directly antagonized by S1PR1 [163]. The authors proposed that S1PR1 may prevent the hyperactivation of LSS/VEGF-C signaling to halt the sprouting of lymphatic vessels. Moreover, they showed that S1PR1 controls the cytoskeletal and membrane localization of claudin-5 by inhibiting RhoA activity. Although it is not clear how LSS enhances VEGF-C signaling, it is plausible that LSS boosts the interaction between VEGFR3 and its coreceptor NRP2 or molecules such as VEGFR2 and integrin-α9. On the other hand, LSS may control the expression or localization of kinases or phosphatases modulating the phosphorylation of VEGFR3 or downstream signaling molecules.
Altogether, these findings indicate that synergistic effects between biomechanical and chemical cues control lymphatic biology and functions, and more studies will be needed to uncover novel synergies between FSS and unexplored biochemical factors, such as chemokines and cytokines.
Mechanotransduction of fluid flow-induced shear stress in lymphatic vessels
Mechanotransduction is the process by which cells translate mechanical information into biological responses. The mechanotransduction of several shear stress responses in ECs, such as cell alignment, has been shown to require VEGFR2. However, the study of Bayens et al. [149] on differences in flow sensitivity between BEC and LEC revealed that VEGFR2 expression levels were comparable, unlike VEGFR3 expression, which was increased in LECs. FSS also stimulates activation of VEGFR3, as indicated by increased phosphorylation [149]. In vivo, augmentation of interstitial fluid volume mechanically stretches LECs, enhances VEGFR3 activation and induces LEC proliferation in a β1 integrin-dependent manner [164] (Fig. 2B) This is consistent with the role of some β1 integrins, such as α5β1, in mechanotransduction [165] and their capacity to associate with VEGFR3 and trigger its activation [166, 167]. Integrins have also been postulated to connect the extracellular environment with intracellular signaling by linking the extracellular matrix with the actin cytoskeleton inside LECs [165]. Furthermore, integrins have been implicated in lymph valve formation in vivo [168] and in lymphangiogenesis during pathological processes such as inflammation, tumor growth, and wound healing [169,170,171,172].
Several groups have focused their efforts on identifying flow-induced transcription factors mediating FSS responses in lymphatic vessels. An in vitro study showed that LECs become elongated and aligned under 4 dynes/cm2 LSS, while they were more cuboidal under 4 dynes/cm2 ¼ Hz OSS and that these alterations in LEC alignment and cytoskeleton reorganization were triggered by the transcription factors Forkhead box protein C2 (FOXC2) and PROX1 [147] (Fig. 2B). FOXC2 is essential for the maturation of lymphatic vessels and lymphatic valve development and PROX-1 functions as the master regulator of lymphatic development [107]. Specifically, OSS, but not LSS, upregulates FOXC2, whereas PROX1 is not affected. Loss of PROX1 abolishes the LEC response to OSS while enhancing cell elongation and alignment in response to LSS [147]. OSS coordinately induces Cx37 and calcineurin/NFAT activation in a PROX1- and FOXC2-dependent manner in LECs in vitro (Fig. 2B). Foxc2- deficient LECs exhibit disrupted cell‒cell junctions and a disorganized cytoskeleton, leading to an abnormal response to shear stress, cell hyperproliferation, and apoptosis. Moreover, this aberrant proliferation of Foxc2-deficient LECs in response to OSS is mediated by increased YAP/TAZ signaling [147, 173]. YAP and TAZ are transcription factors that were previously shown to regulate cell responses to mechanical cues such as stiff ECM, stretching and/or shear stress [174]. Thus, These findings suggest the essential role of FOXC2 in stabilizing collecting vessel quiescence through the coordination of cell‒cell junction maturation and responses to shear stress in regions of disturbed flow, such as valves. Another in vitro shear stress study confirms the effect of FSS on the upregulation of FOXC2, CX37, ITGA9, and GATA2 [175], which are known to be essential for valve-forming LECs in vivo [168, 176,177,178] (Fig. 2B). Notably, a recent study identified FOXP2 as a new flow-induced transcriptional regulator of collecting lymphatic vessel and valve development and a downstream effector of the flow-responsive FOXC2/NFATc1 pathway [179] (Fig. 2B).
An in vivo study identified FAT tumor suppressor homolog 4 (FAT4), a target gene of GATA2, as a key player in shear stress-dependent polarization of LECs [180] (Fig. 2B). Though FAT4 is clearly important for transducing laminar flow-induced signals in LECs, the mechanisms by which it transduces mechanical signals in endothelial cells remains to be elucidated. A study by Choi D et al. [181] investigated the molecular mechanisms underlying flow-induced lymphatic growth. They showed in vitro that LSS enhances the intracellular calcium level in LECs through the early signal and mediator of laminar flow, ORAI1 (calcium release activated calcium modulator 1), a pore subunit of the calcium release-activated calcium channel. ORAI1 activates key transcription regulators, including Klf-2 and Klf-4, which in turn upregulate VEGF-C, VEGF-A, and FGFR3, promoting LEC proliferation and survival, and concurrently downregulated the cell cycle inhibitor p57 (Fig. 2B). These in vitro findings were further validated in mice depleted of ORAI1, Klf-2 or Klf-4, which exhibited a reduction in lymphatic vessel density and development [181]. The same group further demonstrated that calcium-loaded calmodulin forms a protein complex with Klf2 (a master regulator of shear stress responses), and Prox1 binds to the promoters of Dtx1 and Dtx3L and activates their gene expression [182] (Fig. 2B). In turn, the Dtx1 and Dtx3L proteins form a Notch E3 ligase complex, which decreases Notch activity and promotes lymphatic sprouting [182] (Fig. 2B). Consistent with the role of calcium and ORAI1 in the early response to flow, a study showed that the magnitude of shear stress modulates the intracellular calcium dynamics in cultured LECs, and calcium mobilization is reduced through blockade of calcium release-activated calcium channels [183].
Mechanosensing of fluid flow-induced shear stress in lymphatic vessels
As reviewed above, FSS is an essential regulator of the stabilization of mature lymphatics, adult lymphangiogenesis and biological functions of lymphatics. To date, two potential shear stress-sensing mechanisms have been identified in lymphatics. The complex involving mechanosensory receptors at the cell‒cell junctions on the LEC surface including PECAM, VE- cadherin, VEGFR2 and VEGFR3 has been proposed as a mechanosensing mechanism of shear stress in lymphatics (Fig. 2B). Such mechanosensing complexes have been well characterized in BECs [184, 185] and shown to trigger cell proliferation by activating the phosphatidylinositol-3- kinase (PI3K)/Akt pathway and cytoskeleton reorganization in response to FSS [186, 187] (Fig. 2B).
The involvement of PECAM-1 in lymphatic mechanosensing was revealed in mice with PECAM deficiency, which led to defective lymphatic remodeling, affecting processes such as valve formation [188]. Notably, VE-cadherin distribution within the cell membrane in BECs to has been shown be dependent on the spatial flow characteristics [189], but whether this is also observed in LECs remains to be demonstrated. VE-cadherin acts as an adaptor by binding to VEGFR2 and VEGFR3 [184], which are expressed on LECs and known to contribute to lymphatic development and adult lymphangiogenesis [107]. Consistent with the role of VEGFR3 as a flow sensor, VEGFR3 expression in LECs changes in response to flow characteristics [115], and as discussed above, it is a key determinant of flow sensitivity in ECs [149]. Thus, considering the individual contributions of PECAM, VE-cadherin, VEGFR2 and VEGFR3 to lymphatic remodeling, stabilization, and flow sensitivity, it is possible that together they form a mechanosensing complex in LECs to respond to FSS. However, further investigations are required to validate this mechanosensing mechanism.
The molecular sensor that senses flow-induced mechanical force in LECs and activates ORAI1-mediated calcium entry has remained unknown until the recent follow-up study by Choi et al. [190] which identified Piezo-1 (Piezo type mechanosensitive ion channel component 1) as an upstream mechanosensor of ORAI1 that triggers the mechanotransduction signal controlling lymphatic expansion in response to external physical stimuli (Fig. 2B). Piezo1 is sensitive to alterations in membrane tension [191] and has been reported to be essential for sensing fluid shear stress by BECs [192]. Similarly, Piezo-1 has been reported to be mechanically activated in LECs and lymphatic vessels and to serve as a mechanical force sensor controlling lymphatic valve development [193] and maintenance in adult lymphatics [194]. In a later study by Choi et al. [194] depletion of Piezo1 in cultured LECs significantly reduced the OSS-induced upregulation of lymphatic valve signature genes, including GATA-2 and Foxc2, while its overexpression in LECs or activation using the chemical agonist Yoda-1 upregulated the lymphatic valve genes in the absence of OSS. In their more recent study, Choi et al. [190] reported that Piezo1 overexpression alone recapitulates the laminar flow-induced upregulation of OraI1 downstream genes, such as Dtx1, Dtx3L, and Klf2, and downregulation of Notch. OraI1 stimulation alone was sufficient for LEC mechanotransduction in the absence of laminar fluid flow or Piezo1 activation. These findings demonstrate that Piezo-1 is an upstream activator of OraI1 in the laminar flow-activated mechanotransduction pathway in LECs. The authors also showed that Piezo-1 is essential for the maintenance of lymphatic vessels in adult mice, as evidenced by a reduction in mesenteric and dermal lymphatics after specific deletion of Piezo-1 in lymphatics postnatally [190]. Moreover, Piezo-1 exhibited prolymphangiogenic properties in animal models when activated by the agonist Yoda-1, and these properties translated into therapeutic benefits in an animal model of surgically induced lymphedema [190]. The discovery of the essential role of Piezo-1 in lymphatic development and maintenance also provides some mechanistic explanation for the generalized lymphatic dysplasia and dysfunction observed in patients with mutations in Piezo-1 [195, 196].
Concluding remarks
Considering the vital functions that the lymphatic vessels play and the increasing list of their contributions to human diseases, it is imperative to improve our understanding of how these vessels sense and respond to environmental changes encompassing not only biochemical but also mechanical signals. As described in this review, recent research on biomechanical signals further supports the concept that lymphatic vessels are not “inert tubes” but rather highly plastic vasculature structures capable of responding to biomechanical changes and adapting their properties, such as permeability and contractibility, to changes in their surrounding microenvironment. However, many questions remain unresolved, including how biomechanical forces contribute to controlling lymphangiogenesis in pathological conditions such as cancer and metabolic and inflammatory diseases. However, identifying the specific role of biomechanical signals distinct from that of biochemical signals might be challenging because numerous downstream molecules are shared by both signals. It would also be interesting to investigate whether alterations in mechanosensing and/or mechanotransduction in lymphatics are associated with diseases that exhibit altered fluid flow. To this end, new tools may have to be developed to allow the visualization and accurate measurement of lymph flow. Although evidence supports the control of lymphatic functions by FSS, it is likely that lymphatic vessels are exposed to more than one biomechanical cue at a given time, particularly in a pathological setting, where the matrix composition or stiffness is expected to change. Thus, we need to understand how lymphatic vessels integrate multiple biomechanical signals and respond to them to avoid potential deleterious effects.
More research is needed to further understand mechanotransduction and mechanosensing in lymphatic vessel during homeostasis and pathological conditions, and the results of such research may lead to the discovery of new promising therapeutic approaches. As an example, targeting the mechanical force sensor, Piezo-1 has shown encouraging preclinical results for the treatment of secondary or primary lymphedema by promoting the growth of functional lymphatic vessels.
References
Petrova TV, Koh GY. Biological functions of lymphatic vessels. Science. 2020;369. https://doi.org/10.1126/science.aax4063.
Oliver G, Kipnis J, Randolph GJ, Harvey NL. The lymphatic vasculature in the 21(st) century: novel functional roles in homeostasis and disease. Cell. 2020;182:270–96. https://doi.org/10.1016/j.cell.2020.06.039.
Balint L, Jakus Z. Mechanosensation and mechanotransduction by lymphatic endothelial cells act as important regulators of lymphatic development and function. Int J Mol Sci. 2021;22. https://doi.org/10.3390/ijms22083955.
Geng X, Ho YC, Srinivasan RS. Biochemical and mechanical signals in the lymphatic vasculature. Cell Mol Life Sci. 2021;78:5903–23. https://doi.org/10.1007/s00018-021-03886-8.
Sabine A, Saygili Demir C, Petrova TV. Endothelial cell responses to biomechanical forces in lymphatic vessels. Antioxid Redox Signal. 2016;25:451–65. https://doi.org/10.1089/ars.2016.6685.
Gordon E, Schimmel L, Frye M. The importance of mechanical forces for in vitro endothelial cell biology. Front Physiol. 2020;11:684. https://doi.org/10.3389/fphys.2020.00684.
Solari E, Marcozzi C, Negrini D, Moriondo A. Lymphatic vessels and their surroundings: how local physical factors affect lymph flow. Biology (Basel). 2020;9. https://doi.org/10.3390/biology9120463.
Rockson SG. Lymphedema. Am J Med. 2001;110:288–95.
Azhar SH, Lim HY, Tan BK, Angeli V. The unresolved pathophysiology of lymphedema. Front Physiol. 2020;11:137. https://doi.org/10.3389/fphys.2020.00137.
Rockson SG. Advances in lymphedema. Circ Res. 2021;128:2003–16. https://doi.org/10.1161/CIRCRESAHA.121.318307.
Choe K, Jang JY, Park I, Kim Y, Ahn S, Park DY, et al. Intravital imaging of intestinal lacteals unveils lipid drainage through contractility. J Clin Invest. 2015;125:4042–52. https://doi.org/10.1172/JCI76509.
Jiang X, Tian W, Nicolls MR, Rockson SG. The lymphatic system in obesity, insulin resistance, and cardiovascular diseases. Front Physiol. 2019;10:1402. https://doi.org/10.3389/fphys.2019.01402.
Cifarelli V, Eichmann A. The intestinal lymphatic system: functions and metabolic implications. Cell Mol Gastroenterol Hepatol. 2019;7:503–13. https://doi.org/10.1016/j.jcmgh.2018.12.002.
McDonald DM. Tighter lymphatic junctions prevent obesity. Science. 2018;361:551–2. https://doi.org/10.1126/science.aau5583.
Zhang F, Zarkada G, Han J, Li J, Dubrac A, Ola R, et al. Lacteal junction zippering protects against diet-induced obesity. Science. 2018;361:599–603. https://doi.org/10.1126/science.aap9331.
Escobedo N, Proulx ST, Karaman S, Dillard ME, Johnson N, Detmar M, et al. Restoration of lymphatic function rescues obesity in Prox1- haploinsufficient mice. JCI Insight. 2016;1. https://doi.org/10.1172/jci.insight.85096.
Blum KS, Karaman S, Proulx ST, Ochsenbein AM, Luciani P, Leroux JC, et al. Chronic high-fat diet impairs collecting lymphatic vessel function in mice. PLoS One. 2014;9:e94713 https://doi.org/10.1371/journal.pone.0094713.
Harvey NL, Srinivasan RS, Dillard ME, Johnson NC, Witte MH, Boyd K, et al. Lymphatic vascular defects promoted by Prox1 haploinsufficiency cause adult-onset obesity. Nat Genet. 2005;37:1072–81.
Escobedo N, Oliver G. The lymphatic vasculature: its role in adipose metabolism and obesity. Cell Metab. 2017;26:598–609. https://doi.org/10.1016/j.cmet.2017.07.020.
Nanjee MN, Cooke CJ, Wong JS, Hamilton RL, Olszewski WL, Miller NE. Composition and ultrastructure of size subclasses of normal human peripheral lymph lipoproteins: quantification of cholesterol uptake by HDL in tissue fluids. J Lipid Res. 2001;42:639–48.
Randolph GJ, Miller NE. Lymphatic transport of high-density lipoproteins and chylomicrons. J Clin Invest. 2014;124:929–35. https://doi.org/10.1172/JCI71610.
Reichl D, Simons LA, Myant NB, Pflug JJ, Mills GL. The lipids and lipoproteins of human peripheral lymph, with observations on the transport of cholesterol from plasma and tissues into lymph. Clin Sci Mol Med. 1973;45:313–29. https://doi.org/10.1042/cs0450313.
Reichl D, Miller NE. Pathophysiology of reverse cholesterol transport. Insights from inherited disorders of lipoprotein metabolism. Arteriosclerosis. 1989;9:785–97. https://doi.org/10.1161/01.atv.9.6.785.
Martel C, Li W, Fulp B, Platt AM, Gautier EL, Westerterp M, et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J Clin Invest. 2013;123:1571–9. https://doi.org/10.1172/JCI63685.
Lim HY, Thiam CH, Yeo KP, Bisoendial R, Hii CS, McGrath KC, et al. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab. 2013;17:671–84. https://doi.org/10.1016/j.cmet.2013.04.002.
Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res. 2009;50:S189–194. https://doi.org/10.1194/jlr.R800088-JLR200.
Wu JJ, Wagner AM. Verruciform xanthoma in association with milroy disease and leaky capillary syndrome. Pediatr Dermatol. 2003;20:44–7.
Woolling KR, Jenkins RE, Dolan PA, Evans PV. Localized xanthomas in lymphedema praecox. JAMA: J Am Med Assoc. 1970;211:1372–4.
Goldrick RB, Ahrens EH Jr. Unilateral chylous lymphedema and xanthomatosis: a study of factors governing the flow of intestinal lymph. Am J Med. 1964;37:610–22.
Romaní J, Luelmo J, Sáez A, Yébenes M, Sábat M, Fernández-Chico N, et al. Localized xanthomas associated with primary lymphedema. Pediatr Dermatol. 2012;29:113–4. https://doi.org/10.1111/j.1525-1470.2011.01686.x.
Berger BW, Kantor I, Maier HS. Xanthomatosis and lymphedema. Arch Dermatol. 1972;105:730–1.
Yeo KP, Lim HY, Thiam CH, Azhar SH, Tan C, Tang Y, et al. Efficient aortic lymphatic drainage is necessary for atherosclerosis regression induced by ezetimibe. Sci Adv. 2020;6. https://doi.org/10.1126/sciadv.abc2697.
Lemole GM. The role of lymphstasis in atherogenesis. Ann Thorac Surg. 1981;31:290–3. https://doi.org/10.1016/s0003-4975(10)60949-6.
Rademakers T, van der Vorst EP, Daissormont IT, Otten JJ, Theodorou K, Theelen TL, et al. Adventitial lymphatic capillary expansion impacts on plaque T cell accumulation in atherosclerosis. Sci Rep. 2017;7:45263. https://doi.org/10.1038/srep45263.
Vuorio T, Nurmi H, Moulton K, Kurkipuro J, Robciuc MR, Ohman M, et al. Lymphatic vessel insufficiency in hypercholesterolemic mice alters lipoprotein levels and promotes atherogenesis. Arterioscler Thromb Vasc Biol. 2014;34:1162–70. https://doi.org/10.1161/ATVBAHA.114.302528.
Milasan A, Jean G, Dallaire F, Tardif JC, Merhi Y, Sorci-Thomas M, et al. Apolipoprotein A-I modulates atherosclerosis through lymphatic vessel-dependent mechanisms in mice. J Am Heart Assoc. 2017;6. https://doi.org/10.1161/JAHA.117.006892.
Mackay CR. T-cell memory: the connection between function, phenotype and migration pathways. Immunol Today. 1991;12:189–92. https://doi.org/10.1016/0167-5699(91)90051-T.
Mackay CR, Marston WL, Dudler L. Naive and memory T cells show distinct pathways of lymphocyte recirculation. J Exp Med. 1990;171:801–17. https://doi.org/10.1084/jem.171.3.801.
Yawalkar N, Hunger RE, Pichler WJ, Braathen LR, Brand CU. Human afferent lymph from normal skin contains an increased number of mainly memory/effector CD4(+) T cells expressing activation, adhesion and co-stimulatory molecules. Eur J Immunol. 2000;30:491–7.
Bromley SK, Thomas SY, Luster AD. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat Immunol. 2005;6:895–901. https://doi.org/10.1038/ni1240.
Bromley SK, Yan S, Tomura M, Kanagawa O, Luster AD. Recirculating memory T cells are a unique subset of CD4+ T cells with a distinct phenotype and migratory pattern. J Immunol. 2013;190:970–6. https://doi.org/10.4049/jimmunol.1202805.
Tomura M, Honda T, Tanizaki H, Otsuka A, Egawa G, Tokura Y, et al. Activated regulatory T cells are the major T cell type emigrating from the skin during a cutaneous immune response in mice. J Clin Invest. 2010;120:883–93. https://doi.org/10.1172/JCI40926.
Arasa J, Collado-Diaz V, Halin C. Structure and immune function of afferent lymphatics and their mechanistic contribution to dendritic cell and T cell trafficking. Cells. 2021;10. https://doi.org/10.3390/cells10051269.
Tan Y, Tey HL, Chong SZ, Ng LG. Skinny deeping: uncovering immune cell behavior and function through imaging techniques. Immunol Rev. 2022;306:271–92. https://doi.org/10.1111/imr.13049.
Jackson DG. Leucocyte trafficking via the lymphatic vasculature- mechanisms and consequences. Front Immunol. 2019;10:471. https://doi.org/10.3389/fimmu.2019.00471.
Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5:617–28. https://doi.org/10.1038/nri1670.
Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, et al. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity. 2004;21:279–88. https://doi.org/10.1016/j.immuni.2004.06.014.
Saeki H, Moore AM, Brown MJ, Hwang ST. Cutting edge: secondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J Immunol. 1999;162:2472–5.
Förster R, Schubel A, Breitfeld D, Kremmer E, Renner-Müller I, Wolf E, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. https://doi.org/10.1016/s0092-8674(00)80059-8.
Russo E, Teijeira A, Vaahtomeri K, Willrodt AH, Bloch JS, Nitschké M, et al. Intralymphatic CCL21 promotes tissue egress of dendritic cells through afferent lymphatic vessels. Cell Rep. 2016;14:1723–34. https://doi.org/10.1016/j.celrep.2016.01.048.
Weber M, Hauschild R, Schwarz J, Moussion C, de Vries I, Legler DF, et al. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science. 2013;339:328–32. https://doi.org/10.1126/science.1228456.
Tal O, Lim HY, Gurevich I, Milo I, Shipony Z, Ng LG, et al. DC mobilization from the skin requires docking to immobilized CCL21 on lymphatic endothelium and intralymphatic crawling. J Exp Med. 2011;208:2141–53. https://doi.org/10.1084/jem.20102392.
Johnson LA, Jackson DG. The chemokine CX3CL1 promotes trafficking of dendritic cells through inflamed lymphatics. J Cell Sci. 2013;126:5259–70. https://doi.org/10.1242/jcs.135343.
Kabashima K, Shiraishi N, Sugita K, Mori T, Onoue A, Kobayashi M, et al. CXCL12-CXCR4 engagement is required for migration of cutaneous dendritic cells. Am J Pathol. 2007;171:1249–57. https://doi.org/10.2353/ajpath.2007.070225.
Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, Hirsch K, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. 2008;453:51–5. https://doi.org/10.1038/nature06887.
Arasa J, et al. Upregulation of VCAM-1 in lymphatic collectors supports dendritic cell entry and rapid migration to lymph nodes in inflammation. J Exp Med. 2021;218. https://doi.org/10.1084/jem.20201413.
Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med. 2006;203:2763–77. https://doi.org/10.1084/jem.20051759.
Menning A, Höpken UE, Siegmund K, Lipp M, Hamann A, Huehn J. Distinctive role of CCR7 in migration and functional activity of naive- and effector/memory-like Treg subsets. Eur J Immunol. 2007;37:1575–83. https://doi.org/10.1002/eji.200737201.
Randolph GJ, Ivanov S, Zinselmeyer BH, Scallan JP. The lymphatic system: integral roles in immunity. Annu Rev Immunol. 2017;35:31–52. https://doi.org/10.1146/annurev-immunol-041015-055354.
Ulvmar MH, Werth K, Braun A, Kelay P, Hub E, Eller K, et al. The atypical chemokine receptor CCRL1 shapes functional CCL21 gradients in lymph nodes. Nat Immunol. 2014;15:623–30. https://doi.org/10.1038/ni.2889.
Rantakari P, Auvinen K, Jäppinen N, Kapraali M, Valtonen J, Karikoski M, et al. The endothelial protein PLVAP in lymphatics controls the entry of lymphocytes and antigens into lymph nodes. Nat Immunol. 2015;16:386–96. https://doi.org/10.1038/ni.3101.
Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–60. https://doi.org/10.1038/nature02284.
Pham TH, Baluk P, Xu Y, Grigorova I, Bankovich AJ, Pappu R, et al. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J Exp Med. 2010;207:17–27. https://doi.org/10.1084/jem.20091619.
Czeloth N, Bernhardt G, Hofmann F, Genth H, Forster R. Sphingosine-1- phosphate mediates migration of mature dendritic cells. J Immunol. 2005;175:2960–7. https://doi.org/10.4049/jimmunol.175.5.2960.
Rathinasamy A, Czeloth N, Pabst O, Forster R, Bernhardt G. The origin and maturity of dendritic cells determine the pattern of sphingosine 1-phosphate receptors expressed and required for efficient migration. J Immunol. 2010;185:4072–81. https://doi.org/10.4049/jimmunol.1000568.
Garnier L, Gkountidi AO, Hugues S. Tumor-associated lymphatic vessel features and immunomodulatory functions. Front Immunol. 2019;10:720. https://doi.org/10.3389/fimmu.2019.00720.
Johnson LA, Banerji S, Lawrance W, Gileadi U, Prota G, Holder KA, et al. Dendritic cells enter lymph vessels by hyaluronan-mediated docking to the endothelial receptor LYVE-1. Nat Immunol. 2017;18:762–70. https://doi.org/10.1038/ni.3750.
Olszewski WL. The innate reaction of the human skin lymphatic system to foreign and self-antigens. Lymphat Res Biol. 2005;3:50–57. https://doi.org/10.1089/lrb.2005.3.50.
Cohen JN, Guidi CJ, Tewalt EF, Qiao H, Rouhani SJ, Ruddell A, et al. Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J Exp Med. 2010;207:681–8. https://doi.org/10.1084/jem.20092465.
Fletcher AL, Lukacs-Kornek V, Reynoso ED, Pinner SE, Bellemare-Pelletier A, Curry MS, et al. Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J Exp Med. 2010;207:689–97. https://doi.org/10.1084/jem.20092642.
Tewalt EF, Cohen JN, Rouhani SJ, Guidi CJ, Qiao H, Fahl SP, et al. Lymphatic endothelial cells induce tolerance via PD-L1 and lack of costimulation leading to high-level PD-1 expression on CD8 T cells. Blood. 2012;120:4772–82. https://doi.org/10.1182/blood-2012-04-427013.
Hirosue S, Vokali E, Raghavan VR, Rincon-Restrepo M, Lund AW, Corthésy-Henrioud P, et al. Steady-state antigen scavenging, cross-presentation, and CD8+ T cell priming: a new role for lymphatic endothelial cells. J Immunol. 2014;192:5002–11. https://doi.org/10.4049/jimmunol.1302492.
Rouhani SJ, Eccles JD, Riccardi P, Peske JD, Tewalt EF, Cohen JN, et al. Roles of lymphatic endothelial cells expressing peripheral tissue antigens in CD4 T-cell tolerance induction. Nat Commun. 2015;6:6771. https://doi.org/10.1038/ncomms7771.
Dubrot J, Duraes FV, Potin L, Capotosti F, Brighouse D, Suter T, et al. Lymph node stromal cells acquire peptide-MHCII complexes from dendritic cells and induce antigen-specific CD4(+) T cell tolerance. J Exp Med. 2014;211:1153–66. https://doi.org/10.1084/jem.20132000.
Dubrot J, Duraes FV, Harlé G, Schlaeppi A, Brighouse D, Madelon N, et al. Absence of MHC-II expression by lymph node stromal cells results in autoimmunity. Life Sci Alliance. 2018;1:e201800164. https://doi.org/10.26508/lsa.201800164.
Mendoza A, Fang V, Chen C, Serasinghe M, Verma A, Muller J, et al. Lymphatic endothelial S1P promotes mitochondrial function and survival in naive T cells. Nature. 2017;546:158–61. https://doi.org/10.1038/nature22352.
Lukacs-Kornek V, et al. Regulated release of nitric oxide by nonhematopoietic stroma controls expansion of the activated T cell pool in lymph nodes. Nat Immunol. 2011;12:1096–104. https://doi.org/10.1038/ni.2112.
Christiansen AJ, Dieterich LC, Ohs I, Bachmann SB, Bianchi R, Proulx ST, et al. Lymphatic endothelial cells attenuate inflammation via suppression of dendritic cell maturation. Oncotarget. 2016;7:39421–35. https://doi.org/10.18632/oncotarget.9820.
Podgrabinska S, Kamalu O, Mayer L, Shimaoka M, Snoeck H, Randolph GJ, et al. Inflamed lymphatic endothelium suppresses dendritic cell maturation and function via Mac-1/ICAM-1-dependent mechanism. J Immunol. 2009;183:1767–79. https://doi.org/10.4049/jimmunol.0802167.
Stacker SA, Williams SP, Karnezis T, Shayan R, Fox SB, Achen MG. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat Rev Cancer. 2014;14:159–72. https://doi.org/10.1038/nrc3677.
Doeden K, Ma Z, Narasimhan B, Swetter SM, Detmar M, Dadras SS. Lymphatic invasion in cutaneous melanoma is associated with sentinel lymph node metastasis. J Cutan Pathol. 2009;36:772–80. https://doi.org/10.1111/j.1600-0560.2008.01166.x.
Dadras SS, Paul T, Bertoncini J, Brown LF, Muzikansky A, Jackson DG, et al. Tumor lymphangiogenesis: a novel prognostic indicator for cutaneous melanoma metastasis and survival. Am J Pathol. 2003;162:1951–60. https://doi.org/10.1016/S0002-9440(10)64328-3.
Tobler NE, Detmar M. Tumor and lymph node lymphangiogenesis-impact on cancer metastasis. J Leukoc Biol. 2006;80:691–6. https://doi.org/10.1189/jlb.1105653.
Renyi-Vamos F, Tovari J, Fillinger J, Timar J, Paku S, Kenessey I, et al. Lymphangiogenesis correlates with lymph node metastasis, prognosis, and angiogenic phenotype in human non-small cell lung cancer. Clin Cancer Res. 2005;11:7344–53. https://doi.org/10.1158/1078-0432.CCR-05-1077.
Takanami I. Lymphatic microvessel density using D2-40 is associated with nodal metastasis in non-small cell lung cancer. Oncol Rep. 2006;15:437–42.
Barresi V, Reggiani-Bonetti L, Di Gregorio C, De Leon MP, Barresi G. Lymphatic vessel density and its prognostic value in stage I colorectal carcinoma. J Clin Pathol. 2011;64:6–12. https://doi.org/10.1136/jcp.2010.083550.
Doekhie FS, Morreau H, de Bock GH, Speetjens FM, Dekker-Ensink NG, Putter H, et al. Sialyl Lewis X expression and lymphatic microvessel density in primary tumors of node-negative colorectal cancer patients predict disease recurrence. Cancer Microenviron. 2008;1:141–51. https://doi.org/10.1007/s12307-008-0014-3.
Matsumoto K, Nakayama Y, Inoue Y, Minagawa N, Katsuki T, Shibao K, et al. Lymphatic microvessel density is an independent prognostic factor in colorectal cancer. Dis Colon Rectum. 2007;50:308–14. https://doi.org/10.1007/s10350-006-0792-y.
Ran S, Volk L, Hall K, Flister MJ. Lymphangiogenesis and lymphatic metastasis in breast cancer. Pathophysiology. 2010;17:229–51. https://doi.org/10.1016/j.pathophys.2009.11.003.
Das S, Ladell DS, Podgrabinska S, Ponomarev V, Nagi C, Fallon JT, et al. Vascular endothelial growth factor-C induces lymphangitic carcinomatosis, an extremely aggressive form of lung metastases. Cancer Res. 2010;70:1814–24. https://doi.org/10.1158/0008-5472.CAN-09-3675.
Ma Q, Dieterich LC, Ikenberg K, Bachmann SB, Mangana J, Proulx ST, et al. Unexpected contribution of lymphatic vessels to promotion of distant metastatic tumor spread. Sci Adv. 2018;4:eaat4758. https://doi.org/10.1126/sciadv.aat4758.
Dieterich LC, Detmar M. Tumor lymphangiogenesis and new drug development. Adv Drug Deliv Rev. 2016;99:148–60. https://doi.org/10.1016/j.addr.2015.12.011.
Lund AW, Duraes FV, Hirosue S, Raghavan VR, Nembrini C, Thomas SN, et al. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep. 2012;1:191–9. https://doi.org/10.1016/j.celrep.2012.01.005.
Kimura T, Sugaya M, Oka T, Blauvelt A, Okochi H, Sato S. Lymphatic dysfunction attenuates tumor immunity through impaired antigen presentation. Oncotarget. 2015;6:18081–93. https://doi.org/10.18632/oncotarget.4018.
Lund AW, Wagner M, Fankhauser M, Steinskog ES, Broggi MA, Spranger S, et al. Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J Clin Invest. 2016;126:3389–402. https://doi.org/10.1172/JCI79434.
Fankhauser M, et al. Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci Transl Med. 2017;9. https://doi.org/10.1126/scitranslmed.aal4712.
Dieterich LC, Ikenberg K, Cetintas T, Kapaklikaya K, Hutmacher C, Detmar M. Tumor-associated lymphatic vessels upregulate PDL1 to inhibit T-cell activation. Front Immunol. 2017;8:66. https://doi.org/10.3389/fimmu.2017.00066.
Lane RS, Femel J, Breazeale AP, Loo CP, Thibault G, Kaempf A, et al. IFNgamma-activated dermal lymphatic vessels inhibit cytotoxic T cells in melanoma and inflamed skin. J Exp Med. 2018;215:3057–74. https://doi.org/10.1084/jem.20180654.
Mondor I, Baratin M, Lagueyrie M, Saro L, Henri S, Gentek R, et al. Lymphatic endothelial cells are essential components of the subcapsular sinus macrophage niche. Immunity. 2019;50:1453–66.e1454. https://doi.org/10.1016/j.immuni.2019.04.002.
Oliver G. Lymphatic vasculature development. Nat Rev Immunol. 2004;4:35–45. https://doi.org/10.1038/nri1258.
Brakenhielm E, Alitalo K. Cardiac lymphatics in health and disease. Nat Rev Cardiol. 2019;16:56–68. https://doi.org/10.1038/s41569-018-0087-8.
Klaourakis K, Vieira JM, Riley PR. The evolving cardiac lymphatic vasculature in development, repair and regeneration. Nat Rev Cardiol. 2021;18:368–79. https://doi.org/10.1038/s41569-020-00489-x.
Tammela T, Petrova TV, Alitalo K. Molecular lymphangiogenesis: new players. Trends Cell Biol. 2005;15:434–41. https://doi.org/10.1016/j.tcb.2005.06.004.
Srinivasan RS, Oliver G. Prox1 dosage controls the number of lymphatic endothelial cell progenitors and the formation of the lymphovenous valves. Genes Dev. 2011;25:2187–97. https://doi.org/10.1101/gad.16974811.
Prevo R, Banerji S, Ferguson DJ, Clasper S, Jackson DG. Mouse LYVE-1 is an endocytic receptor for hyaluronan in lymphatic endothelium. J Biol Chem. 2001;276:19420–30. https://doi.org/10.1074/jbc.M011004200.
Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med. 2007;204:2349–62. https://doi.org/10.1084/jem.20062596.
Tammela T, Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 2010;140:460–76. https://doi.org/10.1016/j.cell.2010.01.045.
Choi I, Lee S, Hong YK. The new era of the lymphatic system: no longer secondary to the blood vascular system. Cold Spring Harb Perspect Med. 2012;2:a006445 https://doi.org/10.1101/cshperspect.a006445.
Schulte-Merker S, Sabine A, Petrova TV. Lymphatic vascular morphogenesis in development, physiology, and disease. J Cell Biol. 2011;193:607–18. https://doi.org/10.1083/jcb.201012094.
Leak LV, Burke JF. Fine structure of the lymphatic capillary and the adjoining connective tissue area. Am J Anat. 1966;118:785–809. https://doi.org/10.1002/aja.1001180308.
Schmid-Schonbein GW. The second valve system in lymphatics. Lymphat Res Biol. 2003;1:25–9. https://doi.org/10.1089/15396850360495664.
Danussi C, Spessotto P, Petrucco A, Wassermann B, Sabatelli P, Montesi M, et al. Emilin1 deficiency causes structural and functional defects of lymphatic vasculature. Mol Cell Biol. 2008;28:4026–39. https://doi.org/10.1128/MCB.02062-07.
Potente M, Makinen T. Vascular heterogeneity and specialization in development and disease. Nat Rev Mol Cell Biol. 2017;18:477–94. https://doi.org/10.1038/nrm.2017.36.
Yang Y, Oliver G. Development of the mammalian lymphatic vasculature. J Clin Invest. 2014;124:888–97. https://doi.org/10.1172/JCI71609.
Norrmén C, Ivanov KI, Cheng J, Zangger N, Delorenzi M, Jaquet M, et al. FOXC2 controls formation and maturation of lymphatic collecting vessels through cooperation with NFATc1. J Cell Biol. 2009;185:439–57. https://doi.org/10.1083/jcb.200901104.
Scallan JP, Zawieja SD, Castorena-Gonzalez JA, Davis MJ. Lymphatic pumping: mechanics, mechanisms and malfunction. J Physiol. 2016;594:5749–68. https://doi.org/10.1113/JP272088.
Alitalo K. The lymphatic vasculature in disease. Nat Med. 2011;17:1371–80. doi nm.2545 [pii]10.1038/nm.2545.
Kim H, Kataru RP, Koh GY. Regulation and implications of inflammatory lymphangiogenesis. Trends Immunol. 2012;33:350–6. https://doi.org/10.1016/j.it.2012.03.006.
Chen L, Hamrah P, Cursiefen C, Zhang Q, Pytowski B, Streilein JW, et al. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J Am Soc Nephrol. 2004;15:603–12.
Fogt F, Pascha TL, Zhang PJ, Gausas RE, Rahemtulla A, Zimmerman RL. Lymphatic endothelial progenitor cells contribute to de novo lymphangiogenesis in human renal transplants. Nat Med. 2006;12:230–4. https://doi.org/10.1038/nm1340.
Cursiefen C, et al. Inhibition of hemangiogenesis and lymphangiogenesis after normal-risk corneal transplantation by neutralizing VEGF promotes graft survival. Invest Ophthalmol Vis Sci. 2004;45:2666–73. https://doi.org/10.1167/iovs.03-138045/8/2666.
Dietrich T, et al. Inhibition of inflammatory lymphangiogenesis by integrin alpha5 blockade. Am J Pathol. 2007;171:361–72. S0002-9440(10)61969-4 [pii].
Chen L, et al. Vascular endothelial growth factor receptor-3 mediates induction of corneal alloimmunity. Nat Med. 2004;10:813–5. https://doi.org/10.1038/nm1078nm1078.
Zhang Q, Lu Y, Proulx ST, Guo R, Yao Z, Schwarz EM, et al. Proliferation of D2-40-expressing intestinal lymphatic vessels in the lamina propria in inflammatory bowel disease. Int J Mol Med. 2004;13:211–4.
Kaiserling E, Krober S, Geleff S. Lymphatic vessels in the colonic mucosa in ulcerative colitis. Lymphology. 2003;36:52–61.
Pedica F, Ligorio C, Tonelli P, Bartolini S, Baccarini P. Lymphangiogenesis in Crohn’s disease: an immunohistochemical study using monoclonal antibody D2-40. Virchows Arch. 2008;452:57–63. https://doi.org/10.1007/s00428-007-0540-2.
Geleff S, Schoppmann SF, Oberhuber G. Increase in podoplanin-expressing intestinal lymphatic vessels in inflammatory bowel disease. Virchows Arch. 2003;442:231–7. https://doi.org/10.1007/s00428-002-0744-4.
Zhang Q, Lu Y, Proulx ST, Guo R, Yao Z, Schwarz EM, et al. Increased lymphangiogenesis in joints of mice with inflammatory arthritis. Arthritis Res Ther. 2007;9:R118. https://doi.org/10.1186/ar2326.
Kunstfeld R, Hirakawa S, Hong YK, Schacht V, Lange-Asschenfeldt B, Velasco P, et al. Distribution of lymphatic vessels in normal and arthritic human synovial tissues. Ann Rheum Dis. 2003;62:1227–9.
Wilkinson LS, Edwards JC. Demonstration of lymphatics in human synovial tissue. Rheumatol Int. 1991;11:151–5.
Yao LC, Baluk P, Feng J, McDonald DM. Steroid-resistant lymphatic remodeling in chronically inflamed mouse airways. Am J Pathol. 2010;176:1525–41. https://doi.org/10.2353/ajpath.2010.090909.
Baluk P, Tammela T, Ator E, Lyubynska N, Achen MG, Hicklin DJ, et al. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest. 2005;115:247–57.
Farnsworth RH, Karnezis T, Maciburko SJ, Mueller SN, Stacker SA. Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood. 2004;104:1048–57.
Kajiya K, Kunstfeld R, Detmar M, Chung JH. Reduction of lymphatic vessels in photodamaged human skin. J Dermatol Sci. 2007;47:241–3. https://doi.org/10.1016/j.jdermsci.2007.05.003.
Sugaya M, Kuwano Y, Suga H, Miyagaki T, Ohmatsu H, Kadono T, et al. Lymphatic dysfunction impairs antigen-specific immunization, but augments tissue swelling following contact with allergens. J Invest Dermatol. 2012;132:667–76. https://doi.org/10.1038/jid.2011.349.
Norrmen C, Tammela T, Petrova TV, Alitalo K. Biological basis of therapeutic lymphangiogenesis. Circulation. 2011;123:1335–51. https://doi.org/10.1161/CIRCULATIONAHA.107.704098
Farnsworth RH, Karnezis T, Maciburko SJ, Mueller SN, Stacker SA. The interplay between lymphatic vessels and chemokines. Front Immunol. 2019;10:518. https://doi.org/10.3389/fimmu.2019.00518.
Sainz-Jaspeado M, Claesson-Welsh L. Cytokines regulating lymphangiogenesis. Curr Opin Immunol. 2018;53:58–63. https://doi.org/10.1016/j.coi.2018.04.003.
Planas-Paz L, Lammert E. Mechanical forces in lymphatic vascular development and disease. Cell Mol Life Sci. 2013;70:4341–54. https://doi.org/10.1007/s00018-013-1358-5.
Dixon JB, Greiner ST, Gashev AA, Cote GL, Moore JE, Zawieja DC. Lymph flow, shear stress, and lymphocyte velocity in rat mesenteric prenodal lymphatics. Microcirculation. 2006;13:597–610. https://doi.org/10.1080/10739680600893909.
Swartz MA, Fleury ME. Interstitial flow and its effects in soft tissues. Annu Rev Biomed Eng. 2007;9:229–56. https://doi.org/10.1146/annurev.bioeng.9.060906.151850.
Levick JR, Michel CC. Microvascular fluid exchange and the revised Starling principle. Cardiovasc Res. 2010;87:198–210. https://doi.org/10.1093/cvr/cvq062.
Rahbar E, Akl T, Cote GL, Moore JE Jr, Zawieja DC. Lymph transport in rat mesenteric lymphatics experiencing edemagenic stress. Microcirculation. 2014;21:359–67. https://doi.org/10.1111/micc.12112.
Swartz MA, Boardman KC Jr. The role of interstitial stress in lymphatic function and lymphangiogenesis. Ann N. Y Acad Sci. 2002;979:197–210. https://doi.org/10.1111/j.1749-6632.2002.tb04880.x.
Ng CP, Helm CL, Swartz MA. Interstitial flow differentially stimulates blood and lymphatic endothelial cell morphogenesis in vitro. Microvasc Res. 2004;68:258–64. https://doi.org/10.1016/j.mvr.2004.08.002.
Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA. Endothelial fluid shear stress sensing in vascular health and disease. J Clin Invest. 2016;126:821–8. https://doi.org/10.1172/JCI83083.
Sabine A, Agalarov Y, Maby-El Hajjami H, Jaquet M, Hägerling R, Pollmann C, et al. Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic-valve formation. Dev Cell. 2012;22:430–45. https://doi.org/10.1016/j.devcel.2011.12.020.
Breslin JW, Kurtz KM. Lymphatic endothelial cells adapt their barrier function in response to changes in shear stress. Lymphat Res Biol. 2009;7:229–37. https://doi.org/10.1089/lrb.2009.0015.
Baeyens N, et al. Vascular remodeling is governed by a VEGFR3-dependent fluid shear stress set point. Elife. 2015;4. https://doi.org/10.7554/eLife.04645.
Baeyens N, Mulligan-Kehoe MJ, Corti F, Simon DD, Ross TD, Rhodes JM, et al. Syndecan 4 is required for endothelial alignment in flow and atheroprotective signaling. Proc Natl Acad Sci USA. 2014;111:17308–13. https://doi.org/10.1073/pnas.1413725111.
Miteva DO, Rutkowski JM, Dixon JB, Kilarski W, Shields JD, Swartz MA. Transmural flow modulates cell and fluid transport functions of lymphatic endothelium. Circ Res. 2010;106:920–31. https://doi.org/10.1161/CIRCRESAHA.109.207274.
Gasheva OY, Zawieja DC, Gashev AA. Contraction-initiated NO-dependent lymphatic relaxation: a self-regulatory mechanism in rat thoracic duct. J Physiol. 2006;575:821–32. https://doi.org/10.1113/jphysiol.2006.115212.
Liao S, Cheng G, Conner DA, Huang Y, Kucherlapati RS, Munn LL, et al. Impaired lymphatic contraction associated with immunosuppression. Proc Natl Acad Sci USA. 2011;108:18784–9. https://doi.org/10.1073/pnas.1116152108.
Torrisi JS, Hespe GE, Cuzzone DA, Savetsky IL, Nitti MD, Gardenier JC, et al. Inhibition of inflammation and iNOS improves lymphatic function in obesity. Sci Rep. 2016;6:19817. https://doi.org/10.1038/srep19817.
Bohlen HG, Wang W, Gashev A, Gasheva O, Zawieja D. Phasic contractions of rat mesenteric lymphatics increase basal and phasic nitric oxide generation in vivo. Am J Physiol Heart Circ Physiol. 2009;297:H1319–28. https://doi.org/10.1152/ajpheart.00039.2009.
Kornuta JA, Nepiyushchikh Z, Gasheva OY, Mukherjee A, Zawieja DC, Dixon JB. Effects of dynamic shear and transmural pressure on wall shear stress sensitivity in collecting lymphatic vessels. Am J Physiol Regul Integr Comp Physiol. 2015;309:R1122–34. https://doi.org/10.1152/ajpregu.00342.2014.
Kawai Y, Yokoyama Y, Kaidoh M, Ohhashi T. Shear stress-induced ATP-mediated endothelial constitutive nitric oxide synthase expression in human lymphatic endothelial cells. Am J Physiol Cell Physiol. 2010;298:C647–655. https://doi.org/10.1152/ajpcell.00249.2009.
Helm CL, Fleury ME, Zisch AH, Boschetti F, Swartz MA. Synergy between interstitial flow and VEGF directs capillary morphogenesis in vitro through a gradient amplification mechanism. Proc Natl Acad Sci USA. 2005;102:15779–84. https://doi.org/10.1073/pnas.0503681102.
Kim S, Chung M, Jeon NL. Three-dimensional biomimetic model to reconstitute sprouting lymphangiogenesis in vitro. Biomaterials. 2016;78:115–28. https://doi.org/10.1016/j.biomaterials.2015.11.019.
Yoon CM, Hong BS, Moon HG, Lim S, Suh PG, Kim YK, et al. Sphingosine-1-phosphate promotes lymphangiogenesis by stimulating S1P1/Gi/PLC/Ca2+ signaling pathways. Blood. 2008;112:1129–38. https://doi.org/10.1182/blood-2007-11-125203.
Surya VN, Michalaki E, Huang EY, Fuller GG, Dunn AR. Sphingosine 1-phosphate receptor 1 regulates the directional migration of lymphatic endothelial cells in response to fluid shear stress. J R Soc Interface. 2016;13. https://doi.org/10.1098/rsif.2016.0823.
Ostrowski MA, Huang NF, Walker TW, Verwijlen T, Poplawski C, Khoo AS, et al. Microvascular endothelial cells migrate upstream and align against the shear stress field created by impinging flow. Biophys J. 2014;106:366–74. https://doi.org/10.1016/j.bpj.2013.11.4502.
Geng X, et al. S1PR1 regulates the quiescence of lymphatic vessels by inhibiting laminar shear stress-dependent VEGF-C signaling. JCI Insight. 2020;5. https://doi.org/10.1172/jci.insight.137652.
Planas-Paz L, Strilić B, Goedecke A, Breier G, Fässler R, Lammert E. Mechanoinduction of lymph vessel expansion. EMBO J. 2012;31:788–804. https://doi.org/10.1038/emboj.2011.456.
Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol. 2010;2:a005066. https://doi.org/10.1101/cshperspect.a005066.
Zhang X, Groopman JE, Wang JF. Extracellular matrix regulates endothelial functions through interaction of VEGFR-3 and integrin alpha5beta1. J Cell Physiol. 2005;202:205–14. https://doi.org/10.1002/jcp.20106.
Galvagni F, et al. Endothelial cell adhesion to the extracellular matrix induces c-Src- dependent VEGFR-3 phosphorylation without the activation of the receptor intrinsic kinase activity. Circ Res. 2010;106:1839–48. https://doi.org/10.1161/CIRCRESAHA.109.206326.
Bazigou E, Xie S, Chen C, Weston A, Miura N, Sorokin L, et al. Integrin-alpha9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Dev Cell. 2009;17:175–86. https://doi.org/10.1016/j.devcel.2009.06.017.
Hong YK, Lange-Asschenfeldt B, Velasco P, Hirakawa S, Kunstfeld R, Brown LF, et al. VEGF-A promotes tissue repair-associated lymphatic vessel formation via VEGFR-2 and the alpha1beta1 and alpha2beta1 integrins. FASEB J. 2004;18:1111–3. https://doi.org/10.1096/fj.03-1179fje.
Kajiya K, Hirakawa S, Ma B, Drinnenberg I, Detmar M. Hepatocyte growth factor promotes lymphatic vessel formation and function. EMBO J. 2005;24:2885–95. https://doi.org/10.1038/sj.emboj.7600763.
Chen L, Huq S, Gardner H, de Fougerolles AR, Barabino S, Dana MR. Very late antigen 1 blockade markedly promotes survival of corneal allografts. Arch Ophthalmol. 2007;125:783–8. https://doi.org/10.1001/archopht.125.6.783.
Garmy-Susini B, Avraamides CJ, Schmid MC, Foubert P, Ellies LG, Barnes L, et al. Integrin alpha4beta1 signaling is required for lymphangiogenesis and tumor metastasis. Cancer Res. 2010;70:3042–51. https://doi.org/10.1158/0008-5472.CAN-09-3761.
Sabine A, Bovay E, Demir CS, Kimura W, Jaquet M, Agalarov Y, et al. FOXC2 and fluid shear stress stabilize postnatal lymphatic vasculature. J Clin Invest. 2015;125:3861–77. https://doi.org/10.1172/JCI80454.
Shen Z, Stanger BZ. YAP regulates S-phase entry in endothelial cells. PLoS One. 2015;10:e0117522. https://doi.org/10.1371/journal.pone.0117522.
Sweet DT, Jiménez JM, Chang J, Hess PR, Mericko-Ishizuka P, Fu J, et al. Lymph flow regulates collecting lymphatic vessel maturation in vivo. J Clin Invest. 2015;125:2995–3007. https://doi.org/10.1172/JCI79386.
Petrova TV, Karpanen T, Norrmén C, Mellor R, Tamakoshi T, Finegold D, et al. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat Med. 2004;10:974–81. https://doi.org/10.1038/nm1094.
Kazenwadel J, Secker GA, Liu YJ, Rosenfeld JA, Wildin RS, Cuellar-Rodriguez J, et al. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood. 2012;119:1283–91. https://doi.org/10.1182/blood-2011-08-374363.
Kanady JD, Dellinger MT, Munger SJ, Witte MH, Simon AM. Connexin37 and Connexin43 deficiencies in mice disrupt lymphatic valve development and result in lymphatic disorders including lymphedema and chylothorax. Dev Biol. 2011;354:253–66. https://doi.org/10.1016/j.ydbio.2011.04.004.
Hernández Vásquez MN, Ulvmar MH, González-Loyola A, Kritikos I, Sun Y, He L, et al. Transcription factor FOXP2 is a flow-induced regulator of collecting lymphatic vessels. EMBO J. 2021;40:e107192. https://doi.org/10.15252/embj.2020107192.
Betterman KL, Sutton DL, Secker GA, Kazenwadel J, Oszmiana A, Lim L, et al. Atypical cadherin FAT4 orchestrates lymphatic endothelial cell polarity in response to flow. J Clin Invest. 2020;130:3315–28. https://doi.org/10.1172/JCI99027.
Choi D, Park E, Jung E, Seong YJ, Hong M, Lee S, et al. ORAI1 activates proliferation of lymphatic endothelial cells in response to laminar flow through Kruppel-like factors 2 and 4. Circ Res. 2017;120:1426–39. https://doi.org/10.1161/CIRCRESAHA.116.309548.
Choi D, Park E, Jung E, Seong YJ, Yoo J, Lee E, et al. Laminar flow downregulates Notch activity to promote lymphatic sprouting. J Clin Invest. 2017;127:1225–40. https://doi.org/10.1172/JCI87442.
Jafarnejad M, Cromer WE, Kaunas RR, Zhang SL, Zawieja DC, Moore JE Jr. Measurement of shear stress-mediated intracellular calcium dynamics in human dermal lymphatic endothelial cells. Am J Physiol Heart Circ Physiol. 2015;308:H697–706. https://doi.org/10.1152/ajpheart.00744.2014.
Coon BG, Baeyens N, Han J, Budatha M, Ross TD, Fang JS, et al. Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J Cell Biol. 2015;208:975–86. https://doi.org/10.1083/jcb.201408103.
Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–31. https://doi.org/10.1038/nature03952.
Fleming I, Fisslthaler B, Dixit M, Busse R. Role of PECAM-1 in the shear- stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J Cell Sci. 2005;118:4103–11. https://doi.org/10.1242/jcs.02541.
Conway DE, Breckenridge MT, Hinde E, Gratton E, Chen CS, Schwartz MA. Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr Biol. 2013;23:1024–30. https://doi.org/10.1016/j.cub.2013.04.049.
Wang Y, Baeyens N, Corti F, Tanaka K, Fang JS, Zhang J, et al. Syndecan 4 controls lymphatic vasculature remodeling during mouse embryonic. Dev Dev. 2016;143:4441–51. https://doi.org/10.1242/dev.140129.
Miao H, Hu YL, Shiu YT, Yuan S, Zhao Y, Kaunas R, et al. Effects of flow patterns on the localization and expression of VE- cadherin at vascular endothelial cell junctions: in vivo and in vitro investigations. J Vasc Res. 2005;42:77–89. https://doi.org/10.1159/000083094.
Choi D, Park E, Yu RP, Cooper MN, Cho IT, Choi J, et al. Piezo1-regulated mechanotransduction controls flow-activated lymphatic expansion. Circ Res. 2022;131:e2–21. https://doi.org/10.1161/CIRCRESAHA.121.320565.
Cox CD, Bavi N, Martinac B. Origin of the force: the force-from-lipids principle applied to Piezo Channels. Curr Top Membr. 2017;79:59–96. https://doi.org/10.1016/bs.ctm.2016.09.001.
Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, et al. Piezo1 integration of vascular architecture with physiological force. Nature. 2014;515:279–82. https://doi.org/10.1038/nature13701.
Nonomura K, Lukacs V, Sweet DT, Goddard LM, Kanie A, Whitwam T, et al. Mechanically activated ion channel PIEZO1 is required for lymphatic valve formation. Proc Natl Acad Sci USA. 2018;115:12817–22. https://doi.org/10.1073/pnas.1817070115.
Choi D, Park E, Jung E, Cha B, Lee S, Yu J, et al. Piezo1 incorporates mechanical force signals into the genetic program that governs lymphatic valve development and maintenance. JCI Insight. 2019;4. https://doi.org/10.1172/jci.insight.125068.
Lukacs V, Mathur J, Mao R, Bayrak-Toydemir P, Procter M, Cahalan SM, et al. Impaired PIEZO1 function in patients with a novel autosomal recessive congenital lymphatic dysplasia. Nat Commun. 2015;6:8329. https://doi.org/10.1038/ncomms9329.
Fotiou E, Martin-Almedina S, Simpson MA, Lin S, Gordon K, Brice G, et al. Novel mutations in PIEZO1 cause an autosomal recessive generalized lymphatic dysplasia with non-immune hydrops fetalis. Nat Commun. 2015;6:8085. https://doi.org/10.1038/ncomms9085.
Acknowledgements
This work was supported by NUHS internal funding and the Ministry of Education (Project 594 ID: MOE2019-T2-1-136) to VA and the National Medical Research Council (Y-IRG grant) to HYL.
Author information
Authors and Affiliations
Contributions
VA helped write the manuscript, and HYL helped write the manuscript and generated the figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Angeli, V., Lim, H.Y. Biomechanical control of lymphatic vessel physiology and functions. Cell Mol Immunol 20, 1051–1062 (2023). https://doi.org/10.1038/s41423-023-01042-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-023-01042-9
