
Abstract
For a long time, hydrogen sulfide (H2S) has been considered a toxic compound, but recent studies have found that H2S is the third gaseous signaling molecule which plays a vital role in physiological and pathological conditions. Currently, a large number of studies have shown that H2S mediates apoptosis through multiple signaling pathways to participate in cancer occurrence and development, for example, PI3K/Akt/mTOR and MAPK signaling pathways. Therefore, the regulation of the production and metabolism of H2S to mediate the apoptotic process of cancer cells may improve the effectiveness of cancer treatment. In this review, the role and mechanism of H2S in cancer cell apoptosis in mammals are summarized.
Similar content being viewed by others


Facts
-
To date, H2S has been shown to play an important role in tumors of various tissue origins.
-
H2S promotes cancer progression in most tissues through multiple mechanisms.
-
Overexpression or knockdown of genes encoding H2S-producing enzymes, inhibitors of the enzymes, and H2S-releasing reagents have been validated in cancer cells and animal models of xenograft tumors.
-
It has been reported that inhibition of H2S promotes apoptosis of cancer cells through a number of mechanisms.
Open Questions
-
H2S promotes the development of many cancer cells, including colorectal, ovarian, lung, breast and kidney. While it has opposite effects on hepatocellular carcinoma and glioma.
-
What are the mechanisms by which different concentrations of H2S exhibit different physiological and pathological functions?
-
The current detection technology of H2S concentration in tissues and cells is still immature.
Introduction
There are currently three well-known gasotransmitters: carbon monoxide (CO), nitric oxide (NO), and hydrogen sulfide (H2S). Endogenous H2S as the third gaseous signaling molecule is produced through non-enzymatic and enzymatic desulfhydration. Non-enzymatic process is mainly through the decomposition of inorganic substances [1]. H2S mainly comes from different substrates catalyzed by cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST) [2]. The main source of H2S is based on S-adenosine homocysteine as a substrate, catalyzed by two pyridoxal 5’-phosphate-dependent enzymes, CBS and CSE, which are localized in the cytoplasm. 3-MST, which is localized in the cytoplasm and mitochondria, is another enzyme that catalyzes the production of H2S using mercaptopyruvate as a substrate. Mercaptopyruvate is produced by cysteine aminotransferase (CAT) using L-cysteine or D-amino acid oxidase (DAO) using D-cysteine [3]. In the organism, H2S is either metabolized directly or stored in the form of bound sulfane sulfur and acid-labile sulfur to maintain a dynamic equilibrium. Its metabolism is mainly through a series of metabolic enzymes such as mitochondrial sulfide:quinone oxidoreductase (SQR) and persulfide dioxygenase (ETHE1), which eventually produce sulfates and are excreted in the urine or through respiration [4] (Fig. 1). The metabolism of H2S in mammals is also involved in redox reactions, binding to heme-containing metalloproteins or post-translational modifications of proteins, and also shows the role of gasotransmitter [5,6,7,8] (Fig. 2).

In mammals, H2S is produced endogenously from cysteine, serine, homocysteine and other substrates primarily through the actions of three major enzymes. Non-enzymatic pathways: gut microbes as well as polysulfide-derived H2S. Metabolism of H2S: H2S can be oxidized in the mitochondria or metabolized by methylation in the cytoplasm. H2S is first oxidized by SQR in the mitochondria to form a persulfide. This persulfide is further oxidized by ETHE1 to produce SO32−, which is then converted to SO42− and S2O32− by sulfite oxidase and rhodan oxidase and excreted in the urine or by respiration. H2S can also be removed by binding to metalloproteins to form sulfheme. α-KG α-ketoglutaric acid, 3-MST 3-mercaptopyruvate sulfurtransferase, CAT cysteine aminotransferase, CBS cystathionine beta-synthase, CSE cystathionine gamma-lyase, CysS-SH cysteine persulfide, DAO D-amino acid oxidase, GSH glutathione, SAH S-adenosylhomocysteine, SAM S-adenosyl methionine, THF tetrahydrofolic acid.

Promotion or inhibition of signaling pathways, post-translational modification of proteins, activation or shutdown of ion channels, and participation in mitochondrial metabolism. Akt protein kinase B, ERK extracellular signal-regulated kinase, JNK C-Jun N-terminal kinase, MAPK mitogen-activated protein kinase, NF-κB nuclear factor-kappa B, Nrf2 nuclear factor erythroid-2 related factor 2, PI3K phosphoinositide 3-kinase, STAT3 signal transducer and activator of transcription 3, TRPV1 transient receptor potential vanilloid 1.
Three enzymes that produce H2S are reported to be differentially expressed in tumor tissues of different tissue origins [9] and are involved in regulating tumorigenesis and progression (Table 1). Studies have shown that its role in tumors generally exhibits a biphasic bell-shaped effect [10]. Endogenous or low concentrations of exogenous H2S promote cancer cell development by stimulating angiogenesis, increasing mitochondrial bioenergy, and antioxidant. While the donation of H2S at higher concentration prevails the suppressive bioenergetic and cytotoxic effects (Fig. 3). The H2S concentration above a certain threshold plays an anti-cancer role by inducing apoptosis, DNA damage, and inhibiting the cell cycle [11,12,13,14,15,16,17,18].

DATS diallyl trisulfide, DADS diallyl disulfide, GYY4137 morpholin-4-ium 4-methoxyphenyl(morpholino) phosphinodithioate), SPRC S-propargyl-cysteine.
Apoptosis is a form of programmed cell death that results in the orderly and efficient removal of damaged cells to ensure a homeostatic balance between the rate of cell formation and cell death. However, the disruption of this balancing function can contribute to an abnormal cell growth/proliferation or autoimmune disorders. Apoptosis is considered a vital component of various processes including normal cell turnover, proper development and functioning of the immune system, hormone-dependent atrophy, embryonic development and chemical-induced cell death. Deregulation in apoptotic cell death machinery is one of the hallmarks of cancer [19,20,21]. The concept of apoptosis is first introduced as a barrier to cancer by Kerr et al. in 1972 [22]. Tumors are new organisms formed when the cells of local tissues lose the normal regulation of growth under the action of various carcinogenic factors, leading to their clonal abnormal proliferation. This is due to the accumulation of many genetic and epigenetic changes within the cell, expressed in the accumulation of chromosomal or molecular aberrations, which leads to genetic instability. The simultaneous interaction of environmental factors, exogenous factors, and individual genetic instability factors can contribute to tumorigenesis. Among these factors, individual characteristics play the most significant role. [23, 24]. The human oncogenic gene p53, for example, encodes the P53 protein as a transcription factor, which is frequently mutated in human cancers and is associated with early apoptosis [25].
Escaped apoptosis is widely recognized as a prominent hallmark of cancer cells [26]. There are multiple ways in which cancer cells can reduce apoptosis or enhance apoptosis resistance. In general, the mechanisms by which cells evade apoptosis can be broadly classified as: 1) disrupted balance of pro- and anti-apoptotic proteins, 2) reduced caspase function, and 3) impaired the signal transduction of death receptor [27]. Given the importance of apoptosis in tumorigenesis and progression, targeting apoptosis-related pathways in cancer cells have emerged as promising approaches for the treatment of various tumors [28].
There are three main apoptotic pathways: intrinsic, extrinsic and endoplasmic reticulum (ER) pathways [29]. The caspase family is the core member in initiating and executing apoptosis [30]. Those factors that initiate apoptosis are: caspase 8, caspase 9 and caspase 10, the members that execute apoptosis are: caspase 3, caspase 6 and caspase 7. Caspase 8 and caspase 10 are the promoters of extrinsic apoptosis, and caspase 9 is the promoter of intrinsic apoptosis. Caspase 3 is the key enzyme of apoptosis, and its activation indicates the beginning of apoptosis execution. Other major factors involved in apoptosis are: 1. Cytochrome C (Cyt C), Cyt C binds with Apaf-1 protein in the presence of ATP to form an apoptotic complex that recruits and activates pro-caspase 9. Cyt C is also engaged in the caspase non-dependent pathway that promotes chromatin condensation and mediates apoptosis [31]. 2. Smac/Diablo and HtrA2/Omi inhibit the release of inhibitory apoptosis protein (IAP) and indirectly facilitates the release of apoptosis execution protein caspase 3/6/7. 3. Caspase activation induces the translocation of EndoG in addition to AIF into the cytosol, which leads to the subsequent characteristic features of apoptosis, including chromatin condensation and nuclear fragmentation [32,33,34].
-
I.
Intrinsic apoptosis of mitochondrial pathway: DNA damage, growth factor deficiency, ROS and other endogenous stress cause the Bad/Bak proteins (respectively pro-apoptotic members of the Bcl-2 family) to form a complex, which is inserted into the mitochondrial outer membrane, changes the permeability of the mitochondrial membrane, and leads to the change of membrane potential. In addition, the Bax/Bak-mediated mitochondrial outer-membrane permeabilization is sufficient to induce the release of Cyt C, Smac/Diablo and HtrA2/Omi. While downstream caspase activation is required for the release of EndoG and AIF. Once released, Cyt C, interaction with the apoptosis protease-activating factor 1 (Apaf-1), triggers the initiator caspase 9 activation, which leads to the subsequent characteristic features of apoptosis, including chromatin condensation and nuclear fragmentation.
-
II.
Extrinsic apoptosis by the cell surface death receptor (e.g. FasL, TARIL) pathway. After the ligand binds to the corresponding cell surface death receptor, it recruits intracellular apoptosis-associated proteins to form the death-inducing signaling complex (DISC), which contains a junction molecule and pro-caspase 8, then activates caspase 8, causing the onset of the downstream apoptotic cascade [35, 36].
-
III.
Apoptosis of the ER pathway: The ER is the dominant site of protein synthesis and also engaged in the regulation of intracellular calcium homeostasis. When the protein is misfolded or unfolded in the cell, prolonged unfolded protein response (UPR) occurs, the transmembrane proteins PERK, IRE1 and ATF6 on the ER are responsible for protein repair. Apoptosis is triggered when the ER pressure induced by the UPR is too high [37]. ER stress activates inositol 1,4,5-trisphosphate receptor (IP3R) and/or ryanodine receptor (RyR), inducing Ca2+ release from ER, causing an imbalance in intracellular Ca2+ homeostasis, which directly induces the production of pro-apoptotic proteins of the Bcl-2 family and activates calpain, then activates csapase 9 through the activation of capase 12 [38]. In organisms, Ca2+ acts as a second messenger, responsible for intracellular signaling to trigger physiological changes such as apoptosis. It has also been reported that in the early stages of apoptosis, the released Cyt C binds to IP3R, causing Ca2+ release and inducing the calcium-dependent apoptosis [39].
There have been a number of reports on the involvement of H2S in the regulation of physiological apoptosis in the organism.
H2S affects the mitochondrial pathway of apoptosis, and at non-toxic concentrations, H2S accumulation in mitochondria induces mitochondria-dependent apoptosis by inducing Bax translocation, mPTP formation, and release of Cyt C [40]. H2S induces apoptosis through oxidative stress-triggered mitochondrial pathway in zebrafish at embryonic and larval stages [41]. Invasive bacteria in the oral cavity cause apoptosis of human pulp stem cells (HPSCs) by producing large amounts of H2S and activating the mitochondrial pathway [42]. However, under certain conditions, H2S also exhibits an inhibitory effect on apoptosis, it has been reported that H2S inhibits the mitochondrial K+atp/MAPK-mediated pro-apoptotic pathway [43]. In chondrocytes, NaHS-derived H2S may antagonize IL-3β-induced inflammation and apoptosis associated with mitochondrial dysfunction by inhibiting the PI3K/Akt/NF-κB and MAPK signaling pathways, respectively [44].
H2S affects TNF receptor family-mediated exogenous apoptosis: CSE-catalyzed production of endogenous H2S induces sulfurylation of Cys38 of the NF-κB subunit p65, which inhibits TNF-α-induced apoptosis [45].
Role of H2S in the apoptotic pathway of ER stress: In cervical cancer cell line Hela, morpholin-4-ium 4-methoxyphenyl(morpholino) phosphinodithioate (GYY4137), a slow-releasing H2S donor induces apoptosis by stimulating ER stress, causing up-regulation of IP3R1 and IP3R2 expression on the ER, leading to intracellular Ca2+ overload [46]. It has also been reported that H2S exerts a protective effect by inducing ER stress in alveolar epithelial cells in the early stages of acute lung injury in rats [47], the exact mechanism of which is unclear.
H2S and its producing enzymes are involved in three apoptotic pathways and play an important role in tumorigenesis and development, this could be dependent on the dose of this gaseous mediator and possibly on the differences in sensitivity of various cancer cell types to the impact of this molecule. The main objective of this review is to explore the role of H2S and its producing enzymes in tumorigenesis and development by mediating apoptosis.
The roles of H2S in apoptosis in different tumors
GYY4137 and sodium hydrosulfide (NaHS) act as H2S donors and are able to release H2S when water-soluble. Li et al. [48] screen seven different human cancer cell lines (HeLa, HCT-116, Hep G2, HL-60, MCF-7, MV4-11 and U2OS) and normal human lung fibroblasts (IMR90, WI-38) and apply GYY4137 and NaHS, respectively, find that H2S donors specifically cause partial G2/M arrest and promote apoptosis. Another H2S donor sodium sulfide (Na2S) has also been used to selectively upregulate ROS levels in glioblastoma (GBM) cell lines (T98G and U87) to promote apoptosis and enhance their sensitivity to radiotherapy. But the effects were not observed in normal human brain microvascular endothelial cells (hCMEC/D3) [49]. NaHS induces apoptosis in C6 glioma cells by activating p38/MAPK and p53 signaling pathways in an endogenous manner [50]. In addition, the roles of H2S in tumor cells of different tissue origins are highlighted below.
Lung cancer
Non-small cell lung cancer (NSCLC) A549 and 95D highly express CBS, CSE, and 3-MST compared to normal lung epithelial cell lines, induce high intracellular concentrations of H2S, and promote angiogenesis and epithelial-mesenchymal transition [51], which is mainly associated with the regulation of hypoxia-inducible factor-1α (HIF-1α) stimulation of vascular endothelial growth factor expression by intracellular highly concentration of H2S. Silencing of H2S-generating enzymes using siRNAs promote apoptosis, and related inhibitors of the enzymes also inhibit the growth of transplanted tumors in nude mice. CBS expression is down-regulated in A549/DDP cisplatin-resistant lung cancer cell line. After exogenous supplementation with H2S using NaHS (800 μM), the expression of p53 and p21, as well as apoptosis-associated proteins caspase 3 and Bax are upregulated in drug-resistant cells, which promotes apoptosis and enhances the sensitivity to cisplatin [52].
Esophageal carcinoma
Esophageal cancer is one of the most aggressive cancers among all gastrointestinal malignancies [53]. The current main treatment for esophageal cancer is still surgical resection, but it is highly invasive and has high postoperative complications and mortality. Lei et al. demonstrate that exogenous high concentration of H2S (400 μM NaHS) induces cancer cell proliferation, anti-apoptosis, angiogenesis and cell migration in EC109 esophageal cells by activating the HSP90 pathway[54]. The heat shock protein HSP90 acts as a molecular chaperone that promotes the folding of ab initio synthesized or misfolded proteins, relieves the stress of UPRs in the ER, and promotes cell survival [55]. Lei et al. also find that exogenous high concentration of H2S (500 μM NaHS) may significantly reduces cell apoptosis by activating the JAK2/STAT3 signaling pathway, upregulating Bcl-2 and downregulating caspase 3, caspase 9, caspase 12, and Bax [56]. Current studies on the specific mechanism of action of H2S in esophageal cancer are scarce.
Gastric cancer
Zhang et al. prove that [57] the expression levels of CSE and CBS proteins are significantly up-regulated in gastric cancer compared to neighboring non-cancerous tissues. Later, the same group reports exogenous administration of NaHS in human gastric cancer cell line SGC-7901 can promote Bax expression and induce apoptosis via mitochondrial pathway. In gastric cancer SGC-7901 cells, SPRC enhances the expression and enzymatic activity of CSE, which in turn acts as a substrate for CSE to cleave and produce H2S, and promotes apoptosis by activating the MAPK pathway to upregulate the expression of p53 and Bax [58]. Two specific inhibitors of CBS and CSE, aminooxyacetic acid (AOAA) and DL-propargylglycine (PAG), enhance the stronger anticancer effect of 3,3'-Diindolylmethane (DIM) in gastric cancer BGC-823 and SGC-7901 cells [59]. Zhu et al. use cBioPortal to analyze TCGA gastric cancer patients and find a significant association between CBS mutations and PI3K/AKT/mTOR pathway activation [60]. Further validation in gastric cancer cell lines reveals that knockdown of CBS results in excessive activation of PI3K/Akt signaling pathway and promotes oncogenic transformation.
Hepatocellular carcinoma
Compared to normal hepatocyte line L02, human hepatocellular carcinoma cell line (e.g. PLC/PRF/5) shows significantly higher H2S and increased overexpression levels of CSE and CBS [61, 62], at low concentrations, 25-100 μM NaHS promotes the proliferation of hepatocellular carcinoma (HCC) cell lines, while at high concentrations, 800–1000 μM NaHS induces HCC apoptosis. High concentrations of NaHS promote the expression of PTEN, a tumor suppressor protein that inhibits PI3K/Akt signaling [63]. The overexpression of PTEN may lead to the inactivation of Akt/ERK pathway and promote apoptosis. Administration of 500 μM NaHS to PLC/PRF/5 hepatoma cells for 24 h significantly increases the expression of CSE, CBS and induces NF-κB activation, which in turn causes an increase in the expression of downstream pro-proliferative signaling molecules COX-2 and MMP-2 [62]. CSE is over-expressed in hepatoma HepG2 and PLC/PRF/5 cells, inhibition of the CSE/H2S axis causes elevated intracellular ROS, upregulated p53/p21 expression, and decreased Bcl-2/Bax ratio, as well as activates JNK/MAPK, which together promote the mitochondrial apoptotic pathway in HCC cells [64]. Jia et al. [65] find that knockdown of CBS in HCC cells causes an increase in intracellular ROS and induces apoptosis of cancer cells in a mitochondria-dependent manner. Zhou et al. [66] reveal that reduced CBS expression in HCC is associated with poor prognosis in HCC, and downregulation of CBS activates the IL-6/STAT3 signaling pathway, which directly inhibits apoptosis and induces infiltration of Treg cells in the tumor microenvironment. In addition, miR-24-3p is shown to be an upstream suppressor of CBS in HCC. Another endogenous H2S-producing enzyme, 3-MST, is down-regulated in the expression level. Overexpression of 3-MST inhibits proliferation and induces apoptosis, and 3-MST overexpression is also shown to significantly inhibit tumor growth in a nude mouse tumor allograft model, 3-MST silencing using siRNA then significantly promotes tumor growth. In addition, HCC models are more readily induced in 3-MST knockout mice, upregulation of 3-MST expression mainly causes dysregulation of intracellular ROS homeostasis and inhibits the proliferation-related AKT/FOXO3a/Rb signaling pathway [67].
Pancreatic carcinoma
In pancreatic cancer Capan-2 cells, diallyl trisulfide (DATS), an active component of garlic that can release H2S, induces apoptosis by down-regulating Bcl-2, Akt and cyclin D1 protein levels, and up-regulating Bax, Fas, p53 and cyclin B protein levels [68].
Colorectal cancer
Colorectal cancer (CRC) is the third most prevalent tumor and the second leading cause of cancer death worldwide [69]. In human intestinal lumen, there are many microorganisms involved in H2S production and metabolism, and H2S produced via microbial metabolic reactions can easily penetrate into the biofilm covering the colonic cells and epithelial cell membranes [70]. The production and metabolism of H2S in intestinal lumen act on CRC progression [71, 72]. It has been demonstrated that NO, CO and H2S, which are endogenously produced in colon cancer cells as gaseous signaling transmitters in the organism, can inhibit the proliferation of cancer cells at higher or lower concentrations, and it has been further verified that these gas signaling molecules promote apoptosis in colon cancer cells mainly by inhibiting the cGMP/VASP pathway, the Akt and ERK1/2/MAPK signaling pathways [73]. In human metastatic CRC (mCRC) cells, activation of the permeable channel transient receptor potential vanilloid 1 (TRPV1) by NaSH induces extracellular Ca2+ inward flow and subsequent activates the reverse mode Na+/Ca2+ (NCX) exchanger, resulting in sustained intracellular Ca2+ overload, which in turn induces apoptosis [74]. The expression level of cysteine-rich matricellular protein 61 (Cyr61) is higher in CRC tissues and cell lines than in normal colonic mucosa, and Cyr61 is known as an angiogenic inducer that promotes tumor growth and angiogenesis [75]. Cyr61 promotes cell migration, invasion and metastasis in CRC, and high expression of Cyr61 is associated with poor prognosis [76, 77]. Polysulfide and 3-MST-derived H2S promote CRC development and progression via persulfidation of Recombinant Specificity Protein 1 (Sp1) and activation of p38/MAPK to induce high expression of Cyr61, while apoptosis of cancer cells increases after application of HMPSNE to inhibit 3-MST enzyme [78]. It is known that the CBS promoter contains an Sp1 binding site, and Sp1 is essential in the control of CBS transcription [79], In colon cancer cells which devoid of p53, the chemotherapeutic drug 5-FU can induce ribosomal protein L3 (rpL3) as proapoptotic factor. RpL3 can inhibit CBS via binding with Sp1 and/or acting on post-translational. Ultimately leading to reduces H2S synthesis and induces Cyt C release, resulting in apoptosis of CRC cells by the mitochondrial pathway [80]. P53 is known as a tumor suppressor protein and causes tumorigenesis when it is mutated [81, 82]. P53 has a central role in the response to cellular stress, blocking the cell cycle by inducing p21 expression when cell growth is uncontrolled [83]. When damage cannot be repaired, p53 induces high expression of apoptotic proteins (e.g. BAX) to promote apoptosis [84, 85]. Caco-2 is one of the cancer cell lines that lacking p53 protein expression, and the apoptosis activation mechanism is significantly different from other CRC cell lines such as HT-29 [86, 87], apoptosis is promoted mainly by activating the extrinsic apoptotic pathway. GYY4137 induces apoptosis by blocking the cell cycle in human CRC Caco-2 cell line [88]. In other CRC cell lines (e.g. HCT116, SW620, DLD1), GYY4137 also increases the sensitivity of CRC cells to paclitaxel to promote apoptosis [89]. S-adenosyl-L-methionine (SAM), a metabolic activator at low to moderate levels of CBS, produces H2S as an endogenous pro-growth and bioenergetic factor in early stages of colon carcinogenesis, but high doses of SAM downregulate CBS expression and inhibit cancer cell bioenergetics and proliferation [90]. Yue et al. [91] also find that application of AOAA in colon cancer cells can inhibit endogenous H2S levels, disrupt cellular antioxidant capacity, increase intracellular ROS levels, upregulate p53 expression, induce apoptosis in a mitochondria-dependent manner, and enhance the sensitivity of colon cancer cells to oxaliplatin.
Breast cancer
Inhibition of three enzymes that produce H2S in breast cancer (BC) cells significantly promotes apoptosis in the mitochondrial pathway [15], the mechanism is that lower intracellular levels of H2S inhibit phosphorylation of the PI3K/Akt/mTOR signaling pathway. The same team find that in nasopharyngeal carcinoma cells, inhibition of endogenous H2S induces the increase of intracellular ROS, activation of p38/MAPK and JNK/MAPK signaling pathways that contribute to apoptosis, and inhibit cell proliferation via blocking ERK1/2 MAPK [92].
Urogenital cancer
Renal cell carcinoma (RCC) are the most common solid lesions in the kidney, accounting for approximately 90% of all renal malignancies [93]. There are three main subtypes of RCC: clear-cell RCC (ccRCC 70–80%), papillary RCC (pRCC types I and II, 10–15%) and chromophobe RCC (4–5%). Breza. Jr et al. [94] find that the expression of H2S-producing enzymes are downregulated in tumor tissues compared with non-tumor tissues, the levels of CBS and CSE are lower with the higher grade of ccRCC, blocking with AOAA and PAG reveals prevention of apoptosis induction.
Female reproductive system cancer
Sanjib Bhattacharyya et al. [95] find that CBS is overexpressed in primary epithelial ovarian cancer and ovarian cancer cell lines, while knockdown of CBS disruptes intracellular energy metabolism, induces elevated ROS, promotes apoptosis and increases numbers of cancer cells killed by cisplatin via reduce NF-κB activity. Meanwhile, knockdown of CBS also significantly inhibites the growth of nude mouse tumor xenografts and enhances the inhibition of cisplatin. A team find that in ovarian cancer cell line A2780 cells, GYY4137 induces ER stress under hypoxic conditions, leading to apoptosis in a Ca2+-dependent manner [96]. Moreover, in ovarian cancer cell line A2780, GYY4137 induces intracellular acidification by uncoupling the highly expressed sodium-hydrogen exchanger 1 (NHE1) from sodium-calcium exchanger 1 (NCX1) [97]. It has been reported that GYY4137 upregulates NCX1 and β1, β3 adrenergic receptors in the cervical cancer cell line HeLa cells, NCX1 and β1, β3 adrenergic receptors can form a complex to enhance signaling in response to apoptosis [98].
Hematologic neoplasms
Chronic myeloid leukemia (CML) is a malignant cancer that originates from hematopoietic stem cells [99], the critical causative event in CML is the formation of the Philadelphia chromosome, a product of a chromosomal translocation that brings together the ABL gene on chromosome 9 and the BCR gene on chromosome 22. ABL is a proto-oncogene, and after binding to the BCR gene, the downstream pathway is continuously activated, resulting in secondary abnormal leukocyte proliferation [100]. BCR-ABL induces intracellular ROS accumulation, enhances PI3K signaling, activates the transcription factor NF-κB and mediates cell transformation [101]. In CML K562 cells, CBS expression is upregulated and inhibition of CBS by shRNA or AOAA induced apoptosis in the mitochondrial pathway. In K562 cells inhibiting CBS, NF-κB activity is significantly downregulated and NF-κB failed to induce the expression of downstream target genes, causing the accumulation of ROS in cells leading to the activation of JNK and apoptosis [102].
Melanoma
Two of the most frequently deregulated pathways in melanoma are MAPK/ERK and PI3K/Akt [103]. These two pathways play important roles in melanoma development and progression and are involved in the mechanism of resistance to the targeted therapy [104]. In A375 melanoma cells, either CSE overexpression or exogenous H2S donors (e.g. DATS, GYY4137) promotes apoptosis in the mitochondrial pathway, and the pro-apoptotic effect is associated with the inhibition of NF-κB downstream-related pro-survival pathways, such as reduction of the expression of c-FLIP, XIAP and Bcl-2, and inhibition of AKT and ERK1/2 signaling pathways [105]. In addition, in melanoma cells A375 and SK-MEL-28, Xiao et al. [106] find that NaHS also induces apoptosis and autophagy by inhibiting the PI3K/Akt/mTOR signaling pathway. The pleckstrin homology like domain family A member 1 (PHLDA1) is a member of three PHLDA genes. PHLDA1 can inhibit tumor development by suppressing the Akt signaling pathway [107]. H2S produced by bacteria in the oral cavity promotes apoptosis of cancer cells by inhibiting the expression of PHLDA1 in the tongue cancer cell line SCC-1 cells [108].
Novel synthetic H2S releasing drug
In addition, for the sake of improving the pharmacological/therapeutical profiles of the clinical drugs, H2S-releasing moieties are introduced to the parent drugs.
H2S-releasing NSAIDs
Nonsteroidal anti-inflammatory drugs (NSAIDs) are a class of medications used to treat pain, fever, and other inflammatory processes [109]. Aspirin [110] is the most commonly used NSAIDs that can inhibit cyclooxygenase (COX) activity. Cyclooxygenase-2 (COX-2) is frequently expressed in various tumors and plays a role not only in promoting tumor development but also elevating the resistance to chemotherapy and radiotherapy [111]. Aspirin exerts its anticancer effects via inhibition of COX, interference with proliferative pathways, cancer-related inflammation, and antiplatelet-driven pro-carcinogenic activity [112]. However, there is a risk of gastrointestinal and intracranial hemorrhage and other adverse effects associated with the long-term application of large amounts of aspirin. H2S-releasing nonsteroidal anti-inflammatory drugs (HS-NSAIDs) are an emerging class of compounds with significant anti-inflammatory properties [113]. It generally consists of a covalent linkage between traditional NSAIDs and H2S-releasing fractions. The HS-NSAIDs has a stronger apoptosis-inducing effect compared to traditional NSAIDs in human colon, breast, pancreatic, prostate, lung, and leukemia cancer cell lines [114]. Growing evidence indicates that HS-NSAIDs are more effective in suppressing tumors of multiple tissue types origin with less toxic side effects [115,116,117,118]. For example, the mechanism of action of NOSH-aspirin in inhibiting pancreatic cancer cells: FoxM1 promotes a transcription factor that can regulate a network of genes associated with mitosis, NOSH-aspirin inhibits FoxM1 expression via upregulation of ROS levels and p53 expression, and induction of cell cycle arrest and apoptosis [119]. HA-ADT can inhibit the proliferation, migration and invasion of human esophageal cancer cells more effectively than NaHS or GYY4137, mainly by inhibiting the PKB/Akt/mTOR pathway [120]. The thioredoxin reduction system, consisting of thioredoxin (Trx), thioredoxin reductase (TrxR), thioredoxin peroxidase (TPx), involves in cell proliferation, redox state and apoptosis, and frequently upregulate in malignancies, and can regulate the expression of transcription factor NF-κB or apoptosis signal-regulating kinase 1 (ASK-1) [121,122,123]. The nuclear factor NF-κB family of eukaryotic transcription factors plays an important role in regulation of immune responses, embryonic and cellular lineage development, apoptosis, cell cycle progression, inflammation and tumorigenesis [124,125,126]. HS-ASA induces apoptosis by inhibiting NF-κB and TrxR activity in MDA-MB-231 cells, the model of triple-negative breast cancer cells [127]. Aberrant activation of NF-κB is also present in CRC, and H2S-releasing naproxen induces HT-29 apoptosis by inhibiting the activation of NF-κB and TrxR [128]. ATB-346 is a novel H2S-NSAID that promotes apoptosis in human melanoma cells by inhibiting the activation of NF-κB and Akt [129]. ADT-OH inhibits the activation of NF-κB in B16F10 melanoma cells, reduces the expression of downstream target genes of NF-κB pathway (e.g. XIAP, Bcl-2), and induces cell apoptosis via mitochondrial pathway. In addition, ADT-OH can also elevate intracellular FADD levels and induce apoptosis in the exogenous pathway by downregulating the expression of Makorin ring finger protein 1 (MKRN1), the E3 ubiquitin ligase of FADD [130]. Valproic acid (VPA), a short-branched fatty acid, has been shown to inhibit chromatin remodeling class I histone deacetylase (HDAC I) which is involved in tumor development. A novel drug, ACS2, coupled by H2S releasing fraction and VPA, induces apoptosis by mitochondrial pathway in NSCLC as well as inhibits cancer cell invasion and metastasis by down-regulating matrix metalloproteinase-1 expression and enhances the sensitivity of lung cancer cells to cisplatin [131].
Nanoemulsions for H2S release and probes for H2S sensing
BAD-NE, synthesized from BSA/α-linolenic acid (ALA)/diallyl disulfide (DADS), is a nanoemulsion capable of releasing H2S, which has been shown to induce cell cycle arrest by promoting p21 expression and apoptosis by activating the ERK1/2 signaling pathway in both MCF-7 breast cancer and HuT 78 T-cell lymphoma cells [132]. A novel H2S-sensing bifunctional fluorescent probe for H2S-sensing based on naphthalimide peptide coupling, can be activated by endogenous H2S in cancer cells and could serve as a diagnostic molecule for high H2S-expressing cancer cells and induce apoptosis at the same time [133]. CPC is also a novel H2S probe that reacts with endogenous H2S to reduce H2S concentration, decrease the glutathionylation of Keap1 at Cys434, and increase the interaction between Keap1 and Nrf2, thereby inhibiting nuclear translocation of Nrf2, suppressing autophagy and promoting apoptosis [134].
Apoptosis induced by natural organic H2S donors in cancer cells
There are many natural organic H2S donors in natural world, such as the rocket or broccoli plant family [135]. Natural sulfides are mainly derived from garlic, diallyl sulfide (DAS), DADS, and DATS. It has been reported that garlic-derived organic polysulfides induce H2S production in a thiol-dependent manner in erythrocytes [136]. DATS reacts rapidly with GSH and releases H2S via thiol-disulfide exchange. DADS releases trace amounts of H2S via a slow reaction with GSH via the α-carbon nucleophilic substitution pathway [137,138,139,140,141] (Fig. 4). Garlic derivatives such as DATS and DADS are not only H2S donors, but also form a positive feedback loop for H2S production by acting with the H2S/CBS or H2S/CSE axis [142,143,144]. There are many studies related to the promotion of cancer cells by natural sources of H2S. They are summarized in the following Table 2.

DATS and DADS are two main active ingredients in garlic. DATS reacts rapidly with GSH to release H2S via thiol−disulfide exchange followed by allyl perthiol reduction by GSH. DADS only releases a minute amount of H2S via a sluggish reaction with GSH through an α-carbon nucleophilic substitution pathway. DATS diallyl trisulfide, DADS diallyl disulfide, GSH glutathione.
Acetyl deacylasadisulfide (ADA) is also a naturally occurring H2S donor. ADA inhibits the PI3K/Akt pathway and its downstream target NF-κB in melanoma cells, reduces the expression of the anti-apoptotic proteins c-FLIP, XIAP and Bcl-2, and promotes the activation of caspase-3 and PARP to induce apoptosis [145]. 12b is a novel synthetic H2S donor derived from natural ent-kaurane diterpenoid oridonin derivatives that can induce apoptosis in a variety of cancer cells (e.g. HepG2, HCT-116 and K562 cells) via extrinsic and intrinsic apoptotic pathways [146]. Another experiment demonstrates that in a variety of human cancer cell lines 12b inhibits the cell cycle and induces the mitochondrial apoptotic pathway via the release of H2S [147]. Erucin, a diet-derived H2S donor, can inhibit the proliferation and metastasis by suppressing calmodulin and the transcription factor expression during epithelial-to-mesenchymal transition (EMT) of melanoma cells [148]. Activating mutation of KRAS in pancreatic cancer cells AsPC-1 leads to hyperphosphorylation of ERK1/2 kinase, causing proliferation and growth of pancreatic cancer cells [149]. Erucic acid can directly induce apoptosis by releasing H2S and inhibit cell proliferation by inhibiting the phosphorylation of ERK1/2 in AsPC-1 cells.
H2S as a shield
It has been shown that CSE promotes intra-mitochondrial H2S production through translocation to mitochondria that can maintain mitochondrial production of ATP under hypoxic condition [150]. Suitable concentrations of H2S in the mammalian colon can serve as a substrate for the mitochondrial oxidative respiratory chain [151]. In particular, 3-MST modulates H2S production in mitochondria and is involved in complementing and balancing the bioenergetic role of Krebs cycle-derived electron donors [152]. Some teams have demonstrated that the CSE/H2S axis promotes mitochondrial biogenesis in hepatocytes by enhancing the expression and activity of PGC-1α [153]. CBS expression is upregulated in colon cancer cells. CBS produces H2S at low to moderate levels in the early stage of cancer development, which acts as a bioenergetic factor for cancer cell growth and promotes vasorelaxation generation, supporting tumor growth and proliferation [12, 90]. There is a positive feedback loop between H2S and nicotinamide phosphoribosyl transferase (Nampt) that regulates the dedifferentiation of cancer cells and helps them recover from potentially lethal damage [154]. Bhattacharyya S et al. find that CBS co-localizes with mitochondrial markers in ovarian cancer, and silencing CBS inhibits mitochondrial respiration and ATP synthesis, while increases ROS production. Mechanistically, silencing CBS reduces H2S production, severely decreases cellular GSH levels, activates tumor suppressor p53 and inhibits NF-κB [95]. H2S ameliorates hypoxia and oxidative stress-induced injury because of its ability to scavenge intracellular ROS and increase GSH level [155,156,157]. Exogenous application of 100 μM NaHS to pancreatic β cells inhibits cytokine-induced ROS production [158]. H2S is a vital endothelium-derived hyperpolarizing factor (EDHF) which activates ATP-sensitive, medium and small conductance potassium channels through cysteine S-sulfhydration, leading to hyperpolarization and vasorelaxation of vascular endothelial and smooth muscle cells [159]. CBS silencing inhibits tumor growth and neointimal density in rat models of colon and ovarian cancer, and low concentrations or endogenous H2S can promote tumor growth by promoting angiogenesis [12, 95]. An exogenous slow-releasing H2S donor, GYY4137 inhibits TNF-α-mediated endothelial cell death by S-sulfhydration of pro-caspase 3 leading to down-regulation of its activity [160]. Oral squamous cell carcinoma (SCC) is applied with low concentrations of NaHS, p-Akt and p-ERK1/2 expression is up-regulated in the NaHS-treated group compared with untreated controls, and induces more SCC cells to enter the S phase to promote cell proliferation [161]. 50–200 μM NaHS also induces proliferation of human colon cancer cells by inducing Akt and ERK phosphorylation, inhibiting p21 expression and NO production [162].
Some studies have investigated the protective mechanisms of H2S in different cancer cells. Jingfu Chen [163] demonstrates that the p38 MAPK/ERK1/2-COX-2 pathway is involved in NaHS-induced proliferation and anti-apoptosis in C6 glioma cells. H2S not only inhibits bronchial epithelial cell apoptosis through modulation of ER stress, but also inhibits NOD-like receptor pyrin domain containing protein 2 inflammatory vesicle formation via the Nrf2-dependent pathway [164, 165]. In the HCC cell line PLC/PRF/5, exogenous application of 500 μM NaHS inhibits caspase-3 production and activates the NF-κB pathway to promote cell proliferation [62]. It also inhibits apoptosis by activating STAT3/COX-2 signaling pathway [166]. In esophageal cancer EC109 cells, application of 400 μM NaHS significantly inhibits apoptosis by activating NF-κB and p38 MAPK/ERK1/2-COX-2 signaling pathways [54]. Similarly, 500 μM NaHS acts on multiple myeloma cells for 24 h can promote cell proliferation and migration by upregulating p-Akt expression [167].
Discussion
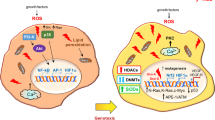
In summary, we know that H2S is a substance with small molecular mass that can freely penetrate lipid membranes and has the redox property, it exerts an anti-apoptotic effect in cancer cells via a variety of mechanisms (Fig. 5). Firstly, H2S, at low concentrations, scavenges ROS via its inherent reducing property and by elevating intracellular levels of GSH through the activation of cysteine/cystine transporter protein. Furthermore, H2S can directly activate K+ATP channel, induce vascular smooth muscle relaxation and promote local blood supply to tumors. Mustafa et al. report that H2S activates glyceraldehyde 3-phosphate dehydrogenase (GAPDH) via S-sulfhydration under hypoxic conditions, which is involved in glucose metabolism, and increases the enzymatic activity of GAPDH to promote anaerobic metabolism [168]. Lower concentrations of H2S also stimulate oxidative phosphorylation and increase ATP production by donating electrons to the mitochondrial electron transport chain. In addition to acting as a signaling molecule, H2S regulates apoptosis in cancer cells via S-sulfhydration of amino acid residues of certain protein molecules in signaling pathways. For example, ERK, JNK, and p38 [169,170,171,172], which control numerous pathophysiological processes, ERK regulates cell growth and differentiation, as well as JNK and p38 play important roles in inflammation and apoptosis. The PI3K/Akt pathway is another important intracellular signaling pathway that is significantly associated with tumorigenesis, cancer progression and drug resistance [173, 174]. It also responds to intra- and extra-cellular signals to promote metabolism, proliferation, cell survival, growth, and angiogenesis [175]. Mutations in the PI3K/Akt pathway are common phenomena in human cancers, and the two most frequently mutated genes are phosphatase and tensin homolog and PI3K-alpha. This pathway is able to inactivate the pro-apoptotic factors Bad and pro-caspase 9 and inhibit the expression of the death ligand FasL [176]. H2S directly sulfurizes the upstream and downstream of the p53 signaling pathway, NF-κB, and Keap1 to inhibit cell proliferation and induce apoptosis. H2S not only directly regulates various intracellular signaling pathways but also participates in the regulation of tumor microenvironment [177, 178]. Therefore, there is an urgent need for more effective methods to detect the concentration of H2S in cancer cells. The H2S level is then used as a guide for the development of novel H2S-releasing drugs, as well as the inhibitors/activators of H2S-producing enzymes. In conclusion, H2S can be applied in cancer treatment by identifing the mechanism of H2S in the apoptotic process of cancer cells.

Cyt C cytochrome C, Fas fas cell surface death receptor, Fasl fas ligand, Smac second mitochondria-derived activator of caspases, JNK C-Jun N-terminal kinase, MAPK mitogen activated protein kinase, NF-κB nuclear factor-kappa B, PI3K phosphoinositide 3-kinase, Akt protein kinase B, ERs endoplasmic reticulum stress, ROS reactive oxygen species.
References
Ishigami M, Hiraki K, Umemura K, Ogasawara Y, Ishii K, Kimura H. A source of hydrogen sulfide and a mechanism of its release in the brain. Antioxid Redox Signal. 2009;11:205–14. https://doi.org/10.1089/ars.2008.2132.
Kimura H. Hydrogen sulfide (H(2)S) and polysulfide (H(2)S(n)) signaling: the first 25 years. Biomolecules. 2021;11, https://doi.org/10.3390/biom11060896.
Shibuya N, Koike S, Tanaka M, Ishigami-Yuasa M, Kimura Y, Ogasawara Y, et al. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat Commun. 2013;4:1366. https://doi.org/10.1038/ncomms2371.
Kabil O, Banerjee R. Enzymology of H2S biogenesis, decay and signaling. Antioxid Redox Signal. 2014;20:770–82. https://doi.org/10.1089/ars.2013.5339.
Nagy P. Mechanistic chemical perspective of hydrogen sulfide signaling. Methods Enzymol. 2015;554:3–29. https://doi.org/10.1016/bs.mie.2014.11.036.
Ono K, Akaike T, Sawa T, Kumagai Y, Wink DA, Tantillo DJ, et al. Redox chemistry and chemical biology of H2S, hydropersulfides, and derived species: implications of their possible biological activity and utility. Free Radic Biol Med. 2014;77:82–94. https://doi.org/10.1016/j.freeradbiomed.2014.09.007.
Cortese-Krott MM, Koning A, Kuhnle GGC, Nagy P, Bianco CL, Pasch A, et al. The reactive species interactome: evolutionary emergence, biological significance, and opportunities for redox metabolomics and personalized medicine. Antioxid Redox Signal. 2017;27:684–712. https://doi.org/10.1089/ars.2017.7083.
Shi Y, Carroll KS. Activity-based sensing for site-specific proteomic analysis of cysteine oxidation. Acc Chem Res. 2020;53:20–31. https://doi.org/10.1021/acs.accounts.9b00562.
Khattak S, Rauf MA, Khan NH, Zhang QQ, Chen HJ, Muhammad P, et al. Hydrogen sulfide biology and its role in cancer. Molecules. 2022;27. https://doi.org/10.3390/molecules27113389.
Yang CT, Chen L, Xu S, Day JJ, Li X, Xian M. Recent development of hydrogen sulfide releasing/stimulating reagents and their potential applications in cancer and glycometabolic disorders. Front Pharmacol. 2017;8:664. https://doi.org/10.3389/fphar.2017.00664.
Nicholls P, Marshall DC, Cooper CE, Wilson MT. Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem Soc Trans. 2013;41:1312–6. https://doi.org/10.1042/bst20130070.
Szabo C, Coletta C, Chao C, Módis K, Szczesny B, Papapetropoulos A, et al. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci USA. 2013;110:12474–9. https://doi.org/10.1073/pnas.1306241110.
Wen YD, Wang H, Kho SH, Rinkiko S, Sheng X, Shen HM, et al. Hydrogen sulfide protects HUVECs against hydrogen peroxide induced mitochondrial dysfunction and oxidative stress. PLoS ONE. 2013;8:e53147. https://doi.org/10.1371/journal.pone.0053147.
Libiad M, Vitvitsky V, Bostelaar T, Bak DW, Lee HJ, Sakamoto N, et al. Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J Biol Chem. 2019;294:12077–90. https://doi.org/10.1074/jbc.RA119.009442.
Khan NH, Wang D, Wang W, Shahid M, Khattak S, Ngowi EE, et al. Pharmacological inhibition of endogenous hydrogen sulfide attenuates breast cancer progression. Molecules. 2022;27. https://doi.org/10.3390/molecules27134049.
Hellmich MR, Coletta C, Chao C, Szabo C. The therapeutic potential of cystathionine β-synthetase/hydrogen sulfide inhibition in cancer. Antioxid Redox Signal. 2015;22:424–48. https://doi.org/10.1089/ars.2014.5933.
Wu D, Li J, Zhang Q, Tian W, Zhong P, Liu Z, et al. Exogenous hydrogen sulfide regulates the growth of human thyroid carcinoma cells. Oxid Med Cell Longev. 2019;2019:6927298. https://doi.org/10.1155/2019/6927298.
Li L, He Z, Zhu Y, Shen Q, Yang S, Cao S. Hydrogen sulfide suppresses skin fibroblast proliferation via oxidative stress alleviation and necroptosis inhibition. Oxid Med Cell Longev. 2022;2022:7434733. https://doi.org/10.1155/2022/7434733.
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. https://doi.org/10.1080/01926230701320337.
Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D’Orazi G. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging. 2016;8:603–19. https://doi.org/10.18632/aging.100934.
Obeng E. Apoptosis (programmed cell death) and its signals - a review. Braz J Biol. 2021;81:1133–43. https://doi.org/10.1590/1519-6984.228437.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57. https://doi.org/10.1038/bjc.1972.33.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–d947. https://doi.org/10.1093/nar/gky1015.
Lewandowska AM, Rudzki M, Rudzki S, Lewandowski T, Laskowska B. Environmental risk factors for cancer - review paper. Ann Agric Environ Med. 2019;26:1–7. https://doi.org/10.26444/aaem/94299.
Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32:5129–43. https://doi.org/10.1038/onc.2012.640.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. https://doi.org/10.1016/j.cell.2011.02.013.
Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin cancer Res. 2011;30:87. https://doi.org/10.1186/1756-9966-30-87.
Plati J, Bucur O, Khosravi-Far R. Dysregulation of apoptotic signaling in cancer: molecular mechanisms and therapeutic opportunities. J Cell Biochem. 2008;104:1124–49. https://doi.org/10.1002/jcb.21707.
Adams JM. Ways of dying: multiple pathways to apoptosis. Genes Dev. 2003;17:2481–95. https://doi.org/10.1101/gad.1126903.
Van De Water TR, Lallemend F, Eshraghi AA, Ahsan S, He J, Guzman J, et al. Caspases, the enemy within, and their role in oxidative stress-induced apoptosis of inner ear sensory cells. Otol Neurotol. 2004;25:627–32. https://doi.org/10.1097/00129492-200407000-00035.
Nur EKA, Gross SR, Pan Z, Balklava Z, Ma J, Liu LF. Nuclear translocation of cytochrome c during apoptosis. J Biol Chem. 2004;279:24911–4. https://doi.org/10.1074/jbc.C400051200.
Ow YP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol. 2008;9:532–42. https://doi.org/10.1038/nrm2434.
Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–32. https://doi.org/10.1038/nrm2952.
Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835–40. https://doi.org/10.1007/s10495-006-0525-7.
Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–33. https://doi.org/10.1038/sj.onc.1207232.
Thorburn A. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) pathway signaling. J Thorac Oncol. 2007;2:461–5. https://doi.org/10.1097/JTO.0b013e31805fea64.
Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–80. https://doi.org/10.1038/sj.cdd.4401378.
Iurlaro R, Muñoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016;283:2640–52. https://doi.org/10.1111/febs.13598.
Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat cell Biol. 2003;5:1051–61. https://doi.org/10.1038/ncb1063.
Murphy B, Bhattacharya R, Mukherjee P. Hydrogen sulfide signaling in mitochondria and disease. FASEB J. 2019;33:13098–125. https://doi.org/10.1096/fj.201901304R.
Liu Y, Chen Q, Li Y, Bi L, Lin S, Ji H, et al. Hydrogen sulfide-induced oxidative stress mediated apoptosis via mitochondria pathway in embryo-larval stages of zebrafish. Ecotoxicol Environ Saf. 2022;239:113666. https://doi.org/10.1016/j.ecoenv.2022.113666.
Kobayashi C, Yaegaki K, Calenic B, Ishkitiev N, Imai T, Ii H, et al. Hydrogen sulfide causes apoptosis in human pulp stem cells. J Endod. 2011;37:479–84. https://doi.org/10.1016/j.joen.2011.01.017.
Hu LF, Lu M, Wu ZY, Wong PT, Bian JS. Hydrogen sulfide inhibits rotenone-induced apoptosis via preservation of mitochondrial function. Mol Pharmacol. 2009;75:27–34. https://doi.org/10.1124/mol.108.047985.
Wang B, Shao Z, Gu M, Ni L, Shi Y, Yan Y, et al. Hydrogen sulfide protects against IL-1β-induced inflammation and mitochondrial dysfunction-related apoptosis in chondrocytes and ameliorates osteoarthritis. J Cell Physiol. 2021;236:4369–86. https://doi.org/10.1002/jcp.30154.
Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, et al. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol cell. 2012;45:13–24. https://doi.org/10.1016/j.molcel.2011.10.021.
Lencesova L, Hudecova S, Csaderova L, Markova J, Soltysova A, Pastorek M, et al. Sulphide signalling potentiates apoptosis through the up-regulation of IP3 receptor types 1 and 2. Acta Physiol. 2013;208:350–61. https://doi.org/10.1111/apha.12105.
Liu ZW, Wang HY, Guan L, Zhao B. Regulatory effects of hydrogen sulfide on alveolar epithelial cell endoplasmic reticulum stress in rats with acute lung injury. World J Emerg Med. 2015;6:67–73. https://doi.org/10.5847/wjem.j.1920-8642.2015.01.012.
Lee ZW, Zhou J, Chen CS, Zhao Y, Tan CH, Li L, et al. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS ONE. 2011;6:e21077. https://doi.org/10.1371/journal.pone.0021077.
Xiao AY, Maynard MR, Piett CG, Nagel ZD, Alexander JS, Kevil CG, et al. Sodium sulfide selectively induces oxidative stress, DNA damage, and mitochondrial dysfunction and radiosensitizes glioblastoma (GBM) cells. Redox Biol. 2019;26:101220. https://doi.org/10.1016/j.redox.2019.101220.
Zhao L, Wang Y, Yan Q, Lv W, Zhang Y, He S. Exogenous hydrogen sulfide exhibits anti-cancer effects though p38 MAPK signaling pathway in C6 glioma cells. Biol Chem. 2015;396:1247–53. https://doi.org/10.1515/hsz-2015-0148.
Wang M, Yan J, Cao X, Hua P, Li Z. Hydrogen sulfide modulates epithelial-mesenchymal transition and angiogenesis in non-small cell lung cancer via HIF-1α activation. Biochem Pharmacol. 2020;172:113775. https://doi.org/10.1016/j.bcp.2019.113775.
Ma Y, Yan Z, Deng X, Guo J, Hu J, Yu Y, et al. Anticancer effect of exogenous hydrogen sulfide in cisplatin‑resistant A549/DDP cells. Oncol Rep. 2018;39:2969–77. https://doi.org/10.3892/or.2018.6362.
Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381:400–12. https://doi.org/10.1016/s0140-6736(12)60643-6.
Lei Y, Zhen Y, Zhang W, Sun X, Lin X, Feng J, et al. Exogenous hydrogen sulfide exerts proliferation, anti-apoptosis, angiopoiesis and migration effects via activating HSP90 pathway in EC109 cells. Oncol Rep. 2016;35:3714–20. https://doi.org/10.3892/or.2016.4734.
Hoter A, El-Sabban ME, Naim HY. The HSP90 family: structure, regulation, function, and implications in health and disease. Int J Mol Sci. 2018;19. https://doi.org/10.3390/ijms19092560.
Lei YY, Feng YF, Zeng B, Zhang W, Xu Q, Cheng F, et al. Exogenous H(2)S promotes cancer progression by activating JAK2/STAT3 signaling pathway in esophageal EC109 cells. Int J Clin Exp Pathol. 2018;11:3247–56.
Zhang L, Qi Q, Yang J, Sun D, Li C, Xue Y, et al. An anticancer role of hydrogen sulfide in human gastric cancer cells. Oxid Med Cell Longev. 2015;2015:636410. https://doi.org/10.1155/2015/636410.
Ma K, Liu Y, Zhu Q, Liu CH, Duan JL, Tan BK, et al. H2S donor, S-propargyl-cysteine, increases CSE in SGC-7901 and cancer-induced mice: evidence for a novel anti-cancer effect of endogenous H2S? PLoS ONE. 2011;6:e20525. https://doi.org/10.1371/journal.pone.0020525.
Ye F, Li X, Sun K, Xu W, Shi H, Bian J, et al. Inhibition of endogenous hydrogen sulfide biosynthesis enhances the anti-cancer effect of 3,3’-diindolylmethane in human gastric cancer cells. Life Sci. 2020;261:118348. https://doi.org/10.1016/j.lfs.2020.118348.
Zhu H, Chan KT, Huang X, Cerra C, Blake S, Trigos AS, et al. Cystathionine-β-synthase is essential for AKT-induced senescence and suppresses the development of gastric cancers with PI3K/AKT activation. eLife. 2022;11. https://doi.org/10.7554/eLife.71929.
Wu D, Li M, Tian W, Wang S, Cui L, Li H, et al. Hydrogen sulfide acts as a double-edged sword in human hepatocellular carcinoma cells through EGFR/ERK/MMP-2 and PTEN/AKT signaling pathways. Sci Rep. 2017;7:5134. https://doi.org/10.1038/s41598-017-05457-z.
Zhen Y, Pan W, Hu F, Wu H, Feng J, Zhang Y, et al. Exogenous hydrogen sulfide exerts proliferation/anti-apoptosis/angiogenesis/migration effects via amplifying the activation of NF-κB pathway in PLC/PRF/5 hepatoma cells. Int J Oncol. 2015;46:2194–204. https://doi.org/10.3892/ijo.2015.2914.
Lee MS, Jeong MH, Lee HW, Han HJ, Ko A, Hewitt SM, et al. PI3K/AKT activation induces PTEN ubiquitination and destabilization accelerating tumourigenesis. Nat Commun. 2015;6:7769. https://doi.org/10.1038/ncomms8769.
Pan Y, Ye S, Yuan D, Zhang J, Bai Y, Shao C. Hydrogen sulfide (H2S)/cystathionine γ-lyase (CSE) pathway contributes to the proliferation of hepatoma cells. Mutat Res. 2014;763-764:10–18. https://doi.org/10.1016/j.mrfmmm.2014.03.002.
Jia H, Ye J, You J, Shi X, Kang W, Wang T. Role of the cystathionine β-synthase/H2S system in liver cancer cells and the inhibitory effect of quinolone-indolone conjugate QIC2 on the system. Oncol Rep. 2017;37:3001–9. https://doi.org/10.3892/or.2017.5513.
Zhou YF, Song SS, Tian MX, Tang Z, Wang H, Fang Y, et al. Cystathionine β-synthase mediated PRRX2/IL-6/STAT3 inactivation suppresses Tregs infiltration and induces apoptosis to inhibit HCC carcinogenesis. Journal for Immunotherapy of Cancer. 2021;9. https://doi.org/10.1136/jitc-2021-003031.
Li M, Song X, Jin Q, Chen Y, Zhang J, Gao J, et al. 3-Mercaptopyruvate sulfurtransferase represses tumour progression and predicts prognosis in hepatocellular carcinoma. Liver Int. 2022;42:1173–84. https://doi.org/10.1111/liv.15228.
Ma HB, Huang S, Yin XR, Zhang Y, Di ZL. Apoptotic pathway induced by diallyl trisulfide in pancreatic cancer cells. World J Gastroenterol. 2014;20:193–203. https://doi.org/10.3748/wjg.v20.i1.193.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49. https://doi.org/10.3322/caac.21660.
Mathai JC, Missner A, Kügler P, Saparov SM, Zeidel ML, Lee JK, et al. No facilitator required for membrane transport of hydrogen sulfide. Proc Natl Acad Sci USA. 2009;106:16633–8. https://doi.org/10.1073/pnas.0902952106.
Blachier F, Andriamihaja M, Larraufie P, Ahn E, Lan A, Kim E. Production of hydrogen sulfide by the intestinal microbiota and epithelial cells and consequences for the colonic and rectal mucosa. Am J Physiol Gastrointest Liver Physiol. 2021;320:G125–35. https://doi.org/10.1152/ajpgi.00261.2020.
Moon JY, Kye BH, Ko SH, Yoo RN. Sulfur metabolism of the gut microbiome and colorectal cancer: the threat to the younger generation. Nutrients. 2023;15, https://doi.org/10.3390/nu15081966.
Oláh G, Módis K, Törö G, Hellmich MR, Szczesny B, Szabo C. Role of endogenous and exogenous nitric oxide, carbon monoxide and hydrogen sulfide in HCT116 colon cancer cell proliferation. Biochem Pharmacol. 2018;149:186–204. https://doi.org/10.1016/j.bcp.2017.10.011.
Faris P, Ferulli F, Vismara M, Tanzi M, Negri S, Rumolo A, et al. Hydrogen sulfide-evoked intracellular Ca(2+) signals in primary cultures of metastatic colorectal cancer cells. Cancers. 2020;12. https://doi.org/10.3390/cancers12113338.
Babic AM, Kireeva ML, Kolesnikova TV, Lau LF. CYR61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc Natl Acad Sci USA. 1998;95:6355–60. https://doi.org/10.1073/pnas.95.11.6355.
Xie L, Song X, Lin H, Chen Z, Li Q, Guo T, et al. Aberrant activation of CYR61 enhancers in colorectal cancer development. J Exp Clin Cancer Res. 2019;38:213. https://doi.org/10.1186/s13046-019-1217-9.
Huang X, Xiang L, Li Y, Zhao Y, Zhu H, Xiao Y, et al. Snail/FOXK1/Cyr61 signaling axis regulates the epithelial-mesenchymal transition and metastasis in colorectal cancer. Cell Physiol Biochem. 2018;47:590–603. https://doi.org/10.1159/000490015.
Ascenção K, Lheimeur B, Szabo C. Regulation of CyR61 expression and release by 3-mercaptopyruvate sulfurtransferase in colon cancer cells. Redox Biol. 2022;56:102466. https://doi.org/10.1016/j.redox.2022.102466.
Maclean KN, Kraus E, Kraus JP. The dominant role of Sp1 in regulating the cystathionine beta-synthase -1a and -1b promoters facilitates potential tissue-specific regulation by Kruppel-like factors. J Biol Chem. 2004;279:8558–66. https://doi.org/10.1074/jbc.M310211200.
Pagliara V, Saide A, Mitidieri E, d’Emmanuele di Villa Bianca R, Sorrentino R, Russo G, et al. 5-FU targets rpL3 to induce mitochondrial apoptosis via cystathionine-β-synthase in colon cancer cells lacking p53. Oncotarget. 2016;7:50333–48. https://doi.org/10.18632/oncotarget.10385.
Bennett MR. Mechanisms of p53-induced apoptosis. Biochem Pharmacol. 1999;58:1089–95. https://doi.org/10.1016/s0006-2952(99)00153-7.
Luo Q, Beaver JM, Liu Y, Zhang Z. Dynamics of p53: a master decider of cell fate. Genes. 2017;8. https://doi.org/10.3390/genes8020066.
Georgakilas AG, Martin OA, Bonner WM. p21: a two-faced genome guardian. Trends Mol Med. 2017;23:310–9. https://doi.org/10.1016/j.molmed.2017.02.001.
Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–908. https://doi.org/10.1038/sj.onc.1208615.
Kanapathipillai, M. Treating p53 mutant aggregation-associated cancer. Cancers. 2018;10. https://doi.org/10.3390/cancers10060154.
Reyes-Zurita FJ, Rufino-Palomares EE, Medina PP, Leticia García-Salguero E, Peragón J, Cascante M, et al. Antitumour activity on extrinsic apoptotic targets of the triterpenoid maslinic acid in p53-deficient Caco-2 adenocarcinoma cells. Biochimie. 2013;95:2157–67. https://doi.org/10.1016/j.biochi.2013.08.017.
Reyes-Zurita FJ, Rufino-Palomares EE, García-Salguero L, Peragón J, Medina PP, Parra A, et al. Maslinic acid, a natural triterpene, induces a death receptor-mediated apoptotic mechanism in Caco-2 p53-deficient colon adenocarcinoma cells. PLoS ONE. 2016;11:e0146178. https://doi.org/10.1371/journal.pone.0146178.
Sakuma S, Minamino S, Takase M, Ishiyama Y, Hosokura H, Kohda T, et al. Hydrogen sulfide donor GYY4137 suppresses proliferation of human colorectal cancer Caco-2 cells by inducing both cell cycle arrest and cell death. Heliyon. 2019;5:e02244. https://doi.org/10.1016/j.heliyon.2019.e02244.
Kajsik M, Chovancova B, Liskova V, Babula P, Krizanova O. Slow sulfide donor GYY4137 potentiates effect of paclitaxel on colorectal carcinoma cells. Eur J Pharmacol. 2022;922:174875. https://doi.org/10.1016/j.ejphar.2022.174875.
Módis K, Coletta C, Asimakopoulou A, Szczesny B, Chao C, Papapetropoulos A, et al. Effect of S-adenosyl-L-methionine (SAM), an allosteric activator of cystathionine-β-synthase (CBS) on colorectal cancer cell proliferation and bioenergetics in vitro. Nitric Oxide. 2014;41:146–56. https://doi.org/10.1016/j.niox.2014.03.001.
Yue T, Zuo S, Bu D, Zhu J, Chen S, Ma Y, et al. Aminooxyacetic acid (AOAA) sensitizes colon cancer cells to oxaliplatin via exaggerating apoptosis induced by ROS. J Cancer. 2020;11:1828–38. https://doi.org/10.7150/jca.35375.
Wang DY, Zhang J, Li HX, Zhang YX, Jing MR, Cai CB, et al. Inhibition of endogenous hydrogen sulfide production suppresses the growth of nasopharyngeal carcinoma cells. Mol Carcinog. 2023. https://doi.org/10.1002/mc.23513.
Ljungberg B, Albiges L, Abu-Ghanem Y, Bedke J, Capitanio U, Dabestani S, et al. European Association of Urology Guidelines on Renal Cell Carcinoma: the 2022 update. Eur Urol. 2022;82:399–410. https://doi.org/10.1016/j.eururo.2022.03.006.
Breza J Jr., Soltysova A, Hudecova S, Penesova A, Szadvari I, Babula P, et al. Endogenous H(2)S producing enzymes are involved in apoptosis induction in clear cell renal cell carcinoma. BMC Cancer. 2018;18:591. https://doi.org/10.1186/s12885-018-4508-1.
Bhattacharyya S, Saha S, Giri K, Lanza IR, Nair KS, Jennings NB, et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS ONE. 2013;8:e79167. https://doi.org/10.1371/journal.pone.0079167.
Lencesova L, Vlcek M, Krizanova O, Hudecova S. Hypoxic conditions increases H2S-induced ER stress in A2870 cells. Mol Cell Biochem. 2016;414:67–76. https://doi.org/10.1007/s11010-016-2659-4.
Szadvari I, Hudecova S, Chovancova B, Matuskova M, Cholujova D, Lencesova L, et al. Sodium/calcium exchanger is involved in apoptosis induced by H(2)S in tumor cells through decreased levels of intracellular pH. Nitric Oxide. 2019;87:1–9. https://doi.org/10.1016/j.niox.2019.02.011.
Markova J, Hudecova S, Soltysova A, Sirova M, Csaderova L, Lencesova L, et al. Sodium/calcium exchanger is upregulated by sulfide signaling, forms complex with the β1 and β3 but not β2 adrenergic receptors, and induces apoptosis. Pflug Arch. 2014;466:1329–42. https://doi.org/10.1007/s00424-013-1366-1.
Cortes J, Pavlovsky C, Saußele S. Chronic myeloid leukaemia. Lancet. 2021;398:1914–26. https://doi.org/10.1016/s0140-6736(21)01204-6.
Hughes T, Branford S. Molecular monitoring of BCR-ABL as a guide to clinical management in chronic myeloid leukaemia. Blood Rev. 2006;20:29–41. https://doi.org/10.1016/j.blre.2005.01.008.
Stein SJ, Baldwin AS. NF-κB suppresses ROS levels in BCR-ABL(+) cells to prevent activation of JNK and cell death. Oncogene. 2011;30:4557–66. https://doi.org/10.1038/onc.2011.156.
Wang D, Yang H, Zhang Y, Hu R, Hu D, Wang Q, et al. Inhibition of cystathionine β-synthase promotes apoptosis and reduces cell proliferation in chronic myeloid leukemia. Signal Transduct Target Ther. 2021;6:52. https://doi.org/10.1038/s41392-020-00410-5.
Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. https://doi.org/10.1016/j.cell.2012.06.024.
Ascierto PA, Kirkwood JM, Grob JJ, Simeone E, Grimaldi AM, Maio M, et al. The role of BRAF V600 mutation in melanoma. J Transl Med. 2012;10:85. https://doi.org/10.1186/1479-5876-10-85.
Panza E, De Cicco P, Armogida C, Scognamiglio G, Gigantino V, Botti G, et al. Role of the cystathionine γ lyase/hydrogen sulfide pathway in human melanoma progression. Pigment cell melanoma Res. 2015;28:61–72. https://doi.org/10.1111/pcmr.12312.
Xiao Q, Ying J, Qiao Z, Yang Y, Dai X, Xu Z, et al. Exogenous hydrogen sulfide inhibits human melanoma cell development via suppression of the PI3K/AKT/ mTOR pathway. J Dermatol Sci. 2020;98:26–34. https://doi.org/10.1016/j.jdermsci.2020.02.004.
Chen Y, Takikawa M, Tsutsumi S, Yamaguchi Y, Okabe A, Shimada M, et al. PHLDA1, another PHLDA family protein that inhibits Akt. Cancer Sci. 2018;109:3532–42. https://doi.org/10.1111/cas.13796.
Murata T, Sato T, Kamoda T, Moriyama H, Kumazawa Y, Hanada N. Differential susceptibility to hydrogen sulfide-induced apoptosis between PHLDA1-overexpressing oral cancer cell lines and oral keratinocytes: role of PHLDA1 as an apoptosis suppressor. Exp Cell Res. 2014;320:247–57. https://doi.org/10.1016/j.yexcr.2013.10.023.
Ghlichloo I, Gerriets V. StatPearls. StatPearls Publishing Copyright © 2023, StatPearls Publishing LLC.; 2023.
Amann R, Peskar BA. Anti-inflammatory effects of aspirin and sodium salicylate. Eur J Pharmacol. 2002;447:1–9. https://doi.org/10.1016/s0014-2999(02)01828-9.
Hashemi Goradel N, Najafi M, Salehi E, Farhood B, Mortezaee K. Cyclooxygenase-2 in cancer: a review. J Cell Physiol. 2019;234:5683–99. https://doi.org/10.1002/jcp.27411.
Elwood P, Protty M, Morgan G, Pickering J, Delon C, Watkins J. Aspirin and cancer: biological mechanisms and clinical outcomes. Open Biol. 2022;12:220124. https://doi.org/10.1098/rsob.220124.
Wallace JL. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol Sci. 2007;28:501–5. https://doi.org/10.1016/j.tips.2007.09.003.
Chattopadhyay M, Kodela R, Nath N, Dastagirzada YM, Velázquez-Martínez CA, Boring D, et al. Hydrogen sulfide-releasing NSAIDs inhibit the growth of human cancer cells: a general property and evidence of a tissue type-independent effect. Biochem Pharmacol. 2012;83:715–22. https://doi.org/10.1016/j.bcp.2011.12.018.
Chattopadhyay M, Kodela R, Olson KR, Kashfi K. NOSH-aspirin (NBS-1120), a novel nitric oxide- and hydrogen sulfide-releasing hybrid is a potent inhibitor of colon cancer cell growth in vitro and in a xenograft mouse model. Biochem Biophys Res Commun. 2012;419:523–8. https://doi.org/10.1016/j.bbrc.2012.02.051.
Kodela, R, Chattopadhyay, M & Kashfi, K Synthesis and biological activity of NOSH-naproxen (AVT-219) and NOSH-sulindac (AVT-18A) as potent anti-inflammatory agents with chemotherapeutic potential. MedChemComm. 2013;4. https://doi.org/10.1039/c3md00185g.
Kashfi K. Utility of nitric oxide and hydrogen sulfide-releasing chimeras as anticancer agents. Redox Biol. 2015;5:420. https://doi.org/10.1016/j.redox.2015.09.030.
Kashfi K, Chattopadhyay M, Kodela R. NOSH-sulindac (AVT-18A) is a novel nitric oxide- and hydrogen sulfide-releasing hybrid that is gastrointestinal safe and has potent anti-inflammatory, analgesic, antipyretic, anti-platelet, and anti-cancer properties. Redox Biol. 2015;6:287–96. https://doi.org/10.1016/j.redox.2015.08.012.
Chattopadhyay M, Kodela R, Santiago G, Le TTC, Nath N, Kashfi K. NOSH-aspirin (NBS-1120) inhibits pancreatic cancer cell growth in a xenograft mouse model: Modulation of FoxM1, p53, NF-κB, iNOS, caspase-3 and ROS. Biochem Pharmacol. 2020;176:113857. https://doi.org/10.1016/j.bcp.2020.113857.
Duan SF, Zhang MM, Zhang X, Liu W, Zhang SH, Yang B, et al. HA-ADT suppresses esophageal squamous cell carcinoma progression via apoptosis promotion and autophagy inhibition. Exp Cell Res. 2022;420:113341. https://doi.org/10.1016/j.yexcr.2022.113341.
Zhang J, Li X, Han X, Liu R, Fang J. Targeting the thioredoxin system for cancer therapy. Trends Pharmacol Sci. 2017;38:794–808. https://doi.org/10.1016/j.tips.2017.06.001.
Bian M, Fan R, Zhao S, Liu W. Targeting the Thioredoxin System as a Strategy for Cancer Therapy. J Med Chem. 2019;62:7309–21. https://doi.org/10.1021/acs.jmedchem.8b01595.
Jastrząb A, Skrzydlewska E. Thioredoxin-dependent system. Application of inhibitors. J Enzym Inhib Med Chem. 2021;36:362–71. https://doi.org/10.1080/14756366.2020.1867121.
Chen F, Castranova V, Shi X. New insights into the role of nuclear factor-kappaB in cell growth regulation. Am J Pathol. 2001;159:387–97. https://doi.org/10.1016/s0002-9440(10)61708-7.
Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death—a new approach to cancer therapy. J Clin Investig. 2005;115:2625–32. https://doi.org/10.1172/jci26322.
Inoue J, Gohda J, Akiyama T, Semba K. NF-kappaB activation in development and progression of cancer. Cancer Sci. 2007;98:268–74. https://doi.org/10.1111/j.1349-7006.2007.00389.x.
Chattopadhyay M, Kodela R, Nath N, Barsegian A, Boring D, Kashfi K. Hydrogen sulfide-releasing aspirin suppresses NF-κB signaling in estrogen receptor negative breast cancer cells in vitro and in vivo. Biochem Pharmacol. 2012;83:723–32. https://doi.org/10.1016/j.bcp.2011.12.019.
Kodela R, Nath N, Chattopadhyay M, Nesbitt DE, Velázquez-Martínez CA, Kashfi K. Hydrogen sulfide-releasing naproxen suppresses colon cancer cell growth and inhibits NF-κB signaling. Drug Des Dev Ther. 2015;9:4873–82. https://doi.org/10.2147/dddt.S91116.
De Cicco P, Panza E, Ercolano G, Armogida C, Sessa G, Pirozzi G, et al. ATB-346, a novel hydrogen sulfide-releasing anti-inflammatory drug, induces apoptosis of human melanoma cells and inhibits melanoma development in vivo. Pharmacol Res. 2016;114:67–73. https://doi.org/10.1016/j.phrs.2016.10.019.
Cai F, Xu H, Cao N, Zhang X, Liu J, Lu Y, et al. ADT-OH, a hydrogen sulfide-releasing donor, induces apoptosis and inhibits the development of melanoma in vivo by upregulating FADD. Cell Death Dis. 2020;11:33. https://doi.org/10.1038/s41419-020-2222-9.
Tesei A, Brigliadori G, Carloni S, Fabbri F, Ulivi P, Arienti C, et al. Organosulfur derivatives of the HDAC inhibitor valproic acid sensitize human lung cancer cell lines to apoptosis and to cisplatin cytotoxicity. J Cell Physiol. 2012;227:3389–96. https://doi.org/10.1002/jcp.24039.
Ciocci M, Iorio E, Carotenuto F, Khashoggi HA, Nanni F, Melino S. H2S-releasing nanoemulsions: a new formulation to inhibit tumor cells proliferation and improve tissue repair. Oncotarget. 2016;7:84338–58. https://doi.org/10.18632/oncotarget.12609.
Singh N, Sharma S, Singh R, Rajput S, Chattopadhyay N, Tewari D, et al. A naphthalimide-based peptide conjugate for concurrent imaging and apoptosis induction in cancer cells by utilizing endogenous hydrogen sulfide. Chem Sci. 2021;12:16085–91. https://doi.org/10.1039/d1sc04030h.
Li N, Wang J, Zang X, Wang Z, Zhang T, Zhao B, et al. H(2)S probe CPC inhibits autophagy and promotes apoptosis by inhibiting glutathionylation of Keap1 at Cys434. Apoptosis. 2021;26:111–31. https://doi.org/10.1007/s10495-020-01652-y.
Martelli A, Citi V, Testai L, Brogi S, Calderone V. Organic isothiocyanates as hydrogen sulfide donors. Antioxid Redox Signal. 2020;32:110–44. https://doi.org/10.1089/ars.2019.7888.
Benavides GA, Squadrito GL, Mills RW, Patel HD, Isbell TS, Patel RP, et al. Hydrogen sulfide mediates the vasoactivity of garlic. Proc Natl Acad Sci USA. 2007;104:17977–82. https://doi.org/10.1073/pnas.0705710104.
Liang D, Wu H, Wong MW, Huang D. Diallyl trisulfide is a fast H2S donor, but diallyl disulfide is a slow one: the reaction pathways and intermediates of glutathione with polysulfides. Org Lett. 2015;17:4196–9. https://doi.org/10.1021/acs.orglett.5b01962.
Chen LY, Chen Q, Zhu XJ, Kong DS, Wu L, Shao JJ, et al. Diallyl trisulfide protects against ethanol-induced oxidative stress and apoptosis via a hydrogen sulfide-mediated mechanism. Int Immunopharmacol. 2016;36:23–30. https://doi.org/10.1016/j.intimp.2016.04.015.
Cai YR, Hu CH. Computational study of H(2)S release in reactions of diallyl polysulfides with thiols. J Phys Chem B. 2017;121:6359–66. https://doi.org/10.1021/acs.jpcb.7b03683.
Zhang F, Jin H, Wu L, Shao J, Zhu X, Chen A, et al. Diallyl trisulfide suppresses oxidative stress-induced activation of hepatic stellate cells through production of hydrogen sulfide. Oxid Med Cell Longev. 2017;2017:1406726. https://doi.org/10.1155/2017/1406726.
Dutta S, Mahalanobish S, Saha S, Ghosh S, Sil PC. Natural products: an upcoming therapeutic approach to cancer. Food Chem Toxicol. 2019;128:240–55. https://doi.org/10.1016/j.fct.2019.04.012.
Tsai CY, Wen SY, Shibu MA, Yang YC, Peng H, Wang B, et al. Diallyl trisulfide protects against high glucose-induced cardiac apoptosis by stimulating the production of cystathionine gamma-lyase-derived hydrogen sulfide. Int J Cardiol. 2015;195:300–10. https://doi.org/10.1016/j.ijcard.2015.05.111.
Xu S, Pan J, Cheng X, Zheng J, Wang X, Guan H, et al. Diallyl trisulfide, a H(2) S donor, inhibits cell growth of human papillary thyroid carcinoma KTC-1 cells through a positive feedback loop between H(2) S and cystathionine-gamma-lyase. Phytother Res. 2020;34:1154–65. https://doi.org/10.1002/ptr.6586.
Rose P, Moore PK, Zhu YZ. Garlic and gaseous mediators. Trends Pharmacol Sci. 2018;39:624–34. https://doi.org/10.1016/j.tips.2018.03.009.
De Cicco P, Panza E, Armogida C, Ercolano G, Taglialatela-Scafati O, Shokoohinia Y, et al. The hydrogen sulfide releasing molecule acetyl deacylasadisulfide inhibits metastatic melanoma. Front Pharmacol. 2017;8:65. https://doi.org/10.3389/fphar.2017.00065.
Li H, Mu J, Sun J, Xu S, Liu W, Xu F, et al. Hydrogen sulfide releasing oridonin derivatives induce apoptosis through extrinsic and intrinsic pathways. Eur J Med Chem. 2020;187:111978. https://doi.org/10.1016/j.ejmech.2019.111978.
Li H, Gao X, Huang X, Wang X, Xu S, Uchita T, et al. Hydrogen sulfide donating ent-kaurane and spirolactone-type 6,7-seco-ent-kaurane derivatives: design, synthesis and antiproliferative properties. Eur J Med Chem. 2019;178:446–57. https://doi.org/10.1016/j.ejmech.2019.06.016.
Maresca DC, Conte L, Romano B, Ianaro A, Ercolano G Antiproliferative and proapoptotic effects of erucin, a diet-derived H(2)S donor, on human melanoma cells. Antioxidants. 2022;12. https://doi.org/10.3390/antiox12010041.
Citi V, Piragine E, Pagnotta E, Ugolini L, Di Cesare Mannelli L, Testai L, et al. Anticancer properties of erucin, an H(2) S-releasing isothiocyanate, on human pancreatic adenocarcinoma cells (AsPC-1). Phytother Res. 2019;33:845–55. https://doi.org/10.1002/ptr.6278.
Fu M, Zhang W, Wu L, Yang G, Li H, Wang R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc Natl Acad Sci USA. 2012;109:2943–8. https://doi.org/10.1073/pnas.1115634109.
Goubern M, Andriamihaja M, Nübel T, Blachier F, Bouillaud F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007;21:1699–706. https://doi.org/10.1096/fj.06-7407com.
Módis K, Coletta C, Erdélyi K, Papapetropoulos A, Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013;27:601–11. https://doi.org/10.1096/fj.12-216507.
Untereiner AA, Fu M, Módis K, Wang R, Ju Y, Wu L. Stimulatory effect of CSE-generated H2S on hepatic mitochondrial biogenesis and the underlying mechanisms. Nitric Oxide. 2016;58:67–76. https://doi.org/10.1016/j.niox.2016.06.005.
Ostrakhovitch EA, Akakura S, Sanokawa-Akakura R, Goodwin S, Tabibzadeh S. Dedifferentiation of cancer cells following recovery from a potentially lethal damage is mediated by H2S-Nampt. Exp Cell Res. 2015;330:135–50. https://doi.org/10.1016/j.yexcr.2014.09.027.
Ali MY, Ping CY, Mok YY, Ling L, Whiteman M, Bhatia M, et al. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br J Pharmacol. 2006;149:625–34. https://doi.org/10.1038/sj.bjp.0706906.
Liu YY, Nagpure BV, Wong PT, Bian JS. Hydrogen sulfide protects SH-SY5Y neuronal cells against d-galactose induced cell injury by suppression of advanced glycation end products formation and oxidative stress. Neurochem Int. 2013;62:603–9. https://doi.org/10.1016/j.neuint.2012.12.010.
Lee ZW, Low YL, Huang S, Wang T, Deng LW. The cystathionine γ-lyase/hydrogen sulfide system maintains cellular glutathione status. Biochem J. 2014;460:425–35. https://doi.org/10.1042/bj20131434.
Taniguchi S, Kang L, Kimura T, Niki I. Hydrogen sulphide protects mouse pancreatic β-cells from cell death induced by oxidative stress, but not by endoplasmic reticulum stress. Br J Pharmacol. 2011;162:1171–8. https://doi.org/10.1111/j.1476-5381.2010.01119.x.
Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res. 2011;109:1259–68. https://doi.org/10.1161/circresaha.111.240242.
Diaz Sanchez L, Sanchez-Aranguren L, Wang K, Spickett CM, Griffiths HR, Dias IHK. TNF-α-mediated endothelial cell apoptosis is rescued by hydrogen sulfide. Antioxidants. 2023;12. https://doi.org/10.3390/antiox12030734.
Ma Z, Bi Q, Wang Y. Hydrogen sulfide accelerates cell cycle progression in oral squamous cell carcinoma cell lines. Oral Dis. 2015;21:156–62. https://doi.org/10.1111/odi.12223.
Cai WJ, Wang MJ, Ju LH, Wang C, Zhu YC. Hydrogen sulfide induces human colon cancer cell proliferation: role of Akt, ERK and p21. Cell Biol Int. 2010;34:565–72. https://doi.org/10.1042/cbi20090368.
Zhen Y, Zhang W, Liu C, He J, Lu Y, Guo R, et al. Exogenous hydrogen sulfide promotes C6 glioma cell growth through activation of the p38 MAPK/ERK1/2-COX-2 pathways. Oncol Rep. 2015;34:2413–22. https://doi.org/10.3892/or.2015.4248.
Lin F, Liao C, Sun Y, Zhang J, Lu W, Bai Y, et al. Hydrogen sulfide inhibits cigarette smoke-induced endoplasmic reticulum stress and apoptosis in bronchial epithelial cells. Front Pharmacol. 2017;8:675. https://doi.org/10.3389/fphar.2017.00675.
Jia G, Yu S, Sun W, Yang J, Wang Y, Qi Y, et al. Hydrogen sulfide attenuates particulate matter-induced emphysema and airway inflammation through Nrf2-dependent manner. Front Pharmacol. 2020;11:29. https://doi.org/10.3389/fphar.2020.00029.
Zhen Y, Wu Q, Ding Y, Zhang W, Zhai Y, Lin X. et al. Exogenous hydrogen sulfide promotes hepatocellular carcinoma cell growth by activating the STAT3-COX-2 signaling pathway. Oncol Lett. 2018;15:6562–70.https://doi.org/10.3892/ol.2018.8154.
Zheng D, Chen Z, Chen J, Zhuang X, Feng J, Li J. Exogenous hydrogen sulfide exerts proliferation, anti-apoptosis, migration effects and accelerates cell cycle progression in multiple myeloma cells via activating the Akt pathway. Oncol Rep. 2016;36:1909–16. https://doi.org/10.3892/or.2016.5014.
Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, et al. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. https://doi.org/10.1126/scisignal.2000464.
Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–49. https://doi.org/10.1038/sj.onc.1207556.
Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–2. https://doi.org/10.1126/science.1072682.
Morrison DK. MAP kinase pathways. Cold Spring Harbor perspectives in biology. 2012;4. https://doi.org/10.1101/cshperspect.a011254.
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–83. https://doi.org/10.1210/edrv.22.2.0428.
Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment. Annu Rev Med. 2016;67:11–28. https://doi.org/10.1146/annurev-med-062913-051343.
Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405. https://doi.org/10.1016/j.cell.2017.04.001.
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. https://doi.org/10.1038/nrg1879.
Kandasamy K, Srivastava RK. Role of the phosphatidylinositol 3’-kinase/PTEN/Akt kinase pathway in tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in non-small cell lung cancer cells. Cancer Res. 2002;62:4929–37.
Machado-Neto JA, Cerqueira ARA, Veríssimo-Filho S, Muscará MN, Costa SKP, Lopes LR. Hydrogen sulfide signaling in the tumor microenvironment: implications in cancer progression and therapy. Antioxid Redox Signal. 2023. https://doi.org/10.1089/ars.2021.0256.
Yue T, Li J, Zhu J, Zuo S, Wang X, Liu Y, et al. Hydrogen sulfide creates a favorable immune microenvironment for colon cancer. Cancer Res. 2023;83:595–612. https://doi.org/10.1158/0008-5472.Can-22-1837.
Yin X, Zhang J, Li X, Liu D, Feng C, Liang R, et al. DADS suppresses human esophageal xenograft tumors through RAF/MEK/ERK and mitochondria-dependent pathways. Int J Mol Sci. 2014;15:12422–41. https://doi.org/10.3390/ijms150712422.
Yin X, Zhang R, Feng C, Zhang J, Liu D, Xu K, et al. Diallyl disulfide induces G2/M arrest and promotes apoptosis through the p53/p21 and MEK-ERK pathways in human esophageal squamous cell carcinoma. Oncol Rep. 2014;32:1748–56. https://doi.org/10.3892/or.2014.3361.
Hong YS, Ham YA, Choi JH, Kim J. Effects of allyl sulfur compounds and garlic extract on the expression of Bcl-2, Bax, and p53 in non small cell lung cancer cell lines. Exp Mol Med. 2000;32:127–34. https://doi.org/10.1038/emm.2000.22.
Xiao D, Zeng Y, Hahm ER, Kim YA, Ramalingam S, Singh SV. Diallyl trisulfide selectively causes Bax- and Bak-mediated apoptosis in human lung cancer cells. Environ Mol Mutagen. 2009;50:201–12. https://doi.org/10.1002/em.20431.
Li W, Tian H, Li L, Li S, Yue W, Chen Z, et al. Diallyl trisulfide induces apoptosis and inhibits proliferation of A549 cells in vitro and in vivo. Acta Biochim Biophys Sin. 2012;44:577–83. https://doi.org/10.1093/abbs/gms033.
Nakagawa H, Tsuta K, Kiuchi K, Senzaki H, Tanaka K, Hioki K, et al. Growth inhibitory effects of diallyl disulfide on human breast cancer cell lines. Carcinogenesis. 2001;22:891–7. https://doi.org/10.1093/carcin/22.6.891.
Lei XY, Yao SQ, Zu XY, Huang ZX, Liu LJ, Zhong M, et al. Apoptosis induced by diallyl disulfide in human breast cancer cell line MCF-7. Acta Pharmacol Sin. 2008;29:1233–9. https://doi.org/10.1111/j.1745-7254.2008.00851.x.
Lee BC, Park BH, Kim SY, Lee YJ. Role of Bim in diallyl trisulfide-induced cytotoxicity in human cancer cells. J Cell Biochem. 2011;112:118–27. https://doi.org/10.1002/jcb.22896.
Na HK, Kim EH, Choi MA, Park JM, Kim DH, Surh YJ. Diallyl trisulfide induces apoptosis in human breast cancer cells through ROS-mediated activation of JNK and AP-1. Biochem Pharmacol. 2012;84:1241–50. https://doi.org/10.1016/j.bcp.2012.08.024.
Chandra-Kuntal K, Lee J, Singh SV. Critical role for reactive oxygen species in apoptosis induction and cell migration inhibition by diallyl trisulfide, a cancer chemopreventive component of garlic. Breast Cancer Res Treat. 2013;138:69–79. https://doi.org/10.1007/s10549-013-2440-2.
Hahm ER, Singh SV. Diallyl trisulfide inhibits estrogen receptor-α activity in human breast cancer cells. Breast Cancer Res Treat. 2014;144:47–57. https://doi.org/10.1007/s10549-014-2841-x.
Kwon KB, Yoo SJ, Ryu DG, Yang JY, Rho HW, Kim JS, et al. Induction of apoptosis by diallyl disulfide through activation of caspase-3 in human leukemia HL-60 cells. Biochem Pharmacol. 2002;63:41–47. https://doi.org/10.1016/s0006-2952(01)00860-7.
Tan H, Ling H, He J, Yi L, Zhou J, Lin M, et al. Inhibition of ERK and activation of p38 are involved in diallyl disulfide induced apoptosis of leukemia HL-60 cells. Arch Pharmacal Res. 2008;31:786–93. https://doi.org/10.1007/s12272-001-1227-0.
Wang H, Sun N, Li X, Li K, Tian J, Li J. Diallyl trisulfide induces osteosarcoma cell apoptosis through reactive oxygen species-mediated downregulation of the PI3K/Akt pathway. Oncol Rep. 2016;35:3648–58. https://doi.org/10.3892/or.2016.4722.
Park EK, Kwon KB, Park KI, Park BH, Jhee EC. Role of Ca(2+) in diallyl disulfide-induced apoptotic cell death of HCT-15 cells. Exp Mol Med. 2002;34:250–7. https://doi.org/10.1038/emm.2002.35.
Yang JS, Chen GW, Hsia TC, Ho HC, Ho CC, Lin MW, et al. Diallyl disulfide induces apoptosis in human colon cancer cell line (COLO 205) through the induction of reactive oxygen species, endoplasmic reticulum stress, caspases casade and mitochondrial-dependent pathways. Food Chem Toxicol. 2009;47:171–9. https://doi.org/10.1016/j.fct.2008.10.032.
Wu PP, Liu KC, Huang WW, Chueh FS, Ko YC, Chiu TH, et al. Diallyl trisulfide (DATS) inhibits mouse colon tumor in mouse CT-26 cells allograft model in vivo. Phytomedicine. 2011;18:672–6. https://doi.org/10.1016/j.phymed.2011.01.006.
Yu CS, Huang AC, Lai KC, Huang YP, Lin MW, Yang JS, et al. Diallyl trisulfide induces apoptosis in human primary colorectal cancer cells. Oncol Rep. 2012;28:949–54. https://doi.org/10.3892/or.2012.1882.
Kim HJ, Kang S, Kim DY, You S, Park D, Oh SC, et al. Diallyl disulfide (DADS) boosts TRAIL-Mediated apoptosis in colorectal cancer cells by inhibiting Bcl-2. Food Chem Toxicol. 2019;125:354–60. https://doi.org/10.1016/j.fct.2019.01.023.
Zhou C, Mao XP, Guo Q, Zeng FQ. Diallyl trisulphide-induced apoptosis in human melanoma cells involves downregulation of Bcl-2 and Bcl-xL expression and activation of caspases. Clin Exp Dermatol. 2009;34:e537–43. https://doi.org/10.1111/j.1365-2230.2009.03594.x.
Wang HC, Yang JH, Hsieh SC, Sheen LY. Allyl sulfides inhibit cell growth of skin cancer cells through induction of DNA damage mediated G2/M arrest and apoptosis. J Agric food Chem. 2010;58:7096–103. https://doi.org/10.1021/jf100613x.
Nakagawa C, Suzuki-Karasaki M, Suzuki-Karasaki M, Ochiai T, Suzuki-Karasaki Y. The mitochondrial Ca(2+) overload via voltage-gated Ca(2+) entry contributes to an anti-melanoma effect of diallyl trisulfide. Int J Mol Sci. 2020;21. https://doi.org/10.3390/ijms21020491.
Lu HF, Sue CC, Yu CS, Chen SC, Chen GW, Chung JG. Diallyl disulfide (DADS) induced apoptosis undergo caspase-3 activity in human bladder cancer T24 cells. Food Chem Toxicol. 2004;42:1543–52. https://doi.org/10.1016/j.fct.2003.06.001.
Wang YB, Qin J, Zheng XY, Bai Y, Yang K, Xie LP. Diallyl trisulfide induces Bcl-2 and caspase-3-dependent apoptosis via downregulation of Akt phosphorylation in human T24 bladder cancer cells. Phytomedicine. 2010;17:363–8. https://doi.org/10.1016/j.phymed.2009.07.019.
Shin DY, Kim GY, Hwang HJ, Kim WJ, Choi YH. Diallyl trisulfide-induced apoptosis of bladder cancer cells is caspase-dependent and regulated by PI3K/Akt and JNK pathways. Environ Toxicol Pharmacol. 2014;37:74–83. https://doi.org/10.1016/j.etap.2013.11.002.
Wen J, Zhang Y, Chen X, Shen L, Li GC, Xu M. Enhancement of diallyl disulfide-induced apoptosis by inhibitors of MAPKs in human HepG2 hepatoma cells. Biochem Pharmacol. 2004;68:323–31. https://doi.org/10.1016/j.bcp.2004.03.027.
Ji C, Ren F, Ma H, Xu M. The roles of p38MAPK and caspase-3 in DADS-induced apoptosis in human HepG2 cells. J Exp Clin cancer Res. 2010;29:50. https://doi.org/10.1186/1756-9966-29-50.
Xiao D, Choi S, Johnson DE, Vogel VG, Johnson CS, Trump DL, et al. Diallyl trisulfide-induced apoptosis in human prostate cancer cells involves c-Jun N-terminal kinase and extracellular-signal regulated kinase-mediated phosphorylation of Bcl-2. Oncogene. 2004;23:5594–606. https://doi.org/10.1038/sj.onc.1207747.
Xiao D, Lew KL, Kim YA, Zeng Y, Hahm ER, Dhir R, et al. Diallyl trisulfide suppresses growth of PC-3 human prostate cancer xenograft in vivo in association with Bax and Bak induction. Clin Cancer Res : Off J Am Assoc Cancer Res. 2006;12:6836–43. https://doi.org/10.1158/1078-0432.Ccr-06-1273.
Xiao D, Singh SV. Diallyl trisulfide, a constituent of processed garlic, inactivates Akt to trigger mitochondrial translocation of BAD and caspase-mediated apoptosis in human prostate cancer cells. Carcinogenesis. 2006;27:533–40. https://doi.org/10.1093/carcin/bgi228.
Kim YA, Xiao D, Xiao H, Powolny AA, Lew KL, Reilly ML, et al. Mitochondria-mediated apoptosis by diallyl trisulfide in human prostate cancer cells is associated with generation of reactive oxygen species and regulated by Bax/Bak. Mol Cancer Ther. 2007;6:1599–609. https://doi.org/10.1158/1535-7163.Mct-06-0754.
Chen WC, Hsu SS, Chou CT, Kuo CC, Huang JK, Fang YC, et al. Effect of diallyl disulfide on Ca2+ movement and viability in PC3 human prostate cancer cells. Toxicol In Vitro. 2011;25:636–43. https://doi.org/10.1016/j.tiv.2010.12.015.
Borkowska A, Sielicka-Dudzin A, Herman-Antosiewicz A, Wozniak M, Fedeli D, Falcioni G, et al. Diallyl trisulfide-induced prostate cancer cell death is associated with Akt/PKB dephosphorylation mediated by P-p66shc. Eur J Nutr. 2012;51:817–25. https://doi.org/10.1007/s00394-011-0260-x.
Shin DY, Kim GY, Lee JH, Choi BT, Yoo YH, Choi YH. Apoptosis induction of human prostate carcinoma DU145 cells by diallyl disulfide via modulation of JNK and PI3K/AKT signaling pathways. Int J Mol Sci. 2012;13:14158–71. https://doi.org/10.3390/ijms131114158.
Jiang X, Zhu X, Huang W, Xu H, Zhao Z, Li S, et al. Garlic-derived organosulfur compound exerts antitumor efficacy via activation of MAPK pathway and modulation of cytokines in SGC-7901 tumor-bearing mice. Int Immunopharmacol. 2017;48:135–45. https://doi.org/10.1016/j.intimp.2017.05.004.
Jiang XY, Zhu XS, Xu HY, Zhao ZX, Li SY, Li SZ, et al. Diallyl trisulfide suppresses tumor growth through the attenuation of Nrf2/Akt and activation of p38/JNK and potentiates cisplatin efficacy in gastric cancer treatment. Acta Pharmacol Sin. 2017;38:1048–58. https://doi.org/10.1038/aps.2016.176.
Das A, Banik NL, Ray SK. Garlic compounds generate reactive oxygen species leading to activation of stress kinases and cysteine proteases for apoptosis in human glioblastoma T98G and U87MG cells. Cancer. 2007;110:1083–95. https://doi.org/10.1002/cncr.22888.
Karmakar S, Banik NL, Patel SJ, Ray SK. Garlic compounds induced calpain and intrinsic caspase cascade for apoptosis in human malignant neuroblastoma SH-SY5Y cells. Apoptosis. 2007;12:671–84. https://doi.org/10.1007/s10495-006-0024-x.
Wallace GCT, Haar CP, Vandergrift WA 3rd, Giglio P, Dixon-Mah YN, Varma AK, et al. Multi-targeted DATS prevents tumor progression and promotes apoptosis in ectopic glioblastoma xenografts in SCID mice via HDAC inhibition. J Neuro Oncol. 2013;114:43–50. https://doi.org/10.1007/s11060-013-1165-8.
Pagliei B, Aquilano K, Baldelli S, Ciriolo MR. Garlic-derived diallyl disulfide modulates peroxisome proliferator activated receptor gamma co-activator 1 alpha in neuroblastoma cells. Biochem Pharmacol. 2013;85:335–44. https://doi.org/10.1016/j.bcp.2012.11.007.
Hwang JS, Lee YY, Lee DH, Kwon KH. DATS sensitizes glioma cells to TRAIL-mediated apoptosis by up-regulation of death receptor 5 via ROS. Food Chem Toxicol. 2017;106:514–21. https://doi.org/10.1016/j.fct.2017.05.056.
Shin HA, Cha YY, Park MS, Kim JM, Lim YC. Diallyl sulfide induces growth inhibition and apoptosis of anaplastic thyroid cancer cells by mitochondrial signaling pathway. Oral Oncol. 2010;46:e15–8. https://doi.org/10.1016/j.oraloncology.2009.10.012.
Pan J, Zhang L, Xu S, Cheng X, Yu H, Bao J, et al. Induction of apoptosis in human papillary-thyroid-carcinoma BCPAP cells by diallyl trisulfide through activation of the MAPK signaling pathway. J Agric Food Chem. 2018;66:5871–8. https://doi.org/10.1021/acs.jafc.8b02243.
Zheng J, Cheng X, Xu S, Zhang L, Pan J, Yu H, et al. Diallyl trisulfide induces G2/M cell-cycle arrest and apoptosis in anaplastic thyroid carcinoma 8505C cells. Food Funct. 2019;10:7253–61. https://doi.org/10.1039/c9fo00646j.
Wu PP, Chung HW, Liu KC, Wu RS, Yang JS, Tang NY, et al. Diallyl sulfide induces cell cycle arrest and apoptosis in HeLa human cervical cancer cells through the p53, caspase- and mitochondria-dependent pathways. Int J Oncol. 2011;38:1605–13. https://doi.org/10.3892/ijo.2011.973.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81802718, 81670088), the Training Program for Young Backbone Teachers of Institutions of Higher Learning in Henan Province, China (No. 2020GGJS038), and the Foundation of Science & Technology Department of Henan Province, China (Nos. 222102310490, 222102310495).
Author information
Authors and Affiliations
Contributions
DDW, XYJ and QYJ conceived and supervised the study. WG, YFL, YXZ, YW, YQJ, HY and XYL drafted the manuscript and prepared the figures. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, W., Liu, YF., Zhang, YX. et al. The potential role of hydrogen sulfide in cancer cell apoptosis. Cell Death Discov. 10, 114 (2024). https://doi.org/10.1038/s41420-024-01868-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-024-01868-w
