
Abstract
Members of the solute carrier organic anion transporting polypeptide (OATPs) family function as transporters for a large variety of amphipathic organic anions including endogenous metabolites and clinical drugs, such as bile salts, steroids, thyroid hormones, statins, antibiotics, antivirals, and anticancer drugs. OATP1B1 plays a vital role in transporting such substances into the liver for hepatic clearance. FDA and EMA recommend conducting in vitro testing of drug–drug interactions (DDIs) involving OATP1B1. However, the structure and working mechanism of OATPs still remains elusive. In this study, we determined cryo-EM structures of human OATP1B1 bound with representative endogenous metabolites (bilirubin and estrone-3-sulfate), a clinical drug (simeprevir), and a fluorescent indicator (2′,7′-dichlorofluorescein), in both outward- and inward-open states. These structures reveal major and minor substrate binding pockets and conformational changes during transport. In combination with mutagenesis studies and molecular dynamics simulations, our work comprehensively elucidates the transport mechanism of OATP1B1 and provides the structural basis for DDI predictions involving OATP1B1, which will greatly promote our understanding of OATPs.
Similar content being viewed by others


Introduction
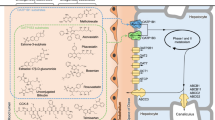
Encoded by genes of SLCO1 solute carrier family (formerly called SLC21), the members of the organic anion transporting polypeptides (OATPs) family transport a wide range of amphipathic organic anions across the plasma membrane, including endogenous and exogenous substances, such as bile salts, steroids, thyroid hormones, and clinical drugs.1,2 Eleven members of human OATPs have been identified and categorized into 6 subfamilies based on their amino acid composition, namely OATP1–6 (Supplementary information, Fig. S1). Among these OATPs, each member exhibits distinct but partially overlapping substrate specificity, which varies according to their specific tissue distribution.3 OATP1B1, encoded by SLCO1B1 and formerly known as SLC21A6, LST-1, OATP-C, or OATP2, is specifically expressed on the basolateral membrane of hepatocytes.4 OATP1B1 plays a vital role in mediating the uptake of endogenous compounds such as bile acids, bilirubin, steroid conjugates, and thyroid hormones, as well as a large variety of clinical drugs including statins, antibiotics, antivirals, and anticancer medications for hepatic clearance (Fig. 1a).5,6,7 Dysfunction of OATP1B1 can lead to statin-induced myopathy8 and hyperbilirubinemia,9 as exemplified by Rotor syndrome, a type of hereditary hyperbilirubinemia.10

a Schematic representation of the biological functions of OATP1B1. b Chemical structures of the four compounds used in this study. c The topology of OATP1B1. NTD and CTD are colored in gold and marine, respectively. EL3–4, EL5–6 and EL9–10 are colored in green, cyan and purple, respectively. Dashed lines show the flexible regions in our structures. d, e Side views of cryo-EM reconstructions (d) and cartoon representations (e) of human OATP1B1 in apo state and bound with bilirubin, E3S, simeprevir and DCF, colored in cyan, green, magenta, and yellow, respectively. OATP1B1 is shown with NTD and CTD colored in gold and marine, respectively.
Numerous studies have emphasized the central role of OATP1B1 in drug–drug interactions (DDIs).5,11,12,13 Food and Drug Administration and European Medicines Agency recommend conducting in vitro testing to evaluate potential OATP1B1 interactions.14,15 Many drugs with significant clinical implications act as substrates and/or inhibitors of OATP1B1.5 For instance, in clinical practice, combination therapy involving lipid-lowering statins16 (e.g., atorvastatin) and various drugs, such as antihypertensive medications, antibiotics, and antiviral agents, is commonly employed. However, certain medications, such as gemfibrozil, cyclosporine A, rifampicin, and ritonavir, can suppress the function of OATP1B1, leading to excessive systemic exposure of the affected drug, including statins.11 The elevation of statin blood levels can potentially lead to fatal myopathy and rhabdomyolysis.17,18
The energy coupling transport mechanism of OATPs is still not fully understood. The transport of OATPs is independent of Na+, K+, Cl− gradients, membrane potential and ATP levels.19,20,21 OATPs are generally believed to act as organic anion exchangers, coupled with the efflux of anions, such as HCO3− and glutathione (GSH), but direct evidence was only provided in rat Oatp1a1.22,23 However, transport of OATP1B1/OATP1B3 was most probably demonstrated to occur through bidirectional facilitated diffusion, which was unaffected by GSH.21 Moreover, evidence has been presented for pH-dependence in transport stimulation for most OATPs with the exception of OATP1C1.24,25
To elucidate the transport mechanism of OATP1B1, we determined the structures of OATP1B1 bound with representative endogenous substrates (bilirubin and estrone-3-sulfate (E3S)), a clinical drug (simeprevir), as well as a fluorescent indicator (2′,7′-dichlorofluorescein (DCF)). These structures reveal unexpected major and minor substrate binding pockets and conformational changes between outward-open and inward-open states. Together with mutagenesis functional studies and molecular dynamics (MD) simulations, our work elucidates the transport mechanism of OATP1B1, and provides the structural basis for DDI predictions involving OATP1B1, which will promote the understanding of OATPs.
Results
Structure determination
Human OATP1B1 was expressed in Expi293F cells and purified using tandem affinity and size exclusion chromatography. To investigate the transporting mechanism of OATP1B1, we supplemented the protein solution with a saturating concentration of endogenous substrates, bilirubin and E3S, and a clinical drug, simeprevir, during cryo-sample preparation. To better explain the binding mode of the fluorescent probe, DCF, which is employed in the following transport assays, we also prepared the cryo-sample of OATP1B1–DCF (Fig. 1b). The purified OATP1B1 protein alone or supplemented with ligands was then subjected to cryo-electron microscopy (cryo-EM) single particle analysis. For simplicity, OATP1B1 bound with each ligand is hereafter referred to OATP1B1–B (bilirubin), OATP1B1–E (E3S), OATP1B1–S (simeprevir) and OATP1B1–D (DCF).
We determined the structures of OATP1B1-apo/B/S/D in outward-open state at resolutions ranging from 2.9 Å to 3.7 Å (Fig. 1d). The particle dataset of OATP1B1–E contained a mixture of outward-open, inward-open, and occluded states, namely OATP1B1-Eou, OATP1B1–Ein and OATP1B1–Eoc, respectively. After extensive trials, we successfully obtained the inward-open structure of OATP1B1–Ein at a resolution of 3.2 Å (Fig. 1d; Supplementary information, Fig S5), with OATP1B1–Eou and OATP1B1–Eoc resolved at moderate to low resolutions. For details of cryo-EM data acquisition and processing, please refer to Materials and methods (Supplementary information, Figs. S2–S6 and Table S1). The ligand model building was based on the EM densities in a combination of ligand-free docking and MD simulations (Supplementary information, Fig. S7 and Table S2). The relatively small and rigid substances, E3S and DCF, revealed stable binding with OATP1B1, while larger ligands, bilirubin and simeprevir, exhibited more flexibility during MD simulations (Supplementary information, Videos S1–S4).
Overall architecture
The overall architecture of OATP1B1 is composed of the transmembrane domain (TMD) and extracellular loops (ELs) with both N- and C-terminal ends situated in the cytoplasm. The TMD follows the canonical MFS fold26 and consists of 12 transmembrane helices (TMs) organized into an N-terminal domain (NTD) and a C-terminal domain (CTD), exhibiting a 2-fold pseudo symmetry around an axis perpendicular to the membrane plane (Fig. 1c). The NTD and CTD are connected by an intracellular helix (ICH) (Fig. 2a, b).

a Overall architecture of outward-open OATP1B1-apo. NTD and CTD are colored in gold and marine, respectively. b Overall architecture of inward-open OATP1B1–Ein. c Multiple disulfide bonds formed by conserved cysteines within EL9–10 are shown as yellow sticks. A fragment of EL3–4 termed as N-lariat, is attached to EL9–10 via a disulfide bond. d Electropositive residues along the translocation path of outward-open OATP1B1-apo. e Slab view showing the conservation level of OATP1B1-apo using ConSurf. f Sliced view of the surface electrostatic potential of OATP1B1-apo. g Top view of key residues in the central cavity of outward-open OATP1B1-apo, forming electropositive, electronegative, and hydrophobic parts. Residues are shown as stick and colored based on ConSurf analysis.
The largest extracellular loop, EL9–10, features several β sheets and pairs of disulfide bonds, known as the Kazal domain27 (Fig. 2c). The Kazal domain is an evolutionarily conserved protein domain, usually indicative of serine protease inhibitors,28 but its function in OATPs still remains elusive. Within EL9–10, two glycosylation sites (N503 and N516) were observed to be covalently linked to N-acetylglucosamine sugar moieties (Fig. 2a, b). In both outward- and inward-open states of OATP1B1, EL3–4 associates with the Kazal domain through a conserved disulfide bond C142–C463, designated as N-lariat (Fig. 2c). MD simulations of the outward-open states with EL3–4 disconnected resulted in abnormally wider opening (data not shown), indicating that the N-lariat potentially regulates the maximum opening extent and acts as an extracellular halter connecting the extracellular halves.
Substrate entrance and central cavity environment
The translocation path is lined with multiple basic residues (Fig. 2d). An extracellular electropositive opening, enclosed by EL5–6, the Kazal domain and N-terminus of TM8, potentially functions as an entrance for organic anion substrates to access the central cavity (Supplementary information, Fig. S8a, b). Most of the positively-charged residues are distributed at this indicated entrance rather than in the cavity itself, implying that selectivity for organic anions might be achieved by partially filtering out cations at the cavity entrance. Besides, in the outward-open OATP1B1-apo/B/S/D, lipid-like EM densities were trapped in the V-shaped lateral opening formed by the two halves between TM2 and TM11 (Supplementary information, Fig. S8c), which might serve as another potential access for hydrophobic substrates, as described in OCT3.29
The interface between NTD and CTD of OATP1B1 forms a conserved cavity in which substrates bind (Fig. 2e). The central cavity exhibits evident features of both hydrophobicity and electro-positivity, wherein TMs 5, 7, 8, and 10 contribute to the hydrophobic side while TMs 1, 2, 4, and 11 form the charged side partly localized oppositely to the hydrophobic side (Figs. 2f, g, and 3c). Positively-charged residues, K41/K49/R580, along with modestly conserved aromatic residues, i.e., Y352, F356, and F386, create an ideal environment for selecting and translocating amphipathic organic anion substrates within the central cavity. Of note, all three basic residues participate in generating charge pairs, K41–E185, K49–D70 and highly conserved R580–E74 (Figs. 2e, g and 3c, d).

a Superimposition of outward-open structures of OATP1B1-apo/B/S/D. b Bilirubin and simeprevir occupy the major pocket and DCF occupies the minor pocket, which is superimposed and presented in the central cavity of OATP1B1–D. c Top view of outward-open OATP1B1-apo showing key residues in the major pocket and minor pocket. Electropositive residues, electronegative residues, hydrophobic residues, and hydrophilic residues are colored in magenta, yellow, gray and cyan, respectively. d Sequence alignment of key residues in the central cavity of OATP1B1 among SLCO family transporters. e–g Local ligand density representations and extracellular views for substrate binding sites of OATP1B1–B (e), OATP1B1–S (f) and OATP1B1–D (g) in outward-open state.
Substrate binding sites in outward-open state
Bilirubin is a representative endogenous substrate of OATP1B1, which is a kind of biologically toxic metabolite produced as a byproduct of heme degradation in red blood cells. Impaired elimination of bilirubin can lead to hyperbilirubinemia, which is closely associated with multiple disorders and reflects abnormal liver function.30 As for simeprevir, a direct-acting antiviral drug for the treatment of hepatitis C virus infection, it not only acts as a substrate for OATP1B1 but also functions as an inhibitor.31,32 Simeprevir can lead to clinical DDIs, as exemplified by an approximately 2-fold increase in plasma when administered concomitantly with atorvastatin or rosuvastatin.32 DCF is a fluorescein derivative and has been previously identified to be a good substrate and probe to assess the transport activity of OATP1B1.33,34,35,36,37
Structures of OATP1B1 bound with bilirubin, simeprevir and DCF, adopt similar outward-open conformations as that of OATP1B1-apo (Fig. 3a). The root mean square deviation between OATP1B1-apo and each of the ligand-bound states is 0.51 Å (apo vs bilirubin), 0.46 Å (apo vs simeprevir), and 0.89 Å (apo vs DCF), respectively. Typically, MFS transporters engage TMs 1, 4, 7, 10 (known as A helices) to bind ligands.26,38 However, OATP1B1 also employs B helices to bind substances, i.e., with TMs 5, 7, 8, and 11 to bind with bilirubin, TMs 1, 5, 7, 8, and 11 to bind with simeprevir, and TMs 7, 8, and 10 to bind with DCF.
Based on the ligand binding sites, two distinct pockets were observed within the translocation pathway. Bilirubin and simeprevir occupy the major pocket located in the central cavity, while DCF binds to the minor pocket extending into the NTD (Fig. 3b). The binding of these ligands primarily relies on interactions with hydrophobic residues opposite to the charged side in the central cavity (Fig. 3c, e–g; Supplementary information, Fig. S7). B/S/D are all closely attached to F356 and Y352 around the middle of TM7, further clamped by F386 from TM8, acting as a core binding hub. The EM densities of ligands of B/S/D were observed to clearly adhere to F356 (Fig. 3e–g). However, no obviously direct associations were observed between ligands and the positively-charged residues in the central cavity.
The multiple residues interacting with bilirubin are mostly derived from the CTD, enabling bilirubin to closely associate with the hydrophobic side in the central cavity. Comparably, simeprevir engages in more interactions with residues from both the NTD and CTD, wherein F356 forms hydrophobic packing with the quinoline group of simeprevir. In vitro studies have shown that simeprevir can inhibit the transport of multiple substrates of OATP1B1.31,39 The characteristic of larger spatial occupation and more complex interactions that simeprevir presented, is in accord with those of previously characterized OATP1B1 inhibitors manifesting significantly large molecular weight and high lipophilicity.40,41
Different from the binding mode of bilirubin and simeprevir, the main body of DCF extends towards a minor hydrophilic pocket within the interior of the CTD by forming hydrogen-bond interactions with N544 and S548. Meanwhile, residues from the core binding hub, Y352, and F356 form hydrophobic packing with the xanthen and benzofuran groups of DCF, respectively (Fig. 3g; Supplementary information, Fig. S7d). The co-existence of major and minor pockets is consistent with the assumption that multiple substrate binding sites are in OATP1B1.13,42
We further performed DCF uptake inhibition assay with mutagenesis to validate the ligand poses built in the structures of OATP1B1–B/S. Consistent with the poses that we built, K49A, F360A, and I383A exhibited substrate-dependent inhibition despite slight variations in their IC50 values for bilirubin and simeprevir, respectively (Supplementary information, Fig. S9).
E3S-bound OATP1B1 adopts an inward-facing conformation
Compared to lipophilic estrone and estradiol, the estrogen ester and conjugate, E3S, is unable to permeate through cell membranes and is transported mainly via OATPs, which has been widely used as a prototypical model substrate of OATP1B1.43,44,45 Inhibitors of OATPs are of interest in treating estrogen-associated cancers.46 In our study, the cryo-EM structures of OATP1B1–E unexpectedly revealed three different conformations: inward-open state with high resolution, outward-open and occluded states with moderate to low resolutions (Fig. 4a). Densities of E3S were found in all three conformations at similar sites (Supplementary information, Fig. S10). The EM density of E3S in OATP1B1–Ein is unambiguously resolved, allowing us to discern the pose of the E3S with high confidence (Fig. 4d; Supplementary information, Fig. S7b).

a Three different conformations were captured when OATP1B1 is supplemented with E3S. Shown here are cryo-EM reconstructions (upper) and representative 2D class averages (lower) of OATP1B1 bound to E3S that correspond to inward-open, outward-open, and occluded conformations, respectively. 2D class average boxed in red shows the representative view in each conformation. Owing to the moderate to low resolutions of the outward-open and occluded conformations, no structural model was assigned. b Cartoon representation of OATP1B1–Ein showing the binding site of E3S in the overall structure. c Slab view of the electrostatic surface of E3S binding site, wherein E3S binds in the minor pocket. d Local EM density map of E3S in OATP1B1–Ein contoured at 5 σ. e Expanded view of E3S binding site in OATP1B1–Ein. f Inhibition of OATP1B1 transport by E3S (mean ± SEM, n = 6). n = 6 represents six biologically independent experiments for each cell line.
The inward-open structure of OATP1B1–Ein opens its central substrate cavity towards the cytoplasm (Fig. 4b, c). Like DCF, E3S also binds at the minor pocket, with its sulfate group extending into the interior of the CTD (Fig. 4b, c). F356 forms hydrophobic packing with the estrone group of E3S, together with Y352 and F386, enclosing the hydrophobic body of E3S (Fig. 4e; Supplementary information, Fig. S7b). Additionally, residues from the minor pocket, Y625, Y422, Q541 and N544 form hydrogen-bond interactions with the sulfate group of E3S. Mutagenesis of Q541A exhibited a significantly varied IC50 value for E3S, supporting the essential role of Q541 for the binding of E3S (Fig. 4f). Accordingly, Q541A led to an evident decrease of DCF uptake activity (Fig. 5h; Supplementary information, Fig. S12). Of particular note, like DCF, the residues involved in the binding of E3S are basically derived from the CTD.

a Side view of the structural alignment of the outward-open and inward-open structures of OATP1B1 relative to the membrane norm. b Extracellular view showing key residues and three pairs of salt bridges in the central cavity. Four compounds in this study together occupy both the major pocket and minor pocket. Overall structures are aligned relative to membrane norm. c, d Extracellular views of four compounds with overall structures aligned by CTD (c) and four individual compounds (d), showing three key residues in substrate binding sites, Y352, F356 and F386. e The distances between center of mass of ligands and sidechains of key residues forming the central binding hub, Y352, F356 and F386 in 500-ns MD simulations, showing the stable interactions between the three residues and ligands. f The distances between sidechain of residues in each pair of salt bridge in 500-ns MD simulations of all the structures in our study, showing the stable interactions within salt bridges. g Schematic summary of key residues involved in the binding of four compounds in our study, showing orthosteric and opportunistic sites. h DCF uptake activity assay showing reduced activity of OATP1B1 mutants regarding residues within major and minor pockets. The activity was normalized to the wild-type (WT) transporter (mean ± SEM, n = 6). Control indicates empty cell control. Surface expression levels are shown in western blotting for control, WT and mutants. i Liposome assay of [3H]-E3S uptake showing reduced activity of OATP1B1 mutants regarding three key residues forming the central binding hub. The activity was normalized to the WT transporter (mean ± SEM, n = 3). Control indicates empty liposome control.
Structural determinants for broad substrate specificity
The ligands, bilirubin and simeprevir, occupy the central cavity in the major pocket in the outward-open state, while DCF and E3S are located at the minor pocket in the outward- and inward-open conformations, respectively (Fig. 5a, b). Superposition of OATP1B1-apo/B/S/D/E structures reveals rearrangements of Y352 and F356 from TM7 upon binding of different ligands. By undergoing a vertical rotation of the side chain of Y352, smaller substrates, i.e., DCF/E3S, can be accommodated in the minor pocket towards the interior of the CTD (Fig. 5c, d).
Each of the ligands (B/S/E/D) exhibits distinct but partially overlapping binding sites with OATP1B1. The hydrophobic mass centers of these substrates are all associated at the core binding sites, composed of three aromatic residues, Y352, F356 and F386 (Fig. 5b–d). [3H]-E3S liposome assay and cell-based DCF uptake assay confirmed the essential roles of these binding hub residues in transport, especially Y352 and F356 (Fig. 5h, i). The stable binding of ligands to Y352, F356 and F386 during 500-ns MD simulations in three replicates further indicates the critical role of these residues in substrate binding (Fig. 5e; Supplementary information, Fig. S11a–d and Videos S1–S4). We term the binding hub formed by the three aromatic residues as the orthosteric site. Additionally, the central cavity possesses a significant spatial volume, allowing large compounds to bind effectively within the primary pocket. Each of the ligands also possesses their own special binding sites in the major or minor pocket, which we term as opportunistic sites, as recently described in OCT147 (Fig. 5g).
OATP1B1-interacting substrates and drugs are structurally diverse with only nebulous similarities, i.e., greater hydrophobicity, higher complexity, and more ringed structures.48 The orthosteric site of OATP1B1 might play a pivotal role in hydrophobic packing with the fairly hydrophobic compounds. The combination of the orthosteric site and the opportunistic site in both major and minor pockets, might collectively contribute to the substrate poly-specificity of OATP1B1. Similarly, the corresponding residues in the orthosteric site of OATP1B3 are F352 and F356, potentially leading to the similar function and substrate specificity between OATP1B1 and OATP1B3.1 On the contrary, in other OATPs with relatively distinctive substrate range and tissue distribution,3 the three aromatic residues are less conserved and mainly substituted by smaller hydrophobic residues (Fig. 3d; Supplementary information, Fig. S1), indicating that the residues in the orthosteric site may determine the substrate specificity of different OATPs.
Of note, despite OATP1B1 substrates being primarily organic anions, no apparent interactions were observed between ligands and the basic residues, K41/K49/K580, within the cavity of OATP1B1–B/S/E/D. As observed in our structures, MD simulations confirmed the presence of three charge pairs: K41–E185, K49–D70 and K580–E74 in the cavity (Fig. 5f), suggesting that the positively-charged residues might contribute to modulating the electrostatics of the cavity rather than direct interacting with anion substrates. The transport activities of K41A, K49A, and R580A exhibit evident reduction despite decreased surface expression, especially R580, suggesting their vital roles (Fig. 5h). Consistently, previous studies have shown that E74A/Q and R580K led to decreased transport activity of OATP1B1.49,50
Rocker-switch alternating access of transport
The intra- and extracellular gates are formed in the outward- and inward-open state, respectively. In outward-open OATP1B1-apo/B/S/D, the intracellular gate is closed mainly by TM4 and TM10. A conserved salt bridge is formed by the interaction between D198 and K568 (Fig. 6a). Additionally, multiple hydrophobic residues act as barriers, blocking the intracellular central pathway. Particularly, the ICH is attached to the C-terminus of TM2 through hydrophobic interactions between F325 and aromatic residues, F83, Y86 and F87. This attachment potentially contributes to the sealing and maintenance of the intracellular gate in the outward state (Fig. 6a). MD simulations support the stable existence of both D198–K568 salt bridge and Y86–F325 hydrophobic interaction in outward-open OATP1B1-apo/B/S/D (Fig. 6b; Supplementary information, Fig. S11e, f). Mutagenesis of D198A significantly decreased the [3H]-E3S uptake of OATP1B1, indicating its vital role during transport (Fig. 6g). Similarly, mutations of Y86A, D198A and K568A almost completely abolished the transport activity of DCF, as determined by confocal microscopy (Supplementary information, Fig. S12).

a The intracellular gate in the outward-open state. b Distances of residues involved in intracellular gate in outward-open OATP1B1-apo/B/S/D in MD simulations. Upper panel: the distances between center of mass of D198 sidechain carboxyl group and K568 sidechain N atom. Lower panel: the distances between center of mass of Y86 and F325 sidechain. c An extracellular gate and hydrophobic constriction in inward-open OATP1B1–Ein. Residues forming the hydrophobic constriction are shown in gray sphere. The extracellular gate is magnified in the right panel. d Dissociation of latch-like ICH and TM2 during transition from outward-open to inward-open state relative to membrane norm. e Structural comparison between inward-open and outward-open structures relative to CTD and NTD separately. f Rotation of representative TM segments between inward-open and outward-open structures relative to the membrane norm. g Liposome assay of [3H]-E3S uptake showing reduced activity of OATP1B1 mutants regarding key residues in intra- and extracellular gates. The activity was normalized to the WT transporter (mean ± SEM, n = 3). Control indicates empty liposome control. h Rocker-switch alternating access model of OATP1B1 during transport. Conformations of OATP1B1 that are achieved in this study are shown. The red circle represents substrate.
In the inward-open OATP1B1–Ein, accompanying the disappearance of the intracellular gate, an extracellular gate is generated by electrostatic interactions between NTD and CTD (Fig. 6c). Two charge pairs, R57–E364 and E56–K361, originating from TM1 and TM7, respectively, are highly conserved. In addition, electrostatic interactions of R614–E60, E471–R253 and K497–S243 further contribute to the closure of extracellular gate. Mutagenesis of one highly conserved salt bridge, R57A/E364A, resulted in significant decrease in the uptake of [3H]-E3S or DCF, suggesting their essential roles during transport (Fig. 6g; Supplementary information, Fig. S12).
Interestingly, DCF and E3S both bind at the minor pocket, while they arrest OATP1B1 in outward-open and inward-open states, respectively, indicating a mechanism of substrate-induced conformational changes. Connected with TM7 like an elbow, ICH closely associates with TM2 like a latch in the outward-open state (Fig. 6a, d). Upon binding of E3S, the fine-tuning of F356 in the orthosteric site potentially triggers the synergic movement of TM7 and the latch-like ICH, followed by the dissociations of TM2b and ICH and conformational alteration of TM2b from a bent to a straight form, ultimately leading to the opening of intracellular gate and sequentially global conformational changes (Fig. 6d; Supplementary information, Fig. S13). Additionally, the local adjustment of F386 and L378 upon binding of E3S causes a slightly dilated movement of TM8, probably disrupting the hydrophobic interactions of F391–I211 and Y395–L207 (Supplementary information, Fig. S13d). A hydrophobic constriction network was identified above E3S, wherein the estrone group of E3S is engaged in hydrophobic interactions with F356, I378, F224, and I46, potentially facilitating the conformational transition from the outward-open to the inward-open state while DCF induces no such interactions (Fig. 6c; Supplementary information, Fig. S13c).
When we compare the inward- and outward-open states with respect to NTD or CTD separately, the overall structure of each domain remains nearly unchanged (Fig. 6e). In general, substrate translocation by OATP1B1 is achieved through rigid-body rotations of the NTD and CTD that expose the central substrate cavity alternatively to extracellular or cytosolic side of the membrane, conforming to the rocker-switch model of MFS transporters (Fig. 6e, f, h; Supplementary information, Video S5).26,51 It is worth noting that the highly conserved charge pair R580–E74, located around the middle of TMD and originating from two halves, might play a role of acting as a central pivot during conformational changes (Fig. 5f; Supplementary information, Fig. S11g).
Discussion
In this study, we successfully resolved the structures of OATP1B1 in different states, including substrate-free outward-open state, substrate-bound outward-open state, substrate-bound inward-open state, and substrate-bound occluded state at low resolution, adopting rocker-switch model during transport (Fig. 6h). Nevertheless, despite the states we captured, further intermediate states, i.e., outward-occluded or inward-occluded, might exist,51 which needs further investigation.
The energy coupling transport mechanism of OATPs is not fully understood yet. Evidence for pH-dependence in transport stimulation has been shown in most OATPs except for OATP1C1.24,25 The pH-dependence of OATP1B1 seems substrate-dependent, given that the transport of E3S was not affected by pH.21,24 A conserved histidine among OATPs, located at the EL5–6, is replaced by a glutamine in OATP1C1, which was found to diminish its pH-sensitivity of transport.24 The structure of OATP1B1–B/S/E/D in our study revealed that this conserved H115 directly interacts with D236, which might enhance the rigid-body movement of EL5–6 with the NTD transmembrane bundle, partially explaining the potential facilitative function of H115 for the pH-sensitive transport (Supplementary information, Fig. S14). Moreover, considering a series of extracellular charge pairs in the inward-open state of OATP1B1, the pH-dependence might facilitate the opening of the extracellular gate during transport. However, previous transport assays investigating the energetics of OATPs have predominantly employed Xenopus laevis oocytes or human cell lines. A significant issue arises from the lack of in vitro liposome assays to validate the energetics. The in vitro [3H]-E3S liposome assay that we performed indicated that the chemical concentration of substrate can probably act as driving force for the transport of OATP1B1 (Figs. 5i, 6g), in accord with the previously proposed bidirectional facilitated diffusion of OATP1B1/OATP1B3.21 The coupling energy mechanism might not be identical in OATPs and further research is needed to fully understand the driving forces and regulatory factors involved.
The cellular uptake of a broad range of structurally diverse organic anions and cations mainly depends on two SLC transporter families, OATPs and SLC22A superfamilies which contain the organic cation transporters (OCTs) and the organic anion transporters (OATs).52 In general, OCTs transport cations, OATPs transport large and fairly hydrophobic organic anions, and OATs transport the smaller and more hydrophilic organic anions. According to the recently published structures of OCT1–329,47 and rat Oat1,53 the substrate binding pocket is highly electronegative or electropositive, creating a selective pathway for the translocation of organic cations or organic anions, respectively.29,47 Overlapping and distinct binding site, or termed as the orthosteric site and the opportunistic sites, were also observed in OCTs and rat Oat1 when bound to different ligands, consistent with their poly-specific nature of substrate recognition similar to OATP1B1. The central pockets of OATs and OCTs are relatively smaller compared to OATP1B1, explaining why OATPs tend to transport larger substrates while OCTs and OATs mostly transport smaller compounds (Supplementary information, Fig. S15).
In summary, our work presents the major and minor substrate binding sites of OATP1B1 and conformational changes during transport, elucidates the broad substrate specificity and transport mechanism of OATPs and provides crucial structural reference for guiding combination therapy strategies and future drug design to prevent DDIs involving OATPs.
Materials and methods
Protein expression and purification
cDNA of human OATP1B1 (UniProt ID: Q9Y6L6) gene was synthesized and cloned into a pCAG vector containing a C-terminal tandem His and Flag affinity tags. For the expression of OATP1B1 protein, Expi293F cells were used and cultured in SMM 293T-II medium at 37 °C under 5% CO2 at 130 rpm in a Multitron-Pro shaker. When the cell density reached approximately 2.0 × 106 cells/mL, Expi293F cells were transiently transfected with the plasmid and polyethylenimines (PEIs) (Polysciences). The total amount of plasmids for transfection of one liter of cell culture is 2 mg and plasmids were pre-mixed with PEIs in 40 mL fresh medium in a 1:2 (w/w) ratio for 15 min before adding the mixture into cell culture.
48 h after transfection, cells were harvested by centrifugation at 2000× g and resuspended in the buffer containing 25 mM HEPES pH 7.4, 150 mM NaCl, 1.3 μg/mL aprotinin (Amresco), 0.7 μg/mL pepstatin (Amresco), and 5 μg/mL leupeptin (Amresco). The membrane fraction was extracted with 2% (w/v) n-Dodecyl-β-D-Maltopyranoside (DDM, Anatrace) at 4 °C for 2 h. After extraction, the cell lysate underwent centrifugation at 12,000× g for 1 h (JA 25.50, Beckman Coulter) and supernatant was loaded to anti-Flag M2 affinity resin (GenScript), washed in buffer containing 25 mM HEPES pH 7.4, 150 mM NaCl, 0.01% LMNG (w/v), and eluted with buffer containing 25 mM HEPES pH 7.4, 150 mM NaCl, 0.01% LMNG (w/v) and 0.4 mg/mL Flag peptide. The protein sample after elution was concentrated and further purified by size-exclusion chromatography on a Superose 6 10/300 GL column (GE Healthcare) in the above wash buffer. The protein peak fraction was determined by SDS-PAGE and collected for cryo-EM analysis.
Cryo-EM sample preparation and data acquisition
The final concentration of protein samples for cryo-EM sample preparation is ∼5–10 mg/mL. For OATP1B1–B/E/S/D, compounds of bilirubin (TargetMol), estrone sulfate sodium (MedChemExpress), simeprevir (MedChemExpress), and 2’,7’-dichlorofluorescein (Sigma-Aldrich) were added to the protein solution at a saturating concentration of 190 μM, 2.6 mM, 440 μM and 1.2 mM, individually. 4 μL of protein sample with compounds was applied to a glow-discharged Quantifoil R1.2/1.3 300-mesh gold holey carbon grid, blotted for 4–5 s and flash-frozen in liquid ethane cooled by liquid nitrogen with Vitrobot (Mark IV, Thermo Fisher Scientific).
For data acquisition of OATP1B1-apo/B/S, micrographs were acquired on a Titan Krios microscope (Thermo Fisher Scientific) operated at 300 kV with a K3 Summit direct electron detector (Gatan) and a GIF Quantum energy filter. SerialEM54 software was used for automated data collection following standard procedures. A nominal magnification of 105,000× was used for imaging, yielding a pixel size of 0.832 Å on images. The defocus range was set from −1.0 μm to −1.8 μm. Each micrograph was dose-fractionated to 40 frames in a total dose of ∼50 e–/Å2, with total collection of 12,329 and 14,613 and 8266 movie stacks, respectively, for OATP1B1-apo/B/S.
For data acquisition of OATP1B1–E/D, micrographs were acquired on a Titan Krios microscope (Thermo Fisher Scientific) operated at 300 kV with a Falcon 4i Direct Electron Detector (Thermo Fisher Scientific) and a Selectris X energy filter. EPU was used for automated data collection following standard procedures. A nominal magnification of 130,000× was used for imaging, yielding a pixel size of 0.932 Å on images. The defocus range was set from –0.9 μm to –1.5 μm. Each micrograph was recorded in a total dose of ∼50 e–/Å2, with total collection of 13,425 and 9687 movie stacks, respectively for OATP1B1–E/D.
Cryo-EM data processing
The data processing pipelines are shown in Supplementary information, Figs. S2–S6. Motion correction and CTF estimations were performed using cryoSPARC v3.3.2.55
For the data processing of OATP1B1-apo, after several rounds of 2D classification and Ab-initio reconstruction using cryoSPARC, an initial model with clear TMs was obtained. Heterogeneous Refinement, Local refinement and Local CTF refinement yielded an outward-open OATP1B1-apo at the resolution of 3.53 Å. During the data processing, “seed”-facilitated 3D classifications using cryoSPARC and skip-align 3D classification via RELION v3.156 contributed to resolution improvement (Supplementary information, Fig. S2 and Table S1).
The initial models generated from good 2D classes in datasets of OATP1B1–B/S/D were very similar to that of OATP1B1-apo, all in outward-open conformations. Therefore, the cryo-EM structure of OATP1B1-apo was used as initial reference. To eliminate reference bias, initial models were re-generated from good particles using Ab-initio in cryoSPARC in the following data processing. Heterogeneous Refinement, Local refinement and Local CTF refinement were applied. 3D classification improved the density for ligands. Outward-open structures of OATP1B1–B/S/D were achieved at the resolution of 3.73 Å, 3.73 Å, and 2.92 Å, respectively (Supplementary information, Figs. S3, S4, S6 and Table S1).
During the data processing for OATP1B1-E, distinct conformations were observed in the generated initial models from good 2D classes. Data processing using the OATP1B1-apo as the initial reference only resulted in an outward-open OATP1B1–E at the resolution of 4.4 Å. Except for outward-open conformation, the occluded and inward-open conformations of OATP1B1–E were also captured at low resolution. Alternant initial model generation and “seed”-facilitated 3D classifications contributed to reconstructing the inward-open OATP1B1–Ein at the resolution of 3.19 Å (Supplementary information, Fig. S5 and Table S1).
Model building
We first built the atomic model of OATP1B1-apo based on the predicted model generated by SWISS-MODEL57 using Protein Data Bank ID: 7EEB as a starting template. Atomic model building based on the density map of OATP1B1-apo was performed in Coot,58 refined in real space using Phenix.real_space_refine.59 Validation tools in Phenix59 and Molprobity60 were used to guide iterative rounds of model adjustment in Coot and refinement in Phenix. Since the conformations of OATP1B1–B/S/D were very similar to that of OATP1B1-apo, atomic model of OATP1B1-apo was used as initial model for model building of OATP1B1–B/S/D, followed by manual adjustment to fit into density maps. For the model building of OATP1B1–Ein, AlphaFold61,62 predicted model with an inward-open conformation conforms to the density map well, with subsequent modifications in accord with the density map.
We performed molecular free docking and estimated the binding free energies using the molecular mechanics-generalized Born surface area (MM/GBSA) with the Schrödinger Maestro program to determine the ligand binding pose. Then we built the proposed model of the ligands based on the EM density map and surrounding environment using Coot, followed by adjustment with the assistance of Phenix.real_space_refine and MD simulations. All figures were prepared in PyMoL (www.pymol.org) or UCSF ChimeraX.63
MD simulations
MD simulations were conducted for both OATP1B1-apo protein and four protein–ligand complexes, OATP1B1–B/S/E/D. The initial proteins include residues 25–651 for outward-open conformation and residues 26–652 for inward-open conformation. Charged residues were protonated assuming pH 7.4, and the N- and C-termini of proteins were capped with acetyl and N-methyl groups, respectively. To build the system, proteins or protein–ligand complex was firstly embedded in a POPC bilayer (~350 molecules) and then solvated in a water box (128 × 128 × 125 Å3). Finally, 150 mM NaCl was added to neutralize the system. Details of system composition are listed in Supplementary information, Table S2.
MD simulations were performed with GROMACS 2021.4.64 CHARMM36 force field was used for protein, POPC lipid and ions,65,66 and TIP3P model67 was used for water. The force fields of four ligands (bilirubin, simeprevir, E3S, and DCF) were generated by CgenFF program.68,69 After energy minimization by steepest descent algorithm, the systems were simulated in NVT ensemble to relax lipid tail, lipid head and solvent (water and ions) in steps for 1.5 ns. Then, the systems were further relaxed in NPT ensemble. Constraints on (1) lipids and solvent, (2) ligand (if any), (3) protein sidechain and (4) protein Cα atoms were gradually released each for 2 ns. After pre-equilibrations, 500 ns production simulations were run without any constraint. Periodic boundary condition was applied, and the temperature and pressure were kept at 310 K and 1 atm by Nose-Hoover thermostat70,71 and Parrinello-Rahman barostat,72 respectively. Long-range electrostatic interactions were described using the particle mesh Ewald (PME) method73 with a cutoff of 12 Å. Van der Waals interactions were smoothly switched to zero from 10 Å to 12 Å. All bonds involving hydrogen atoms were constrained by the SHAKE algorithm74 to allow a time step of 2 fs. Each system was run for three replicates with different initial velocities and the total simulation time was 7.5 µs.
OATP1B1 transport activity assay using DCF
The uptake activity of OATP1B1 WT and mutants was measured by DCF influx using HEK293T cells. The transport activity of OATP1B1 was determined by DCF uptake of cells overexpressing OATP1B1 proteins. Cells cultured in a 6-well plate were used for transport measurement. To each well, HEK293T cells were transfected with pCAG-OATP1B1-His-Flag constructs, using Lipofectamine 2000 reagent (Invitrogen) following the manufacturer’s instructions and the total amount of DNA per transfection was 2.5 μg. Cells transfected with PBS instead of plasmids were used as blank control. After 48 h, DMEM medium in the dish was aspirated out and cells were washed with pre-warmed Phosphate-Buffered Saline (PBS) for three times. Then, 1 mL pre-warmed uptake buffer (142 mM NaCl, 5 mM KCl, 1 mM KH2PO4, 1.2 mM MgSO4, 1.5 mM CaCl2, 5 mM D-glucose, 12.5 mM HEPES pH 7.4) with 5 μM DCF dissolved in was added to each well. After incubation for 5 min at 37 °C, uptake buffer with DCF was aspirated out and cells were washed with pre-cooling PBS for three times. Then, 500 μL uptake buffer with 2% Triton X-100 was added to each well and incubation lasted 1 h at room temperature. When we performed 96-well plate assays using a BioTek Synergy2 Microplate Reader, 100 μL cell lysate was added to each well to detect the fluorescence intensity of DCF (excitation/emission wavelength at 485 nm/528 nm). All statistical analyses were performed with one-way ANOVA followed by Dunnett’s multiple comparisons test comparing WT with mutants using GraphPad Prism v9.3.1. DCF transport assays were repeated for at least three times.
We also conducted transport activity assay using confocal microscopy with the combination of DCF and mKate.75 HEK293T cells were transfected with pCAG-mKate-OATP1B1-His-Flag constructs, using PEIs in a 1:2 (w/w) ratio. The total amount of DNA per transfection was 2 μg and added in a 20-mm diameter dish with 1.0 × 106 cells/mL density. Procedure of DCF uptake assay is similar to the one described above. After DCF uptake, instead of cell lysis, cells were observed under confocal microscope. OATP1B1-expressing HEK293T cells were identified by detecting mKate fluorescence using a monochromator at 543 nm excitation while the influx amount of DCF was measured by detecting DCF fluorescence using a monochromator at 488 nm excitation and the emitted light measurement ranging from 592 nm to 650 nm, 493 nm to 530 nm, respectively. Fluorescence intensity was monitored by a Leica SP8 confocal microscope. After image acquisition, quantitative image analysis was performed with ImageJ.76 All statistical analyses were performed with one-way ANOVA followed by Dunnett’s multiple comparisons test comparing WT with mutants using GraphPad Prism v9.3.1. DCF transport assays were repeated for at least three times.
Cell surface biotinylation and immunoblot analysis
HEK293T cells were cultured in a 6-well plate transiently transfected with plasmids of OATP1B1 WT and mutants. After 48 h, cells were washed twice with 2 mL of ice-cold PBS and then treated for 1 h at room temperature with 1 mL of sulfo-N-hydroxysuccini- mide-SS-biotin (Thermo Fisher Scientific) (1 mg/mL in PBS). After this, cells were washed for three times with 2 mL of ice-cold PBS containing 100 mM glycine and incubated for 10 min at 4 °C in the same buffer. Then, cells were lysed with 500 μL of lysis buffer (10 mM Tris, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, and 1% Triton X-100, pH 7.4, containing protease inhibitors) for 2 h at 4 °C with shaking. Lysates were centrifuged for 2 min at 10,000× g and the supernatants were incubated with 30 μL of streptavidin-agarose beads (Thermo Fisher Scientific) for 1 h at room temperature under constant agitation. The beads were then centrifuged at 500× g for 1 min, washed for five times with ice-cold lysis buffer, and incubated with 100 μL of 2× Laemmli buffer containing 100 mM dithiothreitol at room temperature for 30 min to recover the cell surface proteins. Samples were separated using SDS-PAGE followed by western blot analysis. All constructs and mutant proteins were detected using anti-Flag-HRP antibody (GNI) (1: 5000 dilution). Then the same membrane was stripped for 1 h at room temperature. Na+/K+-ATPase was used as loading control for normalization and was detected with rabbit anti-Na+/K+-ATPase monoclonal antibody (1:10,000 dilution) (ABclonal) combined with secondary HRP goat anti-rabbit monoclonal antibody (1:10,000 dilution) (ABclonal).
Inhibition of OATP1B1-mediated uptake of DCF by compounds of B/S/E
Cells cultured in a 24-well plate were used for transport measurement. To each well, HEK293T cells were transfected with pCAG-OATP1B1-His-Flag constructs, using Lipofectamine 2000 reagent (Invitrogen) following the manufacturer’s instructions and the total amount of DNA per transfection was 0.6 μg. After 48 h, DMEM medium in the dish was aspirated and cells were washed with PBS for three times. 2.5 μM DCF in uptake buffer was mixed with each of the three compounds in six concentrations of 0.001–100 μM for E3S, simeprevir and 0.005–500 μM for bilirubin, in the total volume of 200 μL. The mixtures were then added to each well, and HEK293T cells with addition of 2.5 μM DCF in uptake buffer without compounds were used as a positive control, assumed to possess 100% activity. HEK293T cells transfected by PBS with addition of 2.5 μM DCF in uptake buffer without compounds were used as a blank control. After incubation for 5 min at 37 °C, uptake buffer with DCF and compounds was aspirated out and cells were washed with pre-cooling uptake buffer for three times. Then, 200 μL uptake buffer with 2% Triton X-100 was added to each well and incubation lasted for 1 h at room temperature. When performing 96-well plate assays using a BioTek Synergy2 Microplate Reader, 100 μL cell lysate was added to each well to detect the fluorescence intensity of DCF (excitation/emission wavelength at 485 nm/528 nm). Six experimental repeats were performed for each concentration, except that 5 experimental repeats were performed for WT in inhibition assay of bilirubin.
The fluorescence intensity of blank control was first deducted from experimental group and positive control. Then the measurements of experimental group were normalized according to positive control for final data analysis performed with nonlinear regression (curve fit) using GraphPad Prism v9.3.1.
[3H]-E3S liposome counterflow assay
Escherichia coli polar lipid extract (Avanti) supplemented with 20% (w%) cholesterol was resuspended to 20 mg/mL in the buffer (20 mM HEPES pH 7.4, 150 mM NaCl) plus 0.2 mM estrone sulfate sodium (MedChemExpress). After 10 cycles of freeze-and-thaw by liquid nitrogen, the liposomes were extruded through 0.4 μm polycarbonate track-etched membranes (GE Healthcare) for 21 times. The liposomes were pre-incubated with 1% n-octyl-β-D-glucoside (β-OG) (Anatrace) for 30 min at 4 °C, then the liposomes were incubated with 200 μg/mL OATP1B1 (WT or mutants) for 1 h at 4 °C. Detergents were removed by incubation overnight with 250 mg/mL Bio-Beads SM2 (Bio-Rad) at 4 °C, and then the supernatant was incubated with 100 mg/mL Bio-Beads SM2 for an additional 1 h. After 5 cycles of freeze-and-thaw and extrusion for 21 times again, the proteoliposomes were collected by ultracentrifugation at 100,000× g for 1 h at 4 °C, washed twice and finally resuspended to 100 mg/mL with buffer for the following counterflow assay.
Each counterflow assay was performed by adding 2 μL proteoliposomes into 97 μL buffer (20 mM HEPES pH 7.4, 150 mM NaCl) and 1 μL Ammonium Salt-[6,7-3H(N)]-Estrone Sulfate (PerkinElmer). Uptake of radio-labeled substrates was terminated at 60 s by rapidly filtering the solution through glass fiber filters (Advantec) and washed with 3 mL ice-cold buffer. The filters were incubated with 0.5 mL Optiphase HISAFE 3 (PerkinElmer) overnight for liquid scintillation counting by MicroBeta2® Microplate Counter (PerkinElmer). All experiments were repeated three times at 25 °C and all statistical analyses were performed with one-way ANOVA followed by Dunnett’s multiple comparisons test comparing WT with mutants using GraphPad Prism v9.3.1. Error bars represent SEM.
Data availability
Atomic coordinates of OATP1B1-apo/B/S/Ein/D have been deposited in the Protein Data Bank (http://www.rcsb.org) under the accession codes 8HNB, 8HNC, 8HNH, 8HND, and 8K6L, respectively. The corresponding electron microscopy maps have been deposited in the Electron Microscopy Data Bank (https://www.ebi.ac.uk/pdbe/emdb/), under the accession codes EMD-34909, EMD-34910, EMD-34913, EMD-34911, and EMD-36922, respectively.
References
Hagenbuch, B. & Stieger, B. The SLCO (former SLC21) superfamily of transporters. Mol. Aspects Med. 34, 396–412 (2013).
Hagenbuch, B. & Meier, P. J. The superfamily of organic anion transporting polypeptides. Biochim. Biophys. Acta 1609, 1–18 (2003).
Hagenbuch, B. & Meier, P. J. Organic anion transporting polypeptides of the OATP/ SLC21 family: phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch 447, 653–665 (2004).
König, J., Cui, Y., Nies, A. T. & Keppler, D. A novel human organic anion transporting polypeptide localized to the basolateral hepatocyte membrane. Am. J. Physiol. Gastrointest. Liver Physiol. 278, G156–G164 (2000).
Niemi, M., Pasanen, M. K. & Neuvonen, P. J. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 63, 157–181 (2011).
Kalliokoski, A. & Niemi, M. Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 158, 693–705 (2009).
Shitara, Y. et al. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 34, 45–78 (2013).
Link, E. et al. SLCO1B1 variants and statin-induced myopathy-a genomewide study. N. Engl. J. Med. 359, 789–799 (2008).
Chiou, W. J. et al. In vitro OATP1B1 and OATP1B3 inhibition is associated with observations of benign clinical unconjugated hyperbilirubinemia. Xenobiotica 44, 276–282 (2014).
van de Steeg, E. et al. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J. Clin. Invest. 122, 519–528 (2012).
Alam, K. et al. Regulation of organic anion transporting polypeptides (OATP) 1B1- and OATP1B3-mediated transport: an updated review in the context of OATP-mediated drug-drug interactions. Int. J. Mol. Sci. 19, 855 (2018).
Yoshida, K., Maeda, K. & Sugiyama, Y. Transporter-mediated drug-drug interactions involving OATP substrates: predictions based on in vitro inhibition studies. Clin. Pharmacol. Ther. 91, 1053–1064 (2012).
Noé, J., Portmann, R., Brun, M. E. & Funk, C. Substrate-dependent drug-drug interactions between gemfibrozil, fluvastatin and other organic anion-transporting peptide (OATP) substrates on OATP1B1, OATP2B1, and OATP1B3. Drug Metab. Dispos. 35, 1308–1314 (2007).
Giacomini, K. M. et al. International transporter consortium commentary on clinically important transporter polymorphisms. Clin. Pharmacol. Ther. 94, 23–26 (2013).
Yee, S. W. et al. Influence of transporter polymorphisms on drug disposition and response: a perspective from the international transporter consortium. Clin. Pharmacol. Ther. 104, 803–817 (2018).
Kee, P. S., Chin, P. K. L., Kennedy, M. A. & Maggo, S. D. S. Pharmacogenetics of Statin-induced myotoxicity. Front. Genet. 11, 575678 (2020).
Neuvonen, P. J., Niemi, M. & Backman, J. T. Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin. Pharmacol. Ther. 80, 565–581 (2006).
Hirota, T., Fujita, Y. & Ieiri, I. An updated review of pharmacokinetic drug interactions and pharmacogenetics of statins. Expert Opin. Drug Metab. Toxicol. 16, 809–822 (2020).
Abe, T. et al. Identification of a novel gene family encoding human liver-specific organic anion transporter LST-1. J. Biol. Chem. 274, 17159–17163 (1999).
Hirano, M., Maeda, K., Shitara, Y. & Sugiyama, Y. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J. Pharmacol. Exp. Ther. 311, 139–146 (2004).
Mahagita, C., Grassl, S. M., Piyachaturawat, P. & Ballatori, N. Human organic anion transporter 1B1 and 1B3 function as bidirectional carriers and do not mediate GSH-bile acid cotransport. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G271–G278 (2007).
Li, L., Lee, T. K., Meier, P. J. & Ballatori, N. Identification of glutathione as a driving force and leukotriene C4 as a substrate for oatp1, the hepatic sinusoidal organic solute transporter. J. Biol. Chem. 273, 16184–16191 (1998).
Satlin, L. M., Amin, V. & Wolkoff, A. W. Organic anion transporting polypeptide mediates organic anion/HCO3- exchange. J. Biol. Chem. 272, 26340–26345 (1997).
Leuthold, S. et al. Mechanisms of pH-gradient driven transport mediated by organic anion polypeptide transporters. Am. J. Physiol. Cell Physiol. 296, C570–C582 (2009).
Varma, M. V. et al. pH-sensitive interaction of HMG-CoA reductase inhibitors (statins) with organic anion transporting polypeptide 2B1. Mol. Pharm. 8, 1303–1313 (2011).
Yan, N. Structural biology of the major facilitator superfamily transporters. Annu. Rev. Biophys. 44, 257–283 (2015).
Lin, S., Ke, M., Zhang, Y., Yan, Z. & Wu, J. Structure of a mammalian sperm cation channel complex. Nature 595, 746–750 (2021).
Schlott, B. et al. Interaction of Kazal-type inhibitor domains with serine proteinases: biochemical and structural studies. J. Mol. Biol. 318, 533–546 (2002).
Khanppnavar, B. et al. Structural basis of organic cation transporter-3 inhibition. Nat. Commun. 13, 6714 (2022).
Sticova, E. & Jirsa, M. New insights in bilirubin metabolism and their clinical implications. World J. Gastroenterol. 19, 6398–6407 (2013).
Furihata, T. et al. Different interaction profiles of direct-acting anti-hepatitis C virus agents with human organic anion transporting polypeptides. Antimicrob. Agents Chemother. 58, 4555–4564 (2014).
Ouwerkerk-Mahadevan, S., Snoeys, J., Peeters, M., Beumont-Mauviel, M. & Simion, A. Drug-drug interactions with the NS3/4A protease inhibitor Simeprevir. Clin. Pharmacokinet. 55, 197–208 (2016).
Izumi, S. et al. Investigation of fluorescein derivatives as substrates of organic anion transporting polypeptide (OATP) 1B1 to develop sensitive fluorescence-based OATP1B1 inhibition assays. Mol. Pharm. 13, 438–448 (2016).
Xiang, Y. et al. Investigation of the interactions between flavonoids and human organic anion transporting polypeptide 1B1 using fluorescent substrate and 3D-QSAR analysis. Biochim. Biophys. Acta Biomembr. 1862, 183210 (2020).
Kiander, W. et al. The effect of single nucleotide variations in the transmembrane domain of OATP1B1 on in vitro functionality. Pharm. Res. 38, 1663–1675 (2021).
Kiander, W. et al. Functional in vitro characterization of SLCO1B1 variants and simulation of the clinical pharmacokinetic impact of impaired OATP1B1 function. Eur. J. Pharm. Sci. 176, 106246 (2022).
Häkkinen, K. et al. Functional characterization of six SLCO1B1 (OATP1B1) variants observed in Finnish individuals with a psychotic disorder. Mol. Pharm. 20, 1500–1508 (2023).
Quistgaard, E. M., Löw, C., Guettou, F. & Nordlund, P. Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat. Rev. Mol. Cell Biol. 17, 123–132 (2016).
Chu, X. et al. In vitro assessment of transporter mediated perpetrator DDIs for several hepatitis C virus direct-acting antiviral drugs and prediction of DDIs with Statins using Static models. AAPS J. 24, 45 (2022).
Lane, T. R. et al. Machine learning models identify new inhibitors for human OATP1B1. Mol. Pharm. 19, 4320–4332 (2022).
Karlgren, M. et al. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug-drug interactions. J. Med. Chem. 55, 4740–4763 (2012).
Tamai, I. et al. Functional characterization of human organic anion transporting polypeptide B (OATP-B) in comparison with liver-specific OATP-C. Pharm. Res. 18, 1262–1269 (2001).
Tamai, I. et al. Molecular identification and characterization of novel members of the human organic anion transporter (OATP) family. Biochem. Biophys. Res. Commun. 273, 251–260 (2000).
Ni, C., Yu, X., Fang, Z., Huang, J. & Hong, M. Oligomerization study of human organic anion transporting polypeptide 1B1. Mol. Pharm. 14, 359–367 (2017).
Zhang, Y. et al. Transport by OATP1B1 and OATP1B3 enhances the cytotoxicity of epigallocatechin 3-O-gallate and several quercetin derivatives. J. Nat. Prod. 76, 368–373 (2013).
Banerjee, N. et al. Estrone-3-sulphate, a potential novel ligand for targeting breast cancers. PLoS One 8, e64069 (2013).
Suo, Y. et al. Molecular basis of polyspecific drug binding and transport by OCT1 and OCT2. Nat. Struct. Mol. Biol. 30, 1001–1011 (2023).
Nigam, A. K. et al. Molecular properties of drugs handled by kidney OATs and liver OATPs revealed by chemoinformatics and machine learning: implications for kidney and liver disease. Pharmaceutics 13, 1720 (2021).
Li, N. et al. Identification of amino acids essential for estrone-3-sulfate transport within transmembrane domain 2 of organic anion transporting polypeptide 1B1. PLoS One 7, e36647 (2012).
Weaver, Y. M. & Hagenbuch, B. Several conserved positively charged amino acids in OATP1B1 are involved in binding or translocation of different substrates. J. Membr. Biol. 236, 279–290 (2010).
Drew, D., North, R. A., Nagarathinam, K. & Tanabe, M. Structures and general transport mechanisms by the major facilitator superfamily (MFS). Chem. Rev. 121, 5289–5335 (2021).
Roth, M., Obaidat, A. & Hagenbuch, B. OATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 165, 1260–1287 (2012).
Parker, J. L., Kato, T., Kuteyi, G., Sitsel, O. & Newstead, S. Molecular basis for selective uptake and elimination of organic anions in the kidney by OAT1. Nat. Struct. Mol. Biol. https://doi.org/10.1038/s41594-023-01039-y (2023). Online ahead of print.
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
He, S. & Scheres, S. H. W. Helical reconstruction in RELION. J. Struct. Biol. 198, 163–176 (2017).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 66, 12–21 (2010).
Varadi, M. et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Abraham, M. J. et al. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Klauda, J. B. et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 (2010).
Mark, P. & Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 105, 9954–9960 (2001).
Vanommeslaeghe, K. & MacKerell, A. D. Jr. Automation of the CHARMM general force field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model 52, 3144–3154 (2012).
Vanommeslaeghe, K., Raman, E. P. & MacKerell, A. D. Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model 52, 3155–3168 (2012).
Hoover, W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A Gen. Phys. 31, 1695–1697 (1985).
Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Ryckaert, J.-P., Ciccotti, G. & Berendsen, H. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Nan, J. et al. Cryo-EM structure of the human sodium-chloride cotransporter NCC. Sci. Adv. 8, eadd7176 (2022).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Acknowledgements
We thank Fudan University for providing cryo-EM facility support and computational facility support. We thank Zhenguo Chen, Hui Zhao, Yulei Ren, and Anqi Dong for technical supports during EM data collection. This work was funded by the National Natural Science Foundation of China (32171196 to Y.Z.), Shanghai Rising-Star Program (21QA1401200 to Y.Z.), the National Key R&D Program of China (2021YFA1300101 to Y.Z.), Start-up funds from Fudan University and Shanghai Fifth People’s Hospital (2021IBSPI03 to Y.Z.), and Shenzhen Natural Science Foundation Project (GXWD20201231105722002-20200831175432002 to H.L.).
Author information
Authors and Affiliations
Contributions
Y.Z. conceived and designed the project. Z.S., W.Z., J.N., Y.Y., W.S., J.W., and F.W. contributed to protein purification. Z.S., X.Y., and W.Z. performed cryo-EM data acquisition. Y.Z., Z.S., and X.Y. contributed to the cryo-EM data processing. X.Y. and Z.S. contributed to model building. H.L. performed MD simulations. X.Y. and Z.S. conducted cell-based DCF uptake assays. X.Y. performed IC50 experiments. Y.X. and Y.Y. prepared liposome assay. Y.Z. and Z.S. wrote the manuscript. All authors analyzed the data and contributed to manuscript preparation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shan, Z., Yang, X., Liu, H. et al. Cryo-EM structures of human organic anion transporting polypeptide OATP1B1. Cell Res 33, 940–951 (2023). https://doi.org/10.1038/s41422-023-00870-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-023-00870-8
This article is cited by
-
Structural insights into human organic cation transporter 1 transport and inhibition
Cell Discovery (2024)
-
Membrane transporters in drug development and as determinants of precision medicine
Nature Reviews Drug Discovery (2024)
