
Abstract
Myasthenia gravis (MG) is a rare autoimmune disease. Although the impact of immune cell disorder in MG has been extensively studied, little is known about the transcriptomes of individual cells. Here, we assessed the transcriptional profiles of 39,243 cells by single-cell sequencing and identified 13 major cell clusters, along with 39 subgroups of cells derived from patients with new-onset myasthenia gravis and healthy controls. We found that B cells, CD4+ T cells, and monocytes exhibited more heterogeneity in MG patients. CD4+ T cells were expanded in MG patients. We reclustered B cells and CD4+ T cells, and predict their essential regulators. Further analyses demonstrated that B cells in MG exhibited higher transcriptional activity towards plasma cell differentiation, CD4+ T cell subsets were unbalanced, and inflammatory pathways of monocytes were highly activated. Notably, we discovered a disease-relevant subgroup, CD180− B cells. Increased CD180− B cells in MG are indicative of a high IgG composition and were associated with disease activity and the anti-AChR antibody. Together, our data further the understanding of the cellular heterogeneity involved in the pathogenesis of MG and provide large cell-type-specific markers for subsequent research.
Similar content being viewed by others

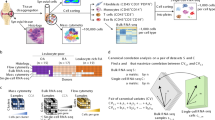

Introduction
Myasthenia gravis (MG) is a rare autoimmune disease characterized by skeletal muscle weakness caused by disrupted neurotransmission at the neuromuscular junction, with a prevalence of 150–250 cases per 1 million1. B and T cell hyperactivity and the autoantibodies secreted by B cells mediate the autoimmune phenotype by responding to muscle neuronal nicotinic receptors2. In 80%-85% of MG patients, the pathogenic antibodies are anti-AChR3. CD4+ T cells, also known as T helper cells, are essential for helping antigen-experienced B cells produce these pathogenic high-affinity antibodies. The innate immune system also plays a significant role in these processes. Dendritic cells (DCs) can function as antigen-presenting cells and induce autoimmunity by promoting the expansion and differentiation of autoreactive T cells4. Autoantibodies produced by B cells can activate myeloid cells and form the proinflammatory milieu, which in turn promotes the disorder of adaptive auto-reactive T or B cells5.
Although major cell types involved in the pathogenic processes of MG are known, key cellular subsets, their transcriptomes characteristics, and the interactions through which they promote MG have remained largely unclear. Studies of the peripheral blood after sorting specific cell subtypes by flow cytometry in bulk also failed to capture the natural transcriptome signatures. Additionally, approximately 10% of MG patients are treatment refractory6, highlighting the need to better understand specific disease-associated pathogenic events. Furthermore, anti-AChR antibodies are markers for diagnosis and disease classification of MG patients but not for disease severity7. Therefore, additional markers are needed for indicating disease severity.
Here, we applied single-cell RNA sequencing (scRNA-seq) to visualize a high-resolution immune landscape of MG patients and compared them to healthy controls (HCs). We first identified 13 major cell groups and assessed the primary changes of these major types of cells. We next analyzed B cells and CD4+ T cells with more granularity due to their important roles in the adaptive immune process and the shift between MG patients and HCs. We characterized their cellular network to better understand their cellular identity and the process of their differentiation.
In summary, our analysis provides insights into the major immune cells in MG and HCs, the possible altered transcriptional differentiation trajectories, the higher connection to plasma cells in MG patients, and cellular cross-talk with potential relevance to pathogenic mechanisms. Moreover, our analysis defines a subset of B cells known as CD180− B cells that clinically coincide with anti-AChR antibody and disease severity. We also showed that immunosuppressive therapy restored CD180− B cells frequency. Finally, we investigated the relationship of major changed cell types and risk genes of MG patients, highlighting the increasing expression of HLA-DRA, HLA-DQA1, HLA-DQB1, and HLA-DPB1 in B cells of Chinese early-onset MG patients. Altogether, these analyses help characterize the cellular pathological mechanism by investigating cell differentiation and cellular interactions, identifying large cell type-based markers and pathways for understanding the pathogenic events that occur in MG to identify new effective therapeutics.
Results
Single-cell survey of major changes in transcriptional profiles between MG patients and healthy controls
To characterize the immune changes in MG patients, we performed scRNA-seq and generated 39,243 high-quality single-cell transcriptomes of PBMCs from 2 early-onset MG (EOMG) patients and 2 HCs using the 10× Genomics platform (Fig. 1a, Supplementary Table S2). MG patients had not undergone immunotherapy treatment (Supplementary Table S1).

a Overview of the scRNA-seq experiment: PBMCs were isolated from two healthy controls and two myasthenia gravis patients. Overall, ~39,243 cells were included in the functional analysis. b Uniform manifold approximation and projection (UMAP) representation of scRNA-seq data showing the seven main cell types: CD8+ T cells, CD4+ T cells, B cells, CD14+ monocytes, FCGR3A+ monocytes, NK cells, and DCs. c Cluster abundance across all samples. d Number of DEGs between MG and HC cells within each cluster projected onto the UMAP. DEG: |log fold change| > 0.5; P-value < 0.05 was calculated using DESeq2. e Quantification of differences between major immune cells between MG patients and HCs. Each dot represents a sub-sample of 500 cells from the principal component analysis space for MG patients and HCs. For the random groups, we sampled 500 cells of sample type. The height of the bar represents the mean values of the subsamples. All comparisons were significant due to 100 replicates of testing, while the mean fold change varied from 1.77-fold (monocytes) to 1.21-fold (CD4+ T cells). P values are from a Wilcoxon rank-sum test comparing the MG vs HC groups to the random selection for each type of major immune cell.
We first partitioned the single-cell profiles into 13 clusters composed of major cell subtypes, including CD8+ T cells, CD4+ T cells, B cells, CD14+ monocytes, FCGR3A+ monocytes, NK cells, and DCs (Fig. 1b, Supplementary Figs. S1, 2) using an unsupervised method. Cell types were identified by known unique signature genes, CD3E (T cells), MS4A1 and CD79A (B cells), CD14 and FCGR3A (monocytes), LILRA4, and FCER1A (DCs), and NKG7 (NK cells) (Supplementary Fig. S1). A small population of platelets was also present after isolating PBMC preparations, which were excluded from further analysis.
We next assessed alterations in transcriptional profiles between MG patients and HCs in two ways. We calculated differentially expressed genes (DEGs) in each cluster (see Materials and Methods) and then projected the numbers of DEGs on UAMP. The DEGs revealed broad transcriptional changes in immune cells, with the most prominent in B cells and CD4+ T cells (Fig. 1d). In addition, we measured the distance between the major types of cells using Bhattacharyya distance8. DCs were not included due to the limited number of cells available. This result revealed large differences in B cells and monocytes (Fig. 1e) between MG patients and HCs, while CD4+ T cells, NK cells, and CD8+ T cells were more similar in this analysis. Actually, B cells exhibited the highest difference with 1.7-fold changes, followed by CD14+ monocytes with 1.67-fold changes (data not shown). Cell proportion analysis revealed that CD4+ cells were significantly expanded in MG patients compared to HCs (Fig. 1c). Overall, we identified the major immune cells and characterized broad changes of transcriptional profiles and cell proportions in MG patients compared to HCs, revealing prominent changes in B cells, CD4+ T cells, and monocytes.
B cell clustering and subgroup analysis
We next bioinformatically separated and reclustered B cells. Clustering of B cells revealed 9 distinct clusters (Fig. 2a). Combined with reported marker genes9,10, we identified naïve B cells (clusters 0 and 3), class-switched memory B cells (clusters 2 and 5), CD27+ memory B cells (clusters 1 and 7), CD27− memory B cells (cluster 6), pre-ASCs (cluster 4), and plasma cells (cluster 8) (Fig. 2b). We next assessed function by gene set enrichment analysis (Fig. 2c), which further supported the cluster annotation. Specifically, GSVA analysis revealed that cluster 6 was associated with cytokines, chemokines, toll-like receptor signaling pathway, and JAK-STAT and MAPK signaling. Cluster 5 was associated with B cell receptor signaling and major histocompatibility complex (MHC) class II antigen presentation. Cluster 4 was enriched in cell cycle set and intrinsic signaling, such as the JAK-STAT and MAPK pathways, implying that they were highly activated (Fig. 2c). This result was also consistent with the GO analysis indicating that clusters 4 and 8 were highly activated. KEGG analysis also showed that clusters 4 and 8 shared similar pathways (Supplementary Fig. S4). Furthermore, cluster 4 highly expressed HOPX, a marker of PrePB10. Thus, we infer that cluster 4 potentially represents a transient state before plasma cell transformation. Class-switched B cells (cluster 2) had undergone antibody class switching and were indicators of B cell activation by antigen stimulation, coincident with their gene enrichment in antigen processing and presentation process in GSVA analysis.

a UMAP plot displaying 4133 B cells from two MG patients and two HCs separated into 9 subtypes. b Violin plots showing key gene markers across B cell subsets. c Gene set enrichment revealed differences in pathway activities and cell identity of naive, memory, switched B cells, and plasma B cells. d B cells were sorted using the DDRTree algorithm and projected onto the different cell states using the color in a. Transcriptional activity was estimated by measuring the ratio between unspliced and spliced mRNAs. The length of the arrow represents the transcriptional activity. e UMAP plots showing the expression of BCL3 and POU2AF1 genes in B cells (top) and the AUC of the estimated regulon activity of the corresponding TFs, predicting the degree of expression regulation of their target genes (bottom). f Heatmap of the AUC scores of expression regulation by transcription factors (regulon activity), as estimated using SCENIC, followed by comparisons between two groups using the t-test. The twenty transcription factors with the highest upregulated expression in MG are shown.
In addition, we comprehensively inferred transcription factor (TF) regulatory networks by SCENIC analysis, highlighted differentiation-associated TFs, and predicted additional markers. As expected, plasma B cells exhibited large transcription factor differences from their B cell predecessors, and the differentiation to plasma B cells is a process that loses B cell identity11. SPI1(PU.1), ETS1, PAX5, SPIB, and BACH2 are transcription factors that represent B cell identities11 and loss of PAX5 implies the ASC differentiation12. As expected, they are expressed in nonplasma cells. STAT3, which functions in IL-21 stimulated B cell differentiation13, are also not expressed on plasma B cells. Plasma cells have high regulon activity of IRF4, Prdm1, and XBP1, as expected. Additional increased TF regulon signatures included CHD2, IRF7, ZBTB18, MYBL2, DDIT3, and ATF6B for plasma cells. Plasma B cells and class-switched memory B cells have high regulon activity of POU2AF1, which is required for GC formation and thus has unique roles in T cell responses14 and generating ASCs15. This implies class-switched memory B cells are an important stage before the terminal ASCs to produce high-affinity antibodies. Pre-ASCs have regulon activity of TBX21, which positively regulates isotype switching to IgG isotypes. Additional possible markers for B cell subsets are provided in the supplement (Supplementary Fig. S6).
Extensive B cell heterogeneity and altered differentiation trajectories
Class-switched B cells were expanded in MG. GSVA analyses revealed that they participate in antigen processing and presentation, supporting that class-switched B cells are highly activated, and their function is increased in MG patients (Fig. 2c, Supplementary Fig. S3).
To characterize transcriptomic changes in B cells between MG patients and HCs, as well as their biological significance, DEGs were calculated (Fig. 3a) and a detailed analysis of the DEGs was performed by the functional annotation of Gene Ontology (GO) (Fig. 3b). Pathways related to antigen processing and presentation and antigen processing and presentation of peptide antigen via MHC class II were upregulated, driven by HLA-DPA1, HLA-DQB1, and HLA-DRB5. HLA-DPA1 and HLA-DQB1 genes, which are also reported predisposing risk genes in Chinese MG patients16. Also upregulated pathways were involved in leukocyte differentiation and activation, with elevated ZFP36L2, IRF1, JUN, JUNB, RELB, CD83, HLA- ZFP36L2, IRF1, JUN, JUNB, RELB, CD83, HLA-DPA1, EZR, DUSP1, and CXCR4 genes. The CXC-chemokine receptor 4 (CXCR4), which is important for the recruitment of ASCs17, was increased in MG patients, implying the increasing homing process of tissues. Overall, antigen presentation, immune response signaling, and differentiation pathways were activated, with cytokine participation contributing to deficits in peripheral immune tolerance.

a Volcano plot showing DEGs between MG patients and HCs. DEG: |log fold change| > 0.5; P value < 0.05 was calculated using DESeq2. b Enrichment analysis of DEGs from B cells between MG patients and HCs (selected among upregulated pathways in MG patients, P value < 0.05). c, d CD180− B cells are increased in MG patients. e, f CD180− B cells are the major IgG secreting cells in MG patients and HCs. CD180− IgG cells are significantly higher than CD180+ IgG cells in MG patients. g Circulating CD180− B cells are associated with disease activity. h Circulating CD180− B cells are associated with titers of anti-AChR antibodies. i Immunotherapy decreases CD180− B cells and is associated with improved disease activity.
To reveal the programming processes with alterations in B cells, we used the Monocle 2 method to construct the differentiation trajectory of B cells from each sample (Fig. 2d). In HCs, trajectory analysis revealed a gradual transition from naïve B cells (cluster 0, 3) to the fate of plasma B cells (cluster 8) or memory B cells (cluster 6), with most cells undergoing differentiation to plasma B cells. In MG patients, while the differentiation pathways were similar, cluster 4 was on average closer to the terminal cluster 8 and seemed to have an aberrant developmental process. Cluster 4 highly expressed BATF, without which B cells fail to induce Aicda and differentiate into plasma B cells18. Cluster 4 also highly expressed TBX21, and a previous study demonstrated that T-bet is expressed by memory pre-ASCs19. RNA velocity further indicated that higher numbers of unspliced RNAs were present in the naïve cells of MG patients (Fig. 2d). Moreover, we observed increased activity towards plasma B cells in MG patients. The above analysis revealed the more highly activated state of naïve B cells and the enhanced propensity for differentiation towards a plasma B cell phenotype in MG patients.
Next, to assess the TFs underlying differences in B cells between MG and HCs, we applied SCENIC analysis (Fig. 2e, f). Among the 20 most elevated TFs, BCL3, and POU2AF1 are associated with differentiation. Eμ-BCL-3 transgenic mice with BCL-3 overexpression led to a hyperactivated state of B cells20. Furthermore, the top enriched BCL3 also serves as a unique factor to control NF-kB activity in the nucleus21, suggesting a role for immune activation and inflammatory regulation. Indeed, the activities of nuclear factor (NF)-κB subunits (RELB) were also highly upregulated in B cells from MG patients. These results also support the prominent feature of MG patients wherein B cells are highly activated and exhibit aberrant differentiation states.
Correlations of B cell subtype-specific signatures with disease activity and autoantibodies
Notably, DEGs revealed decreased expression of the CD180 gene in MG patients compared to HCs (Fig. 3a) (log2Fc = −0.61, adj. P = 2.92E−13). Flow cytometry analysis further supported that the frequencies of CD180 negative B cells were significantly increased in both ocular and generalized MG patients (P = 0.004). The gating strategy is shown in Supplementary Fig. S5. The frequencies of CD180 negative B cells were not different between ocular and generalized MG patients (Fig. 3c, d), suggesting that the increased CD180 negative B cells might be a common pathological mechanism of MG.
Evaluating disease severity by the Quantitative Myasthenia Gravis (QMG) score22,23, the CD180 negative B cells correlated with disease severity (R2 = 0.30, P = 0.025) in MG patients (Fig. 3g). The increased CD180 negative B cells were also associated with anti-AChR autoantibodies (R2 = 0.31, P = 0.018) (Fig. 3h). As anti-AChR antibodies primarily belong to the IgG1 and IgG3 subclass24, we examined the IgG secreting B cells by intracellular flow cytometry. The results showed that though CD180 negative B cells were the major IgG secreting B cells, CD180 positive B cells still stained slightly positive for intracellular IgG and still existed in both MG patients and HCs. However, CD180 negative B cells from MG patients demonstrated significantly increased IgG secreting ability compared to CD180 positive B cells (n = 6, P = 0.031), while no significance was observed in HCs (n = 8, P = 0.313) (Fig. 3e, f).
Assessing treatment efficacy by QMG score25, after immunosuppressive therapy, CD180 negative B cells decreased (P = 0.031), and patients exhibited clinical improvement along with decreased QMG scores (P = 0.036) (Fig. 3i). In summary, we identified a cell group that may have an important role in anti-AChR autoantibody secretion and indicated disease activity.
T cell and NK cell clustering and subgroup analysis
We subgrouped T cells and NK cells into 21 subsets and named them according to reported marker genes. Seven CD4+ T cell clusters (0, 1, 5, 6, 8, 14, 19), 5 CD8+ T cell clusters (3, 11, 12, 13, 15), 3 γδ T cell clusters (4, 7, 10), 1 MK cluster (18), 1 NKT cluster (9), proliferating T cell cluster (20) and 3 NK cell clusters (2, 16, 17) were identified (Fig. 4a).

a UMAP plot displaying T cells and NK cells from two MG patients and two HCs separated into 21 subtypes. b Violin plots showing key gene markers across T cells and NK cells. c Enrichment analysis of DEGs in CD4+ T cells between MG patients and HCs (selected among upregulated pathways in MG patients, P value < 0.05). d CD4+ T cells were sorted using the DDRTree algorithm and projected onto the different cell states using the color in a. e UMAP plots showing expression of the STAT3, JUND, and JUN genes in CD4+ T cells (top) and the AUC of the estimated regulon activity of the corresponding TFs, predicting the degree of expression regulation of their target genes (bottom). f Heatmap of the AUC scores of expression regulation by transcription factors (regulon activity) as estimated using SCENIC, followed by comparisons between two groups using t-test. The top 20 transcription factors with the highest upregulated expression in MG are shown.
In the T cell compartment, the combination of the GSVA analysis results and reported marker genes identified a CD4+ T cell subpopulation of naïve CD4+ cells expressing CCR7 (T4naive, cluster 0), central memory CD4+ T cells (T4cM, cluster 1), effector memory CD4+ T cells (T4eM, cluster 5) with low expression of CCR7, effector CD4+ cells (T4eff, cluster 6) expressing NKG7 and GZMK, regulatory (T4reg, cluster 14) and interferon-activated cells (TIFN, cluster 19) highly expressing ISG15. In addition, cluster 8 highly expressing CCR7 belonged to naïve CD4+ T cells or central memory CD4+ T cells. CD8+ T cells included naïve CD8+ cells (T8naive, clusters 3 and 15) expressing CCR7, central memory CD8+ T cells (T8cM, cluster 12) expressing IL7R, cytotoxic CD8 cells (T8cyto1, clusters 11) expressing GZMH and GZMB, cytotoxic CD8 cells (T8cyto2, cluster 13) expressing GZMK and γδ T cells (clusters 4, 7, 10) expression high levels of TRDC. Additional populations included a small group of megakaryocyte-like cells (MK, cluster 18) expressing PPBP and cluster 20, which represented proliferating cells with high expression of TYMS and MKI67 (Fig. 4b). MG patients exhibited higher proportions of T4cM and T4eM.
We next used SCENIC to identify TF regulatory networks. Naïve CD4+ T cells highly expressed UQCRB, TCF7, CHD2, and MYC. Interferon-activated cells highly expressed IRF1, STAT1, and STAT3. Previous studies have shown that IFN-γ activating STAT1 is important for Th1 differentiation26, and STAT3 is involved in Th17 differentiation and maintenance27. IRF1 can regulate the differentiation and expansion of Th1 and Th1728. Combining the high expression of IL17A in clusters 1, 5, and 19, TIFN may represent the Th1 and Th17 states. HMGB1, which was previously demonstrated to directly enhance immune inhibitory functions of Tregs29, was highly expressed in Tregs. Interestingly, T4eM and T4eff shared similar expression patterns for many TFs, suggesting that the T4eff may be derived from T4eM. Our data characterize the regulators of the CD4+ T cell subgroup, which helps to further investigate the modulation of CD4+ T cell activation and plasticity.
CD4+ T cell heterogeneity
Since CD4+ T cells were significantly expanded in MG patients and given their essential roles in stimulating B cells to produce high-affinity antibodies, we determined the transcriptional changes between MG patients and HCs. DEGs were calculated and followed by enrichment analysis using Metascape (Fig. 4c). The results showed that TNF signaling, NOD-like receptor signaling, IL-17 signaling, T cell receptor signaling, and CD40 pathways were highly enriched in MG patients, indicating that Th17 cells and T helper type 40 (TH40) cells were expanded and exhibited greater antigenic stimulation of peripheral CD4+ T cells in patients with MG compared to HCs. Murine studies highlighted the importance of Th17 cells for their contribution to the loss of B-cell tolerance30.
To investigate the relationships between the different states of CD4+ T cells, differentiation trajectory analysis (Fig. 4d) was performed and showed that naïve T cells and central memory T cells primarily aggregated on the pseudotime backbone. Cytotoxic CD4+ T cells and cluster 8 were primarily located in different directions. The direction to cluster 8 might represent the quiescent state, while the other represents steps to the terminal stages. Interestingly, effector CD4+ T cells exist in both ends of the two directions in myasthenia gravis, while they primarily exist in one direction after the effector memory CD4+ T cells or Treg stage and have more branches in HCs. Previous murine research demonstrated that disequilibrium of the CD4+ helper T-cell subsets promoted the development of EAMG31. Comparing the differentiation progression of CD4+ T cell subsets between MG and HCs, CD4+ T cell subsets except for TIFN showed extensive differences (Supplementary Fig. S7). Our result also indicated that CD4+ T cells from MG patients exhibit imbalance in CD4+ T cell subgroups from their transcriptomes.
We next explored transcription factors (TFs) in CD4+ T cells that might be involved in promoting autoimmunity (Fig. 4e, f). Among the 20 most highly elevated TFs, we observed that regulon activity of STAT3, JUND, and JUN, which are associated with differentiation, was elevated in MG patients. JUND reportedly regulates lymphocyte proliferation and Th cell cytokine expression32. Our results identify key transcription factors associated with changes in CD4+ T subgroups.
Myeloid clustering
11,507 myeloid cells were detected in MG and HCs. Myeloid cells were then sub-grouped into 9 clusters (Fig. 5a, b), including three CD14+ monocytes (clusters 0, 1, and 4) and two CD16+ monocytes (clusters 2 and 3). Three myeloid subsets were defined as DCs, including two conventional DCs (cDCs) highly expressing CLEC9A or FCER1A (clusters 6 and 8) and one plasmacytoid dendritic cells (pDC) highly expressing IRF7 and LILRA4 (cluster 7). We designated one myeloid subset as macrophages according to high expression of CSF3R and ISG15 (cluster 5).

a UMAP plot displaying myeloid cells from two MG patients and two HCs separated into 9 subtypes. b Violin plots showing key gene markers across myeloid cells. c 3D diffusion map displaying the developmental trajectory of myeloid cells. The result shows the possible activation paths of macrophages/cDCs. Cells are colored by their derived clusters. pDCs, plasmacytoid dendritic cells; cDCs, conventional DCs. d Enrichment analysis of DEGs in dendritic cells between MG patients and HCs (selected among upregulated pathways in MG patients, P value < 0.05). e GSVA analysis of CD14+ monocytes between MG patients and HCs. f Dot plot of selected significant paracrine receptor-ligand interactions between CD14+/CD16+ monocytes and cDCs discovered using Cellchat. Commun.Prob., communication probability.
We constructed a diffusion map to depict the developmental trajectory of myeloid cells (Fig. 5c). CD14+ monocytes are the root cell group. The diffusion map of monocytes/macrophages/DCs revealed three developmental paths, possibly corresponding to the macrophage, cDC, and pDC phenotypes, respectively.
Myeloid heterogeneity analysis
CD14+ monocytes, which can further differentiate into CD16+ monocytes, macrophages, or DCs, exhibited large differences between MG patients and HCs using Bhattacharyya distance. DEG analysis of CD14+ monocytes showed that MG patients express high levels of the inflammatory markers S100A4, S100A8, S100A9, S100A10, and S100A12. We found most distinguishing pathway changes between MG patients compared to HCs were inflammatory-relevant pathways, including MAPK family signaling, TNF signaling, TLR4, interferon, and interleukin signaling (Fig. 5d). Furthermore, diseases of metabolism were also enriched in MG patients, such as disorders of glycosylation and carbohydrate metabolism.
As expected, we found that the genes with increased expression in DCs in MG patients were enriched in antigen processing and presentation, cytokine-mediated signaling pathway, and TNF-alpha/nuclear factor k-light-chain-enhancer of activated B cells (Nf-kappa B) signaling (Fig. 5e). Nf-kappa B signaling activation controls the expression of critical co-stimulatory molecules, such as CD80, CD86, and MHC class II, along with pro-inflammatory cytokines, such as TNF-alpha. Our result emphasized the importance of Nf-kappa B signaling in DCs in myasthenia gravis.
MG alters crosstalk between monocyte and cDC subpopulations. CDCs can derive from CD16+ and CD16− monocytes and CD16+ monocytes can derive from CD14+ monocytes. Previous work reported that an estimated 25% of the circulating inflammatory monocytes will differentiate into migrating DCs33, thus we explored cDC, CD14+ monocyte, and CD16+ monocyte receptor–ligand pairing in both MG patients and HCs, which may represent the differentiation of myeloid cells. We found altered receptor–ligand pairing between CD14+ or CD16+ monocytes and cDCs other than pDCs (Fig. 5f), suggesting that the differentiation of circulating monocytes into cDCs might be modulated through TNF signaling and TGF-β signaling and the cDCs derived from monocytes might be with different phenotypes and functional profiles in MG patients.
Global comparison analysis of communications among immune cells
Excluding cell-intrinsic information, scRNA-seq can also indicate putative cell-extrinsic interactions by integrating ligand and receptor information. We used CellChat to investigate the putative interactions between the major types of immune cells in MG versus HCs34.
We identified 13 significant signaling pathways among the 13-cell group by CellChat. The top signaling pathways that are more enriched in MG patients are colored orange. Circos plots were used for visualization for the specific interactions among the 13-cell group. The results showed increasing activities of pathways, including IL2, IL4, CD40, CD70, BMP, RESISTIN, TNF, WNT, and NT, in MG patients (Fig. 6a). In addition, CD4+ T cells had increasing autocrine activity for soluble IL2 and had a massive expansion of interactions with other cell types through the CD40 signaling pathway in MG patients. Another interesting observation is that IL4 pathways from CD4+ T cells to B cells were enriched. Our result proved the T cell also increasingly helping to B cells in peripheral blood except for germinal centre in MG patients. Since the loss of IL-4 was associated with loss of IgG135, the increasing IL-4 might lead to the increasing IgG1 in MG patients. Further analysis showed that the CD70 signaling pathway was most enriched from B cells to CD4+ T cells in MG patients (Fig. 6b). CD16+ monocytes are the prominent influencer controlling TNF signaling, and the TNF signaling pathway was most enriched from CD16+ monocytes to CD14+ monocytes (primarily TNF-TNFRSF1B) (Fig. 6c). These findings are consistent with the known critical roles played by myeloid cells in initiating inflammation. Notably, CellChat predicted that WNT and NT signaling pathways are from a source of both lymphoid cells and myeloid cells, including CD4+ T, CD8+ T, and CD14+ monocytes, and they have similar targets. This reveals that WNT signaling in MG is complex and redundant with multiple ligand sources. CD4+ T cells are the predominant source and mediator for WNT signaling, suggesting their potential roles as a gatekeeper of WNT signaling in MG (Supplementary Fig. S8).

a The significant signaling pathways were ranked based on their differences in overall information flow within the inferred networks between HCs and myasthenia gravis patients. We show the more enriched signaling pathways colored in orange in myasthenia gravis. b Circos plot showing the inferred intercellular communication network across major cell types using the color of b for HCs and MG patients. c Heatmap showing the relative importance of each cell group based on the computed four network centrality measures of the TNF signaling pathway. Information flow refers to the overall communication probability.
Expression of MG-relevant genes and biological pathways
Previous GWAS studies have linked genetic variants to MG; however, the cellar and biological processes of the risk genes are little known due to large variants. We collected large GWAS studies of MG patients and identified variants in MHC class II locus, protein tyrosine phosphatase nonreceptor type 22 (PTPN22), TNFAIP3 interacting protein 1 (TNIP1)36, cytotoxic T-lymphocyte–associated protein 4 gene (CTLA4)37, combining with the predisposing gene of EOMG including HLA-DRA, HLA-DR3, HLA-B8, HLA-DPB1, HLA-DQB1, HLA-DQA1, CD86, AKAP12, VAV1, TNFSF13B (B-cell activating factor, BAFF), and TNF37,38,39,40. We also included the α-subunit of the AChR encoding gene CHRNA141,42, IgG receptor genes (FCGR2A, FCGR3A, and FCGR3B), and cytokines and cytokine receptors genes (TNFB, TNF43, IL1B44, IL1A45, IL1046, IFNG, IL17A, and IL17F47).
Here, we applied transcriptomic atlas to relate the patterns of MG genetic risk with patterns of cell-specific expression (Fig. 7a, b). Some risk genes were not available in our data, and 20 genes were ultimately analyzed. While the human leukocyte antigen (HLA) locus remains the most strongly associated risk factor for MG and is associated with autoantibody expression41, relevant genes, including HLA-DPB1, HLA-DQB1, HLA-DQA1, and HLA-DRA, are expressed predominantly in DCs and B cells. The CHRNA1 gene is expressed mainly in B cells, with a small amount of CHRNA1 expression in CD4+ T cells. A previous study also reported that CHRNA1 and HLA-DQA1 are associated with autoantibody titers48, further emphasizing the importance of B cells. Genes with functions that regulate T cell activation49 are expressed most highly in CD4+ T cells (e.g., CTLA4, IL17A, IL10). Another T cell activation regulatory gene, PTPN22, is highly expressed in CD8+ T cells. IL1B, involved in the modulation of autoimmune inflammation50, is primarily expressed in monocytes and DCs.

a Expression of 20 genes associated with myasthenia gravis by cell type in MG patients. HLA genes have the highest expression in B cells and DCs. b Ternary plot of the 20 risk genes of two MG patients, comparing CD4+ T cells (left), B cells (top), and monocytes (right). The color of each node represents different genes, where the node size represents the average expression of the three cell types.
Several genes associated with immune or inflammatory signaling of autoimmune disease, including VAV138 and TNIP151, are expressed in multiple cell types. TNF, TNFSF13B, and FCGR2A are highly expressed in monocytes, which as previously reported are associated with the autoimmune state of monocytes52. AKAP12, associated with pathway activation, was primarily expressed in monocytes and NK cells. IFNG, FCGR3A, and FCGR3B were primarily expressed in NK cells and monocytes. CD86 was highly expressed in monocytes, DCs, and B cells.
Combined with the DEG analysis, the significantly altered expression of MG risk genes might lead to impaired functions. For example, HLA-DRA was upregulated in B cells, DCs, and CD8+ T cells. HLA-DQA1 was upregulated in B cells, DCs, CD8+ T cells, and monocytes. HLA-DQB1 was upregulated in B cells, DCs, and monocytes. HLA-DPB1 was upregulated in B cells, CD8+ T cells, monocytes, and NK cells. Overall, the HLA class II genes are differentially expressed in B cells of myasthenia gravis, suggesting their function of pathogenic antigen presentation. Further studies are needed to elucidate the functional changes of these specific cells in MG.
Discussion
In this study, we aimed to understand the cellular transcriptional changes in MG patients. We are the first to create a high-resolution atlas and to systematically discuss the cellular heterogeneity and the impaired peripheral tolerance from MG patients. We performed scRNA-seq of 39,243 cells and then annotated their cell type identity, followed by DEG and pathway analysis. At this resolution, we first identified 13 cell clusters and subsequently reclustered them into 39 subsets. We found general differences in monocytes, CD4+ T cells and B cells, while NK cells and CD8+ T cells were more similar between MG and HCs. Based on the 39 clusters, we also compared the differentiation shift and observed aberrant B cell and CD4+ T cell differentiation by monocle2 and RNA velocity. We found large cell type-based DEGs, which might be referenced for investigating the pathological roles of immune-cell subsets. The scenic analysis provides clues for identifying new candidate TFs involved in B cell and T cell dysfunction. Cell communication analysis also provides potential drug targets for treating the disease.
Our approach to transcriptional profiling of the peripheral blood in myasthenia gravis patients is unique because it did not involve presorting. This unbiased method uses the full transcriptome and might provide a more detailed and comprehensive overview. We next noted several key observations. We found that B cell differentiation was highly connected with plasma cells. DEGs and metascape analysis revealed increasing antigen processing and presentation and MHC II molecular expression, including HLA-DRA, HLA-DQA1, HLA-DQB1, and HLA-DPB1, which are also MG-associated risk genes.
Notably, we found that CD180 is decreased in MG patients. We further verified this with an independent cohort by flow cytometry and found a large expansion of CD180− B cells in MG patients, which was correlated with anti-AChR antibodies and disease activity. A similar phenomenon with the expansion of peripheral CD180− B was also observed in SLE53. Our results showed that CD180- B cells were the IgG-secreting B cells both in MG and HCs. We further showed that CD180 negative B cells exhibited increased IgG-secreting ability. Previous studies reported that CD180 knockout mice have one-tenth the serum concentrations of IgG3 than WT mice54, further indicating the importance of the CD180 gene for IgG secretion. In summary, CD180 expression is altered in B cells and might have an important role in autoantibody secretion. CD180− B cells may serve as indicators for the disease activity of MG. In addition, we revealed some TFs may regulate the observed aberrant differentiation and activity of B cells in MG patients, including BCL3, POU2AF1, JUN, RELB, and STAT3. Few of these TFs and their function on B cells were researched in MG, but some TFs were reported in other immune diseases and most of the TFs are associated with antibody-secreting. Bcl-3-deficient Mice have a decreased B cell population, impaired formation of GCs, and severe defects in the production of antigen-specific antibodies55. POU2AF1(Bob1)-sufficient mice lead to abrogated germinal center B cell formation, anti-CII antibody production, and Collagen-induced arthritis development56. Consistent with data from animal studies, Expression of POU2AF1 mRNA was elevated in B from synovial fluid of rheumatoid arthritis patients and was strongly correlated with CD21L, a molecular marker of GCs56. In humans with RelB deficiency, patients suffer from severe immunodeficiency with shortage-specific antibodies due to the halted B cell development and the absent CD27+ memory B cells57. In B6.MRL/lpr mice, STAT3 deficiency led to the defect of plasma cell differentiation and decreased autoantibody production58.
For T cells, we also observed expansion and altered differentiation of CD4+ T cells. The highly enriched IL-17 signaling pathway and CD40 pathway in MG patients suggested the expansion of Th17 and CD40+ CD4+ T cells. Similar to a previous study that showed CD40 can be expressed on CD4+ T cells. Furthermore, CD40+ CD4+ cells are reported to be highly pathogenic and promote broad TCR repertoire expansion in autoimmune type 1 diabetes59,60, which partly explains the increased TCR signaling. Cell communication results revealed increased CD70 signaling between plasma B cells and CD4+ T cells. CD27 is expressed on the majority of T cells and is upregulated with T cell activation, and its ligand CD70 is transiently expressed on activated B cells. CD27 ligation can promote effector cell formation. This might suggest activated B cells also promote CD4+ T subgroup alterations. Moreover, we revealed some TFs may regulate CD4+ T-cell polarization in MG, such as STAT3, JUND, and JUN. CD4STAT3−/− mice are resistant to experimental autoimmune encephalomyelitis and the inability of pathogenic Th17 and Th1 cells61. JUND also promoted Th17 cells polarization, although JUND is not major role compared with JUNB62.
Although previous large studies of pathology in MG focused on adaptive immunity, we observed that monocytes also showed large shifts in MG patients. Apart from enriched antigen processing and presentation activity, DCs also exhibited activated TNF signaling. Cell communication showed increased TNF signaling from CD16+/CD14+ monocytes to cDCs. Monocytes might have functions in mediating the TNF signaling of DCs. TNF signaling plays an important role in inflammatory and autoimmune diseases and participates in biological processes, including cell proliferation, apoptosis, and differentiation. Their altered function might be the cause of the chronic inflammatory reaction. We propose that monocytes also play an essential role in the pathological process of MG.
There are also limitations to our study. First, due to the limitation of RNA capture efficiency, there were undetected genes. For example, some risk genes that are lowly expressed were not identified. Second, our scRNA-seq dataset included only female early-onset MG patients to avoid gender bias, which may not share the same pathological transcriptional changes with other disease subtypes or male patients. Third, clustering based on the expressed mRNA transcriptome may not always be coincidence with clustering by protein surface markers.
In summary, we present a comprehensive single-cell transcriptome atlas of MG. Our study helps better understand the mechanism of MG for basic research, provides indicators of disease activity for clinicians, and provides markers for drug developers.
Materials and methods
Processing of patient samples
Sixty-two MG patients and, seventeen age- and sex-matched HCs were recruited at the Neurology Department of Xiangya Hospital between June 2018 and February 2019. The first cohort including HCs (n = 2) and MG (n = 2), was used for the 10× genomics scRNA-seq. The second cohort, including HCs (n = 15) and MG (n = 60), was used for flow cytometry analysis (Supplementary Table S1). Seven MG patients in the second cohort were followed up after 3 months of immunosuppressant therapy. Participants were excluded based on the following criteria: (1) an ethnic origin other than Han; (2) a history of oral glucocorticoid or immunosuppressant intake; (3) a history of IVIG or plasma therapy; (4) a history of malignant tumor; (5) pregnancy. Diagnosis of MG was based on clinical symptoms, neostigmine test, repetitive nerve electrical stimulation, and serum autoantibody results. Autoantibody results, including AChR and MuSK antibody, were obtained from the DAAN Clinical Laboratory Central (Guangzhou, China). Serum anti-AChR levels greater than 0.45 nmol/L and anti-MuSK levels greater than 0.5 nmol/L were considered positive results. QMG score was used to evaluate disease severity when patients have enrolled and the therapeutic effect after immunotherapy treatment22. All patients and HCs signed informed consent forms. This study was approved by the Ethics Committee of Xiangya Hospital.
Generation and sequencing of single-cell libraries
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood using Ficoll-Paque (TBD, Tianjin, China) according to the manufacturer’s instructions. Cell viability was assessed by flow cytometry (cell viability > 95%). ScRNA-seq libraries were prepared per the Chromium Single Cell 5' library preparation kit user guide (10× Genomics). 90 μL cellular suspension, 40 μL barcoded Single Cell 5' Gel Beads, and 270 μL Partitioning Oil were loaded onto a Chromium Chip A to generate single-cell gel bead-in-emulsions (GEMs). GEM-reverse transcriptions (GEM-RTs) were performed in a Veriti 96-well thermal cycler (Thermo Fisher Scientific). After RT, cDNAs were amplified and cleaned using the SPRIselect Reagent Kit. Indexed sequencing libraries were constructed using the Chromium Single Cell 5' Library Construction Kit (10× Genomics) according to the user guide. Barcoded sequencing libraries were quantified by quantitative PCR on an ABI StepOnePlus Real-Time PCR System (Life Technologies). Pair end single-cell RNA-Seq libraries were sequenced using NovaSeq 6000 (Illumina) with a read length of 150 bp paired-end reads.
ScRNA-seq bioinformatics analysis
Quality control metrics and filtering
For each sample, we processed 10× genomics raw data using the Cell Ranger Single-Cell Software Suite (v 3.1.0) with default parameters. Reads were aligned to the prebuilt human reference genome from the 10× Genomics website (GRCh38 V3.0.0). Then, we mapped the unique molecular identifiers (UMIs) to genes and the barcodes to cells. We further analyzed these metrics using the R (v3.6.3) package Seurat (v 3.1.2). Only genes expressed in at least three cells and cells with a minimum of 200 genes were kept. Low-quality cells meeting one of the following thresholds were further excluded: 1) the number of expressed genes was lower than 500 or larger than 3500; 2) more than 7% of UMIs were mapped to mitochondrial or ribosomal genes. After filtering, we detected 18928 genes in a total of 39,243 cells, as shown in Supplementary Table S2.
Dimensional reduction and visualization
After quality control, variable genes for each four samples were identified using the FindVariableGenes function in Seurat. Then, single-cell transcriptomes data were log-normalized and scaled for further analysis. FindIntegrationAnchors and IntegrateData functions were used to produce a batch-corrected expression matrix63. Principal component analysis (PCA) for dimension reduction was performed with the Seurat function, and 20 PCs were kept for downstream analysis based on the Jackstraw method. Clustering was further performed with the Seurat FindClusters function, and the Uniform manifold approximation and projection (UMAP) was used for visualization. Identities of clusters of cells were manually annotated using known marker genes in published articles with the help of the R package SingleR (v 1.0.1). Sub-clustering of major immune cell clusters was performed in the same workflow.
Differential expression analysis
Cluster-specific marker genes for transcriptional subpopulations were identified by the FindAllMarkers Seurat function with Wilcoxon rank-sum test. For specific cluster comparisons between MG patients and HCs, we used the DESeq2 package (v1.26.0) to detect differentially expressed genes (DEGs)64. Significant DEGs were filtered by |log fold change| > 0.5 and P value < 0.05. Gene ontology and gene-set enrichment analysis from DEGs were performed using Metascape (www.metascape.org)65.
Gene functional annotation
To compare the functional profiles of different clusters, we used the clusterProfiler package (v 3.14.3)66 for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis with significant DEGs. Gene set variation analysis (GSVA) was performed using the GSVA package67 (v 1.32.0). To assess the gene set activity among clusters, we first determined the mean expression of gene sets of each cluster and assessed the log fold-change between clusters. The annotation gene sets were downloaded from C2 (curated gene sets) and C7 (immunologic gene sets) of the molecular signature database (MSigDB, version 7.0). We used Limma package (v 3.42.0)68 to contrast the activity score and then compared pathway activities between MG and HCs.
Quantifying differences between major immune cells in MG versus HCs
After identifying the major immune cells, we also evaluated the differences between lineages derived from MG versus HCs by comparing cell distributions. Bhattacharyya distance was used to measure the distances, and lineages with over 500 cells in each sample of MG and HCs were calculated as reported by Cillo et al.8. More specifically, for the comparison of any given two clusters, we first sampled 500 cells from each individual cluster randomly 100 times. Then, two clusters were mixed up, and 500 cells were randomly sampled twice without replacement, and inter-cluster Bhattacharyya distances were calculated between sampling of individual clusters and normalized with a sampling of mixed clusters.
Regulatory network inference
A single-cell regulatory network for 9 B cell groups and 7 CD4+ T cells was performed with single-cell regulatory network inference and clustering (scenic)69. Specifically, GRNBoost2 (https://github.com/tmoerman/arboreto) in pySCENIC was used to identify gene regulatory networks, and RcisTarget (v 1.6.0) was then used to identify regulatory motifs and to predict candidate target genes. The cell-regulon activity was calculated by AUCell (v 1.8.0). Differences in the AUC between the MG and HC groups were identified by t-test.
Pseudotime-trajectory analysis
We used Monocle2 (v2.14.0)70 to construct the pseudotime-trajectories for B cells and CD4+ T cells. The positive significant marker genes identified by FindAllMarkers Seurat function were used to sort cells in a temporal differentiation order. Dimension reduction was performed using DDRTree from Monocle2.
A diffusion map for myeloid cells was generated using the destiny (v3.0.1) R package71.
RNA velocity analysis
We used velocyto.py (v0.17, https://github.com/velocyto-team/velocyto.py) to quantify spliced and unspliced reads of each 10× bam file generated by cell range with default parameters. Expressed repetitive elements in the GRCh38 reference genome were masked, and the bed file for the masked interval was generated with the UCSC genome browser (https://genome.ucsc.edu/)72. Loom files were then processed with velocyto.R (v0.6 https://github.com/velocyto-team/velocyto.R). We subset B cells for further analysis with the grouping of 20 cells and used Monocle DDRTree embedding for the final RNA velocity plots, and cell to cell distance was calculated with the first 50 PCs. Parameters were set to default unless stated otherwise.
Assessment of receptor/ligand interactions
We use CellChat (v0.0.2)34 to evaluated cell-cell interactions and significant pathways. To identify potential cell-cell interactions that were perturbed or induced in myasthenia gravis patients, we focused on differentially expressed ligands and receptors in CD8+ T cells, CD4+ T cells, B cells, CD14+ monocytes, FCGR3A+ monocytes, NK cells, and DCs. We also evaluated potentially altered interactions between monocytes, including CD14+ and CD16+ monocytes, and DCs, including cDCs and pDCs, based on the reclustered myeloid cells. Briefly, we followed the official workflow, and normalized data were loaded into CellChat. After creating CellChat objects, we used CellChatDB.human and set the Secreted Signaling pathways as the database. Then, default parameters were used to identify putative interaction pairs, and the result were displayed as circos plots.
Flow cytometry
Flow cytometry was used to assess B cell subsets. PBMCs were isolated by standard Ficoll-Paque (TBD, Tianjin, China). Fresh PBMC from the donors were stained with the surface markers of anti-human antibodies, including BB515-labeled anti-CD19 (clone: HIB19, Biolegend), and PE-labeled anti-CD180 (clone: MHR73-11, Biolegend) antibodies, and standard procedures were applied. For intracellular staining, cells were fixed and permeabilized using BD Cytofix/Cytoperm (BD Biosciences) and then incubated with PEcy7-labeled IgG (clone: HP6017, Biolegend). Zombie (Biolegend) was used to exclude dead cells. Cells were subsequently blocked in 2% bovine serum albumin (BD Biosciences) supplemented with human FcX Blocker (Biolegend). 1 × 106 fresh cells were used for every staining, and cells were incubated in staining buffer (0.1% BSA in PBS) for 30 min at 4 °C shielded from light. After staining, cells were washed in 1× PBS and acquired on a Cytek flow cytometer in an hour. Data were analyzed using FlowJo v10.
Statistical analysis
Marker genes for transcriptional subpopulations were identified by FindAllMarkers Seurat function with Wilcoxon rank-sum test. Differences among multiple groups were evaluated by one-way analysis of variance (ANOVA) followed by Newman–Keul post hoc test. Wilcoxon signed-rank test was used for paired data. The paired Wilcox signed-rank test was used to compare differences between paired samples. Correlation analysis was performed using the nonparametric Spearman test. Statistical analysis and graph processing were performed using R (v3.6.3). Data were considered significant at *P < 0.05, **P < 0.01, and ***P < 0.001 unless stated otherwise.
Data availability
The accession numbers for the sequencing raw data in this paper are GSA (Genome Sequence Archive in BIG Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences): HRA000997
References
Carr, A. S., Cardwell, C. R., McCarron, P. O. & McConville, J. A systematic review of population based epidemiological studies in Myasthenia Gravis. BMC Neurol. 10, 46 (2010).
Mantegazza, R. & Cavalcante, P. Diagnosis and treatment of myasthenia gravis. Curr. Opin. Rheumatol. 31, 623–633 (2019).
Lindstrom, J. M., Seybold, M. E., Lennon, V. A., Whittingham, S. & Duane, D. D. Antibody to acetylcholine receptor in myasthenia gravis. Prevalence, clinical correlates, and diagnostic value. Neurology 26, 1054–1059 (1976).
Zhang, Y. et al. Dendritic cells transduced with lentiviral-mediated RelB-specific ShRNAs inhibit the development of experimental autoimmune myasthenia gravis. Mol. Immunol. 46, 657–667 (2009).
Suurmond, J. & Diamond, B. Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J. Clin. Invest. 125, 2194–2202 (2015).
Mantegazza, R. & Antozzi, C. When myasthenia gravis is deemed refractory: clinical signposts and treatment strategies. Ther. Adv. Neurol. Disord. 11, 1756285617749134 (2018).
Gilhus, N. E. & Verschuuren, J. J. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol. 14, 1023–1036 (2015).
Cillo, A. R. et al. Immune landscape of viral- and carcinogen-driven head and neck cancer. Immunity 52, 183–199 (2020).
Hu, Q. et al. Atlas of breast cancer infiltrated B-lymphocytes revealed by paired single-cell RNA-sequencing and antigen receptor profiling. Nat Commun 12, 2186 (2021).
King, H. W. et al. Single-cell analysis of human B cell maturation predicts how antibody class switching shapes selection dynamics. Sci Immunol 6 (2021).
Nutt, S. L., Hodgkin, P. D., Tarlinton, D. M. & Corcoran, L. M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 15, 160–171 (2015).
Nera, K. P. et al. Loss of Pax5 promotes plasma cell differentiation. Immunity 24, 283–293 (2006).
Kwon, H. et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity 31, 941–952 (2009).
Schubart, D. B., Rolink, A., Kosco-Vilbois, M. H., Botteri, F. & Matthias, P. B-cell-specific coactivator OBF-1/OCA-B/Bob1 required for immune response and germinal centre formation. Nature 383, 538–542 (1996).
Corcoran, L. M. et al. Differential requirement for OBF-1 during antibody-secreting cell differentiation. J. Exp. Med. 201, 1385–1396 (2005).
Zhong, H., Zhao, C. & Luo, S. HLA in myasthenia gravis: from superficial correlation to underlying mechanism. Autoimmun. Rev. 18, 102349 (2019).
Jin, W., Luo, Z. & Yang, H. Peripheral B cell subsets in autoimmune diseases: clinical implications and effects of B cell-targeted therapies. J. Immunol. Res. 2020, 9518137 (2020).
Betz, B. C. et al. Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J. Exp. Med. 207, 933–942 (2010).
Stone, S. L. et al. T-bet transcription factor promotes antibody-secreting cell differentiation by limiting the inflammatory effects of IFN-γ on B cells. Immunity 50, 1172–1187 (2019).
Ong, S. T. et al. Lymphadenopathy, splenomegaly, and altered immunoglobulin production in BCL3 transgenic mice. Oncogene 16, 2333–2343 (1998).
Collins, P. E. et al. The IκB-protein BCL-3 controls Toll-like receptor-induced MAPK activity by promoting TPL-2 degradation in the nucleus. Proc. Natl Acad. Sci. USA 116, 25828–25838 (2019).
Jaretzki, A. 3rd et al. Myasthenia gravis: recommendations for clinical research standards. task force of the medical scientific advisory board of the myasthenia gravis foundation of America. Neurology 55, 16–23 (2000).
Garuda, B. R., Vemuri, D., Seepana, G., Sateesh, T. & Aruna Kumari, U. Assessment of disease severity in patients with Myasthenia Gravis – clinical phenotype and immunological correlation. Neurology https://n.neurology.org/content/92/15_Supplement/P5.4-029 (2019).
Rødgaard, A., Nielsen, F. C., Djurup, R., Somnier, F. & Gammeltoft, S. Acetylcholine receptor antibody in myasthenia gravis: predominance of IgG subclasses 1 and 3. Clin. Exp. Immunol. 67, 82–88 (1987).
Narayanaswami, P. et al. International consensus guidance for management of myasthenia gravis: 2020 update. Neurology 96, 114–122 (2021).
Lighvani, A. A. et al. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc. Natl Acad. Sci. USA 98, 15137–15142 (2001).
Chen, Z. et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl Acad. Sci. USA 103, 8137–8142 (2006).
Kano, S. et al. The contribution of transcription factor IRF1 to the interferon-gamma-interleukin 12 signaling axis and TH1 versus TH-17 differentiation of CD4+ T cells. Nat. Immunol. 9, 34–41 (2008).
Wild, C. A. et al. HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int. Immunol. 24, 485–494 (2012).
Schaffert, H. et al. IL-17-producing CD4(+) T cells contribute to the loss of B-cell tolerance in experimental autoimmune myasthenia gravis. Eur. J. Immunol. 45, 1339–1347 (2015).
Mu, L. et al. Disequilibrium of T helper type 1, 2 and 17 cells and regulatory T cells during the development of experimental autoimmune myasthenia gravis. Immunology 128, e826–e836 (2009).
Meixner, A., Karreth, F., Kenner, L. & Wagner, E. F. JunD regulates lymphocyte proliferation and T helper cell cytokine expression. EMBO J. 23, 1325–1335 (2004).
Lyakh, L. A., Sanford, M., Chekol, S., Young, H. A. & Roberts, A. B. TGF-beta and vitamin D3 utilize distinct pathways to suppress IL-12 production and modulate rapid differentiation of human monocytes into CD83+ dendritic cells. J. Immunol. 174, 2061–2070 (2005).
Jin, S. et al. Inference and analysis of cell-cell communication using cellchat. Nat. Commun. 12, 1088 (2021).
Crotty, S. A brief history of T cell help to B cells. Nat. Rev. Immunol. 15, 185–189 (2015).
Gregersen, P. K. et al. Risk for myasthenia gravis maps to a (151) Proion using CellChat-12 production and modulate rapid diff. Ann. Neurol. 72, 927–935 (2012).
Renton, A. E. et al. A genome-wide association study of myasthenia gravis. JAMA Neurol. 72, 396–404 (2015).
Avidan, N. et al. VAV1 and BAFF, via NFκB pathway, are genetic risk factors for myasthenia gravis. Ann. Clin. Transl. Neurol. 1, 329–339 (2014).
Saruhan-Direskeneli, G. et al. Genetic heterogeneity within the HLA region in three distinct clinical subgroups of myasthenia gravis. Clin. Immunol. 166-167, 81–88 (2016).
Horiki, T., Inoko, H., Moriuchi, J., Ichikawa, Y. & Arimori, S. Combinations of HLA-DPB1 and HLA-DQB1 alleles determine susceptibility to early-onset myasthenia gravis in Japan. Autoimmunity 19, 49–54 (1994).
Giraud, M. et al. Genetic control of autoantibody expression in autoimmune myasthenia gravis: role of the self-antigen and of HLA-linked loci. Genes Immun. 5, 398–404 (2004).
Garchon, H. J., Djabiri, F., Viard, J. P., Gajdos, P. & Bach, J. F. Involvement of human muscle acetylcholine receptor alpha-subunit gene (CHRNA) in susceptibility to myasthenia gravis. Proc. Natl Acad. Sci. USA 91, 4668–4672 (1994).
Zelano, G. et al. Tumour necrosis factor beta gene polymorphisms in myasthenia gravis. Eur. J. Immunogenet. 25, 403–408 (1998).
Huang, D., Pirskanen, R., Hjelmström, P. & Lefvert, A. K. Polymorphisms in IL-1beta and IL-1 receptor antagonist genes are associated with myasthenia gravis. J. Neuroimmunol. 81, 76–81 (1998).
Sciacca, F. L. et al. IL-1 genes in myasthenia gravis: IL-1A -889 polymorphism associated with sex and age of disease onset. J. Neuroimmunol. 122, 94–99 (2002).
Alseth, E. H., Nakkestad, H. L., Aarseth, J., Gilhus, N. E. & Skeie, G. O. Interleukin-10 promoter polymorphisms in myasthenia gravis. J. Neuroimmunol. 210, 63–66 (2009).
Yue, Y. X. et al. Association study between IL-17A and IL-17F gene polymorphism and myasthenia gravis in Chinese patients. Neurol. Sci. 37, 123–130 (2016).
Djabiri, F. et al. Association of the AChRalpha-subunit gene (CHRNA), DQA1*0101, and the DR3 haplotype in myasthenia gravis. Evidence for a three-gene disease model in a subgroup of patients. J. Autoimmun. 10, 407–413 (1997).
Schneider, H. et al. Reversal of the TCR stop signal by CTLA-4. Science 313, 1972–1975 (2006).
Sutton, C. E. et al. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31, 331–341 (2009).
Shamilov, R. & Aneskievich, B. J. TNIP1 in autoimmune diseases: regulation of toll-like receptor signaling. J. Immunol. Res. 2018, 3491269 (2018).
Quartuccio, L., Fabris, M. & Ferraccioli, G. B lymphocyte stimulator (BLyS) and monocytes: possible role in autoimmune diseases with a particular reference to rheumatoid arthritis. Reumatismo 56, 143–146 (2004).
You, M. et al. Ligation of CD180 inhibits IFN-α signaling in a Lyn-PI3K-BTK-dependent manner in B cells. Cell. Mol. Immunol. 14, 192–202 (2017).
Nagai, Y. et al. The radioprotective 105/MD-1 complex links TLR2 and TLR4/MD-2 in antibody response to microbial membranes. J. Immunol. 174, 7043–7049 (2005).
Caamaño, J. H. et al. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J. Exp. Med. 187, 185–196 (1998).
Levels, M. J. et al. The transcriptional coactivator Bob1 is associated with pathologic B cell responses in autoimmune tissue inflammation. Arthritis Rheumatol. 69, 750–762 (2017).
Merico, D., Sharfe, N., Hu, P., Herbrick, J.-A. & Roifman, C. M. RelB deficiency causes combined immunodeficiency. LymphoSign J. 2, 147–155 (2015).
Ding, C. et al. STAT3 signaling in B cells is critical for germinal center maintenance and contributes to the pathogenesis of murine models of lupus. J. Immunol. 196, 4477–4486 (2016).
Wagner, D. H. Jr. et al. Expression of CD40 identifies a unique pathogenic T cell population in type 1 diabetes. Proc. Natl Acad. Sci. USA 99, 3782–3787 (2002).
Vaitaitis, G. M., Waid, D. M., Yussman, M. G. & Wagner, D. H. Jr. CD40-mediated signalling influences trafficking, T-cell receptor expression, and T-cell pathogenesis, in the NOD model of type 1 diabetes. Immunology 152, 243–254 (2017).
Liu, X., Lee, Y. S., Yu, C. R. & Egwuagu, C. E. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J. Immunol. 180, 6070–6076 (2008).
Yamazaki, S. et al. The AP-1 transcription factor JunB is required for Th17 cell differentiation. Sci. Rep. 7, 17402 (2017).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019). e1821.
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. Omics: a J. Integr. Biol. 16, 284–287 (2012).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinforma. 14, 7 (2013).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Angerer, P. et al. Destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243 (2016).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Acknowledgements
The authors are grateful for the grants provided by the National Natural Science Foundation of China (81501034 and 81771364), AFM TELETHON (21125) in publishing this paper. We thank Yajun Zhan for every convenience in Flow cytometry testing. We thank Genedenovo Biotechnology Co., Ltd for assisting in sequencing and analyzing.
Author information
Authors and Affiliations
Contributions
W.J. was involved in the design of the study, conduction of the experiments, collection of samples and data, experimental and bioinformatic analysis, interpretation of data, and writing the draft of the manuscript. Q.Y. was involved in the bioinformatic analysis. Y.P., C.Y., and Y.L. were involved in the sample collection. Z.L. was involved in the design of the study. B.X. was involved in the design of the study. L.X. was involved in the design of the study, conduction of the experiments, revising the paper, and providing financial support. H. Y. was involved in the design of the study, revising the manuscript, and providing financial support.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jin, W., Yang, Q., Peng, Y. et al. Single-cell RNA-Seq reveals transcriptional heterogeneity and immune subtypes associated with disease activity in human myasthenia gravis. Cell Discov 7, 85 (2021). https://doi.org/10.1038/s41421-021-00314-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41421-021-00314-w
