
Abstract
Pyroptosis is a form of programmed cell death mediated by gasdermin and is a product of continuous cell expansion until the cytomembrane ruptures, resulting in the release of cellular contents that can activate strong inflammatory and immune responses. Pyroptosis, an innate immune response, can be triggered by the activation of inflammasomes by various influencing factors. Activation of these inflammasomes can induce the maturation of caspase-1 or caspase-4/5/11, both of which cleave gasdermin D to release its N-terminal domain, which can bind membrane lipids and perforate the cell membrane. Here, we review the latest advancements in research on the mechanisms of pyroptosis, newly discovered influencing factors, antitumoral properties, and applications in various diseases. Moreover, this review also provides updates on potential targeted therapies for inflammation and cancers, methods for clinical prevention, and finally challenges and future directions in the field.
Similar content being viewed by others
Introduction
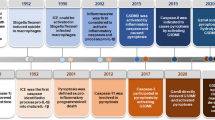
Pyroptosis is a newly discovered programmed cell death regulated by unique sets of critical inflammatory caspases that coordinate biological effects [1, 2]. Scientists first discovered pyroptosis induced by Shigella flexneri in infected macrophages; however, it was misclassified as apoptosis because of the limitations of biotechnology at the time [3]. Later research found that caspase-1 played a very pivotal role in bacteria-induced cell death, which was not observed in other studies [4, 5]. The term “pyroptosis” was first proposed in 2001, which comes from the Greek roots “pyro” and “ptosis”, meaning fever and fall, respectively, to describe this novel cell death in macrophages infected by Salmonella [6].
To date, it has been confirmed that Salmonella [4], Shigella flexneri [5], Listeria [7], Pseudomonas aeruginosa [8], Legionella pneumophila [9], and Yersinia [10, 11] can induce macrophage death induced by caspase-1, not only in mononuclear macrophage cells but also in other cells, such as dendritic cells [12]. Additionally, the stimulus is not limited to conserved PAMPs (pathogen‐associated molecular patterns); some nonbiological stimuli, such as DAMPs (danger/damage-associated molecular patterns), can also activate host pattern recognition receptors to trigger pyroptosis [1, 13, 14]. The definition of pyroptosis has expanded to include cell death triggered by many other caspases, including caspase-4/5/11 [15].
Pyroptosis is executed by gasdermin D (GSDMD); manifests as continuous cell expansion until the cytomembrane ruptures, resulting in the release of cellular contents that activate a strong inflammatory response; and is involved in many pathophysiological processes [16, 17]. The effector of pyroptosis remained unknown until 2015, when Shao Feng et al. discovered that caspase-1/11/4/5 can induce pyroptosis by cleaving a protein called GSDMD to release its N-terminal domain [18, 19]. The N-terminus of GSDMD is highly aggregated and is capable of binding membrane lipids and punching holes in the cell membrane, which leads to changes in cell osmotic pressure and gradual swelling until the final cell membrane ruptures [18, 20, 21]. Pyroptosis shares some aspects of apoptosis, necroptosis and ferroptosis, such as the inducers of stimulation, key components in the process, morphological changes, and cell release (Table 1).
The gasdermin family is classified as pore-forming effector proteins that induce pyroptosis, and gasdermin D is one of six gasdermins found in humans [22, 23]. Gasdermins have strong sequence similarities with the autosomal DFNA5 (dominant nonsyndromic sensory nerve 5) gene that codes for a protein associated with human autosomal dominant nonsyndromic hearing loss [22, 24]. Despite an increase in interest in the mechanisms of gasdermins, their exact biological functions remain poorly understood [22, 25].
After years of searching, comprehensive research methods and analysis tools for pyroptosis have become relatively mature. The key to this field of research is to quantify inflammasome activation and detect the activity of multiple downstream pathways in a consistent and reproducible approach. The inflammasome can be activated by corresponding activators, such as bacteria, viruses, fungi, inflammatory diseases and cancer in vivo [26]. In vitro, bone marrow–derived macrophages (BMDMs) need to first be isolated and then differentiated before activating inflammasomes, which are also triggered by bacteria, viruses, fungi, and ligand-based activators [26]. Several different molecular techniques and assay methods are currently available to monitor multiple components of the inflammasome activation cascade and simultaneously assess inflammasome-activated pyroptosis. The procedures include real-time monitoring of cell status and cell death, microscopy of ASC spots, and LDH (lactate dehydrogenase) assays [26]. Simultaneously, ELISAs are used to determine the release amount of cytokines such as IL-1β and IL-18 from cells into the culture medium, and western blotting is performed to determine caspase-1 and gasdermin D cleavage [26, 27].
Pyroptosis is an important natural immune response that specifically antagonizes infection and endogenous danger signals [1, 28, 29]. Pyroptosis can cause the release of cytokines, promote the activation of macrophages and T lymphocytes, prompt the body to produce a strong inflammatory response, and induce immune phagocytosis [23, 30]. Several immune-mediated diseases in humans have been associated with mutations in components of inflammasome complexes [28]. Therefore, in-depth research is needed to understand the function in inflammation and cancer. This review will provide a comprehensive introduction of the latest progress in comprehending the mechanisms of pyroptosis, newly discovered influencing factors, and applications in treating diseases caused by inflammation. We further summarize the antitumor role of pyroptosis in several types of cancer and its potential applications for the treatment of inflammation and cancer. Finally, we address challenges and future directions in this field.
Basic mechanisms of canonical and noncanonical inflammasomes
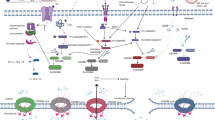
Classic theory states that pyroptosis mainly relies on inflammasomes to activate caspase family proteins to cause various physiological reactions [27, 31]. Inflammasomes are multimeric complexes composed of essential components, wherein they protect host cells against the assault of endogenous danger signals and some pathogens [29, 32]. The pathways by which inflammasomes trigger pyroptosis are traditionally grouped into canonical pathways by caspase-1 activation or noncanonical pathways by caspase-11 or caspase-4/5 activation in mice or human beings, respectively [33, 34]. Canonical inflammasomes are usually formed by sensor proteins called pattern-recognition receptors (PRRs), an adaptor protein (ASC) called apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD), and an inactive pro-caspase-1 [19, 35, 36] (Fig. 1). There are five main types of inflammasomes in canonical pathways: the NLRP3 inflammasome, AIM2 inflammasome, NLRP1 inflammasome, PYRIN inflammasome, and NLRC4 inflammasome [37, 38]. When the PRR is stimulated, inflammasomes produce mature caspase-1, which is different from the noncanonical pathway [13, 39, 40]. Caspase-11 directly recognizes stimuli and is activated by LPS (lipopolysaccharide), which is derived from Gram-negative bacteria such as Escherichia coli [31, 41, 42]. Activated caspase-1/11 can cleave complete GSDMD into two parts, namely, GSDMD-NT (N-terminal domain of GSDMD) and GSDMD-CT (C-terminal domain of GSDMD), but only caspase-1 is competent to induce IL-1β and IL-18 [13, 43, 44] (Fig. 1).

Basic mechanisms of canonical and noncanonical inflammasomes. The mechanisms of pyroptosis are divided into canonical pathways activated by caspase-1 and noncanonical pathways activated by caspase-11/4/5. Activated caspase-1/11 can induce the formation of GSDMD-NT, which contributes to the perforation of the cell membrane and enhances the capability of processing IL-1β and IL-18. Canonical inflammasomes, including NLRP3, AIM2, NLRP1, PYRIN, and NLRC4 inflammasomes, are usually composed of PRR, ASC, and pro-caspase-1. NLRP3 is triggered by endogenous danger signals, such as pore-forming toxins, crystalline structures, extracellular ATP, and RNA. NLRP1 is mainly activated by physiologically relevant factors, including the lethal toxin found in anthrax and muramyl dipeptide. NLRC4 typically responds to bacterial flagellin and PrgJ, which are rod and needle proteins. AIM2 directly binds to cytosolic dsDNA from DNA viruses to activate them. Pyrin activation is caused by exposure to pathogenic toxins. The noncanonical pyroptosis pathway is directly triggered by LPS from extracellular Gram‐negative bacteria
Recent research has identified some upstream signaling sources that activate the different receptors involved in inflammasome formation. Further studies have determined the structure of these sources as large quaternary structure multiprotein complexes. Inflammasomes identified thus far include members of the NLR family (nucleotide binding and oligomerization domains (Nod) and leucine-rich repeat-containing protein (NLR)), the PYHIN family (Pyrin domains and HIN-containing domains) and AIM2 (absent in melanoma 2) [37, 45]. NLR inflammasomes contain the common adaptor ASC on most occasions, but ASC-independent complexes can also exist in certain cases [46].
Different inflammasomes react to a wide array of stimuli. Endogenous danger signals, such as pore-forming toxins, crystalline structures, and extracellular ATP or RNA, can trigger activation of the NLRP3 inflammasome, which decreases the intracellular K+ concentration, promotes the release of cellular lysate from lysosomes, and induces mitochondrial damage [13, 47]. In addition, several pathogens, such as Staphylococcus aureus, and other factors, such as ultraviolet radiation, are also thought to activate the NLRP3 inflammasome [37, 48]. NLRP1 is mainly activated by physiologically relevant factors, including anthrax LeTx (a lethal toxin from Bacillus anthracis), muramyl dipeptide, dsRNA and enteroviral 3 C protease [37, 49, 50]. Humans only have a single NLRP1 gene, while the NLRP1 gene in mice is polymorphic, of which NLRP1b is the main research object [51, 52]. Some research has reported that NLRP1b possesses autoproteolytic activity and that its N‐terminal fragments are required for the subsequent activation of the NLRP1 inflammasome [52, 53]. Other activators of NLRP1 are less understood and require further investigation. The NLRC4 inflammasome has been studied in-depth in a mouse model, wherein it is shown that the expression of NLRC4 is typically induced by bacterial flagellin or PrgJ, both components of the bacterial type III secretion system (T3SS) apparatus [54]. Unlike the aforementioned inflammasomes, NLRC4 is activated by the distinctive receptor NAIP; however, it does not require the adaptor protein ASC to induce pyroptosis [55]. Nevertheless, the presence of ASC can substantially augment NLRC4-mediated pyroptosis [55, 56]. AIM2 is another type of inflammasome that directly binds to cytosolic dsDNA from DNA viruses through HIN-200 domains, unlike NLR inflammasomes [57, 58]. However, AIM2 does not contain CARD domains; therefore, signaling transmission requires the recruitment of ASC for inflammasome activation [59]. The PYRIN inflammasome encoded by the MEFV gene is also formed with ASC and caspase-1, but the activation factors have remained unclear for some time [60, 61]. To date, it has been found that the PYRIN inflammasome is activated by pathogenic toxins, such as cytotoxic TcdB [62, 63] (Fig. 1).
Caspase-11 was largely ignored until recently due to its function in the process of pyroptosis, which has challenged the view of caspase-1 as the sole caspase protein responsible for inflammasome-dependent pyroptosis [64]. Recent studies have confirmed the critical role of LPS derived from Gram‐negative bacteria, which is sensed by transmembrane TLR4 (Toll‐like receptor 4) in caspase‐4/5/11‐dependent pyroptosis, and intracellular LPS can directly bind to caspase-4/5/11 to result in pyroptosis [65,66,67] (Fig. 1).
Emerging mechanisms of essential components in the pyroptosis pathway
Pyroptosis is an extremely complex process in the body, so it is understood that there are many positive or negative regulatory mechanisms controlling the essential components in the process of pyroptosis. Numerous mysteries about the regulatory mechanisms of pyroptosis have been clarified recently.
Novel regulation of NLRP3
Given its involvement in a variety of pathogens, including bacteria, viruses and fungi, the NLRP3 inflammasome has drawn extensive attention from researchers [68]. Recent studies have revealed many exact mechanisms associated with the NLRP3 pathways that can be activated or inhibited by many factors. The NLRP3 receptor protein contains three domains: PYD, NACHT, and LRR [69, 70]. Upon NLRP3 inflammasome assembly, the PYD of NLRP3 interacts with the PYD of ASC to form a homotypic PYD-PYD interaction, while the CARD structure in ASC can recruit and combine with the CARD domain of caspase-1 to generate a similar CARD-CARD interaction [31, 70,71,72]. ASC functions here as a bridge to connect the receptor protein and the effector protein. GSDMD, an essential pyroptosis substrate of inflammatory caspases, is a crucial executor of pyroptosis [1]. Mature caspase-1 cleaves GSDMD to generate approximately 31-kDa N-terminal fragments that initiate pyroptosis and induce the secretion of mature inflammatory cytokines [18, 40, 73] (Fig. 2).

Novel regulation of NLRP3 and gasdermin D. There are many signaling pathways influencing the functions of the NLRP3 inflammasome and gasdermin D to regulate the occurrence of pyroptosis. For example, the PINK1-PARK2 pathway induces the release of dopamine to function on PYD to induce NLRP3 inflammasome activation. In addition, PKM2 selectively activates EIF2AK2 phosphorylation to trigger the NLRP3 inflammasome in macrophages. The kinase NEK7 can combine with NLRP3 to control the activation of the NLRP3 inflammasome, which is suppressed by phosphorylation of PLK4 and is formed by deubiquitinating the Spata2-CYLD complex. DDX3X is coupled with the NACHT domain of full-length NLRP3. The CARD8 inflammasome senses the activities of HIV-1 protease and DPP8/9 inhibitor to regulate NLRP3-caspase-1-dependent pyroptosis. VbP disrupts the interaction of NLRP1 and DPP9 to accelerate the degradation of N-terminal fragments. The cGAS-STING pathway promotes the NF-κB function on caspase-1 to induce pyroptosis, which is suppressed by melatonin. Furthermore, the mTORC1-Ragulator-Rag pathway promotes ROS production to regulate the oligomerization of GSDMD and pore formation. The ESCRT system repairs membrane pores to alleviate pyroptosis and inhibit the production of IL-1β, which is otherwise upregulated by calcium efflux. GPX4 blocks lipid peroxidation to suppress GSDMD activity and inhibits caspase-1/11 to prevent the production of GSDMD-NT. ELANE-mediated GSDMD cleavage produces an active GSDMD-eNT that causes pyroptosis. Finally, IRF7 can physically interact with GSDMD-NT to cause pyroptosis
It is reported that the PINK1-PARK2 pathway is usually considered to function in the pathogenesis of Parkinson’s disease [74]. Additionally, the PINK1-PARK2 pathway can alleviate inflammation by upregulating dopamine, thus downregulating the activation of HIF1α [75]. HIF1α can activate the NLRP3 inflammasome and release abundant HMGB1, a molecular transporter of LPS, into the cytoplasm in a mouse neuroimmune model [75, 76]. EIF2AK2 (eukaryotic translation initiation factor 2 alpha kinase 2) is one of four mammalian kinases involved in the ISR (integrated stress response), and its canonical activation is triggered by dsRNA during viral infection [77]. The M2 isoform of pyruvate kinase (PKM2), a glycolytic regulator, selectively triggers EIF2AK2 phosphorylation to promote NLRP3 inflammasome activation in macrophages, which may be a target for the therapeutic treatment of sepsis [78]. The pharmacological PKM2 inhibitor shikonin and EIF2AK2 inhibitor C16 both effectively suppress EIF2AK2 phosphorylation levels so that caspase-1 enzyme activity is somewhat inhibited in macrophages of septic mice, ultimately promoting a higher survival rate from sepsis in mouse experiments [78,79,80]. The kinase NEK7, which is responsible for cell spindle formation and centrosome separation, is an on-off regulatory protein that binds to NLRP3 protein and regulates the activation of the NLRP3 inflammasome [81, 82]. Research on this mechanism found that the Spata2 and CYLFD genes both play important roles in regulating inflammasome complexes; these genes promote the binding of PLK4 and NEK7 by removing the ubiquitination linked to the centrosome of the kinase PLK4 at position K63, thereby resulting in the phosphorylation of NEK7 at the Ser204 site [83,84,85]. Studies have shown that phosphorylation at the Ser204 site inhibits NEK7 from binding to NLRP3, thereby negatively regulating the activation of the NLRP3 inflammasome [84]. DDX3X is an enzyme that, when mutated, is associated with the development of various cancers, including liver cancer, lung cancer, and pancreatic cancer [86, 87]. DDX3X can combine with the NACHT domain of full-length NLRP3, which influences whether cells survive or are killed via pyroptosis [88]. One study revealed that the formation of stress granules induced by sodium arsenite specifically inhibited the activation of the NLRP3 inflammasome [89]. After LPS stimulation, the authors used affinity purification mass spectrometry to identify any physical interaction between DDX3X and NLRP3 and examined the regulatory mechanism of the packaging of the NLRP3 inflammasome, which may be a candidate for the design of a targeted therapeutic [90] (Fig. 2).
The work stated that CARD8 can trigger pyroptosis to clear infected cells as a sensor for HIV (human immunodeficiency virus) protease, which could be an effective tactic for the elimination of HIV infection [91]. However, HIV can evade the perception of CARD8, as its protease remains inactive before the virus buds in the infected cells [92]. Some drugs can force HIV protease to be expressed prematurely while the virus is still in immune cells, which would then trigger the CARD8 inflammasome to destroy the virus in infected cells [93]. The DPP8/9 inhibitor is also known to induce pyroptosis in human monocytes and macrophages [94, 95]. It has also been shown that the CARD8 protein regulates NLRP3-caspase-1-dependent pyroptosis induced by the DPP8/9 inhibitor, which activates the sensor NLRP1 and directs mature caspase-1 to cause cell death in human myeloid cells [96, 97] (Fig. 2).
NLRP1, an inflammasome sensor whose N-terminus is degraded by the proteasome, mediates the activation of caspase-1 and induces cytokine maturation, which is inserted into the active site of DPP9, and this process requires the presence of full-length NLRP1 [98,99,100]. Val-boroPro (VBP) disrupts the interaction of NLRP1 and DPP9, which promotes the degradation of NLRP1 N-terminal fragments, thereby triggering inflammasome activation [98, 101, 102]. The cGAS-STING signaling pathway is a canonical innate immune system that is known to be closely related to pyroptosis [103]. GSDMD in intestinal macrophages can negatively regulate the cGAS-STING-mediated inflammatory response by perforating the plasma membrane to increase the efflux of potassium ions after the onset of colitis [104, 105]. The cGAS-STING pathway promotes the interaction between NF-κB and caspase-1 to induce pyroptosis, but melatonin inhibits the NF-kB-induced GSDMD pathway to partially inhibit pyroptosis in adipose cells of mice [106, 107] (Fig. 2).
Novel regulation of gasdermin D
Gasdermin D is a well-studied critical effector of pyroptosis, and its regulatory mechanism is mainly focused on the upstream factors that affect its early cleavage, which include pleiotropic pyroptosis-associated complexes that exert pro- or anti-inflammatory effects. However, few studies exist on the factors that influence the formation of cytomembrane pores after GSDMD cleavage. The mTORC1-Ragulator-Rag pathway plays an essential role in membrane pore formation, wherein it promotes reactive oxygen species (ROS) production that will regulate oligomerization of GSDMD and pore formation [108, 109]. The intracellular endosomal sorting complexes required for transport (ESCRT) system is reported to restore membrane pores in the downstream process of pyroptosis without affecting the activation of GSDMD, thus alleviating pyroptosis and inhibiting the release of IL-1β, which is promoted by calcium influx to prevent cell death [110,111,112]. Additionally, glutathione peroxidase 4 (GPX4) can suppress lipid peroxidation to inhibit phospholipase C gamma 1 (PLCG1), thereby preventing the production of GSDMD-NT, which can be improved by the antioxidant activity of vitamin E to promote the survival rate in sepsis [113, 114]. Furthermore, the cleavage of GSDMD in neutrophils is not dependent on caspase proteins but rather on ELANE (neutrophil-specific serine protease, neutrophil elastase), which is a tissue-specific protein complex [115,116,117]. GSDMD cleavage mediated by ELANE produces an active GSDMD-eNT that induces pyroptosis as efficiently as GSDMD-NT, and only its cleavage site is upstream compared with the latter [115]. Moreover, IRF7 (interferon regulatory factor 7), promoted by NF-κB, can physically interact with GSDMD-NT to form a complex that then promotes adipocyte pyroptosis [107, 118] (Fig. 2).
Newly discovered roles of other gasdermins
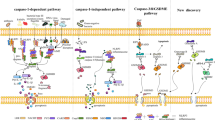
Gasdermins, a conserved protein family, contain gasdermin A, B, C, D, E, and DFNB59, most of which have recently been confirmed to work on pore formation [119, 120]. In addition to GSDMD, there have been many recent studies on the pyroptotic mechanisms of other gasdermin families. Pyroptosis appeared to be only activated by caspase families, but recent research findings have challenged this notion. For example, a recent study revealed that gasdermin can perform the punching function at non-Asp sites via the hydrolysis of the serine protease Granzyme A, a serine protease produced by cytotoxic lymphocytes [121, 122]. Granzyme A can enter target cells through perforin and hydrolyze the Lys229/Lys244 sites of GSDMB to trigger pyroptosis, and GSDMB has tissue-specific expression and is highly upregulated by interferon-γ in epithelial tumor cells [121]. Furthermore, cytotoxic lymphocyte-mediated immunity is dependent on granzymes, which can enhance antitumor immunity and may be a potential candidate targeted site for tumor treatments [121, 123] (Fig. 3).

New roles of other gasdermins. Granzyme A can enter target cells through perforin and can hydrolyze gasdermin B, which is upregulated by interferon-γ. The IpaH7.8 effector secreted by Shigella flexneri allows GSDMB to be used for 26 S proteasome degradation to inhibit Granzyme A-mediated, GSDMB-induced pyroptosis. Another gasdermin, granzyme B, can induce the maturation of caspase-3 to cleave GSDME, where the mechanism of cell death changes from apoptosis to pyroptosis. Under hypoxic conditions, PD-L1 interacts with p-Stat3-Y705 to promote the GSDMC-induced triggering of pyroptosis, which is promoted by TNFα and CHX but prevented by Stat3. TNF-α activates caspase-8 to cleave GSDMC, thus avoiding caspase-8 activation and instead activating caspase-3-induced pyroptosis. Activated GAS cysteine protease SpeB cleaves GSDMA by direct proteolysis after the Gln246 site 134 to form GSDMA pores in the host cell membrane, promoting the pyroptosis of infected cells and ultimately resulting in local inflammation and the clearance of pathogens
GSDMB can also be used as an antibacterial cytolysin, activated by NK cells, in Shigella-infected cells through Granzyme A, and it can self-cleave the N-terminus to perforate the bacterial membrane [124, 125]. Unlike most gasdermin family members, GSDMB demonstrates direct antibacterial activity, wherein it recognizes phospholipids on the membrane of Gram-negative bacteria [126]. However, the IpaH7.8 protein secreted by the intestinal invasive species Shigella flexneri through the expression of the T3SS ubiquitinates GSDMB for 26 S proteasomal degradation and inhibits granzyme A-mediated GSDMB activation to protect Shigella from NK cells, creating a dynamic host‒pathogen conflict [124, 127] (Fig. 3).
Granzyme B is also the most abundant granzyme in cytotoxic granules and is transported to cancer cells, where it cleaves caspase-3 and perforin to induce apoptosis [128, 129]. Granzyme B can induce mature caspase-3 to cleave GSDME and can simultaneously induce pyroptosis independent of caspase by directly cutting GSDME at Asp270, which converts noninflammatory apoptosis into pyroptosis in cells expressing GSDME [130] (Fig. 3).
Recent studies have concentrated on the regulatory mechanisms of tumor pyroptosis, finding that the pyroptosis pathway of tumor cells is completely different from that of macrophage pyroptosis, as evidenced by the finding that the nPD-L1-GSDMC-caspase-8 pathway mediates pyroptosis to lead to tumor death [23, 131]. A large number of PD-L1 molecules enter the cells and nucleus under hypoxic conditions, which is promoted by TNFα and CHX treatment [132]. PD-L1 then interacts with p-Stat3-Y705, which then binds to the GSDMC promoter region to increase the expression of the latter, which can be induced by antibiotic chemotherapeutics. Simultaneously, inhibition of phosphorylated Stat3 can effectively prevent PD-L1 from entering the nucleus [131, 133] (Fig. 3). TNF-α activates caspase-8 to cleave GSDMC at amino acid Asp365 and release the GSDMC amino terminus in tumor cells. The amino terminus is automatically integrated into the cell membrane to form the hole that causes pyroptosis, preventing caspase-8 from recruiting caspase-3 to the apoptosis process, thus switching TNFα-induced apoptosis into pyroptosis [131, 134, 135] (Fig. 3).
Unlike other protein families of GSDMs, the protease that activates GSDMA was not discovered until recently, and its physiological and pathological roles in host immunity have been revealed [136]. Group A Streptococcus (GAS) is a global skin pathogen that can cause a variety of acute infections, such as local suppurative infections and even fatal invasive diseases, with high morbidity and mortality [137]. Following GAS pathogen invasion, the virulence factor of GAS cysteine protease SpeB (streptococcal pyrogenic exotoxin B) is autocatalytically cleaved to produce active protease, which cleaves GSDMA by direct proteolysis after the Gln246 site [136]. Subsequently, cleaved GSDMA-NT forms GSDMA pores in the host cell membrane, promoting pyroptosis of infected cells and ultimately resulting in local inflammation and clearance of pathogens [136]. Simultaneously, GSDMA deficiency or SpeB mutation fails to cleave GSDMA to form activated GSDMA-NT, thus resulting in systemic dissemination and severe multiorgan infection [136] (Fig. 3).
New insights into various factors that influence pyroptosis
Bacteria influence the immune response to manipulate pyroptosis
Shigella, a strain of Gram-negative bacteria, is a common pathogen that causes shigellosis in humans, which is characterized by evasion of immune defense, resulting in hemorrhagic diarrhea and severe intestinal inflammation [138, 139]. Shigella flexneri can suppress the function of caspase-4/11 via the secretion of effector protein OspC3, a member of the OspC family, which mediates a new posttranslational modification called arginine ADP-riboxanation [139,140,141]. OspC3 modifies Arg314/Arg310 of caspase-4/11 to prevent their activation and the cleavage of GSDMD, which in turn inhibits LPS-induced pyroptosis, thus escaping the immune mechanism of the host against intracellular Gram-negative bacteria [140, 142] (Fig. 4A). This process is unique compared with known direct or indirect bacterial inflammasome regulation strategies, which can constitute a new research direction in the field of protein posttranslational modification.

Bacteria influence the immune response to manipulate pyroptosis. Shigella flexneri inhibits caspase-4/11 through the secreted effector protein OspC3 to mediate arginine ADP-riboxanation and block the maturation of caspase-4/11 and consequently GSDMD cleavage. Yersinia secretes YopJ to inhibit TAK1, further activating RIPK1/caspase-8-dependent pyroptosis. Simultaneously, YopJ binds to RIPK1 to function on the Rag-Ragulator complex, causing cleavage and partial activation of GSDMD by caspase-1/11 and cleavage of GSDME by caspase-3/7. Mycobacteria secrete EST12, which binds to RACK1, recruiting UCHL5 to promote deubiquitination of NLRP3 and thus inducing NLRP3 and GSDMD to trigger pyroptosis. Additionally, Brd4 regulates the activation of the NAIP-NLRC4 inflammasome by recruiting PU.1 and IRF8 to initiate the maturation of caspase-1 and activate caspase-8 to cause IEC in cells infected by Salmonella
Unlike Shigella, most pathogens induce pyroptosis after entering host cells. For example, YopJ, a virulence protein secreted by Yersinia, activates the RIPK1-caspase-8 pathway by inhibiting TAK1, further cleaving GSDMD and causing pyroptosis [10, 142]. Any macrophages infected with YopJ are sensed by TLR4, which sends a signal to form a complex consisting of RIP1, caspase-8, and FADD [10, 143, 144]. Under stimulating conditions, RIPK1 and caspase-8 can be recruited to lysosomes that rely on the Rag-Ragulator complex [142, 143]. RagC, in the form of GDP, can bind to RIPK1/caspase-8 to promote cleavage and partial activation of GSDMD via caspase-1/11 and cleavage of GSDME via caspase-3/7, both of which trigger cytomembrane permeabilization when TAK1 is inhibited [10, 144]. Additionally, TAK1 can function on NF-κB and MAPK to increase IL-1β and cFLIPL, respectively, which are essential regulators of cell death and inflammation [145]. When cFLIPL levels are high enough, both caspase-8 and pyroptosis are inhibited, but when the level of cFLIPL is low, caspase-8 is prone to forming homodimers to activate and cleave GSDMD, leading to potassium efflux and inducing NLRP3-dependent pyroptosis [81, 146] (Fig. 4B).
One strategy for combating the pathogenicity of mycobacteria is to maintain their novel mechanism of chronic or persistent infection by reducing their virulence [147]. Studies have discovered a tuberculosis protein called EST12 that is able to induce pyroptosis of macrophages and then binds to the Tyr80 binding side of the activated endogenous host sensor protein RACK1 (the receptor for activated C kinase 1) to form the EST12-RACK1 complex [147,148,149]. The complex recruits the deubiquitinating enzyme UCHL5 to cause Lys48-linked deubiquitination of NLRP3 and subsequently induces GSDMD to cause macrophage pyroptosis and IL-1 secretion [147]. This study demonstrated that RACK1 is an endogenous sensor protein for pathogens and pyroptosis induced by its functions in mycobacterial-induced immunity (Fig. 4C).
It has been revealed that bromodomain-containing protein 4 (Brd4), a significant transcriptional activator in the field of epigenetics, regulates NAIP-NLRC4 inflammasome activation in macrophages under infection with Salmonella [150,151,152]. Brd4 and the macrophage lineage-determining transcription factors PU.1 and IRF8 colocalize on the NARP protein promoter to promote its transcription, induce the maturation of caspase-1 and the cleavage of both GSDMD and pro-IL-1β, ultimately contributing to the infection-mediated inflammatory response and pyroptosis [151, 153]. Simultaneously, the NARP-NLRC4 inflammasome can activate caspase-8 to function in intestinal epithelial cell (IEC) expulsion, which is accompanied by an actin rearrangement in adjacent cells that maintains epithelial integrity [154] (Fig. 4D).
Viral infection triggers or avoids pyroptosis
The inflammasome sensor has the ability to recognize many PAMPs, which indicates that proinflammatory caspases are essential for cell self-defense against pathogens in almost all fields. It has been reported that HIV has a deleterious role in pyroptosis at the time of infection [1]. The CARD-8 inflammasome is a natural alarm system in human immune cells and is a specific protein needed for virus replication and spread that can recognize active HIV protease that cleaves the N-terminus of CARD8 to activate caspase-1 and GSDMD, triggering pyroptosis of infected cells [91, 155, 156]. Unfortunately, HIV can exist in cells for a long time without triggering this alarm, and when the HIV protease is inactive, the CARD8 inflammasome is unable to detect it. Unraveling the mechanism of the CARD8 inflammasome can provide guidance for research on new drugs that can activate HIV protease to cure HIV [93, 157] (Fig. 5).

Virus infection triggers or avoids pyroptosis. CARD8 can recognize active HIV protease that cleaves its N-terminus to activate caspase-1 and GSDMD and can trigger pyroptosis to eliminate cells infected with HIV. Additionally, the SARS-CoV-2 spike protein interacts with ACE2, causing the formation of syncytia that activate caspase-9/3/7 to lead to the cleavage of GSDME. N proteins can function in the ComC pathway, which collaborates with MAC to directly activate the NLRP3 inflammasome. The P2RX7 receptor is mediated by ATP when SARS-CoV-2 enters the airway, leading to NLRP3 activation. RAAS induces angiotensin II to combine with AT1 to trigger the NLRP3 inflammasome in cells infected with SARS-CoV-2. Finally, H7N9 viruses inject ssRNA into the cells to trigger gasdermin E-dependent pyroptosis
The combination of SARS-CoV-2 spike protein and the angiotensin converting enzyme 2 (ACE2) receptor located on the surface of the cells not only mediates the invasion of the virus but also causes adjacent cells to fuse with each other to generate syncytia. Caspase-9 can be activated in syncytia, which in turn activates caspase-3/7 to cleave GSDME, releasing the N-terminal domain to punch holes [158, 159]. In addition to S proteins, the N proteins of SARS-CoV-2 can cleave ComC (complement cascade) to form two segments, the C3a and C5a anaphylatoxins, both of which collaborate with the C5b/C9 membrane attack complex (MAC) to directly activate the NLRP3 inflammasome [160,161,162]. The P2RX7 receptor is mediated by the release of extracellular ATP when SARS-CoV-2 enters the airway, which can lead to NLRP3 activation in macrophages through unknown mechanisms [163, 164]. Furthermore, activation of the renin–angiotensin–aldosterone system (RAAS) causes increased angiotensin II, which combines with the AT1 receptor to activate the NLRP3 inflammasome in kidney, lung, cardiomyocyte, and hematopoietic cells infected by SARS-CoV-2, all of which drives the maturation of downstream caspase-1, the secretion of IL-18 and IL-1β, and consequent pyroptosis [160, 165, 166] (Fig. 5).
The H7N9 virus uses effective replications and injects ssRNA into mouse alveolar epithelial cells, activating gasdermin E-mediated pyroptosis. Then, the contents of the infected cells subsequently trigger a cytokine storm, but the exact components at work are unclear [167, 168]. The ssRNA of the virus activates caspase-3, which cleaves gasdermin E to activate GSDME-NT to form pores in the cell membrane and switches apoptosis to pyroptosis [167, 169] (Fig. 5).
Chemical factors promote or inhibit pyroptosis
Cell fate is closely related to metabolic homeostasis in cells [170]. Recently, it has been discovered that there are many chemical factors in organisms that can affect pyroptosis. For example, α-ketoglutarate (α-KG) penetrates cell membranes and effectively induces tumor pyroptosis, which has a potential antitumor effect on cells [171, 172]. α-KG is converted by the metabolic enzyme MDH1 into another metabolite called L-2-hydroxyglutarate (L-2HG) in an acidic environment, which results in increased ROS levels and induces death receptor-6 (DR6), causing the cell membrane to oxidize, polymerize, and undergo endocytosis to form receptosomes [171, 173]. Endocytic DR6 recruits pro-caspase-8 to the receptosomes through the mediation of the adaptor protein Fas-associated via death domain (FADD) [171, 174]. GSDMC is also recruited to the receptosomes and is then cleaved by mature caspase-8. Finally, the N-terminal of GSDMC perforates the cell membrane and causes pyroptosis [171] (Fig. 6).

Chemical factors promote or inhibit pyroptosis. An example of a chemical factor that affects pyroptosis is α-KG, which is changed into L-2HG to trigger an increase in ROS levels as well as DR6 oxidation and polymerization to induce caspase-8 to cleave GSDMC and trigger pyroptosis. Drugs that enhance ROS production, or iron, result in Tom20 oligomerization and the formation of Bax pores, leading to cytochrome C release, which induces the cleavage of caspase-3/9 to trigger GSDME-dependent pyroptosis. Additionally, DMF interacts with GSDMD at the Cys191/Cys193 position to generate S-(2-succinyl)-cysteine, which prevents the combination of GSDMD and caspase. Galectin-1 induced by GSDMD can promote and enhance LPS-induced pyroptosis. Finally, Mg2+ blocks the influx of Ca2+ into the cells by restraining the ATP-gated calcium channel P2X7 to suppress GSDMD-NT oligomerization
Iron is the critical ingredient of enzymes that produce ROS, such as lipoxygenases (LOX) and the subunits of the mitochondrial electron transport chain (ETC) [175]. Iron-induced production of ROS or drugs induces an increase in ROS in melanoma cells, resulting in the oxidation of Cys13 and Cys21 in Tom20, wherein they oligomerize and accumulate [176]. Iron ions and oligomerized Tom20 trigger the transfer of Bax from the cytoplasm to the mitochondria to form Bax pores, leading to the cleavage of caspase-3/9, the release of cytochrome c, and inhibition of tumor growth and metastasis, which is caused by GSDME that resembles GSDMD [176,177,178] (Fig. 6). In addition to pyroptosis, iron can trigger several types of cell death, such as ferroptosis, iron-induced necroptosis and apoptosis, all of which are consequences of complex signaling pathways in cells [175, 179].
Among the noncanonical mechanisms of pyroptosis, LPS from Gram-negative bacteria directly activates caspase-11 to trigger cleavage of GSDMD, eventually resulting in pyroptosis. However, the succination of dimethyl fumarate (DMF), a Krebs cycle intermediate, can inactivate GSDMD and block pyroptosis [42, 180]. Endogenous and exogenous DMF interacts with GSDMD at essential Cys191/Cys193 sites to become S-(2-succinyl)-cysteine, an irreversible posttranslational modification that prevents the interaction between GSDMD and caspase, thus limiting GSDMD’s ability to be processed, to oligomerize and to perforate the cell membrane [40, 180, 181]. DMF delivery can protect mice from lipopolysaccharide shock by targeting GSDMD as well as alleviating familial Mediterranean fever and experimental autoimmune encephalitis, supporting its potential as a therapeutic drug to treat such diseases [180]. Furthermore, LPS is conducive to secreting and releasing a lectin called galectin-1 that can combine with β-galactoside, but the mechanism of secretion is still not well understood [42, 182, 183]. The secretion of galectin-1 also results from caspase-11-gasdermin D pathway signaling, and it is released into the circulatory system in the course of sepsis, which suggests that galectin-1 can promote and enhance the level of inflammation in the LPS-induced sepsis model [183, 184]. It is possible that galectin-1 may be a candidate for the design of a drug to suppress the cytokine storm during sepsis and can possibly be used as a marker to identify critically ill patients [183, 184] (Fig. 6).
The prerequisite for the function of GSDMD-NT is the influx of calcium ions, which are essential to the membrane localization of GSDMD-NT. However, magnesium ions can act as physiological Ca2+ antagonists and trigger disturbed Ca2+ signaling by causing concentration changes in intracellular and extracellular environments [185,186,187]. Furthermore, Mg2+ blocks the influx of Ca2+ by inhibiting the crucial ATP-gated calcium channel P2X7, as do Cu2+, Zn2+ and Ni2+, and inhibits the process of GSDMD-NT oligomerization and binding to the cell membrane synchronously [187,188,189] (Fig. 6).
Noncoding RNA is associated with the regulation of pyroptosis
Noncoding RNA is a type of RNA that does not encode proteins, such as rRNA, tRNA, siRNA, piRNA, snRNA, microRNA, or lncRNA, which also have a close relationship with pyroptosis [190,191,192]. For example, recent studies have found that overexpression of miR-148a can prevent pyroptosis by inhibiting inflammasomes, thereby improving the symptoms of alcoholic liver injury [193]. Alcohol consumption suppresses the expression of FoxO1, the transcriptional activator of miR-148a, thereby downregulating the expression level of miR-148a in cultured hepatocytes and liver cells of mice [193,194,195]. Furthermore, miR-148a directly inhibits thioredoxin-interacting protein (TXNIP) in such a way that the target gene TXNIP is upregulated, which promotes NLRP3 inflammasome activation, caspase-1 maturation and pyroptosis [193, 196, 197] (Fig. 7).

Noncoding RNA is associated with the regulation of pyroptosis. miR-148a inhibits the expression of TXNIP to suppress NLRP3 inflammasome activation, which is reversed by elevated FoxO1 expression induced by alcohol treatment. Additionally, microRNA-9 functions on ELAVL1 to suppress the expression of TNF-α, which inhibits pyroptosis by preventing NLRP3 inflammasome activation in a hyperglycemic environment. Similarly, MALAT1 downregulates the expression of miR-23c to induce ELAVL1 expression. Finally, the lncRNA Neat1, activated by HIF-2α, activates the NLRP3 inflammasome and directly interacts with caspase-1 p20 subunits to yield mature caspase-1
Another example, microRNA-9, can inhibit the activation of caspase-1 and IL-1β in human cardiomyocytes by interacting with the protein ELAVL1, whose expression, when increased, has a concomitant decrease in a hyperglycemic environment [198, 199]. Ubiquitously expressed ELAVL1 combines and stabilizes the AU-rich element (AUE) of TNF-α mRNA to induce the expression of TNF-α in the cytoplasm, which promotes canonical pyroptosis via NLRP3 inflammasome activation and downstream products, such as caspase-1 and IL-1β, eventually leading to pyroptosis [20, 198, 200]. Similarly, the long noncoding RNA MALAT1 participates in the pyroptosis of renal tubular epithelial cells in diabetic nephropathy by inhibiting the level of miR-23c, whose downregulation promotes the expression of the same target gene ELAVL1 [201] (Fig. 7).
Additionally, lncRNA Neat1, which is transcriptionally activated by hypoxia-induced factor HIF-2α, can directly participate in the assembly and activation of inflammasomes [202, 203]. Upon stimulation with hypoxia, Neat1 will facilitate the activation of the NLRP3 inflammasome and directly interact with the caspase-1 p20 subunit through its 5’ end, and it can combine with pro-caspase-1 to stabilize the aggregation of the complex or with the mature caspase-1 tetramer to increase its stability and enzyme activity during combination [204, 205]. In macrophages, Neat1 promotes the activation and assembly of inflammasomes through activation of the aforementioned pathways, increases the formation of mature caspase-1 tetramers, promotes the maturation and secretion of IL-1β and IL-18, and ultimately induces pyroptosis [204]. Neat1 normally resides in paraspeckles, an irregular substructural region in the chromatin gap of the nucleus, wherein Neat1 dissociates from the nucleus and moves into the cytoplasm to regulate the function of the NLRP3 inflammasome [204, 206, 207] (Fig. 7).
Pyroptosis and antitumor immunity in cancer
Numerous studies have indicated that pyroptosis is closely associated with the development and metastasis of many cancers. Tumorigenesis is regulated by various factors, such as the activity of oncogenes, the immune microenvironment, and especially chronic inflammation [208]. Long-term exposure to an inflammatory environment increases the risk of cancer formation in cells and tissues. Specifically, the cytokine release induced by pyroptosis, such as IL-1 and IL-18, can promote infiltration in tumors, thereby increasing the possibility of tumorigenesis and metastasis [209]. Pyroptosis is a double-edged sword with respect to cancers, as it can either promote or inhibit tumorigenesis. The protumor role has been extensively studied, but the relationship between pyroptosis and anticancer immunity is not fully understood. Currently, we know that pyroptosis occurs in almost all types of cancer; thus, an in-depth study of the relationship between pyroptosis and cancer will broaden our understanding of cancer and inform innovations in cancer prevention and treatment.
Lung cancer
Lung cancer is one of the leading causes of death globally, as evidenced by its high incidence and mortality rate [210, 211]. Polyphyllin VI (PPVI), a saponin isolated from Trillium tschonoskii Maxim, exhibits significant antitumor properties, as it inhibits the proliferation of non-small-cell lung cancer (NSCLC) cells via the ROS/NF-κB/NLRP3/GSDMD signaling pathway [212]. Given the low sensitivity of chemotherapy in NSCLC, this mechanism indicates that PPVI could be a target for a novel NSCLC therapeutic drug in the future [213]. In addition to triggering the nonclassical inflammasome pathway, leading to the pyroptosis of innate immune cells, LPS can also directly cause some tumors to regress [214]; however, the mechanism of LPS’s influence on tumor cells is still unclear, which limits its application. Recent studies have found that a protein called SCGB3A2, predominantly secreted in the lung airways, can combine with LPS, and the resulting SCGB3A2-LPS complex can bind to the cell surface protein syndecan 1, allowing LPS to enter Lewis lung carcinoma (LLC) cells to undergo pyroptosis and thus inhibiting tumor proliferation and growth [215] (Fig. 8).

Pyroptosis and antitumor immunity in cancer. Manipulating pyroptosis to kill tumor cells and inhibit the proliferation, migration, and invasion of tumor cells is a novel research avenue for cancer treatment. Pyroptosis plays antitumor roles in several cancers, including lung, gastric, breast, hepatocellular, and colorectal cancers
Gastric cancer
Gastric cancer is the third most lethal cancer globally because it is often diagnosed at an advanced stage, at which point chemotherapy is the best course of treatment [216]. Chemotherapeutic drugs can “reprogram” apoptosis to pyroptosis through the cleavage of caspase-3-induced GSDME. For example, 5-fluorouracil can induce caspase-3/GSDME-dependent pyroptosis in gastric cancer cells, which may help to elucidate the mechanism of chemotherapeutic treatment of gastric cancer [217]. Furthermore, the expression level of GSDMD in gastric cancer cells is very low compared to that in normal cells [218]. Studies have revealed that diminished expression of GSDMD modulates the expression of cell cycle-related proteins and accelerates the cell cycle S/G2 phase transformation, possibly by activating the STAT3 and ERK1/2 signaling pathways, thereby promoting tumor cell growth [219]. In addition to GSDMD, GSDMA is also downregulated in gastric cancer cells, but GSDMB is highly expressed in most precancerous cancer samples, which may be related to tumor invasion, and both GSDMs are candidates for antitumor drug targets [218, 220] (Fig. 8).
Breast cancer
Breast cancer is a major global public health problem, but identifying effective strategies and interventions to prevent breast cancer remains challenging [221]. At present, many chemotherapeutic drugs play an antitumor role by inducing pyroptosis. For example, cisplatin (DDP) appears to improve complete pathologic response rates and to inhibit tumor growth and metastasis, both in vitro and in vivo, by inducing the NLRP3/caspase-1/GSDMD-mediated pyroptosis pathway in triple-negative breast cancer (TNBC), but the molecular mechanism remains unclear [222, 223]. Recent studies have found that DDP can increase the expression of maternally expressed gene 3 (MEG3), a long noncoding RNA that plays a critical part in the signaling process after DDP treatment [222]. Deletions, mutations, or downregulation of the innate viral nucleotide sensor known as retinoid-induced gene I (RIG-I) were detected in only 1% of clinical breast cancer patients, which suggests that RIG-I agonists are widely applicable for activating innate immunity to treat breast cancer with a high probability of remission [224]. RIG-I agonists trigger upregulation of the expression and mitochondrial localization of RIG-I, thus activating the expression of the proinflammatory transcription factors STAT1 and NF-κB, which in turn induce pyroptosis in breast cancer cells [224]. In addition, RIG-I agonists can augment the level of lymphocyte-recruiting chemokines and type I IFN, thereby increasing the number of tumor lymphocytes while reducing tumor growth and metastasis [224]. These findings are significant for understanding how RIG-I agonists induce tumor cell death and regulate the tumor microenvironment in vivo (Fig. 8).
Hepatocellular carcinoma
Hepatocellular carcinoma (HCC) accounts for 90% of primary liver cancer cases, and its pathophysiology involves the interaction of various factors, such as the susceptibility of a population, viral infection, some liver diseases, the tumor microenvironment, and immune cells, which can serve as prospective therapeutic targets [225]. Sorafenib, a kinase inhibitor, can reduce the major histocompatibility complex class I (MHC-I) expression of HCC tumor cells and is reported to play an antitumor role by inducing the pyroptosis of macrophages [226]. Sorafenib can upregulate caspase-1 activity to induce pyroptosis in MΦs, which triggers the release of proinflammatory cytokines and induces the proliferation and activation of NK cells, ultimately driving the apoptosis of tumor cells [226]. In addition to this indirect effect, pyroptosis exhibits significant antitumor properties in HCC tumor cells directly. For example, cannabidiol (CBD) can trigger an integrative stress response (ISR) and mitochondrial stress in HCC tumor cells, subsequently increasing the activation of ATF4 and its target gene, CHOP, which promotes the expression of Bax protein from the BCL-2 family, to cause caspase-3/caspase-9/GSDME-dependent pyroptosis [227]. Similarly, miltirone may also combat the progression of cancer through the induction of pyroptosis; thus, it is a potential agent that can be used to target GSDME for the treatment of HCC [228]. Miltirone significantly enhances the intracellular accumulation of ROS to repress the phosphorylation of mitogen-activated and extracellular signal-regulated kinase (MEK), which suppresses the activity of extracellular regulated protein kinases ½ (ERK1/2) and triggers BAX/caspase-9/caspase-3/GSDME-dependent pyroptosis [228] (Fig. 8).
Colorectal cancer
Due to population aging and environmental deterioration, colorectal cancer (CRC) is the third most common source of malignant tumors worldwide [225]. The incidence and mortality rates of CRC in China are increasing annually, most recently ranking fourth among cancer deaths [229, 230]. Lobaplatin has a strong antineoplastic effect with few adverse effects and can promote ROS elevation and JNK phosphorylation to induce the Bax/caspase-9/caspase-3-mediated pyroptosis pathway in HT-29 and HCT116 cells [231]. The biological function of lncRNAs in CRC has not been fully elucidated, but recent studies have found that they may be useful biomarkers of cancer and thus may be applicable in the future diagnosis and prognosis determination of CRC. For example, lncRNA RP1-85F18.6 can probably inhibit the activity of GSDMD by increasing the expression of ΔNp63, thus suppressing the pyroptosis of CRC cells to promote the proliferation, metastasis, and cell cycle disruption of CRC cells, ultimately leading to cancer progression, although there is no statistically significant association between lncRNA RP1-85F18.6 and GSDMD expression [232] (Fig. 8).
Prospects for pyroptosis in anticancer therapy
Recent studies have demonstrated the feasibility and clinical potential of utilizing pyroptosis as a mechanism of antitumor immunity, and many researchers are attempting to combine pyroptosis with other tumor treatments to treat cancers by regulating pyroptosis to suppress the proliferation, migration, and invasion of tumor cells.
Chemotherapy
Drug therapy remains one of the most common forms of cancer treatment and is confirmed to elicit tumor cell death by triggering pyroptosis. Shao et al. first reported that chemotherapy drugs, including doxorubicin and topotecan, can treat lung cancer through the cleavage of GSDME by caspase-3, which is usually silenced in cancer cells [178]. Similarly, a series of small molecule targeted inhibitors have also been identified to induce pyroptosis via the caspase-3/GSDME pathway in lung cancer cells, which can be used alongside apoptosis to improve the efficacy of targeted anticancer therapies [233] (Fig. 9). Subsequently, an increasing number of chemical target drugs, reagents, and natural products have been found to cause pyroptosis in various types of cancers, many of which imbue antitumor effects [234,235,236,237,238,239,240,241] (Table 2).

Prospects for pyroptosis in anticancer therapy. The therapeutic feasibility and potential of manipulating pyroptosis as an anticancer therapy have recently been explored. Chemotherapy is still the most common form of cancer treatment, as it elicits tumor cell death by triggering pyroptosis. In addition, some novel target and delivery methods have been developed, such as chimeric antigen receptor engineered T cells (CAR-T), chimeric costimulatory converting receptor (CCCR), immune stimulation, bioorthogonal chemical systems, and epigenetic methods, all of which can effectively induce pyroptosis in tumor cells
Chimeric antigen receptor engineered T cells (CAR-T)/Chimeric costimulatory converting receptor (CCCR)
In the past few years, CAR-T therapy has yielded compelling efficacy in anticancer treatment, especially in B-cell acute lymphoblastic leukemia (B-ALL), which has a complete remission rate of up to 90% [242]. CAR-T cells can induce primary B leukemic cell pyroptosis, which rapidly induces the release of granzyme B to activate caspase-3 cleavage of GSDME, ultimately resulting in pyroptosis [243]. Furthermore, the concentration of granzyme B and perforin triggered by CAR-T therapy rather than existing CD8+ T cells more effectively induces the pyroptosis of cancer cells [243]. This multifaceted therapy, along with other methods to promote the pyroptosis of cancer cells, may be a more efficient way to prevent tumorigenesis than monotherapy. Recently, researchers have designed a novel chimeric costimulatory conversion receptor (CCCR), an NK92 cell containing the modified CCCR comprising the extracellular domain of PD1, which can rapidly induce GSDME-dependent pyroptosis of lung cancer cells and effectively enhance antitumor activity by reversing the immunosuppression of PD1 [244] (Fig. 9).
Immune stimulation
Activating the host’s immune system to recognize and fight abnormal transformation of malignant tumor cells in the body is a canonical cancer treatment. Several studies have confirmed that granzymes activated by NK and CTL cells can trigger tumor cell pyroptosis. Specifically, overexpression of GSDMB in HEK-293T cells that are deficient in endogenous GSDM expression can trigger pyroptosis of the 293 T cells when cocultured with human NK cells, and this response appears to be independent of caspase [121]. Furthermore, perforin, IFN-γ, and some cytokines mediated by immune stimulation can kill tumor cells by inducing pyroptosis [130, 245] (Fig. 9).
Bioorthogonal chemical system through NP-GSDM delivery
The bioorthogonal chemical system is an effective means by which to study the process of cell death, and it has been used to study the effect of pyroptosis on tumor immunity in animal models. Researchers have established a bioorthogonal chemical system consisting of a cancer imaging probe, phenylalanine trifluoroborate (PH-BF3), and nanoparticles (NPs) that can enter tumor cells as a delivery system [246]. When combined with nanoparticle-mediated delivery, PH-BF3 catalyzes desilylation and selectively cleaves the target protein from the nanoparticles, resulting in controlled release of the effector protein, including active GSDMs, that can induce pyroptosis of mouse tumor cells in vivo [246]. One study found that pyroptosis occurred in under 15% of tumor cells, which was sufficient to eliminate 4T1 tumors [246]. This result indicates a possible synergistic effect that may be a promising antitumor immunotherapeutic mechanism (Fig. 9).
Epigenetic methods
The effector protein GSDM is significantly downregulated or inactivated in many types of cancer, which is a main obstacle to the development of anticancer strategies related to pyroptosis. For example, GSDME is absent in breast cancer and colon adenocarcinoma cells due to gene promoter hypermethylation [247]. Epigenetic methods can prominently reverse GSDME silencing by using decitabine (DAC) treatment, which causes epigenetic stimulation to trigger the pyroptosis of tumor cells, thus imparting antitumor action in conjunction with chemotherapy [248]. Furthermore, Zinc Finger DHHC-Type Containing 1 (ZDHHC1) is frequently silenced due to DNA methylation among various cancers, and its restoration can also curb tumor progression by inducing ROS/ER stress-mediated pyroptosis [249]. Unfortunately, no drugs for this epigenetic therapy have been discovered (Fig. 9).
However, the application of pyroptosis to treat cancer in clinical research still presents great challenges. Pyroptosis plays a dual role in cancer, and its ambiguous role is related to the cancer cell type, duration of pyroptosis induction, etc., so improper handling may even promote cancer development [250]. In addition, due to the complexity of pyroptotic pathways, multiple pathways and overlapping components can lead to pyroptosis, so the tumor-specific effect of each pyroptotic molecular component is the overall tumor-specific effect of each pathway, which makes the clinical regulation and utilization of tumor cell pyroptosis difficult [251].
Potential targeted treatment strategies for pyroptosis
The general mechanisms of pyroptosis have been uncovered in the context of many different diseases, which benefits the study of the pathology of these diseases and identifies critical components of pyroptosis, such as the NLRP3 inflammasome, caspase-1, caspase-3 and GSDMD, which play important roles in human health. These mechanisms can contribute to informing us of some targeted drug therapies for a wide variety of diseases.
Mechanisms of pyroptosis inhibition against NLRP3
The NLRP3 inflammasome works on our body’s innate immune system, which recognizes pathogen-related and risk-related molecular patterns, abnormalities or mutations that can contribute to many human inflammatory diseases [252]. Anakinra, an antagonist of the IL-1 receptor, significantly decreases TNF-α, pro-IL-1β, and pro-caspase-1 to suppress pyroptosis, which alleviates severe liver inflammation in mutant NLRP3 knockout mice [253]. It was recently found that the extract of Huaier mushrooms can induce pyroptosis triggered by NLRP3 in vitro and in vivo, thus exhibiting an antineoplastic effect in non-small-cell lung cancer (NSCLC) cells [254]. Atorvastatin reduces the expression of the canonical inflammasome pathway component NLRP3 at the mRNA and protein levels through changes at the level of lncRNA NEXN-AS1 or NEXN to inhibit pyroptosis, providing a new theory on how to reverse atherosclerosis [255]. Additionally, kanglexin, a novel anthraquinone compound, prevents cardiac injury from myocardial infarction (MI) and alleviates cardiac dysfunction by attenuating NLRP3 inflammasome activation and subsequent NLRP3-mediated pyroptosis of myocardial cells, which indicates that kanglexin has the potential to mitigate ischemic heart disease [256]. In addition to kanglexin, there are many other agents associated with cardiovascular diseases, such as glyburide derivatives, colchicine, dapansutrile, melatonin, and liraglutide [257,258,259,260,261,262,263] (Fig. 10).

Potential targeted treatment strategies for pyroptosis. Research on pyroptosis benefits the study of the pathological processes of many different diseases, especially essential components in the process of pyroptosis, such as NLRP3, caspase-1, caspase-3, and gasdermin D, all of which play critical roles in human health and have been developed into many targeted treatment drugs against several inflammatory diseases
Mechanisms of pyroptosis inhibition against caspase-1
Previous studies have confirmed that alkaloids of Dendrobium (DNLA) have neuroprotective effects on oxygen-glucose deprivation/reperfusion injury, wherein they can reduce the expression of caspase-1 to inhibit pyroptosis-associated neuronal death and can prevent cerebral ischemia reperfusion (CIR) impairment in the hippocampal region [264]. Additionally, andrographolide effectively prevents AIM2 from sensing DNA damage caused by chemotherapeutic agents in macrophages, thereby suppressing the maturation of caspase-1 and the release of cytokines, which alleviates pyroptosis-induced lung tissue damage and progressive fibrosis in the later phase of lung diseases [265]. Berberine can reactivate caspase-1 expression and stimulate pyroptosis in HepG2 cells, which inhibits the activity, migration and invasion of hepatocellular carcinoma [266]. Metformin treatment, a new intervention strategy for overcoming chemoradiotherapy tolerance, upregulates the microRNA497-PELP1 signaling pathway, which is responsible for regulating caspase-1-mediated pyroptosis of esophageal cancer cells to kill tumors [267]. In addition to these agents, lncRNA GAS5, a well-known tumor suppressor, inhibits proliferation, colony formation and pyroptosis of ovarian cancer cells, similar to caspase-1-mediated pyroptosis, and can be used as a potential targeted therapeutic treatment [268]. In contrast to the aforementioned agents, AC-YVad-CMK suppresses the levels of proinflammatory cytokines to promote the proliferation and differentiation of alveolar osteoblasts, ultimately alleviating periodontal disease [269, 270]. Treatment with HQH effectively inhibited caspase-1-dependent pyroptosis to improve joint inflammation in juvenile collagen-induced arthritis in rats [271] (Fig. 10).
Mechanisms of pyroptosis inhibition against caspase-3
Recent studies have indicated that lobaplatin eradicates tumor cells by promoting ROS production and the activation of C-Jun positive terminal kinases to drive caspase-3-GSDME-dependent pyroptosis in colon cancer, which has significant implications for clinical application [231]. Doxorubicin, a common chemotherapy drug, triggers caspase-3-dependent pyroptosis, producing antitumor effects in melanoma cell lines [178, 272]. Galangin, a ubiquitous natural flavonoid derived from plants, exerts a distinct antitumor effect against glioblastoma by activating caspase-3 to induce pyroptosis, which can be further improved by repressing LC3B to inhibit autophagy in glioblastoma cells [273]. New information indicates that joint treatment with cisplatin and decitabine can ameliorate breast carcinoma and colon cancer by acting on caspase-3, while cisplatin works in lung cancer and ESCC [248, 274, 275]. As a kind of cancer with the fastest increasing morbidity and mortality, lung cancer has gradually become the most threatening cancer to human health, but there are many drugs acting against caspase-3 or other targeted sites, such as paclitaxel, bleomycin, topotecan, and actinomycin-D, which all influence pyroptosis to treat lung cancer [178, 274] (Fig. 10).
Mechanisms of pyroptosis inhibition against gasdermin D
As the critical effector in the pyroptosis pathway, GSDMD has attracted attention as a candidate for targeted therapies for the treatment of various diseases. Here, we have summarized the knowledge on several compounds that can suppress the GSDMD-mediated pyroptosis pathway to treat inflammatory-related diseases. Chitosan hydrogels can protect bone marrow mesenchymal stem cells during myocardial infarction through inhibition of GSDMD in vascular endothelial cells [276]. Necrosulfonamide, an inhibitor of MLKL, can suppress inflammasome responses to increase the survival rate by inhibiting the oligomerization of GSDMD-NT, which also promotes the proliferation and differentiation of osteoblasts in fracture repair [277,278,279,280]. Disulfiram is commonly used as an alcoholism drug approved by the FDA, but it was recently confirmed to modify Cys191 of GSDMD covalently to inhibit GSDMD-NT oligomerization, which is also applicable to the treatment of sepsis [277, 278]. Similarly, punicalagin may also inhibit GSDMD-NT oligomerization by interfering with membrane fluidity, but its effect in vivo is unknown [281]. Many of the potential drugs described above are versatile, as they can function on several targets. Finally, many new potential treatments that target pyroptosis are being developed to diagnose and treat a variety of inflammatory diseases, including cancers (Fig. 10).
Conclusion and perspectives
Studies on pyroptosis have made great progress in the last decade; therefore, this review was written to summarize some recent developments in understanding the role of pyroptosis in inflammation and cancer, thus illustrating the increasing importance of its functions in many diseases. Here, we describe the most widely studied NLRP3 inflammasome and the new regulatory mechanisms of the gasdermin family. We concluded that activation or inhibition of these important critical factors in the body may be promising for clinical applications, although many unknown mechanisms remain that may have equal value. However, it is obvious that the pyroptosis signaling pathway is involved in chronic inflammation in the development of cellular senescence, some cancers, and certain degenerative diseases.
Pyroptosis plays a critical role in antitumor immune function, specifically the inhibition of tumor growth, although variability in its effectiveness and the possibility of adverse effects limit its clinical application in inflammation and cancer treatment. One of the major challenges of integrating a pyroptosis-based approach to cancer therapy is that, in different cancers, the expression and function of key components involved in the process of pyroptosis are impaired; for example, expression is inhibited or mutations can lead to changes or loss of function. Fortunately, along with advances in molecular genetics, epigenetic targeting, delivery systems and personalized medicine can be used in conjunction with pyroptosis as a novel emerging method in cancer treatment.
Cell metabolism can affect pyroptosis in many ways, for example, the changes in the cell metabolism mode, modification of metabolites, and changes in the intracellular microenvironment, each of which can have a positive or negative effect on pyroptosis regulation and cell fate. For example, the Krebs cycle, a metabolic pathway in aerobic organisms, has recently emerged as the immune metabolic center of macrophages, wherein metabolites are different in inflammatory macrophages and immunomodulatory effects are exerted by modifying target proteins [282, 283]. For example, it has been reported that succination inactivates gasdermin D, which in turn blocks pyroptosis, while itaconate modifies NLRP3 to inhibit inflammasome activation [180, 284]. Regardless, pyroptosis always has some effects on the process of cell metabolism; thus, detecting the changes in metabolites could represent appropriate biomarkers of pyroptosis for monitoring inflammation and cancer. In addition to the Krebs cycle, several other metabolic processes associated with pyroptosis may be useful biomarkers, such as changes in cholesterol metabolism of AIM2 and NLRP3 inflammasomes [285, 286], which could be used to manipulate pyroptosis to design effective therapeutic disease treatments.
Inflammasomes that trigger pyroptosis are generally distributed in the cytoplasm, where there are many signaling molecules. In recent years, several mitochondria-related pyroptosis pathways have been discovered, broadening the mechanisms by which organelles trigger pyroptosis, and are thus of great significance for future drug intervention. Additionally, stress factors selectively activate pyroptosis instead of apoptosis through mitochondrial permeability transition (MPT) via the Apaf-1/caspase-4 protein complex [287]. Direct or indirect interactions between mitochondria and the NLRP3 inflammasome can result in mitochondrial instability, NLRP3 deubiquitination, and linear ubiquitination of ASC, which promote the triggering of pyroptosis by the NLRP3 inflammasome [288, 289]. Similarly, whether other membrane organelles, such as lysosomes, the Golgi apparatus, or endoplasmic reticulum (ER), are involved in pyroptosis remains to be discovered. Lysosomes, whose primary function is the decomposition of cells, have been considered a tool during apoptosis for a long time [290]; however, it was recently confirmed that in corneal stromal cells of macular corneal dystrophy (MCD), lysosomes and autophagosomes can fuse, leading to lysosomal dysfunction and the blockage of autophagic flux, which in turn activates the pyroptosis signaling pathway [291]. The Golgi apparatus and ER are essential for protein synthesis, which is closely associated with the occurrence of pyroptosis; therefore, the nature of the relationship between them warrants further investigation.
Liquid‒liquid phase separation (LLPS) is a prominent research field at present, and existing studies have shown that phase separation is ubiquitous and is the basis for the formation of membraneless compartments and the aggregation of specific molecules in cells, thus forming a certain “order” inside the “chaotic” cells [292,293,294]. It has since been discovered that dsRNA can induce phase separation of the NLRP6 inflammasome to generate an antiviral immune response, representing a breakthrough linking pyroptosis and LLPS [292]. Whether other inflammasomes require LLPS to function is still unknown. However, LLPS allows similar components of pyroptosis pathways to cluster or the sequestration of unrelated molecules to accelerate biological reactions, which is likely to promote the occurrence of pyroptosis. In addition, GSDMD punches holes in the plasma membrane through oligomerization to cause pyroptosis, which may require the help of LLPS.
Pyroptosis is considered a self-defense mechanism of animal cells, with bacteria thought to be the trigger, but new research shows that pyroptosis also occurs in bacterial cells as a defense against phage infection. It is likely that pyroptosis in mammals has a common origin with the ancient bacterial antiphage defense system [295, 296]. Similar to GSDM proteins, the overall structure of bGSDMs in bacteria is largely homologous with the N-terminal structure of GSDMs, and its activation disrupts the bacterial cell membrane, resulting in effective resistance to E. coli phages [295, 297].
Considering the potent proinflammatory properties of pyroptosis, it is not unusual for multiple regulatory mechanisms to influence pyroptosis. From the mechanism of evading inflammasome recognition and destroying inflammasome complex formation or function after pathogen infection to some endogenous or exogenous substances that affect pyroptosis signaling, the regulatory mechanisms of pyroptosis in the body are extremely intricate, which is evidenced by the confirmation that bacteria, viruses, chemical factors, and noncoding RNA, among other factors, can affect the occurrence or process of pyroptosis. Some regulators selectively inhibit complexes in one pyroptosis pathway, while others might function on multiple components and affect multiple pathways simultaneously.
Finally, this review summarizes several potential targeted therapies for diseases related to the four key targeted sites in the pyroptosis pathways and proposes challenges and future directions in this field. To further explore new pyroptosis-based therapeutic targets for the treatment of inflammation and cancer, we must better understand the comprehensive roles of pyroptosis in the formation and proliferation of tumors.
Scientific interest in pyroptosis remains high, although striking studies have revealed many questions regarding the function of inflammasomes in recent years. However, as in all fields, these recent developments raise important new questions. For example, it remains to be seen whether there are other mechanisms that modulate the newly discovered, noncanonical pyroptosis mechanism induced by cytosolic LPS. At present, there is no agreement on exactly how the GSDMD hole is assembled. The formation of many inflammasomes is subject to a variety of stimuli, but some obvious uniformity is not found in these stimuli. The complexities surrounding pyroptosis, including its effective activators and functions in inflammation and cancer, as well as its possible clinical applications, will continue to attract the attention of researchers, especially as drugs targeting pyroptosis are developed and clinically tested.
Data availability
All data generated or analyzed during this study are included in this published article [and its Supplementary Information files].
References
Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017;277:61–75. https://doi.org/10.1111/imr.12534.
Man SM, Kanneganti TD. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol. 2016;16:7–21. https://doi.org/10.1038/nri.2015.7.
Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992; 358: 167–9. https://doi.org/10.1038/358167a0.
Hersh D, et al. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. P Natl Acad Sci USA. 1999;96:2396–401. https://doi.org/10.1073/pnas.96.5.2396.
Hilbi H, Moss JE, Hersh D, Chen Y, Arondel J, Banerjee S, et al. Shigella-induced apoptosis is dependent on Caspase-1 which binds to IpaB. J Biol Chem. 1998;273:32895–900. https://doi.org/10.1074/jbc.273.49.32895.
Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–4. https://doi.org/10.1016/s0966-842x(00)01936-3.
Neiman-Zenevich, J, Stuart, S, Abdel-Nour, M, Girardin, SE & Mogridge, J Listeria monocytogenes and Shigella flexneri Activate the NLRP1B Inflammasome. Infect Immun. 2017;85. https://doi.org/10.1128/IAI.00338-17.
Xu S, Liu X, Liu X, Shi Y, Jin X, Zhang N, et al. Wedelolactone ameliorates Pseudomonas aeruginosa-induced inflammation and corneal injury by suppressing caspase-4/5/11/GSDMD-mediated non-canonical pyroptosis. Exp Eye Res. 2021;211:108750. https://doi.org/10.1016/j.exer.2021.108750.
Schell U, Simon S, Hilbi H. Inflammasome Recognition and Regulation of the Legionella Flagellum. Curr Top Microbiol Immunol. 2016;397:161–81. https://doi.org/10.1007/978-3-319-41171-2_8.
Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci USA. 2018;115:E10888–E10897. https://doi.org/10.1073/pnas.1809548115.
Philip NH, Brodsky IE. Cell death programs in Yersinia immunity and pathogenesis. Front Cell Infect Microbiol. 2012;2:149. https://doi.org/10.3389/fcimb.2012.00149.
Costa Franco M, Marim FM, Alves-Silva J, Cerqueira D, Rungue M, Tavares IP, Oliveira SC. AIM2 senses Brucella abortus DNA in dendritic cells to induce IL-1beta secretion, pyroptosis and resistance to bacterial infection in mice. Microbes Infect. 2019;21:85–93. https://doi.org/10.1016/j.micinf.2018.09.001.
Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–22. https://doi.org/10.1016/j.cell.2014.04.007.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. https://doi.org/10.1016/j.cell.2010.01.022.
Man SM, Kanneganti TD. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol. 2016;16:7–21. https://doi.org/10.1038/nri.2015.7.
Xia X, Wang X, Cheng Z, Qin W, Lei L, Jiang J, et al. The role of pyroptosis in cancer: pro-cancer or pro-“host”? Cell Death Dis. 2019;10:650. https://doi.org/10.1038/s41419-019-1883-8.
Ge X, Li W, Huang S, Yin Z, Xu X, Chen F, et al. The pathological role of NLRs and AIM2 inflammasome-mediated pyroptosis in damaged blood-brain barrier after traumatic brain injury. Brain Res. 2018;1697:10–20. https://doi.org/10.1016/j.brainres.2018.06.008.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. https://doi.org/10.1038/nature15514.
Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci. 2017;42:245–54. https://doi.org/10.1016/j.tibs.2016.10.004.
He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1 beta secretion. Cell Res. 2015;25:1285–98. https://doi.org/10.1038/cr.2015.139.
Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–6. https://doi.org/10.1038/nature18590.
Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20:143–57. https://doi.org/10.1038/s41577-019-0228-2.
Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017;27:673–84. https://doi.org/10.1016/j.tcb.2017.05.005.
Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet. 1998;20:194–7.
Orning P, Lien E, Fitzgerald KA. Gasdermins and their role in immunity and inflammation. J Exp Med. 2019;216:2453–65. https://doi.org/10.1084/jem.20190545.
Tweedell RE, Malireddi RKS, Kanneganti TD. A comprehensive guide to studying inflammasome activation and cell death. Nat Protoc. 2020;15:3284–333. https://doi.org/10.1038/s41596-020-0374-9.
Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6:128. https://doi.org/10.1038/s41392-021-00507-5.
Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–86. https://doi.org/10.1038/nature10759.
Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: A new frontier in cancer. Biomed Pharmacother. 2020;121:109595. https://doi.org/10.1016/j.biopha.2019.109595.
Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 2019;26:99–114. https://doi.org/10.1038/s41418-018-0212-6.
Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 2015;25:308–15. https://doi.org/10.1016/j.tcb.2014.12.009.
de Vasconcelos NM, Lamkanfi M. Recent Insights on Inflammasomes, Gasdermin Pores, and Pyroptosis. Cold Spring Harb Perspect Biol. 2020;12. https://doi.org/10.1101/cshperspect.a036392.
Xu YJ, Zheng L, Hu YW, Wang Q. Pyroptosis and its relationship to atherosclerosis. Clin Chim Acta. 2018;476:28–37. https://doi.org/10.1016/j.cca.2017.11.005.
Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590–604. https://doi.org/10.1038/sj.cdd.4402194.
Kolb R, Liu GH, Janowski AM, Sutterwala FS, Zhang W. Inflammasomes in cancer: a double-edged sword. Protein Cell. 2014;5:12–20. https://doi.org/10.1007/s13238-013-0001-4.
Thi HTH, Hong S. Inflammasome as a Therapeutic Target for Cancer Prevention and Treatment. J Cancer Prev. 2017;22:62–73. https://doi.org/10.15430/Jcp.2017.22.2.62.
Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol. 2012;13:333–42. https://doi.org/10.1038/ni.2237.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26. https://doi.org/10.1016/s1097-2765(02)00599-3.
Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150:606–19. https://doi.org/10.1016/j.cell.2012.07.007.
Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71. https://doi.org/10.1038/nature15541.
Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1 beta in type 2 diabetes. Nat Immunol. 2010;11:897–U1501. https://doi.org/10.1038/ni.1935.
Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–9. https://doi.org/10.1126/science.1240248.
Ramos-Junior ES, Morandini AC. Gasdermin: A new player to the inflammasome game. Biomed J. 2017;40:313–6. https://doi.org/10.1016/j.bj.2017.10.002.
Kayagaki N, Lee BL, Stowe IB, Kornfeld OS, O'Rourke K, Mirrashidi KM, et al. IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci Signal 2019;12. https://doi.org/10.1126/scisignal.aax4917.
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20. https://doi.org/10.1038/nri.2016.58.
Kanneganti TD. Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol. 2010;10:688–98. https://doi.org/10.1038/nri2851.
Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunological Rev. 2015;265:35–52. https://doi.org/10.1111/imr.12286.
Munoz-Planillo R, Franchi L, Miller LS, Nunez G. A Critical Role for Hemolysins and Bacterial Lipoproteins in Staphylococcus aureus-Induced Activation of the Nlrp3 Inflammasome. J Immunol. 2009;183:3942–8. https://doi.org/10.4049/jimmunol.0900729.
Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. P Natl Acad Sci USA. 2008;105:4312–7. https://doi.org/10.1073/pnas.0707370105.
Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, Hornung V. Human NLRP1 is a sensor for double-stranded RNA. Science 2021;371. https://doi.org/10.1126/science.abd0811.
Moayeri M, Sastalla I, Leppla SH. Anthrax and the inflammasome. Microbes Infect. 2012;14:392–400. https://doi.org/10.1016/j.micinf.2011.12.005.
Frew BC, Joag VR, Mogridge J. Proteolytic Processing of Nlrp1b Is Required for Inflammasome Activity. Plos Pathog 2012;8:1002659. https://doi.org/10.1371/journal.ppat.1002659.
Moayeri M, Crown D, Newman ZL, Okugawa S, Eckhaus M, Cataisson C, et al. Inflammasome Sensor Nlrp1b-Dependent Resistance to Anthrax Is Mediated by Caspase-1, IL-1 Signaling and Neutrophil Recruitment. Plos Pathog. 2010;6:1001222. https://doi.org/10.1371/journal.ppat.1001222.
Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. P Natl Acad Sci USA. 2010;107:3076–80. https://doi.org/10.1073/pnas.0913087107.
Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–8. https://doi.org/10.1038/nature02664.
Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1 beta via Ipaf. Nat Immunol. 2006;7:569–75. https://doi.org/10.1038/ni1344.
Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010;11:385–94. https://doi.org/10.1038/ni.1859.
Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–U516. https://doi.org/10.1038/nature07725.
Schattgen SA, Fitzgerald KA. The PYHIN protein family as mediators of host defenses. Immunological Rev. 2011;243:109–18. https://doi.org/10.1111/j.1600-065X.2011.01053.x.
Yu JW, Wu J, Zhang Z, Datta P, Ibrahimi I, Taniguchi S, et al. Cryopyrin and pyrin activate caspase-1, but not NF-kappa B, via ASC oligomerization. Cell Death Differ. 2006;13:236–49. https://doi.org/10.1038/sj.cdd.4401734.
Seshadri S, Duncan MD, Hart JM, Gavrilin MA, Wewers MD. Pyrin levels in human monocytes and monocyte-derived macrophages regulate IL-1 beta processing and release. J Immunol. 2007;179:1274–81. https://doi.org/10.4049/jimmunol.179.2.1274.
Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237–41. https://doi.org/10.1038/nature13449.
Broecker F, Andrae K, Moelling K. Premature Activation of the HIV RNase H Drives the Virus into Suicide: A Novel Microbicide? Aids Res Hum Retrov. 2012;28:1397–403. https://doi.org/10.1089/aid.2012.0067.
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–U146. https://doi.org/10.1038/nature10558.
Yi YS. Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology. 2017;152:207–17. https://doi.org/10.1111/imm.12787.
Ding JJ, Shao F. SnapShot: The Noncanonical Inflammasome. Cell. 2017;168:544–544. https://doi.org/10.1016/j.cell.2017.01.008.
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–92. https://doi.org/10.1038/nature13683.
Zhou RB, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;475:122. https://doi.org/10.1038/nature10156.
Rodgers MA, Bowman JW, Fujita H, Orazio N, Shi M, Liang Q, et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med. 2014;211:1331–45. https://doi.org/10.1084/jem.20132486.
Atianand MK, Rathinam VA, Fitzgerald KA. SnapShot: Inflammasomes. Cell. 2013;153:272. https://doi.org/10.1016/j.cell.2013.03.009.
Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15:738. https://doi.org/10.1038/ni.2919.
Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, et al. Prion-like Polymerization Underlies Signal Transduction in Antiviral Immune Defense and Inflammasome Activation. Cell. 2014;156:1207–22. https://doi.org/10.1016/j.cell.2014.01.063.
Shi P, Tang A, Xian L, Hou S, Zou D, Lv Y, et al. Loss of conserved Gsdma3 self-regulation causes autophagy and cell death. Biochem J. 2015;468:325–36. https://doi.org/10.1042/Bj20150204.
Pickrell AM, Youle RJ. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron. 2015;85:257–73. https://doi.org/10.1016/j.neuron.2014.12.007.
Kang R, Zeng L, Xie Y, Yan Z, Zhou B, Cao L, et al. A novel PINK1-and PARK2-dependent protective neuroimmune pathway in lethal sepsis. Autophagy. 2016;12:2374–85. https://doi.org/10.1080/15548627.2016.1239678.
Andersson U, Tracey KJ. Neural reflexes in inflammation and immunity. J Exp Med. 2012;209:1057–68. https://doi.org/10.1084/jem.20120571.
Kuipers D, Mandemakers W, Lu CS, Olgiati S, Breedveld GJ, Fevga C, et al. EIF2AK2 Missense Variants Associated with Early Onset Generalized Dystonia. Ann Neurol. 2021;89:485–97. https://doi.org/10.1002/ana.25973.
Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun. 2016;7:13280. https://doi.org/10.1038/ncomms13280.
Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014;5:4436. https://doi.org/10.1038/ncomms5436.
Ingrand S, Barrier L, Lafay-Chebassier C, Fauconneau B, Page G, Hugon J. The oxindole/imidazole derivative C16 reduces in vivo brain PKR activation. Febs Lett. 2007;581:4473–8. https://doi.org/10.1016/j.febslet.2007.08.022.
Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. 2016;17:250–8. https://doi.org/10.1038/ni.3333.
He Y, Zeng MY, Yang DH, Metro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354–7. https://doi.org/10.1038/nature16959.
Yang XD, Li W, Zhang S, Wu D, Jiang X, Tan R, et al. PLK4 deubiquitination by Spata2-CYLD suppresses NEK7-mediated NLRP3 inflammasome activation at the centrosome. Embo J. 2020;39:102201. https://doi.org/10.15252/embj.2019102201.
Sun SC. CYLD: a tumor suppressor deubiquitinase regulating NF-kappa B activation and diverse biological processes. Cell Death Differ. 2010;17:25–34. https://doi.org/10.1038/cdd.2009.43.
Hjerpe R, Aillet F, Lopitz-Otsoa F, Lang V, England P, Rodriguez MS. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. Embo Rep. 2009;10:1250–8. https://doi.org/10.1038/embor.2009.192.
Fox D, Man SM. DDX3X: stressing the NLRP3 inflammasome. Cell Res. 2019;29:969–70. https://doi.org/10.1038/s41422-019-0250-8.
Kesavardhana S, Samir P, Zheng M, Malireddi R, Karki R, Sharma BR, et al. DDX3X coordinates host defense against influenza virus by activating the NLRP3 inflammasome and type I interferon response. J Biol Chem. 2021;296:100579. https://doi.org/10.1016/j.jbc.2021.100579.
Samir P, Kanneganti TD. DDX3X Sits at the Crossroads of Liquid-Liquid and Prionoid Phase Transitions Arbitrating Life and Death Cell Fate Decisions in Stressed Cells. DNA Cell Biol. 2020;39:1091–5. https://doi.org/10.1089/dna.2020.5616.
Protter DSW, Parker R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016;26:668–79. https://doi.org/10.1016/j.tcb.2016.05.004.
Samir P, et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature. 2019;573:590. https://doi.org/10.1038/s41586-019-1551-2.
Wang Q, Gao H, Clark KM, Mugisha CS, Davis K, Tang JP, et al. CARD8 is an inflammasome sensor for HIV-1 protease activity. Science. 2021;371:1224. https://doi.org/10.1126/science.abe1707.
Kwong PD, Doyle ML, Casper DJ, Cicala C, Leavitt SA, Majeed S, et al. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature. 2002;420:678–82. https://doi.org/10.1038/nature01188.
Trinite B, Zhang HT, Levy DN. NNRTI-induced HIV-1 protease-mediated cytotoxicity induces rapid death of CD4 T cells during productive infection and latency reversal. Retrovirology 2019;16:17. https://doi.org/10.1186/s12977-019-0479-9.
Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, Wang MS, et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat Chem Biol. 2017;13:46–53. https://doi.org/10.1038/Nchembio.2229.
Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem Biol. 2017;24:507. https://doi.org/10.1016/j.chembiol.2017.03.009.
Johnson DC, et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat Med. 2018;24:1151. https://doi.org/10.1038/s41591-018-0082-y.
Okondo MC, et al. Inhibition of Dpp8/9 Activates the Nlrp1b Inflammasome. Cell Chem Biol. 2018;25:262. https://doi.org/10.1016/j.chembiol.2017.12.013.
Hollingsworth LR, Sharif H, Griswold AR, Fontana P, Mintseris J, Dagbay KB, et al. DPP9 sequesters the C terminus of NLRP1 to repress inflammasome activation. Nature. 2021;592:778–83. https://doi.org/10.1038/s41586-021-03350-4.
Martinon F, Burns K, Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26. https://doi.org/10.1016/S1097-2765(02)00599-3.
Mitchell PS, Sandstrom A, Vance RE. The NLRP1 inflammasome: new mechanistic insights and unresolved mysteries. Curr Opin Immunol. 2019;60:37–45. https://doi.org/10.1016/j.coi.2019.04.015.
Chui AJ, et al. N-terminal degradation activates the NLRP1B inflammasome. Science. 2019;364:82. https://doi.org/10.1126/science.aau1208.
Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science. 2019;364:42. https://doi.org/10.1126/science.aau1330.
Feltham R, Vince JE. Ion Man: GSDMD Punches Pores to Knock Out cGAS. Immunity. 2018;49:379–81. https://doi.org/10.1016/j.immuni.2018.08.026.
Ma C, Yang D, Wang B, Wu C, Wu Y, Li S, et al. Gasdermin D in macrophages restrains colitis by controlling cGAS-mediated inflammation. Sci Adv. 2020;6:6717. https://doi.org/10.1126/sciadv.aaz6717.
Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, et al. Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity. 2018;49:413–26 e415. https://doi.org/10.1016/j.immuni.2018.07.006.
Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, et al. Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity. 2018;49:413–26. https://doi.org/10.1016/j.immuni.2018.07.006.
Liu Z, Gan L, Xu Y, Luo D, Ren Q, Wu S, Sun C. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-kappa B/GSDMD signal in mice adipose tissue. J Pineal Res 2017;63. https://doi.org/10.1111/jpi.12414.
Evavold CL, et al. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell. 2021;184:4495. https://doi.org/10.1016/j.cell.2021.06.028.
Moon JS, Hisata S, Park MA, DeNicola GM, Ryter SW, Nakahira K, et al. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015;12:102–15. https://doi.org/10.1016/j.celrep.2015.05.046.
Ruhl S, et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. 2018;362:956. https://doi.org/10.1126/science.aar7607.
Henne WM, Buchkovich NJ, Emr SD. The ESCRT Pathway. Dev Cell. 2011;21:77–91. https://doi.org/10.1016/j.devcel.2011.05.015.
Waites C, Sheehan P, Silva J, Zhu M, Sotiropoulos I. Roles of the RAB35/ESCRT pathway in synaptic protein degradation and neurodegenerative mechanisms. J Neurochem. 2017;142:44–45.
Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Bio Med. 2003;34:145–69. https://doi.org/10.1016/S0891-5849(02)01197-8.
Kang R, et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe. 2018;24:97. https://doi.org/10.1016/j.chom.2018.05.009.
Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep. 2018;22:2924–36. https://doi.org/10.1016/j.celrep.2018.02.067.
Loison F, Zhu H, Karatepe K, Kasorn A, Liu P, Ye K, et al. Proteinase 3-dependent caspase-3 cleavage modulates neutrophil death and inflammation. J Clin Invest. 2014;124:4445–58. https://doi.org/10.1172/Jci76246.
Adkison AM, Raptis SZ, Kelley DG, Pham CTN. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363–71. https://doi.org/10.1172/Jci200213462.
Wang L, Zhao J, Ren J, Hall KH, Moorman JP, Yao ZQ, et al. Protein phosphatase 1 abrogates IRF7-mediated type I IFN response in antiviral immunity. Eur J Immunol. 2016;46:2409–19. https://doi.org/10.1002/eji.201646491.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. https://doi.org/10.1038/nature15514.
Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family (vol 535, pg 111, 2016). Nature. 2016;540:150. https://doi.org/10.1038/nature20106.
Zhou ZW, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368:965. https://doi.org/10.1126/science.aaz7548.
Ewen CL, Kane KP, Bleackley RC. A quarter century of granzymes. Cell Death Differ. 2012;19:28–35. https://doi.org/10.1038/cdd.2011.153.
Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. 2015;15:388–400. https://doi.org/10.1038/nri3839.
Hansen JM, et al. Pathogenic ubiquitination of GSDMB inhibits NK cell bactericidal functions. Cell. 2021;184:3178. https://doi.org/10.1016/j.cell.2021.04.036.
Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K, et al. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics. 2007;89:618–29. https://doi.org/10.1016/j.ygeno.2007.01.003.
Chao KL, Kulakova L, Herzberg O. Gene polymorphism linked to increased asthma and IBD risk alters gasdermin-B structure, a sulfatide and phosphoinositide binding protein. P Natl Acad Sci USA. 2017;114:E1128–E1137. https://doi.org/10.1073/pnas.1616783114.
Pierce NW, Kleiger G, Shan SO, Deshaies RJ. Detection of sequential polyubiquitylation on a millisecond timescale. Nature. 2009;462:615–U685. https://doi.org/10.1038/nature08595.
Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128. https://doi.org/10.1038/ncomms14128.
Chowdhury D, Lieberman J. Death by a thousand cuts: Granzyme pathways of programmed cell death. Annu Rev Immunol. 2008;26:389–420. https://doi.org/10.1146/annurev.immunol.26.021607.090404.
Zhang ZB, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579:415 https://doi.org/10.1038/s41586-020-2071-9.
Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis (Sep, 10.1038/s41556-020-0575-z, 2020). Nat Cell Biol. 2020;22:1396–1396. https://doi.org/10.1038/s41556-020-00599-1.
Shimizu S, Eguchi Y, Kamiike W, Itoh Y, Hasegawa J, Yamabe K, et al. Induction of apoptosis as well as necrosis by hypoxia and predominant prevention of apoptosis by Bcl-2 and Bcl-X(L). Cancer Res. 1996;56:2161–6.
Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity. 2016;44:303–15. https://doi.org/10.1016/j.immuni.2016.01.014.
Timmer JC, Salvesen GS. Caspase substrates. Cell Death Differ. 2007;14:66–72. https://doi.org/10.1038/sj.cdd.4402059.
Tummers B, Green DR. Caspase-8: regulating life and death. Immunological Rev. 2017;277:76–89. https://doi.org/10.1111/imr.12541.
Deng W, Bai Y, Deng F, Pan Y, Mei S, Zheng Z, et al. Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature. 2022;602:496–502. https://doi.org/10.1038/s41586-021-04384-4.
Cole JN, Barnett TC, Nizet V, Walker MJ. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol. 2011;9:724–36. https://doi.org/10.1038/nrmicro2648.
Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 Protects Against Bacteria That Escape the Vacuole. Science. 2013;339:975–8. https://doi.org/10.1126/science.1230751.
Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, Ashida H, et al. The Shigella OspC3 Effector Inhibits Caspase-4, Antagonizes Inflammatory Cell Death, and Promotes Epithelial Infection. Cell Host Microbe. 2013;13:570–83. https://doi.org/10.1016/j.chom.2013.04.012.
Li ZL, et al. Shigella evades pyroptosis by arginine ADP-riboxanation of caspase-11. Nature. 2021;599:290. https://doi.org/10.1038/s41586-021-04020-1.
Xu Y, et al. A Bacterial Effector Reveals the V-ATPase-ATG16L1 Axis that Initiates Xenophagy. Cell. 2019;178:552. https://doi.org/10.1016/j.cell.2019.06.007.
Chung LK, Park YH, Zheng Y, Brodsky IE, Hearing P, Kastner DL, et al. The Yersinia Virulence Factor YopM Hijacks Host Kinases to Inhibit Type III Effector-Triggered Activation of the Pyrin Inflammasome. Cell Host Microbe. 2016;20:296–306. https://doi.org/10.1016/j.chom.2016.07.018.
Rosadini CV, Zanoni I, Odendall C, Green ER, Paczosa MK, Philip NH, et al. A Single Bacterial Immune Evasion Strategy Dismantles Both MyD88 and TRIF Signaling Pathways Downstream of TLR4. Cell Host Microbe. 2015;18:682–93. https://doi.org/10.1016/j.chom.2015.11.006.
Zheng Z, Deng W, Bai Y, Miao R, Mei S, Zhang Z, et al. The lysosomal Rag-Ragulator complex licenses RIPK1-and caspase-8-mediated pyroptosis by Yersinia. Science. 2021;372:1412. https://doi.org/10.1126/science.abg0269.
Paquette N, Conlon J, Sweet C, Rus F, Wilson L, Pereira A, et al. Serine/threonine acetylation of TGF beta-activated kinase (TAK1) by Yersinia pestis YopJ inhibits innate immune signaling. P Natl Acad Sci USA. 2012;109:12710–5. https://doi.org/10.1073/pnas.1008203109.
Muendlein HI, Jetton D, Connolly WM, Eidell KP, Magri Z, Smirnova I, et al. cFLIP(L) protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science. 2020;367:1379–84. https://doi.org/10.1126/science.aay3878.
Qu Z, Zhou J, Zhou Y, Xie Y, Jiang Y, Wu J, et al. Mycobacterial EST12 activates a RACK1-NLRP3-gasdermin D pyroptosis-IL-1 beta immune pathway. Sci Adv. 2020;6. https://doi.org/10.1126/sciadv.aba4733.
Ruiz Carrillo D, Chandrasekaran R, Nilsson M, Cornvik T, Liew CW, Tan SM, Lescar J. Structure of human Rack1 protein at a resolution of 2.45 A. Acta Crystallogr F. 2012;68:867–72. https://doi.org/10.1107/S1744309112027480.
Lyskov S, Gray JJ. The RosettaDock server for local proteinprotein docking. Nucleic Acids Res. 2008;36:W233–W238. https://doi.org/10.1093/nar/gkn216.
Hargreaves DC, Horng T, Medzhitov R. Control of Inducible Gene Expression by Signal-Dependent Transcriptional Elongation. Cell. 2009;138:129–45. https://doi.org/10.1016/j.cell.2009.05.047.
Dong X, Hu X, Bao Y, Li G, Yang XD, Slauch JM, Chen LF. Brd4 regulates NLRC4 inflammasome activation by facilitating IRF8-mediated transcription of Naips. J Cell Biol. 2021;220. https://doi.org/10.1083/jcb.202005148.
Huang B, Yang XD, Zhou MM, Ozato K, Chen LF. Brd4 Coactivates Transcriptional Activation of NF-kappa B via Specific Binding to Acetylated RelA. Mol Cell Biol. 2009;29:1375–87. https://doi.org/10.1128/Mcb.01365-08.
Karki R, et al. IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell. 2018;173:920. https://doi.org/10.1016/j.cell.2018.02.055.
Rauch I, Deets KA, Ji DX, von Moltke J, Tenthorey JL, Lee AY, et al. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and-8. Immunity. 2017;46:649–59. https://doi.org/10.1016/j.immuni.2017.03.016.
Linder A, Bauernfried S, Cheng Y, Albanese M, Jung C, Keppler OT, et al. CARD8 inflammasome activation triggers pyroptosis in human T cells. Embo J. 2020;39:e105071. https://doi.org/10.15252/embj.2020105071.
Figueiredo A, Moore KL, Mak J, Sluis-Cremer N, de Bethune MP, Tachedjian G. Potent nonnucleoside reverse transcriptase inhibitors target HIV-1 Gag-Pol. Plos Pathog. 2006;2:1051–9. https://doi.org/10.1371/journal.ppat.0020119.
Sparrer KMJ, Kirchhoff F. HIV protease: late action to prevent immune detection. Signal Transduct Tar 2021;6: 157. https://doi.org/10.1038/s41392-021-00588-2.
Ma H, Zhu Z, Lin H, Wang S, Zhang P, Li Y, et al. Pyroptosis of syncytia formed by fusion of SARS-CoV-2 spike and ACE2-expressing cells. Cell Discov. 2021;7:73. https://doi.org/10.1038/s41421-021-00310-0.
Zhang Z, Zheng Y, Niu Z, Zhang B, Wang C, Yao X, et al. SARS-CoV-2 spike protein dictates syncytium-mediated lymphocyte elimination. Cell Death Differ. 2021;28:2765–77. https://doi.org/10.1038/s41418-021-00782-3.
Ratajczak MZ, Kucia M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia. 2020;34:1726–9. https://doi.org/10.1038/s41375-020-0887-9.
Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res. 2020;220:1–13. https://doi.org/10.1016/j.trsl.2020.04.007.
Violi F, Pastori D, Cangemi R, Pignatelli P, Loffredo L. Hypercoagulation and Antithrombotic Treatment in Coronavirus 2019: A New Challenge. Thromb Haemost. 2020;120:949–56. https://doi.org/10.1055/s-0040-1710317.
Freeman TL, Swartz TH. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front Immunol. 2020;11:1518. https://doi.org/10.3389/fimmu.2020.01518.
Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity. 2017;47:15–31. https://doi.org/10.1016/j.immuni.2017.06.020.
Vaduganathan M, Vardeny O, Michel T, McMurray J, Pfeffer MA, Solomon SD. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. N. Engl J Med. 2020;382:1653–9. https://doi.org/10.1056/NEJMsr2005760.
Zhao M, Bai M, Ding G, Zhang Y, Huang S, Jia Z, et al. Angiotensin II Stimulates the NLRP3 Inflammasome to Induce Podocyte Injury and Mitochondrial Dysfunction. Kidney Dis-Basel. 2018;4:83–94. https://doi.org/10.1159/000488242.
Wan X, Li J, Wang Y, Yu X, He X, Shi J, et al. H7N9 virus infection triggers lethal cytokine storm by activating gasdermin E-mediated pyroptosis of lung alveolar epithelial cells. Natl Sci Rev. 2021;9:137. https://doi.org/10.1093/nsr/nwab137.
Chen S, Wang A, Sun L, Liu F, Wang M, Jia R, et al. Immune-Related Gene Expression Patterns in GPV- or H9N2-Infected Goose Spleens. Int J Mol Sci 2016;17:1990. https://doi.org/10.3390/ijms17121990.
Nemeth NM, Brown JD, Stallknecht DE, Howerth EW, Newman SH, Swayne DE. Experimental Infection of Bar-Headed Geese (Anser indicus) and Ruddy Shelducks (Tadorna ferruginea) With a Clade 2.3.2 H5N1 Highly Pathogenic Avian Influenza Virus. Vet Pathol. 2013;50:961–70. https://doi.org/10.1177/0300985813490758.
Ito K, Ito K. Hematopoietic stem cell fate through metabolic control. Exp Hematol. 2018;64:1–11. https://doi.org/10.1016/j.exphem.2018.05.005.
Zhang J, Zhou B, Sun R, Ai Y, Cheng K, Li F, et al. The metabolite alpha-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res. 2021;31:980–97. https://doi.org/10.1038/s41422-021-00506-9.
Morris JP. 4th et al. alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature. 2019;573:595–9. https://doi.org/10.1038/s41586-019-1577-5.
Wang YP, Zhou W, Wang J, Huang X, Zuo Y, Wang TS, et al. Arginine Methylation of MDH1 by CARM1 Inhibits Glutamine Metabolism and Suppresses Pancreatic Cancer. Mol Cell. 2016;64:673–87. https://doi.org/10.1016/j.molcel.2016.09.028.
Schwarzer R, Jiao HP, Wachsmuth L, Tresch A, Pasparakis M. FADD and Caspase-8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL- and GSDMD-Mediated Death of Intestinal Epithelial Cells. Immunity. 2020;52:978 https://doi.org/10.1016/j.immuni.2020.04.002.
Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17. https://doi.org/10.1038/nchembio.1416.
Zhou B, Zhang J, Liu X, Chen H, Ai Y, Cheng K, et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28:1171–85. https://doi.org/10.1038/s41422-018-0090-y.
Ott M, Norberg E, Walter KM, Schreiner P, Kemper C, Rapaport D, et al. The mitochondrial TOM complex is required for tBid/Bax-induced cytochrome c release. J Biol Chem. 2007;282:27633–9. https://doi.org/10.1074/jbc.M703155200.
Wang YP, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99. https://doi.org/10.1038/nature22393.
Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochemical Sci. 2016;41:274–86. https://doi.org/10.1016/j.tibs.2015.11.012.
Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science. 2020;369:1633. https://doi.org/10.1126/science.abb9818.
Blatnik M, Frizzell N, Thorpe SR, Baynes JW. Inactivation of glyceraldehyde-3-phosphate dehydrogenase by fumarate in diabetes - Formation of S-(2-succinyl)cysteine, a novel chemical modification of protein and possible biomarker of mitochondrial stress. Diabetes. 2008;57:41–49. https://doi.org/10.2337/db07-0838.
Sundblad V, Morosi LG, Geffner JR, Rabinovich GA. Galectin-1: A Jack-of-All-Trades in the Resolution of Acute and Chronic Inflammation. J Immunol. 2017;199:3721–30. https://doi.org/10.4049/jimmunol.1701172.
Russo AJ, Vasudevan SO, Méndez-Huergo SP, Kumari P, Menoret A, Duduskar S, et al. Intracellular immune sensing promotes inflammation via gasdermin D-driven release of a lectin alarmin. Nat Immunol. 2021;22:154–U178. https://doi.org/10.1038/s41590-020-00844-7.
Bone RC, et al. American-College of Chest Physicians Society of Critical Care Medicine Consensus Conference - Definitions for Sepsis and Organ Failure and Guidelines for the Use of Innovative Therapies in Sepsis. Crit Care Med. 1992;20:864–74. https://doi.org/10.1097/00003246-199206000-00025.
Ousingsawat J, Wanitchakool P, Schreiber R, Kunzelmann K. Contribution of TMEM16F to pyroptotic cell death. Cell Death Dis. 2018;9:300. https://doi.org/10.1038/s41419-018-0373-8.
de Baaij JHF, Hoenderop JGJ, Bindels RJM. Magnesium in Man: Implications for Health and Disease. Physiol Rev. 2015;95:1–46. https://doi.org/10.1152/physrev.00012.2014.
Bruzzone S, Basile G, Chothi MP, Nobbio L, Usai C, Jacchetti E, et al. Diadenosine Homodinucleotide Products of ADP-ribosyl Cyclases Behave as Modulators of the Purinergic Receptor P2X7. J Biol Chem. 2010;285:21165–74. https://doi.org/10.1074/jbc.M109.097964.
Wang D, Zheng J, Hu Q, Zhao C, Chen Q, Shi P, et al. Magnesium protects against sepsis by blocking gasdermin D N-terminal-induced pyroptosis. Cell Death Differ. 2020;27:466–81. https://doi.org/10.1038/s41418-019-0366-x.
Yang DH, He Y, Munoz-Planillo R, Liu Q, Nunez G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity. 2015;43:923–32. https://doi.org/10.1016/j.immuni.2015.10.009.
Costa FF. Non-coding RNAs: Meet thy masters. Bioessays. 2010;32:599–608. https://doi.org/10.1002/bies.200900112.
Chen S, Luo LM, Chen HD, He CD. The Current State of Research Regarding the Role of Non-Coding RNAs in Cutaneous Squamous Cell Carcinoma. Oncotargets Ther. 2020;13:13151–8. https://doi.org/10.2147/Ott.S271346.
Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15:R17–R29. https://doi.org/10.1093/hmg/ddl046.
Heo MJ, Kim TH, You JS, Blaya D, Sancho-Bru P, Kim SG. Alcohol dysregulates miR-148a in hepatocytes through FoxO1, facilitating pyroptosis via TXNIP overexpression. Gut. 2019;68:708–20. https://doi.org/10.1136/gutjnl-2017-315123.
Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, et al. FoxO1 regulates multiple metabolic pathways in the liver. - Eff gluconeogenic, glycolytic, lipogenic gene Expr J Biol Chem. 2006;281:10105–17. https://doi.org/10.1074/jbc.M600272200.
Liang XM, Hu M, Rogers CQ, Shen Z, You M. Role of SIRT1-FoxO1 Signaling in Dietary Saturated Fat-Dependent Upregulation of Liver Adiponectin Receptor 2 in Ethanol-Administered Mice. Antioxid Redox Sign. 2011;15:425–35. https://doi.org/10.1089/ars.2010.3780.
Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, et al. NLRP3 Gene Silencing Ameliorates Diabetic Cardiomyopathy in a Type 2 Diabetes Rat Model. Plos One 2014;9:104771. https://doi.org/10.1371/journal.pone.0104771.
Park YJ, Yoon SJ, Suh HW, Kim DO, Park JR, Jung H, et al. TXNIP Deficiency Exacerbates Endotoxic Shock via the Induction of Excessive Nitric Oxide Synthesis. Plos Pathog. 2013;9:1003646. https://doi.org/10.1371/journal.ppat.1003646.
Jeyabal P, Thandavarayan RA, Joladarashi D, Suresh Babu S, Krishnamurthy S, Bhimaraj A, et al. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem Bioph Res Co. 2016;471:423–9. https://doi.org/10.1016/j.bbrc.2016.02.065.
Wei N, Xiao L, Xue R, Zhang D, Zhou J, Ren H, et al. MicroRNA-9 Mediates the Cell Apoptosis by Targeting Bcl2l11 in Ischemic Stroke. Mol Neurobiol. 2016;53:6809–17. https://doi.org/10.1007/s12035-015-9605-4.
Abdelmohsen K, Lal A, Kim HH, Gorospe M. Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle. 2007;6:1288–92. https://doi.org/10.4161/cc.6.11.4299.
Li X, Zeng L, Cao C, Lu C, Lian W, Han J, et al. Long noncoding RNA MALAT1 regulates renal tubular epithelial pyroptosis by modulated miR-23c targeting of ELAVL1 in diabetic nephropathy. Exp Cell Res. 2017;350:327–35. https://doi.org/10.1016/j.yexcr.2016.12.006.
Zhang F, Wu L, Qian J, Qu B, Xia S, La T, et al. Identification of the long noncoding RNA NEAT1 as a novel inflammatory regulator acting through MAPK pathway in human lupus. J Autoimmun. 2016;75:96–104. https://doi.org/10.1016/j.jaut.2016.07.012.
Sunwoo H, Dinger ME, Wilusz JE, Amaral PP, Mattick JS, Spector DL. MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 2009;19:347–59. https://doi.org/10.1101/gr.087775.108.
Zhang PF, Cao LM, Zhou RB, Yang XL, Wu M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat Commun. 2019;10:1495. https://doi.org/10.1038/s41467-019-09482-6.
Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–5. https://doi.org/10.1038/nri2725.
Sasaki YTF, Ideue T, Sano M, Mituyama T, Hirose T. MEN epsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. P Natl Acad Sci USA. 2009;106:2525–30. https://doi.org/10.1073/pnas.0807899106.
Imamura K, Imamachi N, Akizuki G, Kumakura M, Kawaguchi A, Nagata K, et al. Long Noncoding RNA NEAT1-Dependent SFPQ Relocation from Promoter Region to Paraspeckle Mediates IL8 Expression upon Immune Stimuli (vol 53, pg 393, 2014). Mol Cell. 2014;54:1055–1055. https://doi.org/10.1016/j.molcel.2014.06.013.
Chang SC, Yang WV. Hyperglycemia, tumorigenesis, and chronic inflammation. Crit Rev Oncol Hematol. 2016;108:146–53. https://doi.org/10.1016/j.critrevonc.2016.11.003.
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow. Lancet. 2001;357:539–45. https://doi.org/10.1016/S0140-6736(00)04046-0.
Romaszko AM, Doboszynska A. Multiple primary lung cancer: A literature review. Adv Clin Exp Med. 2018;27:725–30. https://doi.org/10.17219/acem/68631.
Brody H. Lung cancer. Nature. 2014;513:S1. https://doi.org/10.1038/513S1a.
Teng JF, Mei QB, Zhou XG, Tang Y, Xiong R, Qiu WQ, et al. Polyphyllin VI Induces Caspase-1-Mediated Pyroptosis via the Induction of ROS/NF-kappaB/NLRP3/GSDMD Signal Axis in Non-Small Cell Lung Cancer. Cancers. 2020;12. https://doi.org/10.3390/cancers12010193.
West H, McCleod M, Hussein M, Morabito A, Rittmeyer A, Conter HJ, et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20:924–37. https://doi.org/10.1016/S1470-2045(19)30167-6.
Kocijancic D, Leschner S, Felgner S, Komoll RM, Frahm M, Pawar V, et al. Therapeutic benefit of Salmonella attributed to LPS and TNF-alpha is exhaustible and dictated by tumor susceptibility. Oncotarget. 2017;8:36492–508. https://doi.org/10.18632/oncotarget.16906.
Yokoyama S, Cai Y, Murata M, Tomita T, Yoneda M, Xu L, et al. A novel pathway of LPS uptake through syndecan-1 leading to pyroptotic cell death. Elife. 2018;7. https://doi.org/10.7554/eLife.37854.
Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396:635–48. https://doi.org/10.1016/S0140-6736(20)31288-5.
Wang Y, Yin B, Li D, Wang G, Han X, Sun X. GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem Biophys Res Commun. 2018;495:1418–25. https://doi.org/10.1016/j.bbrc.2017.11.156.
Qiu SQ, Liu J, Xing FY. ‘Hints’ in the killer protein gasdermin D: unveiling the secrets of gasdermins driving cell death. Cell Death Differ. 2017;24:588–96. https://doi.org/10.1038/cdd.2017.24.
Wang WJ, Chen D, Jiang MZ, Xu B, Li XW, Chu Y, et al. Downregulation of gasdermin D promotes gastric cancer proliferation by regulating cell cycle-related proteins. J Dig Dis. 2018;19:74–83. https://doi.org/10.1111/1751-2980.12576.
Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, et al. Distinctive Expression and Function of Four GSDM Family Genes (GSDMA-D) in Normal and Malignant Upper Gastrointestinal Epithelium. Gene Chromosome Canc. 2009;48:261–71. https://doi.org/10.1002/gcc.20636.
Veronesi U, Boyle P, Goldhirsch A, Orecchia R, Viale G. Breast cancer. Lancet. 2005;365:1727–41. https://doi.org/10.1016/S0140-6736(05)66546-4.
Yan H, Luo B, Wu X, Guan F, Yu X, Zhao L, et al. Cisplatin Induces Pyroptosis via Activation of MEG3/NLRP3/caspase-1/GSDMD Pathway in Triple-Negative Breast Cancer. Int J Biol Sci. 2021;17:2606–21. https://doi.org/10.7150/ijbs.60292.
La Belle A, Khatib J, Schiemann WP, Vinayak S. Role of Platinum in Early-Stage Triple-Negative Breast Cancer. Curr Treat Option On 2017;18:68. https://doi.org/10.1007/s11864-017-0506-9.
Elion DL, Jacobson ME, Hicks DJ, Rahman B, Sanchez V, Gonzales-Ericsson PI, et al. Therapeutically Active RIG-I Agonist Induces Immunogenic Tumor Cell Killing in Breast Cancers. Cancer Res. 2018;78:6183–95. https://doi.org/10.1158/0008-5472.Can-18-0730.
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers 2021;7:6. https://doi.org/10.1038/s41572-020-00240-3.
Hage C, Hoves S, Strauss L, Bissinger S, Prinz Y, Pöschinger T, et al. Sorafenib Induces Pyroptosis in Macrophages and Triggers Natural Killer Cell-Mediated Cytotoxicity Against Hepatocellular Carcinoma. Hepatology. 2019;70:1280–97. https://doi.org/10.1002/hep.30666.
Shangguan F, Zhou H, Ma N, Wu S, Huang H, Jin G, et al. A Novel Mechanism of Cannabidiol in Suppressing Hepatocellular Carcinoma by Inducing GSDME Dependent Pyroptosis. Front Cell Dev Biol 2021;9:697832. https://doi.org/10.3389/fcell.2021.697832.
Zhang X, Zhang P, An L, Sun N, Peng L, Tang W, et al. Miltirone induces cell death in hepatocellular carcinoma cell through GSDME-dependent pyroptosis. Acta Pharm Sin B. 2020;10:1397–413. https://doi.org/10.1016/j.apsb.2020.06.015.
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer Statistics in China, 2015. Ca-Cancer J Clin. 2016;66:115–32. https://doi.org/10.3322/caac.21338.
Weitz J, et al. Colorectal cancer. Lancet. 2005;365:153–65. https://doi.org/10.1016/S0140-6736(05)17706-X.
Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J, et al. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019;10:193. https://doi.org/10.1038/s41419-019-1441-4.
Ma YS, Chen YF, Lin CW, Hu G. Biological functions and clinical significance of the newly identified long non-coding RNA RP1-85F18.6 in colorectal cancer. Oncol Rep. 2018;40:2648–58. https://doi.org/10.3892/or.2018.6694.
Zhang Y, Yang H, Sun M, He T, Liu Y, Yang X, et al. Alpinumisoflavone suppresses hepatocellular carcinoma cell growth and metastasis via NLRP3 inflammasome-mediated pyroptosis. Pharm Rep. 2020;72:1370–82. https://doi.org/10.1007/s43440-020-00064-8.
Chen L, Li Q, Zheng Z, Xie J, Lin X, Jiang C, et al. Design and optimize N-substituted EF24 as effective and low toxicity NF-kappa B inhibitor for lung cancer therapy via apoptosis-to-pyroptosis switch. Chem Biol Drug Des. 2019;94:1368–77. https://doi.org/10.1111/cbdd.13514.
Tang Z, Ji L, Han M, Xie J, Zhong F, Zhang X, et al. Pyroptosis is involved in the inhibitory effect of FL118 on growth and metastasis in colorectal cancer. Life Sci. 2020;257:118065. https://doi.org/10.1016/j.lfs.2020.118065.
Zhang Y, Li F, Wang LL, Lou Y. A438079 affects colorectal cancer cell proliferation, migration, apoptosis, and pyroptosis by inhibiting the P2X7 receptor. Biochem Bioph Res Co. 2021;558:147–53. https://doi.org/10.1016/j.bbrc.2021.04.076.
Huang J, Fan P, Liu M, Weng C, Fan G, Zhang T, et al. Famotidine promotes inflammation by triggering cell pyroptosis in gastric cancer cells. Bmc Pharmacol Toxico. 2021;22:62. https://doi.org/10.1186/s40360-021-00533-7.
Deng BB, Jiao BP, Liu YJ, Li YR, Wang GJ. BIX-01294 enhanced chemotherapy effect in gastric cancer by inducing GSDME-mediated pyroptosis. Cell Biol Int. 2020;44:1890–9. https://doi.org/10.1002/cbin.11395.
Li CF, Qiu, JQ, Xue YW. Low-dose Diosbulbin-B (DB) activates tumor-intrinsic PD-L1/NLRP3 signaling pathway mediated pyroptotic cell death to increase cisplatin-sensitivity in gastric cancer (GC). Cell Biosci 2021;11:38. https://doi.org/10.1186/s13578-021-00548-x.
Pizato N, Luzete BC, Kiffer L, Corrêa LH, Santos IO, Assumpção J, et al. Omega-3 docosahexaenoic acid induces pyroptosis cell death in triplenegative breast cancer cells (vol 8, 2018). Sci Rep-Uk. 2018;8:9775. https://doi.org/10.1038/s41598-018-27850-y.
Chen L, Weng B, Li H, Wang H, Li Q, Wei X, et al. A thiopyran derivative with low murine toxicity with therapeutic potential on lung cancer acting through a NF-kappa B mediated apoptosis-to-pyroptosis switch. Apoptosis. 2019;24:74–82. https://doi.org/10.1007/s10495-018-1499-y.
Wang ZG, Wu ZQ, Liu Y, Han WD. New development in CAR-T cell therapy. J Hematol Oncol 2017;10:53. https://doi.org/10.1186/s13045-017-0423-1.
Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. 2020;5. https://doi.org/10.1126/sciimmunol.aax7969.
Lu C, Guo C, Chen H, Zhang H, Zhi L, Lv T, et al. A novel chimeric PD1-NKG2D-41BB receptor enhances antitumor activity of NK92 cells against human lung cancer H1299 cells by triggering pyroptosis. Mol Immunol. 2020;122:200–6. https://doi.org/10.1016/j.molimm.2020.04.016.
Xi G, Gao J, Wan B, Zhan P, Xu W, Lv T, et al. GSDMD is required for effector CD8(+) T cell responses to lung cancer cells. Int Immunopharmacol 2019;74:105713. https://doi.org/10.1016/j.intimp.2019.105713.
Wang QY, et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature. 2020;579:421. https://doi.org/10.1038/s41586-020-2079-1.
Kim MS, Chang X, Yamashita K, Nagpal JK, Baek JH, Wu G, et al. Aberrant promoter methylation and tumor suppressive activity of the DFNA5 gene in colorectal carcinoma. Oncogene. 2008;27:3624–34. https://doi.org/10.1038/sj.onc.1211021.
Fan JX, Deng RH, Wang H, Liu XH, Wang XN, Qin R, et al. Epigenetics-Based Tumor Cells Pyroptosis for Enhancing the Immunological Effect of Chemotherapeutic Nanocarriers. Nano Lett. 2019;19:8049–58. https://doi.org/10.1021/acs.nanolett.9b03245.
Le X, Mu J, Peng W, Tang J, Xiang Q, Tian S, et al. DNA methylation downregulated ZDHHC1 suppresses tumor growth by altering cellular metabolism and inducing oxidative/ER stress-mediated apoptosis and pyroptosis. Theranostics. 2020;10:9495–511. https://doi.org/10.7150/thno.45631.
Wu D, Wei C, Li Y, Yang X, Zhou S. Pyroptosis, a New Breakthrough in Cancer Treatment. Front Oncol. 2021;11:698811. https://doi.org/10.3389/fonc.2021.698811.
Loveless R, Bloomquist R, Teng Y. Pyroptosis at the forefront of anticancer immunity. J Exp Clin Canc Res 2021;40:264. https://doi.org/10.1186/s13046-021-02065-8.
Huang B, Qian Y, Xie S, Ye X, Chen H, Chen Z, et al. Ticagrelor inhibits the NLRP3 inflammasome to protect against inflammatory disease independent of the P2Y(12)signaling pathway. Cell Mol Immunol. 2021;18:1278–89. https://doi.org/10.1038/s41423-020-0444-5.
Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 Inflammasome Activation Results in Hepatocyte Pyroptosis, Liver Inflammation, and Fibrosis in Mice. Hepatology. 2014;59:898–910. https://doi.org/10.1002/hep.26592.
Xie J, et al. Huaier extract suppresses non-small cell lung cancer progression through activating NLRP3-dependent pyroptosis (Retraction of Vol 304, Pg 291, 2021). Anat Rec. 2021;304:2899–2899. https://doi.org/10.1002/ar.24811.
Wu LM, Wu SG, Chen F, Wu Q, Wu CM, Kang CM, et al. Atorvastatin inhibits pyroptosis through the lncRNA NEXN-AS1/NEXN pathway in human vascular endothelial cells. Atherosclerosis. 2020;293:26–34. https://doi.org/10.1016/j.atherosclerosis.2019.11.033.
Bian Y, Li X, Pang P, Hu XL, Yu ST, Liu YN, et al. Kanglexin, a novel anthraquinone compound, protects against myocardial ischemic injury in mice by suppressing NLRP3 and pyroptosis. Acta Pharm Sin. 2020;41:319–26. https://doi.org/10.1038/s41401-019-0307-8.
Smits P. Cardiovascular effects of sulphonylurea derivatives. Diabetologia. 1997;40:S160–S161. https://doi.org/10.1007/s001250051438.
Hoffman WE, Albrecht RF, Jonjev ZS. Glyburide decreases myocardial oxygen pressure in dogs. Acta Anaesth Scand. 2003;47:221–5. https://doi.org/10.1034/j.1399-6576.2003.00039.x.
Andreis A, Imazio M, De Ferrari GM. Colchicine for the treatment of cardiovascular diseases: old drug, new targets. J Cardiovasc Med. 2021;22:1–8. https://doi.org/10.2459/Jcm.0000000000001079.
Toldo S, Mauro AG, Cutter Z, Van Tassell BW, Mezzaroma E, Del Buono MG, et al. The NLRP3 Inflammasome Inhibitor, OLT1177 (Dapansutrile), Reduces Infarct Size and Preserves Contractile Function After Ischemia Reperfusion Injury in the Mouse. J Cardiovasc Pharm 2019;73:215–22. https://doi.org/10.1097/Fjc.0000000000000658.
Hsu CN, Huang LT, Tain YL. Perinatal Use of Melatonin for Offspring Health: Focus on Cardiovascular and Neurological Diseases. Int J Mol Sci. 2019;20. https://doi.org/10.3390/ijms20225681.
Mikhail N. Cardiovascular Effects of Liraglutide. Curr Hypertens Rev. 2019;15:64–69. https://doi.org/10.2174/1573402114666180507152620.
Qiao L, Wu X, Zhang J, Liu L, Sui X, Zhang R, et al. alpha-NETA induces pyroptosis of epithelial ovarian cancer cells through the GSDMD/caspase-4 pathway. Faseb J. 2019;33:12760–7. https://doi.org/10.1096/fj.201900483RR.
Liu D, Dong Z, Xiang F, Liu H, Wang Y, Wang Q, Rao J. Dendrobium Alkaloids Promote Neural Function After Cerebral Ischemia-Reperfusion Injury Through Inhibiting Pyroptosis Induced Neuronal Death in both In Vivo and In Vitro Models. Neurochem Res. 2020;45:437–54. https://doi.org/10.1007/s11064-019-02935-w.
Gao J, Peng S, Shan X, Deng G, Shen L, Sun J, et al. Inhibition of AIM2 inflammasome-mediated pyroptosis by Andrographolide contributes to amelioration of radiation-induced lung inflammation and fibrosis. Cell Death Dis. 2019;10:957. https://doi.org/10.1038/s41419-019-2195-8.
Chu Q, Jiang Y, Zhang W, Xu C, Du W, Tuguzbaeva G, et al. Pyroptosis is involved in the pathogenesis of human hepatocellular carcinoma. Oncotarget. 2016;7:84658–65. https://doi.org/10.18632/oncotarget.12384.
Wang L, Li K, Lin X, Yao Z, Wang S, Xiong X, et al. Metformin induces human esophageal carcinoma cell pyroptosis by targeting the miR-497/PELP1 axis. Cancer Lett. 2019;450:22–31. https://doi.org/10.1016/j.canlet.2019.02.014.
Li J, Yang C, Li Y, Chen A, Li L, You Z. LncRNA GAS5 suppresses ovarian cancer by inducing inflammasome formation. Biosci Rep 2018;38. https://doi.org/10.1042/Bsr20171150.
Yang L, Liu J, Shan Q, Geng G, Shao P. Corrigendum to “High glucose inhibits proliferation and differentiation of osteoblast in alveolar bone by inducing pyroptosis” [Biochem. Biophys. Res. Commun. 522(2) (2020) 471-8]. Biochem Biophys Res Commun. 2020;528:404 https://doi.org/10.1016/j.bbrc.2020.05.039.
Wang S, Yang M, Yang X, Xu L, Ke S, Ding X, et al. Endothelial pyroptosis underlies systemic inflammatory response following radiofrequency ablation of hepatic hemangiomas. Scand J Clin Lab Inv. 2019;79:619–28. https://doi.org/10.1080/00365513.2019.1689428.
He T, Xu X, Zhang XY, Shen P, Ling JY, Han YX, et al. Effectiveness of Huai Qi Huang Granules on Juvenile Collagen-induced Arthritis and Its Influence on Pyroptosis Pathway in Synovial Tissue. Curr Med Sci. 2019;39:784–93. https://doi.org/10.1007/s11596-019-2106-3.
Yu P, Wang HY, Tian M, Li AX, Chen XS, Wang XL, et al. Eukaryotic elongation factor-2 kinase regulates the cross-talk between autophagy and pyroptosis in doxorubicin-treated human melanoma cells in vitro. Acta Pharm Sin. 2019;40:1237–44. https://doi.org/10.1038/s41401-019-0222-z.
Kong Y, Feng Z, Chen A, Qi Q, Han M, Wang S, et al. The Natural Flavonoid Galangin Elicits Apoptosis, Pyroptosis, and Autophagy in Glioblastoma. Front Oncol. 2019;9:942. https://doi.org/10.3389/fonc.2019.00942.
Zhang CC, Li CG, Wang YF, Xu LH, He XH, Zeng QZ, et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis. 2019;24:312–25. https://doi.org/10.1007/s10495-019-01515-1.
Wu M, Wang Y, Yang D, Gong Y, Rao F, Liu R, et al. A PLK1 kinase inhibitor enhances the chemosensitivity of cisplatin by inducing pyroptosis in oesophageal squamous cell carcinoma. Ebiomedicine. 2019;41:244–55. https://doi.org/10.1016/j.ebiom.2019.02.012.
Liu Y, Li P, Qiao C, Wu T, Sun X, Wen M, et al. Chitosan Hydrogel Enhances the Therapeutic Efficacy of Bone Marrow-Derived Mesenchymal Stem Cells for Myocardial Infarction by Alleviating Vascular Endothelial Cell Pyroptosis. J Cardiovasc Pharm. 2020;75:75–83. https://doi.org/10.1097/Fjc.0000000000000760.
Li S, Wu Y, Yang D, Wu C, Ma C, Liu X, et al. Gasdermin D in peripheral myeloid cells drives neuroinflammation in experimental autoimmune encephalomyelitis. J Exp Med. 2019;216:2562–81. https://doi.org/10.1084/jem.20190377.
Hu JJ, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21:736. https://doi.org/10.1038/s41590-020-0669-6.
Zhang, JL & Wei, KH Necrosulfonamide reverses pyroptosis-induced inhibition of proliferation and differentiation of osteoblasts through the NLRP3/caspase-1/GSDMD pathway. Exp Cell Res. 2021;405:112648. https://doi.org/10.1016/j.yexcr.2021.112648.
Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol. 2018;3. https://doi.org/10.1126/sciimmunol.aat2738.
Martín-Sánchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J, et al. Inflammasome-dependent IL-1 beta release depends upon membrane permeabilisation. Cell Death Differ. 2016;23:1219–31. https://doi.org/10.1038/cdd.2015.176.
Ryan DG, O’Neill LAJ. Krebs Cycle Reborn in Macrophage Immunometabolism. Annu Rev Immunol, Vol 38. 2020;38:289–313. https://doi.org/10.1146/annurev-immunol-081619-104850.
Murphy MP, O’Neill LAJ. Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell. 2018;174:780–4. https://doi.org/10.1016/j.cell.2018.07.030.
Hooftman A, Angiari S, Hester S, Corcoran SE, Runtsch MC, Ling C, et al. The Immunomodulatory Metabolite Itaconate Modifies NLRP3 and Inhibits Inflammasome Activation. Cell Metab. 2020;32:468. https://doi.org/10.1016/j.cmet.2020.07.016.
Guo C, Chi Z, Jiang D, Xu T, Yu W, Wang Z, et al. Cholesterol Homeostatic Regulator SCAP-SREBP2 Integrates NLRP3 Inflammasome Activation and Cholesterol Biosynthetic Signaling in Macrophages. Immunity. 2018;49:842–56 e847. https://doi.org/10.1016/j.immuni.2018.08.021.
Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell. 2017;171:1057–71 e1011. https://doi.org/10.1016/j.cell.2017.09.029.
Xu WF, et al. Apaf-1 Pyroptosome Senses Mitochondrial Permeability Transition. Cell Metab. 2021;33:424. https://doi.org/10.1016/j.cmet.2020.11.018.
Liu QY, Zhang DY, Hu DY, Zhou XM, Zhou Y. The role of mitochondria in NLRP3 inflammasome activation. Mol Immunol. 2018;103:115–24. https://doi.org/10.1016/j.molimm.2018.09.010.
Wei PH, Yang F, Zheng Q, Tang, WX Li JJ. The Potential Role of the NLRP3 Inflammasome Activation as a Link Between Mitochondria ROS Generation and Neuroinflammation in Postoperative Cognitive Dysfunction. Front Cell Neurosci 2019;13:73. https://doi.org/10.3389/fncel.2019.00073.
Thorburn A. Crosstalk between autophagy and apoptosis: Mechanisms and therapeutic implications. Prog Mol Biol Transl. 2020;172:55–65. https://doi.org/10.1016/bs.pmbts.2020.04.023.
Zheng T, Zhao C, Zhao B, Liu H, Wang S, Wang L, et al. Impairment of the autophagy-lysosomal pathway and activation of pyroptosis in macular corneal dystrophy. Cell Death Discov. 2020;6:85. https://doi.org/10.1038/s41420-020-00320-z.
Shen C, et al. Phase separation drives RNA virus-induced activation of the NLRP6 inflammasome. Cell. 2021;184:5759. https://doi.org/10.1016/j.cell.2021.09.032.
Alberti S, Gladfelter A, Mittag T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell. 2019;176:419–34. https://doi.org/10.1016/j.cell.2018.12.035.
Strom AR, et al. Phase separation drives heterochromatin domain formation. Nature. 2017;547:241. https://doi.org/10.1038/nature22989.
Johnson AG, Wein T, Mayer ML, Duncan-Lowey B, Yirmiya E, Oppenheimer-Shaanan Y, et al. Bacterial gasdermins reveal an ancient mechanism of cell death. Science. 2022;375:221–5. https://doi.org/10.1126/science.abj8432.
Lieberman J, Wu H, Kagan JC. Gasdermin D activity in inflammation and host defense. Sci Immunol. 2019;4. https://doi.org/10.1126/sciimmunol.aav1447.
Jiang S, Zhou Z, Sun Y, Zhang T, Sun L. Coral gasdermin triggers pyroptosis. Sci Immunol. 2020;5. https://doi.org/10.1126/sciimmunol.abd2591.
Funding
This work was supported by a special program from the National Key Research and Development Program of China (2021YFA1101000 to LZ), the Chinese National Natural Science Funds (U20A201376, U20A20393, and 31925013 to LZ; 82041009, 31871405, and 32125016 to FZ; 92169122 and 31701232 to FX, 32025011 to FW, 31822031, 31970664, 31961160725 to JH and TL), the Science and Technology Plan Project of Suzhou (SYS2019020 to FX), the Jiangsu National Science Foundation (BK20180043 to FZ), Natural Science Foundation of Zhejiang Province (LZ19C070001 to FW), and the Key Project of University Natural Science Foundation of Jiangsu Province (19KJA550003 to FZ).
Author information
Authors and Affiliations
Contributions
XW, FX, XZ and YW conceived and drafted the manuscript, drew the figures and summarized the tables. XW, FX, XZ, YW, HY, TL, JH, FW, LZ and FZ discussed the concepts of the manuscript. LZ approved the version to be submitted. XW, FX, XZ and YW contributed equally to this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wei, X., Xie, F., Zhou, X. et al. Role of pyroptosis in inflammation and cancer. Cell Mol Immunol 19, 971–992 (2022). https://doi.org/10.1038/s41423-022-00905-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-022-00905-x
Keywords
This article is cited by
-
16S rRNA seq-identified Corynebacterium promotes pyroptosis to aggravate diabetic foot ulcer
BMC Infectious Diseases (2024)
-
Rab32 facilitates Schwann cell pyroptosis in rats following peripheral nerve injury by elevating ROS levels
Journal of Translational Medicine (2024)
-
Flaxseed oil fraction reverses cardiac remodeling at a molecular level: improves cardiac function, decreases apoptosis, and suppresses miRNA-29b and miRNA 1 gene expression
BMC Complementary Medicine and Therapies (2024)
-
Epigenetic regulation of diverse cell death modalities in cancer: a focus on pyroptosis, ferroptosis, cuproptosis, and disulfidptosis
Journal of Hematology & Oncology (2024)
-
Ferroptosis: a new hunter of hepatocellular carcinoma
Cell Death Discovery (2024)
