
Abstract
Class B1 G protein-coupled receptors (GPCRs) are important regulators of many physiological functions such as glucose homeostasis, which is mainly mediated by three peptide hormones, i.e., glucagon-like peptide-1 (GLP-1), glucagon (GCG), and glucose-dependent insulinotropic polypeptide (GIP). They trigger a cascade of signaling events leading to the formation of an active agonist–receptor–G protein complex. However, intracellular signal transducers can also activate the receptor independent of extracellular stimuli, suggesting an intrinsic role of G proteins in this process. Here, we report cryo-electron microscopy structures of the human GLP-1 receptor (GLP-1R), GCG receptor (GCGR), and GIP receptor (GIPR) in complex with Gs proteins without the presence of cognate ligands. These ligand-free complexes share a similar intracellular architecture to those bound by endogenous peptides, in which, the Gs protein alone directly opens the intracellular binding cavity and rewires the extracellular orthosteric pocket to stabilize the receptor in a state unseen before. While the peptide-binding site is partially occupied by the inward folded transmembrane helix 6 (TM6)–extracellular loop 3 (ECL3) juncture of GIPR or a segment of GCGR ECL2, the extracellular portion of GLP-1R adopts a conformation close to the active state. Our findings offer valuable insights into the distinct activation mechanisms of these three important receptors. It is possible that in the absence of a ligand, the intracellular half of transmembrane domain is mobilized with the help of Gs protein, which in turn rearranges the extracellular half to form a transitional conformation, facilitating the entry of the peptide N-terminus.
Similar content being viewed by others

Introduction
Class B1 G protein-coupled receptors (GPCRs), consisting of 15 members, have a large N-terminal domain involved in recognition of peptide hormones1. Due to their broad physiological functions, class B1 family members are important drug targets for many diseases, including type 2 diabetes, obesity, osteoporosis, migraine, cardiovascular diseases, and short bowel syndrome2. As such, understanding the structural underpinnings of receptor activation is of prime interest. The typically assumed GPCR activation mechanism posits that the agonist binding induces conformational changes of the receptor towards active state, resulting in the opening of an intracellular cavity, where G protein binds3,4. Structures of all 15 class B1 GPCRs in complex with peptide agonists and Gs protein have been determined by cryo-electron microscopy (cryo-EM) in recent years5, providing important insights into hormone recognition and receptor activation of this family.
It has been widely accepted that class B1 GPCRs adopt a two-step model for peptide binding and receptor activation1. However, several recently determined non-peptidic ligand-bound structures of class B1 GPCRs have uncovered intracellular ligand-binding pockets. A prototypical example is PCO371, which could bind within the highly conserved intracellular pocket, directly and selectively interact with Gs protein, activating 7 out of 15 family members6,7. In addition, compound 2, an ago-positive allosteric modulator (ago-PAM), has been found to covalently bond to the intracellular side of transmembrane helix 6 (TM6) and allosterically induce insertion of the N-terminus of extracellular domain (ECD) into the orthosteric binding site of glucagon-like peptide-1 receptor (GLP-1R), thus triggering Gs-biased activation8. Meanwhile, GPCRs can also be activated in the absence of a ligand causing basal signaling9,10.
GLP-1R, glucagon receptor (GCGR) and glucose-dependent insulinotropic polypeptide receptor (GIPR) are validated therapeutic targets for type 2 diabetes and obesity11,12,13. There exist a number of crystal and/or cryo-EM structures of them in inactive, intermediate and active states revealing key information about their interactions with various ligands2,8,14,15, including dual or triple agonist-bound GLP-1R, GCGR or GIPR–Gs complexes16,17,18. Although these structures provide molecular insights into diverse activation mechanisms of these three receptors, it remains unclear whether an extracellular agonist is required for G protein coupling and downstream signal transduction.
Here, we report the cryo-EM structures of GLP-1R, GCGR, and GIPR in complex with Gs protein in the absence of any ligand. Based on multi-perspective structural analyses, we present a transitional state, in which Gs protein directly interacts with the inactive receptor prior to ligand binding and opens the intracellular cavity by breaking the conserved HETY network (H2.50b–E3.50b–T6.42b–Y7.57b) and cytoplasmic polar network (R2.46b–R6.37b–N7.61b–E8.49b). Gs protein thus stabilizes the receptor in a distinct extracellular conformation until agonist binding to the orthosteric site. This process tightens the extracellular pocket and intracellular region allowing Gs protein coupling and full activation of the receptor.
Results
Structures of ligand-free receptor–Gs complexes
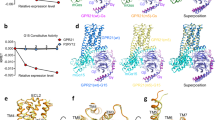
The cryo-EM structure of ligand-free GLP-1R–Gs complex (Supplementary Fig. S1a) was determined at an overall resolution of 2.54 Å (Fig. 1a; Supplementary Figs. S2a and S3a). Structural superimposition shows that this structure is almost identical to that bound by the ago-PAM compound 2 with a Cα root mean square deviation (RMSD) of 0.53 Å (Supplementary Fig. S4), indicating a newly discovered conformational state beyond those of antagonist-bound, apo without G protein, agonist-bound and agonist-bound plus G protein. Following a similar protocol (Supplementary Fig. S1b, c), three-dimensional (3D) structures of the ligand-free GCGR–Gs and GIPR–Gs complexes were obtained at global resolutions of 2.70 Å and 2.86 Å, respectively (Fig. 1c, e; Supplementary Fig. S2b, c). They display canonical structural features of the GPCR–G protein complex and no extra density was observed in the transmembrane domain (TMD) binding pocket, confirming the absence of any ligand (Fig. 1). The density maps clearly exhibit the backbones and most side chains of TMs, extracellular loops (ECLs), intracellular loops (ICLs), the amphipathic helix 8 (H8), and three Gs subunits (Fig. 1; Supplementary Fig. S3). Moreover, the interface residues between the receptor and α5 helix of Gs (GαH5) were solved (Supplementary Figs. S3 and S5) and no ECD density was seen in the three maps (Fig. 1), suggesting that the receptors may not require a specific ECD conformation to engage Gs. Interestingly, an electron density of cylindrical helix shape was found in the GCGR membrane-facing surfaces of TM4 and TM5 that were omitted from modeling due to sequence uncertainty (Fig. 1c).

a, c, e Cryo-EM density maps of the GLP-1R–Gs (a), GCGR–Gs (c), and GIPR–Gs (e) complexes are shown from two viewpoints. GLP-1R, GCGR, GIPR, Gαs, Gβ, Gγ, and Nb35 are shown in orange, pale green, hot pink, slate gray, powder blue, khaki and light gray, respectively. The maps reveal several lipid densities around the receptors consistent with the shape of cholesterol (olive), palmitate (brown) or phosphatidylinositol 4,5-bisphosphate (purple). There is a strong unassigned cylindrical density in the shape of helix (green) dropping down along the TM4 and TM5 of GCGR, tentatively named as chain X (c). b, d, f Structural models of the GLP-1R–Gs (b), GCGR–Gs (d) and GIPR–Gs (f) complexes are constructed from the respective cryo-EM maps and shown in ribbon.
Gs protein coupling by ligand-free receptors
The three ligand-free receptors interact with Gs proteins similarly to those bound by endogenous agonists, suggesting a common mechanism for G protein coupling (Fig. 2). It appears that Gs is able to induce a consensus kink at the middle of TM6 without an agonist. Subsequently, an outward shift of the cytoplasmic end of TM6 (~11.9 Å in GLP-1R (measured at the Cα of R3486.37b), 14.1 Å in GCGR (measured at the Cα of R3466.37b) and 10.9 Å in GIPR (measured at the Cα of R3386.37b)) renders the receptor to form a cavity for Gs coupling (Fig. 2). However, the position of the cytoplasmic end of TM6 in the inactive state (either antagonist-bound or apo without G protein) structures is incompatible with the insertion of the C-terminus of GαH5 (Fig. 2a–c), indicating that Gs coupling is necessary for the kink of TM6. While TM6 conformations are identical among ligand-free and agonist-bound GLP-1R and GCGR structures, the movement of TM6 at intracellular region of GIPR is significantly different (4.5 Å measured at the Cα of R3386.37b and 4.6 Å measured at the Cα of T3436.42b) (Fig. 2c), pointing to a receptor-dependent structural feature.

a–c Comparison of ligand-free and peptide-bound GLP-1R (a), GCGR (b) and GIPR (c) with inactive receptor structures reveals a similar Gs coupling interface in the absence or presence of an agonist. Black arrows show the movements of TM6 and H8 by indicating the distances of Cα atoms of T6.42b, R6.37b, and R/H8.60b residues. d–f Structural comparison of the TMD bundles of GLP-1R (d), GCGR (e) and GIPR (f) indicates the requirement of Gs protein for receptor activation. Conformational changes are shown for the conserved HETY inactive motif (H2.50b–E3.50b–T6.42b–Y7.57b) and cytoplasmic polar network (R2.46b–R6.37b–N7.61b–E8.49b). Black arrows indicate the hallmark conformational changes of TM6. The Gβ and Gγ subunits are omitted for clarity. PDB IDs: 6LN2 (inactive GLP-1R), 5XEZ (inactive GCGR), 6X18 (GLP-1-bound GLP-1R), 6LMK (GCG-bound GCGR), and 7DTY (GIP-bound GIPR). The position of N7.61b in d, e and f refers to N8.47b in GPCRdb numbering.
It is known that two highly conserved polar networks at the cytoplasmic face of class B1 GPCRs, the HETY network (H2.50b–E3.50b–T6.42b–Y7.57b) and the cytoplasmic polar network (R2.46b–R6.37b–N7.61b–E8.49b), lock the base of the receptor in a closed conformation19. G protein binding eliminates the interactions between T6.42b or R6.37b and other three residues within the respective motif, releasing them from tightly packing constraints to participate in Gs coupling (Fig. 2). With respect to ligand-free GLP-1R, the conformational transition induced by Gs coupling resulted in breakup of these two networks to release R1762.46b in the TM2–ICL1 juncture, N4067.61b and E4088.49b in the TM7–H8 juncture as well as Y4027.57b and H1802.50b in the TMD core, to facilitate Gs coupling (Fig. 2d). Superimposition of receptor–Gs complex structures with or without agonist reveals that, although there are limited differences in the overall Gs conformation, the ligand-free receptors make less interactions with Gαs Ras and Gβγ domains (Supplementary Figs. S5–S7). For GLP-1R and GCGR, the key Y391GαH5 side chain is hosted in a pocket defined by R2.46b, Y3.53b and L3.54b, while L393GαH5 and L394GαH5 interact with a set of residues located in proximity to the polar networks, such as L3.54b, I5.57b, and L6.43b, a feature shared by ligand-bound and ligand-free structures (Supplementary Fig. S5a, b). For GIP-bound GIPR, the C-terminal residues of GαH5 (L388, Y391, L393, and L394) insert into a hydrophobic pocket (formed by L2413.54b, L2443.57b, L3215.61b, and L3255.65b), and L394GαH5 makes one hydrogen bond with the side chain of R3386.37b. This phenomenon was not observed in the ligand-free GIPR (Supplementary Fig. S5c), indicating that a conformational rearrangement occurs in the intracellular side upon GIP binding. Of note, E392GαH5 switches toward the TMD core to form multiple hydrogen bonds with L4017.56b, N4067.61b, and N4078.48b in the GLP-1-bound GLP-1R (Supplementary Fig. S5a) and flips downward to make polar interactions with positively charged residues such as K8.48b and N7.61b in the peptide-bound GCGR and GIPR (Supplementary Fig. S5b, c). These polar contacts are significantly reduced in the ligand-free receptor–Gs complexes, likely due to the rotation of the side chain of Y7.57b. Following peptide binding, Y7.57b shifts toward Gs protein-binding face in GCGR and GIPR, while stands still in the case of GLP-1R (Fig. 2d–f). Previous study demonstrated that removal of the hydroxyl group of Y4007.57b in GCGR led to a 40% decrease in maximal cAMP signaling elicited by glucagon20, suggesting a critical role of this conformational change.
Besides TMs, ICL2 and ICL3 of the ligand-free receptors contribute weaker contacts to maintain the receptor–Gs interface compared to those bound by agonist (Supplementary Figs. S6, S7). Unlike the upward conformation in the ligand-free structures, F257ICL2 of GLP-1-bound GLP-1R switches downward and forms intensive stacking interactions with R380GαH5, F376GαH5, and H41GαN. Meanwhile, H8 moves closer to Gβ and makes intensive hydrophobic interactions with V307, A309, and G310 in Gβ subunit (Supplementary Fig. S6). Similar reduced contacts are observed at the ICL2 of GCGR and GIPR, where E260ICL2 of GCGR as well as S251ICL2 and E253ICL2 in GIPR make negligible contacts with G protein in the ligand-free structures. Furthermore, the hydrogen bonds between R380GαH5 and ICL2 with the backbone atoms of L2453.58b, V2463.59b, L2473.60b, and V248ICL2 are also reduced in the ligand-free GIPR structure (Supplementary Fig. S6). The polar residues S251 and E253 in ICL2 form two hydrogen bonds with K34GαN and Q35GαN, and H8 forms hydrogen bonds with Gβ (E4028.53b–R164ICL1–D312Gβ), but these interactions are not observed in the ligand-free GIPR structure (Supplementary Fig. S6). For ICL3, the TM5–ICL3 region only has a limited density in both GLP-1-bound and ligand-free GLP-1R structures, but is clearly visible for those of GCGR and GIPR (Supplementary Fig. S7). In the agonist-bound structures, GαN helices interact with ICL3 through Y385GαH5, but this interaction is not found in the ligand-free GLP-1R and GCGR structures and greatly reduced in that of GIPR (Supplementary Fig. S7).
TM packing induced by Gs coupling
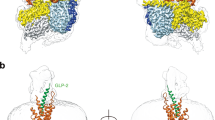
Superimposition of the ligand-free and peptide-bound structures shows that the intracellular half of TMD aligns well, while the extracellular half is structurally divergent and exhibits a receptor-specific conformation in the absence of ligand (Fig. 3a–c). In addition, TM6 kink is the most significant structural feature of ligand-free Gs-coupled receptors distinguished from that of inactive or intermediate states (Supplementary Fig. S8a, b). For GLP-1R, Gs coupling induces a significant outward movement of the extracellular portion of TM bundles (Supplementary Fig. S8a). Upon GLP-1 binding, 6.5 Å, 4.8 Å, and 6.2 Å inward movements of the extracellular portion of TM1, TM6 and TM7 (measured at the Cα atoms of E1381.33b, V3706.59b, and T3787.33b) were seen, respectively. However, in the absence of Gs, the TM1 movement from inactive state to agonist peptide 5-bound state is larger (9.1 Å), while TM6 and TM7 shift outward by 3.2 Å and 11.5 Å, respectively (Fig. 3a). These conformational rearrangements of the ligand-free GLP-1R are in general consistent with those observed in the compound 2-bound GLP-1R–Gs structure8 and the apo-state GLP-1R obtained from structure determination of tirzepatide-bound GLP-1R–Gs complex21 (Supplementary Fig. S8c), except that in our new structure, ECL3 splays out and causes a sharper TM6 kink distinct from the GLP-1-bound active structure as seen from the kink angle (98° and 112° for the ligand-free and GLP-1-bound GLP-1R–Gs structures, respectively, measured at the Cα atoms of D3446.33b–P3586.47b–V3706.59b) (Fig. 3e).

a Structural superimposition of ligand-free and GLP-1-bound GLP-1R–Gs (PDB: 6X18) complexes from side and top views, as well as inactive GLP-1R (PDB: 6LN2) and peptide 5-bound GLP-1R (PDB: 5NX2) from top view, shows different conformational changes induced by agonist binding with or without Gs protein coupling. Gray arrows indicate the movements of TM1, TM6 and TM7 measured by the Cα atoms of E1381.33b, V3706.59b and T3787.33b, respectively. b Structural superimposition of ligand-free and GCG-bound GCGR–Gs (PDB: 6LMK) complexes from side and top views, as well as inactive GCGR (PDB: 5XEZ) and NNC1702-bound GCGR (PDB: 5YQZ) from top view, shows different conformational changes induced by agonist binding with or without Gs protein coupling. Gray arrows indicate the movements of TM1, TM6, and TM7 measured by the Cα atoms of K1361.34b, V3686.59b, and T3767.33b, respectively. c Structural superimposition of ligand-free and GIP-bound GIPR–Gs (PDB: 7DTY) complexes from side and top views shows a unique conformational change in the extracellular region induced by GIP binding. Gray arrows indicate the movements of TM1, TM6 and TM7 measured by the Cα atoms of L1281.30b, V3566.55b, and A3687.33b, respectively. d Superimposition of ligand-free GLP-1R, GCGR and GIPR structures reveals dynamics of the ECLs. The conformational differences are indicated by two-way arrows. e Structures of ligand-free and endogenous ligand-bound receptors are superimposed onto the inactive GLP-1R or GCGR structure, showing different TM6 kink conformations. The kink angle is shown among the Cα atoms of V6.59b–P6.47b–D6.33b in TM6. Gs proteins are omitted for clarity.
Unlike GLP-1R, GCGR in complex with Gs has an almost identical kink angle of TM6 (~120° measured at the Cα of D3426.33b–P3536.47b–V3586.59b) regardless of peptide binding or not (Fig. 3e), indicative of a relatively stable TM6–ECL3–TM7 conformation. In terms of the extracellular portion, TM1 and TM7 in the NNC1702-bound GCGR slightly move outward relative to the inactive-state structure. However, in the presence of Gs, GCG binding causes the extracellular portions of TM1 and TM7 to move inward by 4.8 Å (measured at the Cα of K1361.34b) and 5.3 Å (measured at the Cα of T3767.33b), respectively, accompanied by a 9.1 Å (measured at the Cα of A3666.57b) outward movement of TM6 (Fig. 3b). The ligand-free GIPR displays the most profound structural feature of TM packing, in which the extracellular portion of TM6 and ECL3 fold inward to occupy the TMD pocket. Consequently, the kink in TM6 adopts a unique angle (148° measured at the Cα of D3346.33b–P3486.47b–V3606.59b) different from those of GIP-bound GIPR (113°) and peptide-free GLP-1R and GCGR (Fig. 3e). Upon GIP binding, the extracellular tip of TM6 moves outward by 10.9 Å (measured by the Cα distance of V3566.55b) to facilitate the insertion of GIP. Meanwhile, the extracellular halves of TM1 and TM7 move toward the orthosteric pocket by 10.1 Å (measured at the Cα of L1281.30b) and 8.1 Å (measured at the Cα of A3687.33b), respectively (Fig. 3c). These data imply that Gs coupling rewires the extracellular halves of TMs in a receptor-dependent manner.
At the residue level, the central polar network (K/R2.60b–N3.43b–H6.52b–Q7.49b) located below the peptide-binding pocket and PxxG (P6.47b–L6.48b–L6.49b–G6.50b) motif where the TM6 sharp kink occurs directly or indirectly participates in peptide recognition by switching from loose (ligand-free structure) to compact packing (peptide-bound structure) to tighten the TMD orthosteric pocket (Supplementary Fig. S9). Upon peptide binding, the central polar network is not reconstructed in GLP-1R, but most residues change their conformations and partially pull out of the orthosteric pocket in the case of GCGR. Conformational divergence around the peptide-binding pocket is more evident in GIPR, due to the steric clashes of the bent TM6–ECL3 (Supplementary Fig. S9b). The ligand-free GLP-1R and GCGR deform the PxxG motif in a manner similar to the agonist-bound structures, while the ligand-free GIPR prefers a moderately kinked TM6 that is significantly different from the fully active structures. The inward rotation of the PxxG motif in GLP-1R and GCGR is further stabilized by hydrogen bonds with TM5 (G6.50b–N5.50b) and TM7 (P6.47b–Q7.49b), as well as hydrophobic contacts with F5.54b and V7.53b, a phenomenon not observed in the ligand-free GIPR structure (Supplementary Fig. S9c).
Conformational flexibility of the ECLs
Structural superimposition of the ligand-free Gs-coupled GLP-1R, GCGR and GIPR with those bound by endogenous peptides reveals that the ligand-binding pocket of ligand-free receptor is partially occupied by GCGR ECL2 or GIPR ECL3 but not the ECLs of GLP-1R. In all the three structures, the densities of most ECL1 residues are invisible, whereas ECL2 and ECL3 are well-defined in the EM maps of GLP-1R and GIPR, and largely solved in GCGR except one short segment (D299–G302) (Supplementary Fig. S3). The peptide-binding pocket in the ligand-free GLP-1R exhibits a more opened conformation than those of the other two receptors. This is because of a large outward movement of GLP-1R ECL3 (~10 Å measured at the Cα of V370GLP-1R–V368GCGR, and 21 Å measured at the Cα of V370GLP-1R–V360GIPR), despite that the ECL2 conformation is well maintained (Fig. 3d).
Unlike GLP-1R, N291–N298 segment of ECL2 in the ligand-free GCGR stretches toward the TMD core to partially occupy the orthosteric pocket, which overlaps with the N-terminal segment (T5P–S8P) of GCG (Fig. 4a). This unique ECL2 conformation may result from interactions with chain X (Supplementary Fig. S10a). Superimposition of the ligand-free and GCG-bound GCGR structures reveals that five residues in the TM4, TM5 and ECL2 spatially clash with the cryo-EM density of chain X, including four phenylalanine (F2784.56b, F2894.67b, F303ECL2, and F3095.41b) and one isoleucine I3065.38b (Supplementary Fig. S10a). To avoid this steric hindrance, several aromatic amino acids shift inwards, pushing ECL2 to occupy the peptide-binding pocket (Supplementary Fig. S10b). Specifically, residues F2894.67b, F303ECL2, and F3095.41b flip away from chain X–GCGR interface to create room for chain X binding. This conformational change is propagated to W295ECL2, W3045.36b, and W3055.37b, pushing them to rotate toward the central pore and inducing W2824.60b to shift outward (Supplementary Fig. S10c). Consequently, this “F–W switch” in the vicinity of chain X creates steric clashes with ECL2 and facilitates its stretched conformation. In the ligand-free GCGR, N291–N298 segment of ECL2 points to the TMD core like a “latch”, and is stabilized by hydrogen bonds (N291ECL2–R2253.30b–Q293ECL2), hydrophobic contacts via V2213.26b, M2313.36b and Q2323.37b and a conserved disulfide bond (C294ECL2–C2243.29b) (Fig. 4d). In addition, the movements of TM4 and TM5 causes the formation of two additional residue pairs under the ECL2, one hydrogen bond (Q2323.37b–K2864.64b) and several hydrophobic contacts (I2353.40b–W3045.36b), to lock ECL2 in the orthosteric pocket. However, in the GCG-bound structure, ECL2 mainly interacts with the peptide N-terminus through D299–G302 segment (Fig. 4d). Recent studies have shown that several class A orphan GPCRs are self-activated by ECL2 that directly penetrates to the orthosteric binding pocket, such as GPR2122, GPR5223, GPR1724, GPR161 and GPR6125. Although the ECL2 of GCGR partially overlaps with the peptide-binding site, the majority of which is likely disordered owing to the missing cryo-EM density, suggesting its conformational flexibility and non-built-in agonist nature.

a Superimposition of ligand-free and GCG-bound GCGR structures reveals that the GCG-binding pocket is partially occupied by inward moved ECL2 upon G protein coupling. GCG-bound GCGR (PDB: 6LMK) is shown in surface representation. ECL2 residues of ligand-free GCGR are shown in sphere. b Superimposition of ligand-free and GIP-bound GIPR structures reveals that the GIP-binding pocket is partially occupied by inward moved TM6–ECL3 juncture upon G protein coupling. GIP-bound GIPR (PDB: 7DTY) is shown in surface representation. Residues in the TM6–ECL3 juncture of ligand-free GIPR are shown in sphere. c Sequence alignment of ECL2 and ECL3 of GCGR, GLP-1R and GIPR. d Magnified view of the ECL2 within the orthosteric binding pocket (left panel) and its interaction with GCG (right panel). e Magnified view of the TM6–ECL3 juncture in the orthosteric binding pocket (left panel) and its interaction with GIP (right panel). Key interacting residues and their contacts are shown as sticks and dashed lines, respectively.
Instead of ECL2, TM6–ECL3 of ligand-free GIPR folds toward the TMD core and occupies the orthosteric pocket (Fig. 4e; Supplementary Fig. S11). Specifically, TM6–ECL3 is clasped by TM5 and TM7 and stabilized by hydrophobic interactions with TM3, TM5 and ECL2 via L3496.48b, V3526.51b, V3556.54b, P3596.58b and V3606.59b, and by polar contacts with TM1, TM3 and TM7 via E3546.53b, F3576.56b, E363ECL3, Q364ECL3 and A365ECL3. In the GIP-bound GIPR structure, the ECL3 is stabilized by two polar contacts (T5P–E363ECL3 and D9P–R3707.35b) (Fig. 4e). W287ECL2 orientates its side chain from the TM3–TM4 crevice in the GIP–GIPR–Gs structure to occupy the orthosteric site in the ligand-free structure, reflecting the engagement of GIPR ECL2 in the closure of TMD ligand-binding pocket.
Activation-related conformational changes
Inspecting conformational changes of highly conserved residues reveals: (i) several distinct fastening pieces within the central and outer layers that stabilize the receptor in a transitional state; and (ii) similar contacts within the ECL3–TM5 interface and intracellular cavity that respond to peptide binding and may transmit extracellular stimuli to Gs protein (Fig. 5). In the absence of agonist and Gs protein, the receptors are locked in an inactive state, posing an extracellular shutting conformation through interactions between ECD and ECLs and an intracellularly closed conformation through interactions within conserved HETY and cytoplasmic polar networks, thereby preventing peptide binding and Gs coupling (Fig. 5a). Gs coupling will break the bottom polar networks, interfere with movement of the extracellular portions and render the receptor to a transitional state (Fig. 5b). Overall, the transitional conformation is close to the fully active ensemble in the intracellular half, but the extracellular half, especially the peptide-binding site, is stabilized differently. In the ligand-free GLP-1R, Gs coupling rearranges several residues located above the PxxG motif, which form hydrogen bonds (Q7.49b–G6.50b and N3.43b–R2.60b) and π stacking (H6.52b–F7.45b) at the central layer (Fig. 5c), thus maintaining the TMD and ECLs in a conformation close to the active state. However, these interactions are disrupted by marked shifts of TM1, TM6 and TM7 in the ligand-free GCGR and GIPR. Located above the central network is a cluster of hydrophobic packing interactions, stabilizing the orthosteric site in different conformations. For GLP-1R, residues Y1481.43b, F2303.33b, M2333.36b and R3105.40b partially occupy the orthosteric stie and form hydrophobic interactions with K1972.67b, W297ECL2 and L3887.43b, as well as π stacking with W2844.60b. However, the ligand-free GCGR mainly composes the structural feature in the TM4–ECL2–TM5 region, where hydrophobic interactions (W3045.36b–I2353.40b and M2313.36b–V1912.64b) and hydrogen bonds (Q2323.37b–K2864.64b and Q293ECL2–N291ECL2/R2253.30b) form. Notably, in the ligand-free GIPR, the TM6–ECL3 juncture is sandwiched by the TM5–ECL2 juncture and the extracellular portion of TM7, weaving an intensive interaction network by a hydrogen bond between Q2203.33b and R1902.67b, and several hydrophobic contacts (R3005.40b–V3556.54b/V3566.55b, W2744.60b–Q2243.37b, and W287ECL2–L1942.71b/V3606.59b) (Fig. 5d).

a Superimposition of inactive GLP-1R and GCGR structures shows an extracellular shutting conformation (yellow shadow) and intracellular closed conformation (gray shadow), thus intercepting peptide binding and Gs coupling. b Superimposition of ligand-free GLP-1R, GCGR and GIPR structures shows that Gs coupling directly causes the formation of an intracellular cavity and stabilizes the intracellular half of TMD, whereas the extracellular portion of the receptor is stabilized in the central and outer layers. c, d Magnified views of different contacting modes in the central (c) and outer (d) layers of GLP-1R, GCGR and GIPR. Key interacting residues are shown as ball-and-sticks, and the interactions are shown as dashed lines. e Superimposition of endogenous ligand-bound GLP-1R, GCGR and GIPR structures shows that peptide binding stabilizes the TM6–ECL3–TM7 conformation and rewires Gs protein. The right panel shows magnified views of the M/K5.33b–R5.40b–T/D/EECL3 and L6.48b–Y7.57b–N7.61b–E8.49b–E392GαH5 interfaces. Key interacting residues are shown as ball-and-sticks. PDB IDs: 6LN2 (inactive GLP-1R), 5XEZ (inactive GCGR), 6X18 (GLP-1-bound GLP-1R), 6LMK (GCG-bound GCGR) and 7DTY (GIP-bound GIPR). The position of N7.61b in e refers to N8.47b in GPCRdb numbering.
Upon peptide binding, M/K5.33b and R5.40 switch toward ECL3 to tighten the orthosteric pocket by interactions within the TM5–ECL3 interface, which was observed in all three structures (Fig. 5e). The distinct fastening pieces within the central and outer layers of the ligand-free receptors are also reshaped to a similar conformation, stabilizing the active state by interactions with peptide N-terminus as reported previously15,26,27. Obviously, agonist binding can enhance Gs coupling, as seen from the increased interface area and strengthened interactions (Supplementary Figs. S5–S7), suggesting that peptide binding not only tightens the pocket, but also transmits extracellular signal to Gs protein. Structural comparison shows that the conserved residues L6.48b, Y7.57b, N7.61b and E8.49b relocate E392GαH5 to stabilize Gs protein coupling (Fig. 5e). Thus, despite the different conformational changes in the orthosteric pocket, activation signal is transduced from the extracellular to intracellular sides through L6.48b–Y7.57b–N7.61b–E8.49b–E392GαH5 motif, where L6.48b pushes Y7.57b to a similar position, transmitting to N7.61b and E8.49b to respond to Gs residue E392 (Fig. 5e), thus fully activating the receptor. Collectively, upon agonist binding, the receptor in a transitional state may rapidly initiate residue rearrangement without the need of large TM movement to activate signal transduction.
Discussion
GLP-1R, GCGR and GIPR are essential regulators of glucose homeostasis and therapeutic targets for type 2 diabetes and obesity. It has been widely accepted that class B1 GPCRs adopt a two-step model for ligand binding and receptor activation28. However, this may not apply to small-molecule modulators that bind to a small part of the peptide-binding pocket or even to an allosteric site, thus bypassing the reorganization of ECD, such as PCO371, a small-molecule agonist bound within the intracellular pocket of 7 class B1 GPCRs to directly interact with Gs protein6,7. These atypical activation processes indicate that intracellular signal transducers may have opportunity to engage the receptor without extracellular stimuli, inducing basal activity. A previous study has demonstrated that constitutive cAMP signaling from GLP-1R, in the absence of GLP-1 or GCG, contributed to glucose-induced insulin secretion, indicating the physiological relevance of basal signaling activity29.
The three cryo-EM structures of ligand-free Gs-coupled GLP-1R, GCGR and GIPR presented here expanded our knowledge of G protein coupling and receptor activation from three aspects. First, the ligand-free receptors successfully promote a large outward movement of the cytoplasmic half of TM6 and reorganize the HETY and cytoplasmic polar networks to open an intracellular crevice for G protein coupling. Even though the overall G protein coupling interface is similar between the ligand-free and peptide-bound receptors, the former has weaker contacts with Gs. Second, the extracellular halves of TMs 1, 6, and 7 of the ligand-free receptors are all structurally different from those of peptide-bound receptors in a receptor-specific manner. For instance, the ligand-free GIPR adopts a moderately kinked TM6 different from the fully active GIPR and the ligand-free GLP-1R or GCGR. Third, the TMD orthosteric pocket is partially occupied by ECLs in the ligand-free Gs-coupled GCGR (via the inward stretched ECL2) and GIPR (via the inward folded TM6–ECL3) but not GLP-1R. Such special ECL2 and ECL3 conformations are not seen in other class B1 GPCRs. These results provide valuable structural evidence for the existence of a ligand-free and Gs-coupled transitional state distinct from those reported previously, i.e., inactive (antagonist-bound and apo without G protein) and active (agonist-bound and agonist-bound plus G protein-coupled).
Structural superimposition of the ligand-free and Gs-coupled receptor with that bound by peptide agonist reveals that agonist binding or G protein coupling individually can cause differential packing in a receptor-dependent manner but not to the extent shown in the fully active state20. Specifically, peptidic agonist (NNC0712 for GCGR and peptide 5 for GLP-1R) binding reshapes the orthosteric pocket but cannot appropriately reorganize the central polar network to propagate extracellular signals30,31. As a result, the sharp kink in TM6 could not be formed and the intracellular crevice is still closed. In comparison, the structures of ligand-free and Gs-coupled GLP-1R, GCGR and GIPR suggest that Gs protein successfully not only opens the intracellular binding cavity without agonist action, but also rewires the extracellular orthosteric pocket to stabilize the receptor in a transitional state, ready for agonist binding. We noted that ECLs are constitutively flexible without agonist binding, thereby posing a conformation different from those observed in both active and inactive states. Although ECL3 of the ligand-free GLP-1R swings out of the peptide-binding pocket, the extracellular portion of TMD is maintained in a conformation close to the active state, because several residues located at the orthosteric site form an interaction network to stabilize the TMD core. However, GIPR has a uniquely folded TM6–ECL3 juncture covering the orthosteric site, and GCGR orthosteric site is partially occupied by ECL2, locking the receptors in a transitional state. Peptide binding tightens the orthosteric pocket and triggers conformational changes of L6.48b–Y7.57b–N7.61b–E8.49b–E392GαH5 motif, thereby transmitting signal from the extracellular to intracellular sides. Thus, despite different molecular features of the transitional receptors, agonist binding can reshape them to a similar conformational state that is required for receptor activation.
It is also possible that in the absence of ligand, the intracellular half of TMD can be independently mobilized by Gs protein, which in turn rearranges the extracellular half to form a transitional conformation. The receptor in this state, without an extracellular shutting conformation due to flexible ECD, may simultaneously mobilize the orthosteric pocket and ECLs (e.g., outward-splayed ECL3 of GLP-1R, inward-stretched ECL2 of GCGR and inward-folded ECL3 of GIPR) to facilitate the entry of the peptide N-terminus, leading to downstream signaling events. Previous studies indicate that agonist-bound intermediate and fully active states possess a higher activation energy barrier for GCGR than the class A GPCR adrenergic receptor20. It is therefore possible that the “transitional state” presented in our study could be a Gs pre-coupled status to cope with the energy barrier required for the opening of the intracellular end of TM6. This “Gs-first” feature may allow more rapid activation than “agonist-first” model. Our findings would certainly broaden the overall understanding of activation mechanisms of class B1 GPCRs and provide insights into the development of better therapeutics targeting GLP-1R, GCGR and GIPR.
Materials and methods
Constructs
The human GLP-1R, GCGR and GIPR were cloned into pFastBac vector and modified with their native signal sequences replaced by the HA signal peptide to facilitate receptor expression. The NanoBiT tethering strategy was used for the complexes as described previously32, in which the receptor C-terminus was directly attached to LgBiT subunit followed by a TEV protease cleavage site and a double maltose binding protein (MBP) tag. To improve the thermostability of GCGR–Gs complex, 45 residues (H433–F477) were truncated and the affinity tag HPC4 was added to the receptor C-terminus. For GIPR, the residue T345 was mutated to phenylalanine, a BRIL fusion protein was added to the N-terminus with 2GSA linker, and 45 amino acids (Q422–C466) at C-terminus were truncated. These modifications did not alter receptor pharmacology15,18. An engineered Gs construct (G112) was used for expression and purification of the GLP-1R–Gs or GCGR–Gs complexes33. A dominant-negative Gαs (DNGαs) with 9 mutations (S54N, G226A, E268A, N271K, K274D, R280K, T284D, I285T and A366S) were generated by site-directed mutagenesis to stabilize interactions with the βγ subunits in the GIPR–Gs complex34. Rat Gβ1 was cloned with a C-terminal SmBiT (peptide 86 or HiBiT, Promega) connected to a 15-amino acid polypeptide linker35. The modified rat Gβ1 and bovine Gγ2 were both cloned into pFastBac vector. Nanobody 35 (Nb35) with a C-terminal 6× His-tag was cloned into the expression vector (pET28a) and used to limit G protein dissociation by binding at Gαs–Gβ interface36.
Complex expression and purification
Spodoptera frugiperda (Sf9) or High Five insect cells (Expression Systems) were grown in ESF 921 serum-free medium (Expression Systems) at 27 °C and 120 rpm. The Bac-to-Bac Baculovirus Expression System (Invitrogen) was used to generate high-titer recombinant baculovirus for GLP-1R, GCGR, GIPR, and three Gs subunits. P0 viral stock was produced by transfecting 5 μg recombinant bacmids into Sf9 cells (2.5 mL, density of 1 × 106 cells/mL) followed by 96 h incubation and then used to produce P1 and P2 baculoviruses. Cell culture was grown to a density of 3 × 106 cells/mL and then infected with four separate baculoviruses, including the receptor and three Gs subunits. Complex formation and purification were performed as described previously14. For GLP-1R, the GLP-1R-LgBiT-2MBP, G112, Gβ1-HiBiT and Gγ2 were co-expressed by infecting Sf9 cells at a ratio of 1:1:1:1. For GCGR, the GCGR-LgBiT-2MBP, G112, His6-tagged Gβ1-HiBiT and Gγ2 were co-expressed by infecting Sf9 cells at a ratio of 1:3:3:3. For GIPR–Gs complex, BRIL-GIPR(T345F)-LgBiT-2MBP, DNGαs (with 9 mutations), Gβ1-HiBiT and Gγ2 were co-expressed at a ratio of 1:3:3:3 in High Five cells. After 48 h incubation at 27 °C, the cells were collected by centrifugation and stored at –80 °C until use.
Cell pellets were lysed in a buffer containing 20 mM HEPES, pH 7.4, 50 mM NaCl and 2 mM MgCl2 supplemented with EDTA-free protease inhibitor cocktail (Bimake). Cell membranes were then collected by ultracentrifugation at 4 °C, 65,000× g for 35 min. A buffer consisting of 20 mM HEPES, 100 mM NaCl, pH 7.4, 10 mM MgCl2, 1 mM MnCl2, 100 μM TCEP, 25 mU/mL apyrase (Sigma-Aldrich), 15 μg/mL Nb35, protease inhibitor cocktail and 10% glycerol was used to resuspend the collected membranes. After incubating for 1 h at room temperature (RT), the complexes were solubilized from membrane using 0.5% (w/v) lauryl maltose neopentyl glycol (LMNG, Anatrace) and 0.03% (w/v) cholesteryl hemisuccinate (CHS, Anatrace) for 2 h at 4 °C. The supernatant was collected by centrifugation at 65,000× g for 30 min at 4 °C and then reacted with amylose resin for 2 h at 4 °C. After packing, the resin was washed with 20 column volumes of buffer containing 20 mM HEPES, pH 7.4, 100 mM NaCl, 10% (v/v) glycerol, 25 μM TCEP, 5 mM MgCl2, 1 mM MnCl2, 0.03% (w/v) LMNG, 0.01% (w/v) GDN and 0.008% (w/v) CHS. TEV enzyme was added to the resin and kept at 4 °C overnight to remove the 2× MBP tag. The complex was eluted from the resin and concentrated to 500 μL using a 100 kDa MWCO Amicon Ultra Centrifugal Filter. The size exclusion chromatography was carried out by loading the protein sample to Superdex 200 Increase 10/300GL or Superose 6 Increase 10/300GL (Cytiva) to obtain the monomer complex.
Nb35 expression and purification
Nb35 was expressed in the periplasm of E. coli BL21 (DE3) and purified by nickel affinity chromatography as described previously37. Briefly, the Nb35 target gene was transformed into BL21 and grown in a TB culture medium with 100 μg/mL ampicillin, 2 mM MgCl2 and 0.1% (w/v) glucose at 37 °C, 180 rpm. The expression was induced by adding 1 mM IPTG when OD600 reached 0.7–1.2. The cell pellet was collected by centrifugation after overnight incubation. The HiLoad 16/600 Superdex 75 column (Cytiva) was used to separate the monomeric fractions with running buffer containing 20 mM HEPES, pH 7.5 and 100 mM NaCl. The purified Nb35 was flash-frozen in 30% (v/v) glycerol by liquid nitrogen and stored at –80 °C until use.
Cryo-EM data acquisition
Cryo-EM samples were prepared by plunge vitrification in liquid ethane on a Vitrobot Mark IV (ThermoFisher Scientific) with blotting chamber set to 4 °C and 100% humidity. The purified samples at concentrations of 10–20 mg/mL were applied to glow-discharged holey carbon grids (Quantifoil, R1.2/1.3, Au 300 mesh) and blotted for 2.5 s, 3.5 s, and 3 s before plunging for the GLP-1R–Gs–Nb35, GCGR–Gs–Nb35 and GIPR–Gs–Nb35 complexes, respectively. Data were collected on a Titan Krios (ThermoFisher Scientific) 300 kV electron microscope equipped with a Gatan K3 Summit direct electron detector and serial EM3.7 was used to acquire cryo-EM images. The microscope was operated at a nominal magnification of 46,685× in counting mode, corresponding to a pixel size of 1.071 Å. The total exposure time was set to 7.2 s with intermediate frames recorded every 0.2 s, resulting in an accumulated dose of 80 e/Å2 fractionated into a movie stack of 36 frames with defocus range of –1.2 μm to –2.2 μm. Totally, 4741 movies for GLP-1R−Gs, 5553 movies for GCGR−Gs and 6517 movies for GIPR−Gs complexes were collected.
Cryo-EM data processing
The collected data were processed by cryoSPARC (v3.2.0) as summarized in Supplementary Fig. S2. Motion correction and CTF estimation for micrographs were done by patch motion correction and patch CTF estimation, respectively. Micrographs under 5 Å CTF resolution were cut off by Curate Exposures, and particles were auto-picked by template picker referenced from a previously published map of GLP-1R−Gs complex (EMBD code: EMD-30867)8. Auto-picking yielded particle projections that were subjected to two rounds of reference-free 2D classification to discard false-positive particles or particles categorized in poorly defined classes. After 2D classifications, particles from better classes were selected and classified by hetero refinement into 6 classes using Ab-initio volume generated from 200,000 particles as reference model. Particles from the class with the clearest model were selected for further 3D classification where soft masks around Gs subunits and ECD were used in focused classifications to improve the density in these regions. After several rounds of hetero refinement, 3D reconstruction was performed by NU-refinement, and the final map was improved to 2.54 Å for GLP-1R–Gs, 2.70 Å for GCGR–Gs and 2.86 Å for GIPR–Gs complexes. Reported resolution is based on the gold-standard Fourier shell correlation using the 0.143 criterion.
Model building and refinement
The models of the GLP-1R–Gs, GCGR−Gs and GIPR–Gs complexes were built based on the cryo-EM structures of GLP-1–GLP-1R–Gs (PDB: 6X18)27, glucagon–GCGR–Gs (PDB: 6LMK)26 and GIP–GIPR–Gs (PDB: 7DTY)15, respectively. The models were docked into relevant cryo-EM density maps using UCSF Chimera v1.1538, followed by iterative manual adjustment and rebuilding in COOT v0.9.239. Real-space refinement was performed using Phenix v1.19.140. The final refinement statistics were validated using the module comprehensive validation (cryo-EM) in Phenix v1.19.140. The final refinement statistics are provided in Supplementary Table S1. Structural figures were prepared using UCSF Chimera X and PyMOL v2.1 (https://pymol.org/2/).
Data availability
The atomic coordinates and the electron microscopy maps have been deposited in the Protein Data Bank (PDB) under the accession codes: 8WG7 (GLP-1R–Gs–Nb35 complex), 8WG8 (GCGR–Gs–Nb35 complex) and 8WA3 (GIPR–Gs–Nb35 complex), and Electron Microscopy Data Bank (EMDB) with the accession codes: EMD-37504 (GLP-1R–Gs–Nb35 complex), EMD-37505 (GCGR–Gs–Nb35 complex) and EMD-37390 (GIPR–Gs–Nb35 complex). All relevant data are available from the authors and/or included in the manuscript or Supplementary information.
References
Cao, C., Zhang, H., Yang, Z. & Wu, B. Peptide recognition, signaling and modulation of class B G protein-coupled receptors. Curr. Opin. Struct. Biol. 51, 53–60 (2018).
Cary, B. P. et al. New insights into the structure and function of class B1 GPCRs. Endocr. Rev. 44, 492–517 (2023).
An, K., Zhu, X. & Bai, C. The nature of functional features of different classes of G-protein-coupled receptors. Biology 11, 1839 (2022).
Hauser, A. S. et al. GPCR activation mechanisms across classes and macro/microscales. Nat. Struct. Mol. Biol. 28, 879–888 (2021).
Cong, Z. et al. Structural perspective of class B1 GPCR signaling. Trends Pharmacol. Sci. 43, 321–334 (2022).
Kobayashi, K. et al. Class B1 GPCR activation by an intracellular agonist. Nature 618, 1085–1093 (2023).
Zhao, L. H. et al. Conserved class B GPCR activation by a biased intracellular agonist. Nature 621, 635–641 (2023).
Cong, Z. et al. Molecular insights into ago-allosteric modulation of the human glucagon-like peptide-1 receptor. Nat. Commun. 12, 3763 (2021).
Bond, R. A. & Ijzerman, A. P. Recent developments in constitutive receptor activity and inverse agonism, and their potential for GPCR drug discovery. Trends Pharmacol. Sci. 27, 92–96 (2006).
Sadee, W. Ligand-free signaling of G-protein-coupled receptors: physiology, pharmacology, and genetics. Molecules 28, 6375 (2023).
Wan, W. et al. GLP-1R signaling and functional molecules in incretin therapy. Molecules 28, 751 (2023).
Bailey, C. J., Flatt, P. R. & Conlon, J. M. An update on peptide-based therapies for type 2 diabetes and obesity. Peptides 161, 170939 (2023).
Alexiadou, K., Anyiam, O. & Tan, T. Cracking the combination: Gut hormones for the treatment of obesity and diabetes. J. Neuroendocrinol. 31, e12664 (2019).
Cong, Z. et al. Structural basis of peptidomimetic agonism revealed by small- molecule GLP-1R agonists Boc5 and WB4-24. Proc. Natl. Acad. Sci. USA 119, e2200155119 (2022).
Zhao, F. et al. Structural insights into hormone recognition by the human glucose-dependent insulinotropic polypeptide receptor. Elife 10, e68719 (2021).
Zhao, F. et al. Structural insights into multiplexed pharmacological actions of tirzepatide and peptide 20 at the GIP, GLP-1 or glucagon receptors. Nat. Commun. 13, 1057 (2022).
Li, Y. et al. Structural analysis of the dual agonism at GLP-1R and GCGR. Proc. Natl. Acad. Sci. USA 120, e2303696120 (2023).
Chang, R. et al. Cryo-electron microscopy structure of the glucagon receptor with a dual-agonist peptide. J. Biol. Chem. 295, 9313–9325 (2020).
Zhang, Y. et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546, 248–253 (2017).
Hilger, D. et al. Structural insights into differences in G protein activation by family A and family B GPCRs. Science 369, eaba3373 (2020).
Sun, B. et al. Structural determinants of dual incretin receptor agonism by tirzepatide. Proc. Natl. Acad. Sci. USA 119, e2116506119 (2022).
Wong, T. S. et al. Cryo-EM structure of orphan G protein-coupled receptor GPR21. MedComm 4, e205 (2023).
Lin, X. et al. Structural basis of ligand recognition and self-activation of orphan GPR52. Nature 579, 152–157 (2020).
Ye, F. et al. Cryo-EM structure of G-protein-coupled receptor GPR17 in complex with inhibitory G protein. MedComm 3, e159 (2022).
Nie, Y. et al. Specific binding of GPR174 by endogenous lysophosphatidylserine leads to high constitutive G(s) signaling. Nat. Commun. 14, 5901 (2023).
Qiao, A. et al. Structural basis of G(s) and G(i) recognition by the human glucagon receptor. Science 367, 1346–1352 (2020).
Zhang, X. et al. Differential GLP-1R binding and activation by peptide and non-peptide agonists. Mol. Cell 80, 485–500.e7 (2020).
Hoare, S. R. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov. Today 10, 417–427 (2005).
Shuai, H., Xu, Y., Ahooghalandari, P. & Tengholm, A. Glucose-induced cAMP elevation in beta-cells involves amplification of constitutive and glucagon-activated GLP-1 receptor signalling. Acta Physiol. 231, e13611 (2021).
Jazayeri, A. et al. Crystal structure of the GLP-1 receptor bound to a peptide agonist. Nature 546, 254–258 (2017).
Zhang, H. et al. Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 546, 259–264 (2017).
Duan, J. et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 11, 4121 (2020).
Cong, Z. et al. Constitutive signal bias mediated by the human GHRHR splice variant 1. Proc. Natl. Acad. Sci. USA 118, e2106606118 (2021).
Liang, Y. L. et al. Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 555, 121–125 (2018).
Zhou, F. et al. Structural basis for activation of the growth hormone-releasing hormone receptor. Nat. Commun. 11, 5205 (2020).
Rasmussen, S. G. et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011).
Rasmussen, S. G. et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469, 175–180 (2011).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Acknowledgements
The cryo-EM data were collected at the Cryo-Electron Microscopy Research Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences. This work was supported by the National Natural Science Foundation of China (81872915, 82073904, 82273961 to M.-W.W.; 82273985, 82121005, 81973373 to D.H.Y.; 32200576 to Z.T.C.; 82204474 to F.H.Z.); Postdoctoral Science Foundation of China (2022M710806 to Z.T.C.; 2022M713266 to F.H.Z.); Postdoctoral Innovative Talent Support Plan of China (BX20220070 to Z.T.C.); the National Science & Technology Major Project of China — Key New Drug Creation and Manufacturing Program (2018ZX09735-001 to M.-W.W.; 2018ZX09711002-002-005 to D.H.Y.); STI2030-Major Project (2021ZD0203400 to Q.T.Z.); the National Key Basic Research Program of China (2018YFA0507000 to M.-W.W.; 2023YFA1800804 to D.H.Y.); Hainan Provincial Major Science and Technology Project (ZDKJ2021028 to D.H.Y. and Q.T.Z.); Shanghai Municipality Science and Technology Development Fund (21JC1401600 to D.H.Y.) and Program of Shanghai Academic/Technology Research Leader (23XD1400900 to D.H.Y.).
Author information
Authors and Affiliations
Contributions
M.-W.W., D.H.Y. and Q.T.Z. initiated the project and supervised the studies. Z.T.C., F.H.Z. and Y.L. performed research and participated in manuscript preparation; Z.T.C., Q.T.Z., F.H.Z., and Y.L. conducted map calculation, built the models of the complexes and carried out structural analyses; G.L. assisted with data analysis; Y.T.M., X.Y.C., Y.Y.C., S.L. and X.Q.C. performed functional studies; Z.T.C., Q.T.Z. and M.-W.W. wrote the manuscript with inputs from all co-authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cong, Z., Zhao, F., Li, Y. et al. Molecular features of the ligand-free GLP-1R, GCGR and GIPR in complex with Gs proteins. Cell Discov 10, 18 (2024). https://doi.org/10.1038/s41421-024-00649-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41421-024-00649-0
