
Abstract
Ferroptosis is an iron ion-dependent, regulatory cell death modality driven by intracellular lipid peroxidation that plays a key role in the development of HCC. Studies have shown that various clinical agents (e.g., sorafenib) have ferroptosis inducer-like effects and can exert therapeutic effects by modulating different key factors in the ferroptosis pathway. This implies that targeting tumor cell ferroptosis may be a very promising strategy for tumor therapy. In this paper, we summarize the prerequisites and defense systems for the occurrence of ferroptosis and the regulatory targets of drug-mediated ferroptosis action in HCC, the differences and connections between ferroptosis and other programmed cell deaths. We aim to summarize the theoretical basis, classical inducers of ferroptosis and research progress of ferroptosis in HCC cells, clued to the treatment of HCC by regulating ferroptosis network. Further investigation of the specific mechanisms of ferroptosis and the development of hepatocellular carcinoma and interventions at different stages of hepatocellular carcinoma will help us to deepen our understanding of hepatocellular carcinoma, with a view to providing new and more precise preventive as well as therapeutic measures for patients.
Similar content being viewed by others

Facts
-
Ferroptosis plays an extremely important role in the development of hepatocellular carcinoma, and precise targeting of tumor cells to induce ferroptosis is a new way forward in the treatment of cancer.
-
Ferroptosis occurs as a metabolic mode of cell death involving lipid metabolism, amino acid metabolism, iron metabolism, glutathione metabolism, etc., and an imbalance in any of these metabolisms affects the extent of cellular ferroptosis.
-
Natural defense mechanisms against ferroptosis exist within the cell, consisting mainly of enzymatic and non-enzymatic antioxidant mechanisms.
-
Studies have shown that many ferroptosis-regulated genes are widely involved in the development of hepatocellular carcinoma, and the resistance problem of sorafenib, the first-line drug in hepatocellular carcinoma clinics, is also inextricably linked to ferroptosis.
Questions
-
What is the ultimate executioner of ferroptosis?
-
What is the normal physiological function of ferroptosis?
-
How to accurately target ferroptosis in tumor cells and reduce the level of ferroptosis in normal cells, and whether there are recognition molecules on the surface of different cells with different degrees of sensitivity to ferroptosis?
-
How ferroptosis and other modes of cell death interact and are regulated in tumor therapy, or how much ferroptosis is involved?
Introduction
Ferroptosis was proposed in 2012 as an iron-dependent, non-apoptotic form of cellular demise instigated by lipid peroxidation [1]. The occurrence of ferroptosis is dependent on the accumulation of iron in cells and lipid peroxidation. Multiple intracellular molecules regulate the onset of ferroptosis by affecting intracellular iron levels and lipid peroxidation status. Cells undergoing ferroptosis exhibit morphological features that are more distinctive from other programmed cell death modalities such as apoptosis, autophagy, necroptosis, and pyroptosis. The main features of ferroptosis and apoptosis, necroptosis, pyroptosis, and autophagy are shown in Table 1. The most predominant morphological changes of ferroptosis include atrophy of mitochondria and reduction of cristae whereas the nucleus usually remains intact. In contrast, apoptosis usually involves fragmentation and margination of chromatin, as well as the production of apoptotic vesicles and plasma membrane vesicles [2]. Programmed cellular necrosis has been shown to occur with cell swelling and the production of plasma membrane fragments that are eventually released from the plasma membrane in the form of vesicles [3]. When cells undergo autophagy, the intracellular lysosomes form a double membrane, While the membrane structure and nuclear morphology are more normal [4]. Another form of cell death is pyroptosis, which is heavily dependent on the gasdermin D protein. Pyroptotic cells swell and expand until the cell membrane ruptures, leading to the release of cellular contents and the activation of a strong inflammatory response [5]. All of these different modes of cell death are significantly different from ferroptosis. By comparing ferroptosis with other forms of cell death, it can help us better understand the mechanism of ferroptosis occurrence and its unique features. In summary, ferroptosis as a new form of programmed cell death is characterized by iron dependence and increased lipid peroxidation. Ferroptosis plays an important role in cell proliferation, differentiation, cycling and senescence [6]. Ferroptosis can be induced to occur by structurally distinct small molecules (e.g., erastin, sulfasalazine, and RSL3) or inhibited by lipophilic antioxidants (e.g., CoQ10, vitamin E, ferrostatins, and liproxstatins). Elevated intracellular iron ion concentrations and deficiency of the antioxidant GSH lead to cellular lipid peroxidation, which results in cellular ferroptosis [7, 8].
The principal regulatory mechanisms orchestrating ferroptosis encompass lipid peroxidation, amino acid metabolism, oxidative stress, iron ion homeostasis, and diverse organelles, all constituting prerequisites for the manifestation of ferroptosis to occur. Naturally, the body is equipped with a ferroptosis defense system to safeguard cells, primarily including (1) Cystine/System Xc−/GSH/GPX4, (2) FSP1-CoQ10-NAD(P)H, (3) DHODH-CoQ10-CoQH2, (4) GCH1/BH4/DHFR, and (5) FSP1/ ESCRT-III [9]. The body maintains a dynamic equilibrium of cellular state by modulating both the promotive and resistive factors of ferroptosis. These essential prerequisites and defense mechanisms for the occurrence of ferroptosis inherently convey that ferroptosis constitutes a metabolic form of cell demise. While the physiological relevance of ferroptosis remains a subject of debate, its inseparable connection with hepatocellular carcinoma is undeniable.
Primary hepatic carcinoma stands as the foremost neoplastic manifestation within the liver, ranking as the fourth principal cause of global cancer-related mortality [10, 11]. According to the Barcelona Clinic Liver Cancer classification, the majority of patients present at an intermediate to advanced disease stage upon diagnosis, foreclosing the prospect of radical surgical intervention [12]. The current first-line drugs for HCC patients are the multi-kinase inhibitors sorafenib and lenvatinib. However, a multicenter randomized controlled clinical trial showed that sorafenib only prolonged the median survival of patients with HCC by 10 months and lenvatinib by 15 months [13]. Unfortunately, despite this, patients still lose the effectiveness of the drug after a few months of treatment due to drug resistance [14,15,16].
Certain investigations have identified a propensity for iron ion accumulation and lipid peroxide overload in HCC. For certain tissue sources, specific cell differentiation states, and certain types of HCC cells, although they are resistant to other types of induced cell death, they are extremely sensitive to ferroptosis inducers. Sorafenib is the most widely studied of these drugs. Some studies have found that sorafenib induces ferroptosis in some tumor cells and that ferroptosis regulatory proteins also affect the sensitivity of HCC to the sorafenib [17]. Secondly, in the tumor microenvironment, immune cells and their factors affect the susceptibility of tumor cells to ferroptosis [18], while ferroptosis of tumor cells triggers the host to mount an anti-tumor immune response [19]. Therefore, targeting ferroptosis in tumor cells would be a promising therapeutic strategy for tumor treatment and combined with immunotherapy to produce coordinated antitumor effects. The following will describe the discovery of ferroptosis targets in HCC in recent years [20].
The regulatory frameworks governing networks of ferroptosis in liver cancer have been discovered to possess a high degree of richness, bordering on extraordinariness. including P53, P62-Keap1-Nrf2-ARE, Rb, MT-1G, S1R, CER, ACSL4, CISD1, HNF4A/HIC1, etc. The mechanisms by which these targets function in the ferroptosis network and how they regulate the drug sensitivity to ferroptosis in HCC cells will be expounded upon in meticulous detail [21].
Prerequisites for ferroptosis
Lipid peroxidation
The cellular membrane is a barrier that prevents extracellular substances from freely entering the cell, it ensures the stability of the intracellular environment, and lipids, as the main components of the cellular membrane, distinguish the biochemical processes inside the cell from the external environment [22]. The composition and content of lipids in the membrane vary depending on the cell type, organelles, and membrane leaflets [23]. Disruption of membrane integrity is essential for the execution of all forms of regulated cell death [24].
The operative molecules of cellular ferroptosis are specific lipid peroxides and peroxyl radicals containing polyunsaturated lipid chains. These lipids can form lipid peroxides by autoxidation or enzymatic catalysis, which are subsequently converted to lipid peroxyl radicals in the presence of divalent iron ions, thereby affecting the fluidity, integrity and stability of the cell membrane [25]. Disruption of cell membrane integrity is the underlying driver of ferroptosis occurrence.
Lipid metabolomics reveals that polyunsaturated fatty acids such as arachidonic acid (AA) or Adrenic Acid (AdA) are the most oxidized lipids during ferroptosis and are regulated by three synthetases [26]. Acyl-CoA synthetase long-chain familymember 4 (ACSL4) catalyzes the conversion of AA or AdA to AA-CoA and AdA-CoA [27]. Subsequently, Lys phosphatidylcholine acyltransferase 3 (LPCAT3) esterifies it to phosphatidylethanolamines (PEs) to form AA-PE and AdA-PE [28], and finally oxidized to PE-AA-OOH and PE-AdA-OOH by polyunsaturated fatty acid lipoxygenase 15 (ALOX15) [29]. And Glutathione peroxidase 4 (GPX4) can convert these lipid peroxides back to the corresponding alcohols, thereby reducing the level of intracytoplasmic lipid peroxides. [30]. Studies also showed that knockdown or inhibition of all three of these synthases inhibited the development of ferroptosis [31]. The excessive accumulation of specific lipid peroxides directly precipitates ferroptosis, provoking disturbances such as cell membrane disruption, cellular swelling and mitochondrial fragmentation, ultimately culminating in the manifestation of ferroptosis.
Since polyunsaturated fatty acids (PUFA) promote ferroptosis and monounsaturated fatty acids (MUFA) inhibit ferroptosis [32], inducing the conversion of monounsaturated fatty acids to polyunsaturated fatty acids to enhance the susceptibility to ferroptosis in hepatocellular carcinoma may be a proven strategy.
Amino acid metabolism
Amino acid metabolism sustains human life by providing essential elements such as proteins, energy substrates, glutathione, and neurotransmitters for human life. It intricately governs ferroptosis by regulating several lipid antioxidant systems. Even before the discovery of ferroptosis, scientists observed that cystine deficiency in the culture medium led to cell death, and that cells could only grow at extremely high densities. Further research found that, cysteine amino acid metabolism is most closely related to ferroptosis, which is inhibited through metabolic pathways such as synthesis of glutathione [33], glutathione peroxidase 4 [34], iron-sulfur cluster [35], and hydrogen sulfide [36]. Methionine can regulate ferroptosis through the sulfur transfer pathway and polyamine synthesis. The mevalonate pathway is involved in the synthesis of selenocysteine and coenzyme Q10 (CoQ10), products that are catalytic centres for GPX4 and reducing agents for the inhibition of lipid peroxidation, respectively [37,38,39,40]. In contrast, glutamine catabolism also promotes ferroptosis induced by cysteine deprivation [41].
In conclusion, amino acid anabolism intricately participates in the complex network governing ferroptosis regulation, where GSH levels may be a hub for the regulation of ferroptosis sensitivity.
Oxidative stress and ferroptosis
Oxidative stress is cellular and tissue damage caused by the production of reactive oxygen species (ROS) in the organism that exceed their scavenging effect [42]. Essentially, ferroptosis is caused by an imbalance in iron-mediated production and scavenging of ROS resulting in cell death due to the accumulation of lethal lipid peroxides [43]. This disequilibrium in the dynamic process is the direct cause of ferroptosis [21]. ROS play a pivotal role in cellular signal transduction and tissue homeostasis [44]. The primary ROS components encompass the anion(O2−), peroxides (H2O2) and LOOH, and free radicals (HO· and LO·). Mitochondria serve as the main source of ROS [45]. Mitochondria produce a large amount of free ROS in the normal metabolism of electron transport chain and cell energy supply, and endogenous ROS can also be released by microsomes, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase, peroxidase, etc. There is a homeostatic ROS scavenging system in the body, which can remove excessive ROS in the body. The body’s clearance of ROS is mainly dependent on the body’s antioxidant system, including Superoxide dismutase (SOD) [46], Catalase (CAT), GPX4, etc. [47].
The significance of oxidative stress in ferroptosis is further demonstrated by the fact that oxidative stress is involved in the whole process of ferroptosis, and the antioxidant system mitigates ferroptosis by scavenging ROS [48, 49].
Iron ion homeostasis
Normal intracellular iron levels play an irreplaceable role in maintaining normal cellular physiological functions. Abnormalities in any of the various aspects of iron metabolism, involving, for example, iron absorption, storage, conversion, utilization, distribution and excretion, can lead to an imbalance in the intracellular iron ion content, thus affecting the overall cellular state and the function of the organism [50,51,52]. As the main form of intracellular iron presence, the relative conversion of ferrous and ferric ions, as the primary forms of intracellular iron, directly or indirectly influences cellular metabolism and viability [8, 51]. The liver, as the largest iron storage organ in the body, plays an irreplaceable role in the regulation of iron metabolism [53, 54]. Meanwhile, the change of iron content in the liver is also closely related to the physiological state of the liver [50, 55].
The main forms of iron present in the body include trivalent iron and divalent iron [56]. Trivalent iron can be reduced to divalent iron by duodenal cytochrome b and can be transported to the duodenal epithelium by divalent metal transporter 1 (DMT1) [57]. transferrin-bound (TF) iron uptake mediated by Transferrin Receptor 1 (TFR1), or non-TF-bound iron uptake mediated by Solute carrier family 39 (SLC39A14) is the major intracellular iron acquisition pathway [58, 59]. Trivalent iron ions can also be converted to divalent ferrous ions by Six-Transmembrane Epithelial Antigen of Prostate 3 (STEAP3) [60], which excretes iron required for normal physiological functions, and the rest is partially transported out of the membrane by ferroportin (FPN) [61, 62], consisting of ferritin heavy chain 1 (FTH1) and ferritin light chain (FTL), and stored in tissues and organs, such as the liver [63]. Additionally, heme degradation and NCOA4-mediated ferritin autophagy can degrade ferritin, releasing iron into the labile iron pool (LIP) [64, 65], subsequently sensitizing cells to iron-induced atrophy via the Fenton response. The intracellular iron content is also influenced by hepcidin, which at low levels rapidly and stably transports iron into the plasma, while at high levels, FPN is inactivated and iron entry into the plasma is restricted [66, 67]. The amount of ferrous ions in the unstable iron pool is a key factor in determining the sensitivity of ferrous ions to iron atrophy. It has also been found that such as SLC39A8 can also exercise the function of iron transport [68].
Cellular ferroptosis, triggered by intracellular iron accumulation, emerges as a pathogenic factor in HCC. Consequently, considering iron chelators to impede ferroptosis may present a promising therapeutic approach for associated diseases [8, 69,70,71,72,73].
Cell organelles and ferroptosis
Various organelles assume significant roles in the development of ferroptosis. Mitochondria, as the epicenter of energy metabolism, intricately regulate amino acid, carbohydrate, and lipid metabolism, influencing ferroptosis. Simultaneously, they modulate ferroptosis through ROS and may impact iron metabolism [74,75,76]. The release of iron ions by lysosomes through the degradation of ferritin mediated by autophagy increases intracellular free iron ion content, thereby promoting ferroptosis [6, 77, 78]. The rich lipid content of the endoplasmic reticulum is the main substrate source of ferroptosis, and the synthesis of fatty acids is mainly completed in the endoplasmic reticulum. Secondly, ferroptosis inducers can activate endoplasmic reticulum stress [79,80,81,82]. Peroxisomes regulate ferroptosis by regulating the antioxidant system. Further studies on lipidomics have found that peroxisomes can also regulate ferroptosis by regulating the synthesis of PUFA-ePL, and the sensitivity of cells to ferroptosis is also closely related to PUFA-ePL [83,84,85].
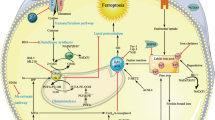
In summary, the regulation of ferroptosis requires the coordinated operation of multiple organelles, so understanding how these organelles interact has implications for the development of clinical drugs for ferroptosis (Fig. 1).

The initiators of ferroptosis include lipid metabolism and iron metabolism. In lipid metabolism, adrenoic acid and arachidonic acid in a series of enzymatic reactions culminate in the production of PUFA to promote ferroptosis. In iron metabolism, the binding of transferrin (TF) and transferrin receptor (TFRC) on the membrane drives extracellular Fe3+ into the cytosol, followed by the reduction of Fe3+ to Fe2+ by STEAP3, which is transported between different organelles under the action of SLC39A14, DMT1, and TRPML1/2, and finally sinks into the Lip. Also, various organelles are extensively involved in ferroptosis; the tricarboxylic acid cycle in mitochondria generates enormous amounts of ROS, and metabolic activity or oxidative stress in the Golgi, endoplasmic reticulum, and lysosomes can trigger ferroptosis.
Ferroptosis’s defense system
Cystine/System Xc-/GSH/GPX4
The Xc− system, a sodium-independent reverse transporter for cystine and glutamate, comprises integral subunits: the light chain, Solute Carrier Family 7 Member 11 (SLC7A11) (also known as xCT) [86], serving as the main functional subunit, and the heavy chain subunit Solute Carrier Family 3, Member 2 (SLC3A2), also known as (4F2hc), acting as the molecular chaperone [87, 88]. This system facilitates the transfer of cystine from the extracellular to intracellular environment in a 1:1 ratio, with rapid conversion of cystine to cysteine upon entry. In the initial step, γ-GCS catalyzes the dehydration condensation of cysteine with glutamate to γ-glutamyl cysteine. In the second step, γ-glutamyl cysteine undergoes dehydration condensation with glycine again to glutathione catalyzed by GS. Where γ-GCS is the rate-limiting enzyme for the reaction and cysteine is the rate-limiting substrate for the reaction [89, 90].
GPX4 is an crucial negative regulator of lipid peroxidation in living organisms [91]. GPX4 protects cells from ferroptosis induced by lipid peroxidation by catalyzing the conversion of harmful intracellular lipid peroxides to harmless lipid alcohol compounds. GPX4 uses GSH as a cofactor to reduce peroxides (e.g., R-OOH) to the corresponding alcohols (R-OH) and also to reduce free of hydrogen peroxide to water, reducing the accumulation of toxic free radicals (e.g., RO-) [49, 92].
Functioning as a cell protector, GPX4 employs reduced GSH as a substrate to act as a cell protector and resist oxidative stress. During the reduction of peroxides (L-OOH) by GPX4, the oxidized GSSG produced can be reduced by glutathione reductase and reduced coenzyme ii (NADPH/H+) to regenerate reduced GSH, leading to the recycling of GSH [93]. Although GPX4 can catalyze the reduction of hydrogen peroxide, organic peroxides and lipid peroxides [94]. However, GPX4 prefers lipid peroxides. Thus, GPX4 is the main defender of cellular ferroptosis in living organisms [95, 96].
FSP1-CoQ10-NAD (P)H
Ferroptosis suppressor protein 1 (FSP1), also identified as Mitochondrial apoptosis-inducing factor 2 (AIFM2) [97], is a flavoprotein oxidoreductase encoded by the AIFM2 gene. Initially discovered in 2002, FSP1 was first considered a P53-responsive gene.
Initially it was thought that ferroptosis was regulated only by GPX4, which reduces hydroperoxides, and by antioxidants, which trap free radicals. However, one study found that certain tumor cells were still able to survive and proliferate after GPX4 deletion. This suggests that there may be a parallel ferroptosis inhibition axis independent of the GPX4 axis.
Two papers published back-to-back in Nature 2019 suggest that FSP1 overexpression in cells significantly protects them from ferroptosis-inducing factors and that this process is not dependent on GPX4 [98, 99]. At the plasma membrane, FSP1 catalyzes the reduction of coenzyme Q10 (also known as ubiquinone) to panthenol-10, a process that requires the involvement of NAD(P)H, a lipid-soluble radical-trapping antioxidant that prevents ferroptosis caused by lipid peroxidation damage, and this biological function is independent of the level of intracellular glutathione [99, 100].
DHODH- CoQ10-CoQH2
Mitochondria are organelles wrapped by dual membranes, the inner and outer membrane, function as the main sites of aerobic respiration, where the inner membrane orchestrates electron transfer, generating large amounts of ROS. Dihydroorotate dehydrogenase (DHODH), located in the inner mitochondrial membrane, is responsible for catalyzing the fourth step of the pyrimidine nucleotide synthesis pathway, namely the oxidation of dihydroorotate acid to orotate acid (OA), while CoQ10 in the inner membrane receives electrons to be reduced to CoQH2 [101].
In addition to the synthesis of pyrimidine nucleotides, DHODH was found to inhibit ferroptosis in mitochondria by producing CoQH2 in the inner mitochondrial membrane because CoQH2 can act as a radical trapping antioxidant to prevent lipid peroxidation and thus inhibit ferroptosis [102].
Further studies revealed that mechanistic studies confirmed that DHODH could only perform its ferroptosis inhibitory function when localized in mitochondria and that this function was dependent on its enzymatic activity. Subsequently, it was found that co-inactivation of DHODH and mitochondrial GPX4 led to mitochondrial lipid peroxidation and that BQR treatment significantly increased the intracellular CoQ10/CoQH2 ratio [103], suggesting that DHODH inhibits mitochondrial lipid peroxidation by reducing CoQ10 to CoQH2 in concert with mitochondrial GPX4. Notably, the cell membrane, composed of cytosolic GPX4 and FSP1, constitutes a distinct ferroptosis defense system [104].
GCH1/BH4/BH2
A GPX4-independent ferroptosis inhibition was identified through a CRISPR genome-wide screen: the GTP Cyclohydrolase 1 (GCH1), GCH1 is another important regulator of ferroptosis. It catalyzes the conversion of dihydrobiopterin (BH2) to tetrahydrobiopterin (BH4). BH4 is a cofactor for aromatic amino acid hydroxylase and other enzymes, and GCH1 mediation is the rate-limiting reaction in the BH4 biosynthesis pathway. BH4 is another radical-trapping antioxidant that can capture free radicals from lipid peroxidation. Its recycling requires the involvement of dihydrofolate reductase (DHFR), and its inhibition of ferroptosis seems to be independent of the action of its cofactor. Ferroptosis was inhibited by the generation of BH4 as a free radical trapping antioxidant, and by GCH1-mediated production of CoQH2 and PLs containing two PUFA tails. However, the specific subcellular compartment in which the GCH1-BH4 system works remains to be determined. BH4 mechanism [105], a free radical-trapping antioxidant whose recycling requires the involvement of the DHFR, and thus if blocked, the DHFR could potentially collaborate with GPX4 inhibitors to induce ferroptosis [106].
FSP1/ESCRT-III
A 2020 study reported that FSP1-deficient tumor cells displayed heightened sensitivity to cell death induced by ferroptosis inducers, but the addition of exogenous CoQ10 did not reverse the cell death induced by FSP1 deficiency [107, 108]. In contrast, investigators have identified a novel mechanism of action of FSP1-mediated inhibition of ferroptosis by the ESCRT that is independent of CoQ10 and its downstream inhibition of lipid peroxidation [109]. Both in vivo and in vitro experiments demonstrated that inhibition of gene expression of the constituent proteins of the ESCRT-III complex significantly enhanced the level of ferroptosis in tumor cells [110]. However, the interaction of FSP1/ESCRT-III and the specific mechanism of action of this pathway still need more studies to support and elucidate (Fig. 2).

(1) Cystine is transported into the cytosol via the cystine/glutamate reverse transporter and metabolized to cysteine. Cysteine and intracellular glutamate are metabolized by glutamate. Cysteine ligase (GCL) catalyses the formation of Y-glutamate cysteine. The latter is catalyzed by glutathione synthetase (GS) and glycine (Gly) to produce glutathione. Glutathione peroxidase 4, assisted by the cofactor glutathione, inhibits the accumulation of phospholipid hydroperoxides (PL00H) and ultimately inhibits ferroptosis. (2) Coenzyme Q10 generates its reduced form CoQ10-H2 in the presence of FSP1. (3) GCH1 promotes BH4 biosynthesis reduces phospholipid hydroperoxide accumulation inhibits ferroptosis. (4) DHODH, localized in the mitochondria, oxidizes dihydroorotate (DHO) to orotate (OA) while transferring electrons to ubiquinone in the inner mitochondrial membrane for reduction to ubiquinol.
Regulators of ferroptosis in HCC
P53
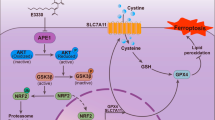
TP53 stands as the most extensively studied and frequently mutated tumor suppressor gene in human tumors [111]. P53 acts as a double-edged sword in ferroptosis. P53 acts as a transcription factor regulating the levels of multiple ferroptosis-related genes to promote or inhibit ferroptosis.
Initially, P53 is involved in the regulation of the ferroptosis process as a transcriptional repressor of SLC7A11, inhibiting cysteine uptake and consequently GSH production and ROS-mediated increase in cellular ferroptosis, a molecular cascade reaction that may contribute to the anti-cancer effects of P53 [112]. This specific induction is mediated by a P53 mutant with an acetylation-deficient phenotype, P53KR [113]. This mutant has a strong inhibitory effect on SLC7A11 but has little inhibitory effect on other known target genes of P53 [114]. Recent studies have also shown that SLC7A11 is highly expressed in tumor cells and thus promotes tumor growth and proliferation in part by suppressing the onset of ferroptosis. Non-synonymous single nucleotide mutations in P53 codon 47, S47, do not affect most P53 functions, but their tumor suppressive effects are impaired, and S47 heterozygous and pure mice are significantly more susceptible to the liver and other cancers, which may be related to impaired negative regulation of SLC7A11 by S47, consequently inhibiting ferroptosis [115,116,117,118,119].
Glutaminase 2 (GLS2) is the key enzyme in the liver that catalyzes the conversion of glutamine to glutamate. One study found that the use of ferroptosis inhibitors and glutaminolysis inhibitors inhibited erastin-induced ferroptosis, resulting in an increase in intracellular GSH and a decrease in intracellular ROS content, thus finding that glutamine and GLS2 are required for the development of cellular ferroptosis. It has also been found that upregulation of GLS2 expression contributes to ferroptosis caused by aerobic glycolysis rather than oxidative phosphorylation [120].
Prostaglandin Endoperoxide Synthase 2 (PTGS2), a key enzyme in the initiation step of prostaglandin (PG) synthesis in organisms, can regulate cellular sensitivity to ferroptosis by regulating the levels of key intracellular membrane phospholipids PE.
When ferroptosis inhibitors such as GLS2 and erastin were applied to P53 wild-type cells, the cells exhibited upregulated PTGS2 gene expression and underwent ferroptosis, but when P53-deficient cells were induced with GLS2 and erastin, the PTGS2 gene expression level was unchanged and ferroptosis did not occur. Therefore, P53 is required for the upregulation of PTGS2 expression and is directly associated with ferroptosis, and PTGS2 has been widely used as a marker of ferroptosis occurrence [121, 122].
Spermidine/spermine N1-acetyltransferase 1 (SAT1) can catalyze the acetylation reaction of spermidine and spermine using CoA. Abnormal polyamine metabolism is closely related to tumors. SAT1 activation can induce ferroptosis, while P53 can induce SAT1 transcriptional expression to promote lipid peroxidation and ROS-induced ferroptosis, and silencing SAT1 diminishes ROS-induced cellular ferroptosis in wild-type P53 cells [123, 124].
dipeptidyl-peptidase-4 (DPP4) is a membrane-bound dimeric peptidase that is widely expressed in different cell types and functions to degrade bioactive peptides. DPP4 can promote a variety of tumors and its abnormal expression is highly correlated with tumor invasion. Plasma membrane-associated DPP4-dependent lipid peroxidation is increased in human hepatocellular carcinoma cells, which brings about cellular ferroptosis. Mechanistically, it was found that P53 can inhibit ferroptosis in hepatocellular carcinoma cells by promoting the entry of DPP4 into the nucleus and forming a DPP4-P53 complex. Dissolution of this complex restores the sensitivity of hepatocellular carcinoma cells to erastin. In the absence of P53, DPP4 can also interact with NOX1 to form a complex that leads to increased lipid peroxidation and ferroptosis, and inhibition of DPP4 activity can significantly suppresses ferroptosis [125,126,127].
A recent study revealed that pretreatment of cells with Nutlin-3 delayed ferroptosis in various tumor cells (including hepatocellular carcinoma cells) and that the inhibition of ferroptosis was mainly dependent on a key target of P53-regulated transcription-CDKN1A (which can encode P21), but the exact mechanism is unclear and may be related to the accumulation of intracellular GSH leading to reduced cellular ferroptosis sensitivity related [128].
In conclusion, the role of P53 in ferroptosis is colorful and more in-depth studies are needed to explore potential therapeutic implications [128, 129].
P62-Keap1-Nrf2-ARE
Nuclear factor erythroid 2-related factor 2 (Nrf2), is a key transcription factor in the antioxidant response. Under non-oxidative stress conditions, Nrf2 protein expression is low because Nrf2 binds to Keap1, mediating ubiquitination modifications that result in rapid proteasomal degradation of Nrf2 [130]. In contrast, under oxidative stress conditions, Cys273 and Cys288 cysteines on Keap1 protein are rapidly oxidized, leading to the loss of Keap1 protein-mediated ubiquitination modifications and the translocation of Nrf2 to the nucleus, where it binds to multiple AREs on DNA and initiates the transcriptional expression of multiple antioxidant proteins [131]. Under oxidative stress, P62 protein can regulate the protein stability of Nrf2 and the transcriptional activity of downstream AREs by directly interacting with KEAP1 [132]. Numerous studies have shown that the P62-Keap1-Nrf2 signaling pathway exerts an inhibitory role in ferroptosis in HCC cells [133].
Upon exposure of HCC cells to specific ferroptosis inducers (e.g., Sorafenib, erastin, etc.), the expression of P62 increases and prevents the degradation of Nrf2 protein by competitively binding to Keap1 protein, and promotes the entry of Nrf2 into the nucleus [134], where nuclear Nrf2 can bind to transcriptional co-activators such as Maf protein and activate a series of downstream ferroptosis-related proteins, especially antioxidant proteins transcription, including NADH Dehydrogenase, Quinone 1 (NQO1), heme oxygenase-1 (HO-1), and FTH1 [135,136,137,138]. NQO1 reduces ubiquinone to ubiquinol and directly inhibits the accumulation of ROS [139]. activation of HO-1 catalyzes the degradation of heme to ferrous iron and increases the amount of free iron, thereby increasing the susceptibility of cells to ferroptosis [140, 141]. FTH1 is a subunit of the major intracellular iron storage protein. When FTH1 is reduced, it causes intracellular iron overload, and iron overload promotes the production of large amounts of ROS by Fenton, consequently promoting cellular ferroptosis [142]. Expression of each of these genes prevents the aggregation of iron-dependent lipid peroxides. Inhibition of Nrf2 expression using shRNA resulted in reduced expression of NQO1, HO1, and FTH1 expression, promoting ferroptosis in erastin- and sorafenib-treated HCC cells [143, 144].
A study found that GSTZ1 can also affect the sensitivity of HCC cells to sorafenib-induced ferroptosis via the Nrf2 pathway. This study found that HCC cells with low GSTZ1 expression were highly tolerant to sorafenib [145]. While overexpression treatment of GSTZ1 in HCC cells could increase the characteristic products of ferroptosis induced by sorafenib, such as MDA and 4-HNE; while treatment with t-BHQ, an activator of Nrf2, could significantly reduce the expression of these products, inhibiting the extent of ferroptosis in HCC cells.
However, the role of Nrf2 in ferroptosis is complex and variable; on the one hand, inhibition of Nrf2 can cause ferroptosis in tumor cells, and on the other hand, activation of Nrf2 can also set off ferroptosis in tumor cells. Tumor cells have increased iron ion content compared to normal cells, and activation of Nrf2 can HO-1 activation, while excessive activation of HO-1 can cause cellular iron overload, induce cellular ferroptosis and exert cytotoxicity.
All these findings suggest that the stability and transcriptional activity of Nrf2 protein are critical in determining the effect of ferroptosis-targeted therapy in HCC cells.
Rb
Rb proteins are essential members of the Rb protein family, which is primarily responsible for the cell cycle and hepatocarcinogenesis [146]. loss of Rb function is a common molecular event in primary HCC. At the animal level, Rb deficiency can also directly contribute to hepatocarcinogenesis [147, 148].
Louandre’s team delved into the interrelationship between Rb proteins and sorafenib-induced ferroptosis in HCC cells in in vivo and in vitro experiments [149]. Their results showed that reducing Rb protein expression using shRNA in the presence of sorafenib treatment increased the amount of ROS in the mitochondria of HCC cells to some extent, which increased the sensitivity of HCC cells to sorafenib-induced ferroptosis, probably because silencing Rb protein increased the expression of GPX4 mRNA in the cells, suggesting that Rb protein silencing resulted in HCC cellular susceptibility to sorafenib may be caused by an imbalance in cellular antioxidant capacity.
Therefore, Rb protein plays a considerable role in the progression of ferroptosis in HCC cells. However, it is not clear how useful the detection of Rb protein levels in HCC tissues is in predicting sorafenib treatment efficacy remains unclear.
MT-1G
Metallothionein, MT is a family of intracellular proteins widely expressed by eukaryotic cells and divided into four subgroups (MT1–MT4) characterized by a common structure and a high cysteine content composition.
It has been discerned that MT1G can be used as a biomarker for altered redox metabolism in HCC cells. MT1G was most significantly upregulated in the presence of sorafenib, and the overall survival rate of HCC patients treated with sorafenib was negatively correlated with MT1 protein expression level in serum. MT-1G can promote the resistance of HCC to sorafenib and inhibit the expression of MT-1G in vivo or in vitro to enhance the anticancer activity of sorafenib. This effect of MT-1G is achieved by blocking the GSH depletion-mediated lipid peroxidation to inhibit ferroptosis in HCC cells, independent of iron ion content.
In a parallel study similarly indicated that sorafenib upregulated MT1G expression in HCC cells. Further studies revealed that MT1G is an additional target gene of Nrf2, and inhibition or knockdown of Nrf2 significantly inhibited MT1G expression and increased ROS production in HCC cells. Consequently, this heightened cellular sensitivity of cells to sorafenib-induced ferroptosis [150].
Thus, MT1G could be both a promising biomarker for predicting sorafenib efficacy and a potential therapeutic target for sensitizing sorafenib efficacy. MT-1G holds promise as a target to overcome the acquired resistance of HCC cells to sorafenib.
S1R
The Sigma 1 receptor (S1R) is a membrane protein found in the central nervous system, liver, lung, and other organs and tumors.
It is commonly understood that S1R modulates ROS through Nrf2, serving as a cell protector in cells. When S1R was knocked out, Nrf2 expression was downregulated and KEAP1 expression was increased. Nrf2 can in turn regulate S1R gene expression, and in sorafenib-treated HCC cells, if Nrf2 is inactivated and intracellular ROS accumulation, S1R expression is upregulated to protect HCC cells from ferroptosis [151]. In addition, ferroptosis inducers such as erastin and Sorafenib significantly upregulated the protein expression level of S1R but not the mRNA level, suggesting a transcription-independent role of S1R in ferroptosis [152].
These findings imply that S1R may regulate ferroptosis by negative feedback through Nrf2-dependent and GPX4-dependent pathways at the same time, but the trigger conditions and the bridge connecting the two pathways remain to be discovered.
CER
CER is a glycoprotein synthesized mainly by the liver and plays a key role in iron metabolism [153]. It has the effect of antioxidant, and has oxidase activity, and can catalyze the oxidation of polyphenol and polyamine substrates.
CER is able to regulate iron metabolism levels in HCC cells to modulate iron toxicity, and when protein levels are reduced, It leads to increased intracellular iron levels and accumulation of lipid peroxides, Then promotes erastin and RSL3-induced iron toxicity, which may be achieved by relying on FPN, The inhibitory effect of CER on ferroptosis was abolished when FPN expression was inhibited by shRNA [154]. This underlines the intricate involvement of CER in the regulation of iron-related cellular processes and its role in mitigating ferroptosis responses in HCC cells.
ACSL4
Ferroptosis proceeds through a series of enzymatic reactions of lipid peroxidation. In a screen for key lipid peroxidases in iron toxicity, Acyl-CoA synthetase long-chain family member 4 (ACSL4) was shown to be perhaps indispensable as a key factor in promoting iron toxicity. It was found that ACSL4 expression in HCC cell lines was positively correlated with sorafenib sensitivity. The use of ACSL4 inhibitors or knockdown of ACSL4 in HCC cells significantly enhanced the anti-HCC efficacy of sorafenib. More importantly, the expression of ACSL4 in HCC cells does not change with sorafenib treatment and has a certain stability, which means that ACLS4 is a good biomarker for predicting the treatment of HCC by sorafenib through ferroptosis pathway [155]. ACSL4 is not only serves as a key executor but also functions as a reliable indicator of iron deposition [27].
CISD1
CDGSH iron-sulfur domain 1 (CISD1) is an iron-containing mitochondrial outer membrane protein that is widely found in mitochondria-rich tissues such as liver and heart [156]. The main role of CISD1 is to regulate iron uptake in mitochondria and mitochondrial respiratory function. CISD1 deficiency leads to iron ion accumulation and subsequent mitochondrial oxidative damage [157].
Yuan et al. found that subferric ion content in HCC cells is augmented by erastin induction, and the expression of CISD1 is also increased simultaneously. Knockdown of CISD1 using shRNA increased the content of subferric ions in the mitochondria of HCC cells, subsequently increasing the extent of lipid peroxidation in the mitochondria and ultimately increasing the sensitivity of HCC cells to erastin-induced ferroptosis [158].
HNF4A/HIC1
Human hepatocyte nuclear factor alpha (HNF4A) and Hypermethylated in cancer 1 (HIC1) are two transcription factors with opposite functions in the regulation of HCC growth [159]. HNF4A is upregulated in HCC and stimulates EGFR-mediated tumor proliferation response, thereby fostering HCC progression [160]. In contrast, HIC1 inhibits tumor growth, migration, and invasion [161, 162].
In studies targeting ferroptosis in HCC, HNF4A was found to transcriptionally regulate such as STMN1, CAPG, and RRM2 under the action of erastin, and to increase GSH production and thus inhibit ferroptosis by promoting PSAT1. In contrast, HIC1 can regulate such as HBA1, NNMT, PLIN4 and promote ferroptosis in HCC by repressing PSAT1 [163]. Therefore, breaking the balance between HNF4A and HIC1 may contribute to the treatment of HCC. (Table 2), (Fig. 3).

Cystine enters the cell through the Xc− system to be converted to GSH, and erastin and Sorafenib inhibit this pathway. While RSL3 can directly target GPX4. P53 regulates ferroptosis by affecting the transcriptional level of downstream SLC7A11, SAT1, GLS2, PTGS2, P21, DPP4, etc. Rb, CISD1 can directly regulate the intracellular ROS level. GSTZ1 and P62 are by affecting the entry of Nrf2 into the nucleus to affect the MT1G, NQO1, HO1, and FTH1 levels to regulate ferroptosis.
Classical ferroptosis inducers in hepatocellular carcinoma
Erastin
A high-throughput screening of synthetic compounds for tumor cell killing identified a compound with RAS-selective activity: erastin. It was found to induce a non-apoptotic cell death. Subsequently, further studies revealed that erastin reduced GSH levels by directly inhibiting Systems Xc− (Fig. 4). The quinazolinone backbone may be the main active moiety for its lethality, and other moieties could improve the inhibitory effect of erastin on Systems Xc−. The mitochondrial voltage-dependent anion system is also a molecular target of erastin. In addition to its ability to directly activate ferroptosis for hepatocellular carcinoma treatment, erastin also enhances the chemotherapeutic effects of certain conventional antitumor agents in hepatocellular carcinoma cell lines. erastin could enhance the clinical efficacy of PD1/L1 by affecting the polarization of tumor-forming associated macrophages [164]. Aspirin induced a significant ferroptosis response in HepG2 and Huh7 cells, which was enhanced by the ferroptosis inducer erastin [134].

Chemical structural formula of erastin, IKE, RSL3, sorafenib, sulfasalazine, and simvastatin.
As a classical ferroptosis inducer, erastin has been used by researchers as a standard reference for testing novel compounds or ferroptosis inducers of existing drugs.
IKE
Although erastin has a good inhibitory effect on Systems Xc−, its low water solubility and metabolic instability seriously affect its in vivo application. Imidazolidinone (IKE), a derivative based on the improved structure of erastin, further improved the aqueous solubility and anticancer properties of the prototype, with three times the solubility of erastin and a 50-fold reduction in the LC50 for tumor cells [165, 166].
RSL3
In a gene-selective screening assay of more than 40,000 compounds related to cell proliferation, two small molecule compounds, RSL3 and RSL5, were screened for high lethality in the presence of oncogenic RAS [167]. Further experiments revealed that RSL3 could induce ferroptosis by directly targeting the inhibition of GPX4 [168], however, it has also been suggested that RSL3 is not an inhibitor of GPX4 but TXNRD1 [169]. the chloroacetamide portion of the RSL3 structure was essential for its bioactivity. RSL3 induced ferroptosis in a wide range of hepatocellular carcinoma cell lines, including all of them, and RSL3 could also enhance the sensitivity of tumors to ferroptosis in the presence of IFN-γ [170].
Sorafenib
Sorafenib is a multikinase inhibitor that has been widely used in patients with clinically advanced hepatocellular carcinoma. Sorafenib can prolong the survival of patients with hepatocellular carcinoma. Previous studies have suggested that the action of sorafenib in the treatment of hepatocellular carcinoma is mainly due to its multikinase inhibitory effect, which leads to the inhibition of cell proliferation, angiogenesis and the promotion of apoptosis of tumor cells. It was found that the toxic effect of sorafenib on hepatocellular carcinoma cells was partly dependent on ferroptosis rather than apoptosis, and this toxic effect was also not dependent on its multikinase inhibitory activity, but induced ferroptosis by inducing iron aggregation and lipid peroxidation stress in hepatocellular carcinoma cells, and that depletion of stored iron in hepatocellular carcinoma cells by use of iron chelating agents could effectively weaken the cytotoxic effect of sorafenib. Further studies revealed that sorafenib induces ferroptosis by inhibiting SLC7A11 transporter activity on the cell membrane, thereby reducing cystine content in hepatocellular carcinoma cells, which in turn leads to insufficient GSH synthesis and diminished GPX4 activity. Hepatocellular carcinoma patients treated with clinical application of sorafenib tend to develop drug resistance quickly, thus affecting the prognosis. As mentioned above, Nrf2, Rb, MT-1G and SIR are involved in the regulation of the sensitivity of ferroptosis induced by sorafenib through the corresponding pathways, thus inducing the emergence of drug resistance. From a new perspective, targeting the relevant inhibitor pathways to induce ferroptosis can effectively improve the drug resistance of sorafenib.
Sulfasalazine
SAS is an anti-inflammatory drug which has been approved by the U.S. Food and Drug Administration for a long time, is a first-line treatment for rheumatoid arthritis. SAS induces ferroptosis by inhibiting the Xc-system in a manner similar to erastin. However, compared to erastin, SAS is much less effective in inducing ferroptosis [171]. SAS has been shown to induce ferroptosis in a variety of tumor cells and may also be used in combination with other liver cancer therapies to improve treatment efficacy.
Iron-containing statins
Classic iron-containing statins include pravastatin sodium, lovastatin, and simvastatin. Statins, chemical inhibitors of HMG-CoA reductase, are a class of lipid-lowering drugs that are effective in reducing the prevalence of cardiovascular disease and mortality [172]. HMG-CoA reductase promotes mevalonate synthesis, and statins can inhibit MVA synthesis to block CoQ10 synthesis, which is a substrate for lipid peroxidation by the ferroptosis inhibitors FSP1 and DHODH, and statins can also directly affect GPX4 activity to promote ferroptosis. In addition, it has been found that atorvastatin-treated tissues have elevated levels of ROS, MDA, and Fe2+ along with decreased expression of GSH and GPX4, suggesting the presence of a ferroptosis response. The suppression of the HMG-CoA reductase by statins leads to decreased expression of glutathione peroxidase 4 (GPX4), a key regulator of lipid peroxidation, which in turn results in lipid ROS production and induction of ferroptosis. Experimental data suggest that statins may act as anti-cancer drugs by enhancing tumor cells’ ferroptosis [173, 174].
Summary and outlook
Ferroptosis, as an emerging mode of cell death, participates in the development of HCC and may play a key role in both the diagnosis prevention treatment and prognosis of HCC. Although ferroptosis has opened a new horizon in the basic field in recent years, translation into specific clinical treatments is yet to be achieved.
It is believed that as ferroptosis-related research progresses and these key questions are answered, targeting ferroptosis to treat HCC, especially in combination with immunotherapy, may overcome immunotherapy resistance and benefit more HCC patients.
References
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87.
Li L, Tong A, Zhang Q, Wei Y, Wei X. The molecular mechanisms of MLKL-dependent and MLKL-independent necrosis. J Mol Cell Biol. 2021;13:3–14.
Debnath J, Gammoh N, Ryan KM. Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol. 2023;24:560–75.
Liu X, Xia S, Zhang Z, Wu H, Lieberman J. Channelling inflammation: gasdermins in physiology and disease. Nat Rev Drug Discov. 2021;20:384–405.
Chen X, Yu C, Kang R, Kroemer G, Tang D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021;28:1135–48.
Xu H, Ye D, Ren M, Zhang H, Bi F. Ferroptosis in the tumor microenvironment: perspectives for immunotherapy. Trends Mol Med. 2021;27:856–67.
Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35:830–49.
Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401–21.
Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127:S35–50.
Foerster F, Gairing SJ, Muller L, Galle PR. NAFLD-driven HCC: Safety and efficacy of current and emerging treatment options. J Hepatol. 2022;76:446–57.
Fujiwara N, Friedman SL, Goossens N, Hoshida Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J Hepatol. 2018;68:526–49.
Xia S, Pan Y, Liang Y, Xu J, Cai X. The microenvironmental and metabolic aspects of sorafenib resistance in hepatocellular carcinoma. EBioMedicine. 2020;51:102610.
Tang W, Chen Z, Zhang W, Cheng Y, Zhang B, Wu F, et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther. 2020;5:87.
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Prim. 2021;7:6.
Mendez-Blanco C, Fondevila F, Garcia-Palomo A, Gonzalez-Gallego J, Mauriz JL. Sorafenib resistance in hepatocarcinoma: role of hypoxia-inducible factors. Exp Mol Med. 2018;50:1–9.
Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W, et al. Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cell Mol Med. 2019;23:4900–12.
Li L, Wang X, Xu H, Liu X, Xu K. Perspectives and mechanisms for targeting ferroptosis in the treatment of hepatocellular carcinoma. Front Mol Biosci. 2022;9:947208.
Huang A, Yang XR, Chung WY, Dennison AR, Zhou J. Targeted therapy for hepatocellular carcinoma. Signal Transduct Target Ther. 2020;5:146.
Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18:280–96.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Harayama T, Riezman H. Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol. 2018;19:281–96.
Yamashita A, Hayashi Y, Nemoto-Sasaki Y, Ito M, Oka S, Tanikawa T, et al. Acyltransferases and transacylases that determine the fatty acid composition of glycerolipids and the metabolism of bioactive lipid mediators in mammalian cells and model organisms. Prog Lipid Res. 2014;53:18–81.
Riegman M, Sagie L, Galed C, Levin T, Steinberg N, Dixon SJ, et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol. 2020;22:1042–8.
Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419–25.
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–8.
Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604–9.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113:E4966–75.
Yang WS, Sriramaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–76.
Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. 2019;26:420–32.e9.
Fujii J, Homma T, Kobayashi S. Ferroptosis caused by cysteine insufficiency and oxidative insult. Free Radic Res. 2020;54:969–80.
Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. 2021;12:1589.
Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551:639–43.
Wang Y, Yu R, Wu L, Yang G. Hydrogen sulfide guards myoblasts from ferroptosis by inhibiting ALOX12 acetylation. Cell Signal. 2021;78:109870.
Manier S, Ingegnere T, Escure G, Prodhomme C, Nudel M, Mitra S, et al. Current state and next-generation CAR-T cells in multiple myeloma. Blood Rev. 2022;54:100929.
Zhang T, Bauer C, Newman AC, Uribe AH, Athineos D, Blyth K, et al. Polyamine pathway activity promotes cysteine essentiality in cancer cells. Nat Metab. 2020;2:1062–76.
Cao J, Chen X, Jiang L, Lu B, Yuan M, Zhu D, et al. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat Commun. 2020;11:1251.
Wang L, Cai H, Hu Y, Liu F, Huang S, Zhou Y, et al. A pharmacological probe identifies cystathionine beta-synthase as a new negative regulator for ferroptosis. Cell Death Dis. 2018;9:1005.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308.
Poljsak B, Suput D, Milisav I. Achieving the balance between ROS and antioxidants: when to use the synthetic antioxidants. Oxid Med Cell Longev. 2013;2013:956792.
Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–3.
Latunde-Dada GO. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. 2017;1861:1893–900.
Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94:909–50.
Wang Y, Branicky R, Noe A, Hekimi S. Superoxide dismutases: dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol. 2018;217:1915–28.
He YJ, Liu XY, Xing L, Wan X, Chang X, Jiang HL. Fenton reaction-independent ferroptosis therapy via glutathione and iron redox couple sequentially triggered lipid peroxide generator. Biomaterials. 2020;241:119911.
Stockwell BR, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. 2020;30:478–90.
Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. 2020;8:586578.
Blaby-Haas CE, Merchant SS. Iron sparing and recycling in a compartmentalized cell. Curr Opin Microbiol. 2013;16:677–85.
Sornjai W, Nguyen Van Long F, Pion N, Pasquer A, Saurin JC, Marcel V, et al. Iron and hepcidin mediate human colorectal cancer cell growth. Chem Biol Interact. 2020;319:109021.
Ibrahim AS. Host cell invasion in mucormycosis: role of iron. Curr Opin Microbiol. 2011;14:406–11.
Chen S, Zhu JY, Zang X, Zhai YZ. The emerging role of ferroptosis in liver diseases. Front Cell Dev Biol. 2021;9:801365.
Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17.
Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6:49.
Vogt AS, Arsiwala T, Mohsen M, Vogel M, Manolova V, Bachmann MF. On iron metabolism and its regulation. Int J Mol Sci. 2021;22:4591.
Yanatori I, Kishi F. DMT1 and iron transport. Free Radic Biol Med. 2019;133:55–63.
Senyilmaz D, Virtue S, Xu X, Tan CY, Griffin JL, Miller AK, et al. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature. 2015;525:124–8.
Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10:822.
Li P-L, Liu H, Chen G-P, Li L, Shi H-J, Nie H-Y, et al. STEAP3 (six-transmembrane epithelial antigen of prostate 3) inhibits pathological cardiac hypertrophy. Hypertension. 2020;76:1219–30.
Li Y, Zeng X, Lu D, Yin M, Shan M, Gao Y. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Hum Reprod. 2021;36:951–64.
Pu B, Lu Y, Chen J, Li S, Zhu N, Wei W, et al. MobileUNet-FPN: a semantic segmentation model for fetal ultrasound four-chamber segmentation in edge computing environments. IEEE J Biomed Health Inf. 2022;26:5540–50.
Tian Y, Lu J, Hao X, Li H, Zhang G, Liu X, et al. FTH1 inhibits ferroptosis through ferritinophagy in the 6-OHDA model of Parkinson’s disease. Neurotherapeutics. 2020;17:1796–812.
Fang Y, Chen X, Tan Q, Zhou H, Xu J, Gu Q. Inhibiting ferroptosis through disrupting the NCOA4-FTH1 interaction: a new mechanism of action. ACS Cent Sci. 2021;7:980–9.
Santana-Codina N, Mancias JD. The role of NCOA4-mediated ferritinophagy in health and disease. Pharmaceuticals. 2018;11:114.
Joachim JH, Mehta KJ. Hepcidin in hepatocellular carcinoma. Br J Cancer. 2022;127:185–92.
Sangkhae V, Nemeth E. Regulation of the iron homeostatic hormone hepcidin. Adv Nutr. 2017;8:126–36.
Zhang R, Witkowska K, Afonso Guerra-Assuncao J, Ren M, Ng FL, Mauro C, et al. A blood pressure-associated variant of the SLC39A8 gene influences cellular cadmium accumulation and toxicity. Hum Mol Genet. 2016;25:4117–26.
Chen K, Jiang X, Wu M, Cao X, Bao W, Zhu LQ. Ferroptosis, a potential therapeutic target in Alzheimer’s disease. Front Cell Dev Biol. 2021;9:704298.
Wang Y, Wu S, Li Q, Lang W, Li W, Jiang X, et al. Epigallocatechin-3-gallate: a phytochemical as a promising drug candidate for the treatment of Parkinson’s disease. Front Pharm. 2022;13:977521.
Tang B, Zhu J, Li J, Fan K, Gao Y, Cheng S, et al. The ferroptosis and iron-metabolism signature robustly predicts clinical diagnosis, prognosis and immune microenvironment for hepatocellular carcinoma. Cell Commun Signal. 2020;18:174.
Xiao Z, Kong B, Fang J, Qin T, Dai C, Shuai W, et al. Ferrostatin-1 alleviates lipopolysaccharide-induced cardiac dysfunction. Bioengineered. 2021;12:9367–76.
Lazaridou M, Christodoulou E, Nerantzaki M, Kostoglou M, Lambropoulou DA, Katsarou A, et al. Formulation and in-vitro characterization of chitosan-nanoparticles loaded with the iron chelator deferoxamine mesylate (DFO). Pharmaceutics. 2020;12:228.
Park MW, Cha HW, Kim J, Kim JH, Yang H, Yoon S, et al. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. 2021;41:101947.
Chen GH, Song CC, Pantopoulos K, Wei XL, Zheng H, Luo Z. Mitochondrial oxidative stress mediated Fe-induced ferroptosis via the NRF2-ARE pathway. Free Radic Biol Med. 2022;180:95–107.
Jang S, Chapa-Dubocq XR, Tyurina YY, St Croix CM, Kapralov AA, Tyurin VA, et al. Elucidating the contribution of mitochondrial glutathione to ferroptosis in cardiomyocytes. Redox Biol. 2021;45:102021.
Li J, Liu J, Xu Y, Wu R, Chen X, Song X, et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy. 2021;17:3361–74.
Wu Y, Zhang S, Gong X, Tam S, Xiao D, Liu S, et al. The epigenetic regulators and metabolic changes in ferroptosis-associated cancer progression. Mol Cancer. 2020;19:39.
Lee YS, Lee DH, Choudry HA, Bartlett DL, Lee YJ. Ferroptosis-induced endoplasmic reticulum stress: cross-talk between ferroptosis and apoptosis. Mol Cancer Res. 2018;16:1073–6.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523.
Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. 2020;27:420–35.
Wang Y, Liao S, Pan Z, Jiang S, Fan J, Yu S, et al. Hydrogen sulfide alleviates particulate matter-induced emphysema and airway inflammation by suppressing ferroptosis. Free Radic Biol Med. 2022;186:1–16.
Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585:603–8.
Fan XE, Xu M, Ren Q, Fan Y, Liu B, Chen J, et al. Downregulation of fatty acid binding protein 4 alleviates lipid peroxidation and oxidative stress in diabetic retinopathy by regulating peroxisome proliferator-activated receptor γ-mediated ferroptosis. Bioengineered. 2022;13:10540–51.
Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17:2054–81.
Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12:599–620.
Kajarabille N, Latunde-Dada GO. Programmed cell-death by ferroptosis: antioxidants as mitigators. Int J Mol Sci. 2019;20:4968.
Lee J, Roh JL. SLC7A11 as a gateway of metabolic perturbation and ferroptosis vulnerability in cancer. Antioxidants. 2022;11:2444.
Jyotsana N, Ta KT, Delgiorno KE. The role of cystine/glutamate antiporter SLC7A11/xCT in the pathophysiology of cancer. Front Oncol. 2022;12:858462.
Qiu Y, Cao Y, Cao W, Jia Y, Lu N. The application of ferroptosis in diseases. Pharm Res. 2020;159:104919.
Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol. 2017;403:143–70.
Niu B, Liao K, Zhou Y, Wen T, Quan G, Pan X, et al. Application of glutathione depletion in cancer therapy: Enhanced ROS-based therapy, ferroptosis, and chemotherapy. Biomaterials. 2021;277:121110.
Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32:920–37.
Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144–52.
Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. 2019;19:e1800311.
Novo N, Ferreira P, Medina M. The apoptosis-inducing factor family: moonlighting proteins in the crosstalk between mitochondria and nuclei. IUBMB Life. 2021;73:568–81.
Doll S, Freitas FP, Shah R, Aldrovandi M, Da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Yang M, Tsui MG, Tsang JKW, Goit RK, Yao KM, So KF, et al. Involvement of FSP1-CoQ(10)-NADH and GSH-GPx-4 pathways in retinal pigment epithelium ferroptosis. Cell Death Dis. 2022;13:468.
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–90.
Ma Y, Zhu Q, Wang X, Liu M, Chen Q, Jiang L, et al. Synthetic lethal screening identifies DHODH as a target for MEN1-mutated tumor cells. Cell Res. 2022;32:596–9.
Wang F, Min J. DHODH tangoing with GPX4 on the ferroptotic stage. Signal Transduct Target Ther. 2021;6:244.
Olsen TK, Dyberg C, Embaie BT, Alchahin A, Milosevic J, Ding J, et al. DHODH is an independent prognostic marker and potent therapeutic target in neuroblastoma. JCI Insight. 2022;7:e153836.
Liu Y, Zhai E, Chen J, Qian Y, Zhao R, Ma Y, et al. m(6) A-mediated regulation of PBX1-GCH1 axis promotes gastric cancer proliferation and metastasis by elevating tetrahydrobiopterin levels. Cancer Commun. 2022;42:327–44.
Wei JL, Wu SY, Yang YS, Xiao Y, Jin X, Xu XE, et al. GCH1 induces immunosuppression through metabolic reprogramming and IDO1 upregulation in triple-negative breast cancer. J Immunother Cancer. 2021;9:e002383.
Marmolejo-Garza A, Dolga AM. PEG out through the pores with the help of ESCRTIII. Cell Calcium. 2021;97:102422.
Dai E, Meng L, Kang R, Wang X, Tang D. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun. 2020;522:415–21.
Pedrera L, Espiritu RA, Ros U, Weber J, Schmitt A, Stroh J, et al. Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 2021;28:1644–57.
Liu J, Kang R, Tang D. ESCRT-III-mediated membrane repair in cell death and tumor resistance. Cancer Gene Ther. 2021;28:1–4.
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, et al. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 2016;17:366–73.
Li Y, Cao Y, Xiao J, Shang J, Tan Q, Ping F, et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 2020;27:2635–50.
Leu JI, Murphy ME, George DL. Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc Natl Acad Sci USA. 2019;116:8390–6.
Luo Y, Gao X, Zou L, Lei M, Feng J, Hu Z. Bavachin induces ferroptosis through the STAT3/P53/SLC7A11 axis in osteosarcoma cells. Oxid Med Cell Longev. 2021;2021:1783485.
Yang Y, Ma Y, Li Q, Ling Y, Zhou Y, Chu K, et al. STAT6 inhibits ferroptosis and alleviates acute lung injury via regulating P53/SLC7A11 pathway. Cell Death Dis. 2022;13:530.
Zeng C, Lin J, Zhang K, Ou H, Shen K, Liu Q, et al. SHARPIN promotes cell proliferation of cholangiocarcinoma and inhibits ferroptosis via p53/SLC7A11/GPX4 signaling. Cancer Sci. 2022;113:3766–75.
Liu G, Wei C, Yuan S, Zhang Z, Li J, Zhang L, et al. Wogonoside attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis through SOCS1/P53/SLC7A11 pathway. Phytother Res. 2022;36:4230–43.
Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. 2010;107:7461–6.
Lee HJ, Kim SR, Jung YJ, Han JA. Cyclooxygenase-2 induces neoplastic transformation by inhibiting p53-dependent oncogene-induced senescence. Sci Rep. 2021;11:9853.
Zhang CY, Tan XH, Yang HH, Jin L, Hong JR, Zhou Y, et al. COX-2/sEH dual inhibitor alleviates hepatocyte senescence in NAFLD mice by restoring autophagy through Sirt1/PI3K/AKT/mTOR. Int J Mol Sci. 2022;23:8267.
Liang T, Xie J, Zhao J, Huang W, Xu Z, Tian P, et al. Novel lnc-HZ03 and miR-hz03 promote BPDE-induced human trophoblastic cell apoptosis and induce miscarriage by upregulating p53/SAT1 pathway. Cell Biol Toxicol. 2021;37:951–70.
Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci USA. 2016;113:E6806–E12.
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20:1692–704.
Kim EM, Jung CH, Kim J, Hwang SG, Park JK, Um HD. The p53/p21 complex regulates cancer cell invasion and apoptosis by targeting Bcl-2 family proteins. Cancer Res. 2017;77:3092–100.
Engeland K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022;29:946–60.
Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. 2019;133:162–8.
Beyfuss K, Hood DA. A systematic review of p53 regulation of oxidative stress in skeletal muscle. Redox Rep. 2018;23:100–17.
He F, Ru X, Wen T. NRF2, a transcription factor for stress response and beyond. Int J Mol Sci. 2020;21:4777.
Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal. 2018;29:1727–45.
Lee DH, Park JS, Lee YS, Han J, Lee DK, Kwon SW, et al. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy. 2020;16:1949–73.
Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–31.
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–84.
Zhan S, Lu L, Pan SS, Wei XQ, Miao RR, Liu XH, et al. Targeting NQO1/GPX4-mediated ferroptosis by plumbagin suppresses in vitro and in vivo glioma growth. Br J Cancer. 2022;127:364–76.
Kim DH, Choi HI, Park JS, Kim CS, Bae EH, Ma SK, et al. Farnesoid X receptor protects against cisplatin-induced acute kidney injury by regulating the transcription of ferroptosis-related genes. Redox Biol. 2022;54:102382.
Yan N, Xu Z, Qu C, Zhang J. Dimethyl fumarate improves cognitive deficits in chronic cerebral hypoperfusion rats by alleviating inflammation, oxidative stress, and ferroptosis via NRF2/ARE/NF-kappaB signal pathway. Int Immunopharmacol. 2021;98:107844.
Dang R, Wang M, Li X, Wang H, Liu L, Wu Q, et al. Edaravone ameliorates depressive and anxiety-like behaviors via Sirt1/Nrf2/HO-1/Gpx4 pathway. J Neuroinflamm. 2022;19:41.
Li S, Zhang Y, Zhang J, Yu B, Wang W, Jia B, et al. Neferine exerts ferroptosis-inducing effect and antitumor effect on thyroid cancer through Nrf2/HO-1/NQO1 inhibition. J Oncol. 2022;2022:7933775.
Wei R, Zhao Y, Wang J, Yang X, Li S, Wang Y, et al. Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int J Biol Sci. 2021;17:2703–17.
Ma H, Wang X, Zhang W, Li H, Zhao W, Sun J, et al. Melatonin suppresses ferroptosis induced by high glucose via activation of the Nrf2/HO-1 signaling pathway in type 2 diabetic osteoporosis. Oxid Med Cell Longev. 2020;2020:9067610.
Kong N, Chen X, Feng J, Duan T, Liu S, Sun X, et al. Baicalin induces ferroptosis in bladder cancer cells by downregulating FTH1. Acta Pharm Sin B. 2021;11:4045–54.
Ren X, Li Y, Zhou Y, Hu W, Yang C, Jing Q, et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol. 2021;46:102122.
Sun J, Zhou C, Zhao Y, Zhang X, Chen W, Zhou Q, et al. Quiescin sulfhydryl oxidase 1 promotes sorafenib-induced ferroptosis in hepatocellular carcinoma by driving EGFR endosomal trafficking and inhibiting NRF2 activation. Redox Biol. 2021;41:101942.
Wang Q, Bin C, Xue Q, Gao Q, Huang A, Wang K, et al. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021;12:426.
Bremner R, Chen D, Pacal M, Livne-Bar I, Agochiya M. The RB protein family in retinal development and retinoblastoma: new insights from new mouse models. Dev Neurosci. 2004;26:417–34.
Hutcheson J, Witkiewicz AK, Knudsen ES. The RB tumor suppressor at the intersection of proliferation and immunity: relevance to disease immune evasion and immunotherapy. Cell Cycle. 2015;14:3812–9.
Chau BN, Wang JY. Coordinated regulation of life and death by RB. Nat Rev Cancer. 2003;3:130–8.
Louandre C, Marcq I, Bouhlal H, Lachaier E, Godin C, Saidak Z, et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356:971–7.
Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64:488–500.
Bai T, Lei P, Zhou H, Liang R, Zhu R, Wang W, et al. Sigma-1 receptor protects against ferroptosis in hepatocellular carcinoma cells. J Cell Mol Med. 2019;23:7349–59.
Vaczi S, Barna L, Harazin A, Meszaros M, Porkolab G, Zvara A, et al. S1R agonist modulates rat platelet eicosanoid synthesis and aggregation. Platelets. 2022;33:709–18.
Warren JJ, Lancaster KM, Richards JH, Gray HB. Inner- and outer-sphere metal coordination in blue copper proteins. J Inorg Biochem. 2012;115:119–26.
Shang Y, Luo M, Yao F, Wang S, Yuan Z, Yang Y. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell Signal. 2020;72:109633.
Feng J, Lu PZ, Zhu GZ, Hooi SC, Wu Y, Huang XW, et al. ACSL4 is a predictive biomarker of sorafenib sensitivity in hepatocellular carcinoma. Acta Pharm Sin. 2021;42:160–70.
Geldenhuys WJ, Leeper TC, Carroll RT. mitoNEET as a novel drug target for mitochondrial dysfunction. Drug Discov Today. 2014;19:1601–6.
Lu T, Li C, Xiang C, Gong Y, Peng W, Chen C. Overexpression of CISD1 Predicts Worse Survival in Hepatocarcinoma Patients. Biomed Res Int. 2022;2022:7823191.
Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun. 2016;478:838–44.
Dill MT, Tornillo L, Fritzius T, Terracciano L, Semela D, Bettler B, et al. Constitutive Notch2 signaling induces hepatic tumors in mice. Hepatology. 2013;57:1607–19.
Niehof M, Borlak J. EPS15R, TASP1, and PRPF3 are novel disease candidate genes targeted by HNF4alpha splice variants in hepatocellular carcinomas. Gastroenterology. 2008;134:1191–202.
Szczepny A, Carey K, Mckenzie L, Jayasekara WSN, Rossello F, Gonzalez-Rajal A, et al. The tumor suppressor Hic1 maintains chromosomal stability independent of Tp53. Oncogene. 2018;37:1939–48.
Ubaid U, Andrabi SBA, Tripathi SK, Dirasantha O, Kanduri K, Rautio S, et al. Transcriptional repressor HIC1 contributes to suppressive function of human-induced regulatory T cells. Cell Rep. 2018;22:2094–106.
Zhang X, Du L, Qiao Y, Zhang X, Zheng W, Wu Q, et al. Ferroptosis is governed by differential regulation of transcription in liver cancer. Redox Biol. 2019;24:101211.
Tang B, Zhu J, Wang Y, Chen W, Fang S, Mao W, et al. Targeted xCT-mediated ferroptosis and protumoral polarization of macrophages is effective against HCC and enhances the efficacy of the anti-PD-1/L1 response. Adv Sci (Weinh). 2023;10:e2203973.
Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol. 2019;26:623–33.e9.
Badgley MA, Kremer DM, Maurer HC, Delgiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368:85–9.
Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–45.
Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, et al. RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Front Pharm. 2018;9:1371.
Cheff DM, Huang C, Scholzen KC, Gencheva R, Ronzetti MH, Cheng Q, et al. The ferroptosis-inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. 2023;62:102703.
Jia M, Qin D, Zhao C, Chai L, Yu Z, Wang W, et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat Immunol. 2020;21:727–35.
Sun S, Guo C, Gao T, Ma D, Su X, Pang Q, et al. Hypoxia enhances glioma resistance to sulfasalazine-induced ferroptosis by upregulating SLC7A11 via PI3K/AKT/HIF-1alpha axis. Oxid Med Cell Longev. 2022;2022:7862430.
Sahebkar A, Foroutan Z, Katsiki N, Jamialahmadi T, Mantzoros CS. Ferroptosis, a new pathogenetic mechanism in cardiometabolic diseases and cancer: Is there a role for statin therapy? Metabolism. 2023;146:155659.
Zhu PF, Wang MX, Chen ZL, Yang L. Targeting the tumor microenvironment: a literature review of the novel anti-tumor mechanism of statins. Front Oncol. 2021;11:761107.
Shu X, Wu J, Zhang T, Ma X, Du Z, Xu J, et al. Statin-induced geranylgeranyl pyrophosphate depletion promotes ferroptosis-related senescence in adipose tissue. Nutrients. 2022;14:4365.
Kashyap D, Garg VK, Goel N. Intrinsic and extrinsic pathways of apoptosis: role in cancer development and prognosis. Adv Protein Chem Struct Biol. 2021;125:73–120.
Su Y, Zhao B, Zhou L, Zhang Z, Shen Y, Lv H, et al. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020;483:127–36.
Liang C, Xi-Xi X, Yun-Xiang S, Qiu-Hua X, Yang-Yong L, Yuan-Sen H, et al. Surfactin inhibits Fusarium graminearum by accumulating intracellular ROS and inducing apoptosis mechanisms. World J Microbiol Biotechnol. 2023;39:340.
Mandal P. Molecular signature of nitric oxide on major cancer hallmarks of colorectal carcinoma. Inflammopharmacology. 2018;26:331–6.
Morana O, Wood W, Gregory CD. The apoptosis paradox in cancer. Int J Mol Sci. 2022;23:1328.
Samir P, Malireddi RKS, Kanneganti TD. The PANoptosome: a deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol. 2020;10:238.
Mohammad RM, Muqbil I, Lowe L, Yedjou C, Hsu HY, Lin LT, et al. Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol. 2015;35:S78–S103.
Tong X, Tang R, Xiao M, Xu J, Wang W, Zhang B, et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J Hematol Oncol. 2022;15:174.
Khoury MK, Gupta K, Franco SR, Liu B. Necroptosis in the pathophysiology of disease. Am J Pathol. 2020;190:272–85.
Cao L, Mu W. Necrostatin-1 and necroptosis inhibition: pathophysiology and therapeutic implications. Pharm Res. 2021;163:105297.
Yu Z, Jiang N, Su W, Zhuo Y. Necroptosis: a novel pathway in neuroinflammation. Front Pharm. 2021;12:701564.
Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol. 2017;12:103–30.
Wei X, Xie F, Zhou X, Wu Y, Yan H, Liu T, et al. Role of pyroptosis in inflammation and cancer. Cell Mol Immunol. 2022;19:971–92.
Feng Y, Li M, Yangzhong X, Zhang X, Zu A, Hou Y, et al. Pyroptosis in inflammation-related respiratory disease. J Physiol Biochem. 2022;78:721–37.
Ruan S, Han C, Sheng Y, Wang J, Zhou X, Guan Q, et al. Antcin A alleviates pyroptosis and inflammatory response in Kupffercells of non-alcoholic fatty liver disease by targeting NLRP3. Int Immunopharmacol. 2021;100:108126.
Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42:245–54.
Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, et al. Autophagy in major human diseases. EMBO J. 2021;40:e108863.
Shi Y, Tao M, Ma X, Hu Y, Huang G, Qiu A, et al. Delayed treatment with an autophagy inhibitor 3-MA alleviates the progression of hyperuricemic nephropathy. Cell Death Dis. 2020;11:467.
Ferreira PMP, Sousa RWR, Ferreira JRO, Militao GCG, Bezerra DP. Chloroquine and hydroxychloroquine in antitumor therapies based on autophagy-related mechanisms. Pharm Res. 2021;168:105582.
Miller DR, Thorburn A. Autophagy and organelle homeostasis in cancer. Dev Cell. 2021;56:906–18.
Onorati AV, Dyczynski M, Ojha R, Amaravadi RK. Targeting autophagy in cancer. Cancer. 2018;124:3307–18.
Hadian K, Stockwell BR. SnapShot: ferroptosis. Cell. 2020;181:1188–e1.
Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med. 2019;133:130–43.
Scarpellini C, Klejborowska G, Lanthier C, Hassannia B, Vanden Berghe T, Augustyns K. Beyond ferrostatin-1: a comprehensive review of ferroptosis inhibitors. Trends Pharm Sci. 2023;44:902–16.
Guo Z, Lin J, Sun K, Guo J, Yao X, Wang G, et al. Deferoxamine alleviates osteoarthritis by inhibiting chondrocyte ferroptosis and activating the Nrf2 pathway. Front Pharm. 2022;13:791376.
Li Q, Liao J, Chen W, Zhang K, Li H, Ma F, et al. NAC alleviative ferroptosis in diabetic nephropathy via maintaining mitochondrial redox homeostasis through activating SIRT3-SOD2/Gpx4 pathway. Free Radic Biol Med. 2022;187:158–70.
Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254–62.
Tang K, Chen Q, Liu Y, Wang L, Lu W. Combination of metformin and sorafenib induces ferroptosis of hepatocellular carcinoma through p62-Keap1-Nrf2 pathway. J Cancer. 2022;13:3234–43.
Acknowledgements
We apologize to all the scientists whose work could not be discussed in the pages of this review. We sincerely thank all our lab partners for their continued help and advice on this manuscript. We want to express our gratitude for the drawing materials provided by BioRender. (https://www.biorender.com/).
Funding
This work was supported by the Major Project of Shanghai Municipal S and T Commission (No. 19401972300), Shandong Province Key R&D Program (Major Science and Technology Innovation Project, 2021CXGC010509), Key Laboratory of Chronic Deficiency Liver Disease of the State Administration of Traditional Chinese Medicine of the People’s Republic of China (20DZ2272200), Shanghai Key Specialty of Traditional Chinese Clinical Medicine (shslczdzk01201).
Author information
Authors and Affiliations
Contributions
YJ and YY conceptualized and developed the design of the article, interpreted relevant literature, and wrote the original manuscript. ZP and CG revised the manuscript. MS was responsible for conceptualization, writing—review & editing, funding acquisition, and validation. All authors have read and approved the final submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, Y., Yu, Y., Pan, Z. et al. Ferroptosis: a new hunter of hepatocellular carcinoma. Cell Death Discov. 10, 136 (2024). https://doi.org/10.1038/s41420-024-01863-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-024-01863-1
