
Abstract
Cutaneous wound healing, an integral part for protection of skin barrier, is a complex biological process and intimately associated with keratinocyte migration. However, mechanisms regulating keratinocyte migration in the process of cutaneous wound repair remain largely unknown. Here, we found that N-acetyltransferase 10 (NAT10) is essential for cutaneous wound repair in an in vivo skin wound healing model—a significant delay of wound repair in Nat10 haploinsufficient mice and a remarkable inhibition of keratinocyte migration by NAT10 knockdown in an in vitro keratinocyte migration model. We further demonstrate that loss of NAT10 expression attenuates the wound-induced IL-6/IL-8 expression through inhibiting NF-κB/p65 activity in keratinocytes. By deeply digging, silencing NAT10 compromises the level of nuclear p65 by facilitating its poly-ubiquitination, thus accelerates its degradation in the nucleus. Notably, we detected a strong positive correlation between the expression of NAT10 and relevant NF-kB/p65-IL6 signaling activity in mouse wound skin tissues. Overall, our study reveals an important role of NAT10 on cutaneous wound repair by potentiating NF-κB/p65-IL-6/8-STAT3 signaling. Targeting NAT10 might be a potential strategy for the treatment of skin wound dysfunctions and related diseases.
Similar content being viewed by others

Introduction
One main function of the skin is to protect internal environment and homeostasis of the body from the invasion of harmful elements in external environment, known as skin barrier [1]. When disrupted by external injury or some certain diseases and in order to rebuild the integrity of skin barrier, skin initiates cutaneous wound healing process, i.e., wound repair, to ensure immediate response and quick recovery [2]. Skin repair disorders may lead to chronic unhealed skin ulcer that seriously affects the life quality, and cause severe defects of other organs and even death under some circumstances [3]. Cutaneous wound healing is composed of several delicately organized procedures. Firstly, tissue injury disrupts blood vessels and extravasation of blood constituents disturbs the regional hemostasis; then, keratinocytes, fibroblasts, and infiltrating inflammatory cells at the site of injury initiate the inflammatory response to remove harmful substances; lastly, accompanying with degradation of extracellular matrix and migration and proliferation of epidermal cells, tissue remodeling starts, and wound healing is thoroughly finished [4]. Notably, keratinocytes basically run through the whole process of skin repair by releasing inflammatory cytokines, self-migrate, and secreting growth factors, which promote proliferation of other skin cells [5]. However, the underlying mechanisms that regulate the function of keratinocyte during skin repair are not well explored.
Acetylation is one of the most commonly discovered post-translation modifications for amounts of proteins, which plays important roles in various physiological and pathological progressions, as well as skin barrier protection [6]. Acetylation-related regulators, for example, acetylases and deacetylases, are involved in the cutaneous wound repair [6]. N-acetyltransferase 10 (NAT10), a member of the general control non-repressible 5 (GCN5)-related N-acetyltransferase (GNAT) family, is the only one so far identified with both acetylase domain and RNA binding domain. Recently, with dozens of novel substrates being discovered, NAT10 is sparkling because of its involvement in multiple biological or pathological processes [7]. For instances, NAT10 directly acetylates MORC2 to sensitize DNA-damaging, which potentiates the efficacy of chemotherapy and radiotherapy in breast cancer [8]; NAT10 is reported as a cellular stress sensor which acetylates p53 and counteracts Mdm2 action, thus eliciting p53 activation under DNA damage-related stress [9]; NAT10 interacts with Che-1 in the anabolism-catabolism transition in response to energy stress [10]. Moreover, inhibiting NAT10 extended the healthy lifespan in an LmnaG609G progeria mouse model, suggesting its intimate link with aging [11]. Recent findings revealed that N4-acetylcytidine (ac4C) modification of tRNA, rRNA, and mRNA is catalyzed by NAT10, improving the stability of RNA and protein translation efficiency [12, 13]. For example, NAT10 has been found to execute ac4C acetylation for mRNA of RUNX2 and COL5A1, which prevents ovariectomy-induced bone loss and promotes gastric cancer metastasis, respectively [14, 15]. Albeit current findings dictate crucial role of NAT10 in cell apoptosis, proliferation, and motility, thus modulating cancer progression, metabolism and aging, whether NAT10 regulates cutaneous wound healing and skin barrier maintaining is largely unknown. In this study, we investigated the potential function of NAT10 in wound repair by using keratinocyte as an in vitro cell model and scratching-wound as an in vivo mouse model. Our results demonstrate that NAT10 is critical for potentiating NF-κB/p65-IL-6 signaling which ensures efficient skin repair, and NAT10 haploinsufficiency elicits apparent delayed skin repair. Our findings highlight NAT10 as a potential target that might be useful for the treatment of disorders associated with skin repair dysfunction.
Results
Nat10 haploinsufficiency delays wound repair in mice
NAT10 regulates multiple physiological and pathological processes [7]. However, the roles of NAT10 in skin barrier, a delicately manipulated procedure, are barely delineated. Here, using a canonical wound healing model, we investigated the functions of NAT10 in skin repair. We generated a Nat10 knockout (KO) allele by using the standard CRISPR-Cas9 methodology (Fig. S1A, B in Supporting Information). After backcross, we successfully obtained heterozygous mice (Fig. S1C in Supporting Information). Notably, consistent with the previous finding [11], the Nat10 KO mice were also embryo lethal, while no apparent difference was observed between the wildtype (WT) and Nat10+/− littermates, which displayed comparable body size and skin morphogenesis (Fig. S1D in Supporting Information).
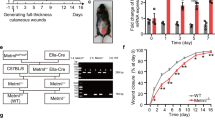
To evaluate whether Nat10 affects wound healing process, a full-thickness 6-mm punch-biopsy wound was made, basing on the previously reported method [16], in the dorsal skins of WT and Nat10+/− mice (Fig. 1A, day 0) and then the wound morphology was recorded at day 0, 1, 3, 5, 7, and 10 after punch. As shown, the wound healing speed of WT (n = 10) and Nat10+/− mice (n = 7) displayed no significant difference at early stage (on day 1 and day 3). However, apparent delayed repair rate was obvious in late period (on day 5 and day 7) in Nat10+/− mice compared with WT littermates (Fig. 1A). Quantification of unhealed skin area affirmed slower repair speed in Nat10+/− mice (Fig. 1B). We next performed histologic analysis by H&E staining of skin biopsies collected at day 5 when apparent morphology difference was first observed. As shown, the average wound diameter in Nat10+/− mice was longer than WT littermates (Fig. 1C, D). Re-epithelialization is a hall marker of successful wound repair [17]. Notably, the wound edges of Nat10+/− mice had less extent of re-epithelialization (indicated by arrows) compared with WT mice (Fig. 1C, F), further consolidating the finding of delayed wound healing in Nat10+/− mice. To complete wound repair, fibrosis is indispensable and considered to play irreplaceable roles because reconstituting fresh skin needs amount of various matrix proteins [18]. Indeed, significant lower expression of fibronectin and α-SMA, in both mRNA and protein levels, was observed in skin samples of wound area derived from Nat10+/− mice when compared with WT mice (Fig. 1F–H), suggesting an accompanied delay of fibrosis. Taken together, these data demonstrate that NAT10 haploinsufficiency in mice compromises wound repair.

A Representative images showing the wound healing process of Nat10+/− mice (n = 7) and their WT littermates (n = 10). Yellow arrows indicate the evident delayed wound repair in Nat10+/− mice. Scale bars, 5 mm. B Wound closure of (A) was quantified and unhealed rate was presented as the percentage of wound compared with the initial wound area size. n.s., no significance. C H&E staining of the wounded skin tissues derived from WT and Nat10+/− mice on day 5 after punching. Black lines, unhealed wound; Arrows, fresh skin with re-epithelialization. Scale bars, 100 μm. D The chart showing quantified wound width basing on the H&E staining images (WT: n = 9, Nat10+/−: n = 11). E Quantification of the re-epithelialization in wounds of both genotypes based on the H&E staining images (WT: n = 9, Nat10+/−: n = 11). F qPCR analysis of Nat10, Fn1 and Acta2 expression in wounded skin tissues collected from WT and Nat10+/− mice, n = 3 for each genotype. Immunoblot analysis of NAT10, FN1 and α-SMA protein expression in wounded skin tissues of WT and Nat10+/− mice at day 5 after wounding (G), and the relative quantification were presented in (H) using ImageJ Plus 6.0 (n = 3 mice for each group). The data are presented as the means ± SEM, * P < 0.05, ** P < 0.01, *** P < 0.001, determined by Student’s t test.
NAT10 knockdown inhibits keratinocyte migration
We next asked how NAT10 affected skin repair. Interestingly, based on the analysis of single cell RNA-sequencing data of skin tissue from a public online tool (The Human Protein Atlas), we found a much higher expression of NAT10 in keratinocyte cells, when comparing with other skin cell types, for example, monocytes and fibroblast cells (Fig. 2A). Indeed, once wound injury is created, skin keratinocyte is one of the earliest activated cell types and its migration is crucial to initiate the following wound repair [5]. Those findings encouraged us to explore the roles of NAT10 in keratinocyte. To that end, NAT10 expression was silenced in a widely-used keratinocyte cell line, i.e., HaCaT cells [19], by RNA interference (RNAi). As expected, both mRNA and protein levels of NAT10 were ablated (Fig. 2B, C). We then set out to analyze the effect of NAT10 silence on keratinocyte migration. As evaluated by trans-well migration assay and scratching-induced wound healing assay, it was clear that downregulation of NAT10 expression in HaCaT cells prominently inhibited cell migration (Fig. 2D, E) and resulted in apparent delay of wound closure (Fig. 2F, G). Meanwhile, NAT10 knockdown elicited little change on cell proliferation and viability when compared with scramble controls (Fig. S2A, B in Supporting Information), emphasizing the major contribution of NAT10 for keratinocyte migration. Moreover, silencing NAT10 impaired wound-induced up-regulation of MMP-2 which is critical for keratinocyte collective migration during wound healing [20] (Fig. 2H). Moreover, the activity of FAK, indicated by the phosphorylation of Y397 and the up-stream regulator of MMP2, was significantly downregulated in NAT10 knockdown cells (Fig. 2I). Taken together, these data suggest that NAT10 modulates wound healing process, at least partially, through potentiating keratinocyte migration.

A Single cell RNA-sequencing analysis showing the expression of Nat10 in various skin cell types. Original data were from public online tool of The Human Protein Atlas. B, C Immunoblot and qPCR analysis showing NAT10 protein and mRNA levels in HaCaT cells with or without knocking down NAT10 expression after 72 h of transfection (n = 3 for each group). Representative images (D) and related quantification (E) showing the migrated cells with or without NAT10 knockdown by using trans-well assay for 24 h. Scale bars, 50 μm. F, G Representative images (F) and related quantitative analysis showing the wound closure of HaCaT cells treated with or without NAT10 siRNA by using wound healing assays for 24 h (n = 6 for each group). Scale bars, 50 μm. H qPCR analysis of MMP2 expression in HaCaT cells with indicated treatments after scratching for two hours (n = 3 for each group). I Immunoblotting analysis of p-FAK (Y397) in HaCaT cells with or without NAT10 knockdown after scratching. Data are presented as the means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, obtained by Student’s t test.
Silencing NAT10 attenuates the wound-induced IL-6/IL-8 expression
We found that both mRNA and protein levels of NAT10 were merely affected in HaCaT cells upon scratch-induced wound, implicating that NAT10 regulating keratinocyte migration is not due to gain-of-expression but possibly imposed by an intrinsic mechanism (Fig. S3A, B in Supporting Information). We further asked how NAT10 affected keratinocyte cell migration-related regulators, for example as wound-induced cytokines, chemokines, matrix metalloproteinases (MMPs) and growth factors released by keratinocytes that are critical for wound-healing process [2]. Interestingly, wound conferred cells expressing cytokines and chemokines mainly peaked at 1–2 h after scratching, MMPs at 2 h, and growth factors at 4 h (Fig. 3A, B). Indeed, those early secreted cytokines are recognized as key triggers for wound repair initiation, such as IL6 [21]. Of note, NAT10 silencing resulted in more apparent decrease of wound-induced IL-6 and IL-8 expression compared with other cytokines (Fig. 3C). Accordingly, wound-induced STAT3 activation (p-STAT3), which is considered as one of the major effectors of IL6 or IL8 signaling [22], was significantly declined by NAT10 knockdown (Fig. 3D). We thus assumed that NAT10 knockdown might suppress keratinocyte migration through downregulation of IL6/IL8-STAT3 activity. In terms of STAT3 activation, IL6 and IL8 possess similar effect [23], we thus used recombinant human IL-6 (rIL6) to perform the rescue experiment. We found that supplementing rIL6 largely restored the ability of cell migration, as assessed by both trans-well and wound-healing assays (Fig. 3E-H). Likewise, rIL6 almost completely recovered the level of p-STAT3 and MMP2 in NAT10 knockdown HaCaT cells (Fig. 3I and Fig. S4 in Supporting Information). Together, these results demonstrate that NAT10 is crucial for keratinocyte cell migration through promoting wound-induced IL-6 expression.

A The schematic diagram described the induction of cytokines, chemokines, MMPs and growth factors expression by scratch-induced wound at different time points. B The heat map showing the production of cytokines, chemokines, MMPs and growth factors at serial time points in keratinocyte cells post scratching. C qPCR analysis of the expression of several key cytokines in HaCaT cells treated with control or NAT10 siRNAs in the scratch-wounded model (n = 3 for each group). n.s., no significance. D Immunoblotting analysis showing level of phosphorylated STAT3 in HaCaT cells treated with control or NAT10 siRNAs in the wound healing model. E, F Representative images (E) and related quantitative analysis showing the trans-well assays (lasting for 24 h) of HaCaT cells with or without NAT10 knockdown in the presence of absence of rIL-6. rIL-6, exogenous recombinant human IL-6 (n = 5 for each group). Scale bars, 50 μm. G, H Representative images and related quantitative analysis showing the in vitro wound healing assay of HaCaT cells (lasting for 16 h) treated with NAT10 siRNA or rIL-6 (n = 3 for each group). Scale bars, 50 μm. I Immunoblot analysis of phosphorylated STAT3 expression in HaCaT cells treated with NAT10 siRNAs and rIL-6 or not. Data are represented as mean ± SEM. * P < 0.05, ** P < 0.01, determined by Student’s t test.
NF-κB/p65 underlies NAT10-mediated IL6 regulation during wound repair
IL-6/IL8 expression is mainly regulated by NF-κB/p65 in various cellular conditions [24, 25]. Moreover, previous studies have demonstrated the activation of NF-κB/P65 during wound-healing [26, 27]. We thus asked whether NAT10 regulates IL6 expression via NF-κB/p65 pathway. Indeed, p65 activity (p-p65) was prominently increased after scratching wound (Ctrl group, Fig. 4A). Silencing NAT10 remarkably attenuated such activation (Fig. 4A, B). To fulfill the function in transcription regulation, phosphorylated p65 translocates into nucleus [28]. Consistently, determined by immunofluorescence staining, we found that the intensity of nuclear p65 was significantly lower in HaCaT cells with silenced NAT10, and the difference was more evident in cells along the wound edge (Fig. 4C, D). To gain more evidence of NAT10 regulating NF-κB/p65 activation, we employed TNF-α, a widely-used stimulator of NF-κB pathway, to assess whether similar effect could be recapitulated. As shown, NAT10 silencing suppressed TNF-α-induced p65 phosphorylation (Fig. 4E), and nuclear accumulation (Fig. 4F, G). If NAT10 regulating IL6 is governed by NF-κB/p65, TNF-α-stimulated IL6 expression would be affected by NAT10 manipulation. Indeed, TNF-α-induced IL6 expression was greatly attenuated in NAT10 silenced HaCaT cells (Fig. 4H). Together, these data implicate a NAT10-NF-κB/p65-IL6 signaling axis in regulating wound repair (Fig. 4I).

A, B Immunoblot (A) analysis of p65 activation in scratch-wounded keratinocyte HaCaT cells treated with control or NAT10 siRNAs. Cells lysates were prepared after scratch for 1 h. Relative levels of p-p65/p65 protein were quantified by ImageJ Plus 7.0 in (B) (n = 3 for each group). C, D Representative images (C) showing immunofluorescence staining of p65 in keratinocyte HaCaT cells treated with control or NAT10 siRNAs post scratch for 1 h. Percentage of nuclear p65 positive cells were calculated (D) (n = 8 for each group). Dot line indicating cells along the wounded edge. Scale bars, 50 μm. Immunoblotting (E) showing p65 activation in HaCaT cells treated with or without NAT10 siRNAs and addition of TNFα for 15 min. F, G Representative images showing immunofluorescence staining of p65 in HaCaT cells treated with or without NAT10 siRNAs and addition of TNFα for 15 min (F).The percentage of nuclear p65-positive cells were calculated (D, n = 6 for each group). Scale bars, 50 μm. H qPCR analysis of IL6 expression in HaCaT cells with or without treatment of TNFα (lasting for 1 h) and NAT10 siRNAs (n = 3 for each group). I Schematic diagram illustrating the mechanism of NAT10 affecting keratinocyte migration through NF-kB/p65-IL6 signaling. Data are represented as mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001, determined by Student’s t test.
Silencing NAT10 compromises nuclear p65 stability via facilitating its poly-ubiquitination
One of the key regards to NF-κB/p65 activation is associated with relevant receptor of TNF-α [29]. However, we found little change of TNFR1 or TNFR2 expression level in HaCaT cells upon NAT10 interference (Fig. S5 in Supporting Information), thus excluding defect in receptor-mediated signal initiation. NF-κB/p65 activation is tightly regulated by the cytoplasm-to-nucleus translocation. We thus asked whether NAT10 mediates this process. To that end, nuclear and cytoplasmic fractions derived from HaCaT cells with or without NAT10 knockdown and TNFα were subjected to immunoblotting. Interestingly, p-p65 and total p65 in the nucleus fraction showed a very apparent change, displaying more extent of decrease by NAT10 knockdown (Fig. 5A–C), while the cytoplasmic portion had less change (Fig. 5A, and Fig. S6A, B in Supporting Information), indicating that NAT10 mainly regulates nuclear p65. This finding seems plausible as NAT10 is a nucleus-resided protein [30]. Additionally, applying phosphatase inhibitor LB-100, the inhibitor of PP2A which dephosphorylates p65 [31, 32], did not markedly affect the p65 nuclear distribution in HaCaT cells with NAT10 knockdown or not (Fig. S6C in Supporting Information), further excluding the possibility of PP2A-mediated regulation of nuclear p65. We next asked whether NAT10 affected p65 nuclear exportation, which is mainly manipulated by nucleocapsid CRM1 [33]. However, after treatment with Leptomycin B (LMB), the CRM1 inhibitor [34] that restrains nuclear p65 exportation, we found that p65 level was yet not apparently restored in NAT10 silenced cells, suggesting the decrease of nuclear p65 by NAT10 knockdown was not due to the change of nuclear p65 exportation (Fig. 5D, E). It should be noted that, on one hand, LMB treatment restrained the export of nuclear p65, on the other hand, cytoplasmic p65 continually translocated into nucleus. Considering the combined effects, we reasoned that p65 nuclear proportion should be underwent much faster turnover upon NAT10 knockdown. It has been revealed the degradation of nuclear p65 is mainly mediated by ubiquitination-mediated proteasome pathway [35, 36]. We thus determined whether the ubiquitination of p65 was affected by NAT10. Indeed, NAT10 knockdown significantly enhanced p65 poly-ubiquitination in the nucleus, indicating that the low level of nuclear p65 is, at least partially, owing to faster degradation (Fig. 5F). Collectively, our results suggest that NAT10 maintains the steady activity of nuclear p65 through restraining its poly-ubiquitination-mediated degradation in nucleus.

A Immunoblotting analysis of cytoplasmic and nuclear p65 activation in HaCaT cells with or without NAT10 siRNAs and TNFα treatment. GAPDH and Lamin B1 marked the cytoplasmic and nuclear protein fraction, respectively. N, nucleus; C, cytoplasm. B, C The relative levels of nuclear p65/Lamin B1 and p-p65/Lamin B1 in (A) were quantified from 3 independent experiments. D, E Immunoblotting (D) showing nuclear p65 levels in Leptomycin B-treated HaCaT cells with control or NAT10 siRNA treatment. The relative level of p65/Lamin B1 was quantified in (E) (n = 3 for each group). F Immunoblotting showing the poly-ubiquitination of nuclear p65 in HaCaT cells with or without NAT10 knockdown. Data are presented as the means ± SEM. * P < 0.05, ** P < 0.01, determined by Student’s t test.
NF-kB/p65-IL6 signaling is positively correlated with NAT10 during wound repair
We next examined the in vivo correlation of NAT10 and NF-kB/p65-IL6 signaling. The levels of p-p65 and p-STAT3 were significantly decreased in wound skin tissues of Nat10+/− mice compared with those of WT mice, detected at day 5 after punching (Fig. 6A-C; Fig. S7A, B). In addition, a greater decline of cytokines that are in the control of NF-kB/p65 signaling, i.e., IL-6, IL-1α and TNF-α, was consistently observed in skin tissues of Nat10+/− mice (Fig. 6D). Such changes remained even to day 9, when skin repair was almost completed in both genotypes (Fig. 6E).

A–C Immunoblotting (A) showing p-p65 and p-STAT3 expression level derived from wounded skin tissues of WT and Nat10+/− mice (n = 3 mice for each group). The indicated quantification was displayed in (B) and (C). D, E qPCR analysis of IL-6, IL-1α, IL-1β and TNFα levels derived from wounded skin tissues of WT and Nat10+/− mice at day 5 (WT: n = 6; Nat10+/−: n = 10) and day 9 (WT: n = 12; Nat10+/−: n = 12) after punching. Data are presented as the means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, determined by Student’s t test.
Discussion
To maintain skin barrier homeostasis which is frequently disrupted by skin wound, skin autonomously activates the cutaneous repair process. Keratinocyte, as one of the important components of the skin, is a determinant factor for adequate and precise wound healing, which requires keratinocyte migrating into the right place at the right time. Dysfunction or impaired keratinocyte migration causes delayed wound healing, which may lead to chronic unhealed skin ulcer, defects of other organs and even death. In our study, we identified NAT10 as a key regulator of keratinocyte migration. Downregulation of NAT10 apparently attenuates wound-induced IL6/IL8 expression, compromises keratinocyte cell migration, and thus significantly delays mouse skin repair in both in vivo and in vitro models. Mechanically, NAT10 potentiates the NF-κB/p65 signaling through restraining nuclear p65 poly-ubiquitination. Our findings highlight NAT10 as a critical regulator of wound repair and skin barrier homeostasis.
Protein acetylation regulates keratinocyte migration during wound healing. It has been shown that the impairment of acetylase P300/CBP-associated factor (PCAF) delays skin repair, and the PCAF activator pentadecylidenemalonate 1b (SPV-106) promotes wound repair [6]. Treatment with Histone deacetylase inhibitor elicits increased wound closure through enrichment of specific subset of macrophages [37]. The sirtuin family, NAD+-dependent and conserved longevity related deacetylases [38], also regulates wound healing. Sirt1 promotes corneal epithelial wound healing through the deacetylation of p53 and enhancing IGFBP3/IGF-1/AKT pathway [39]. NAT10 functions as an acetylase [9]. Our study, to some extent, further emphasizes the great importance of acetylation regulation implicates wound repair process.
NF-κB/p65 pathway is critical for keratinocyte migration, which is activated by phosphorylation of p65 and subsequent translocation to the nucleus to drive the expression of wound healing related genes, such as IL6/IL8. One mechanism leading p65 inactivation is ubiquitination-mediated degradation [35, 36, 40]. For instance, an elegant study has unraveled that after p65 entering the nucleus, PDLIM2, as a nuclear ubiquitin E3 ligase, binds to p65 and promotes its polyubiquitination. In current study, we unravel a novel regulator that NAT10 contributes for the stability of nuclear p65 and thus sustains long-term activation. The absence of NAT10 facilitates the degradation of nuclear p65 via poly-ubiquitination. Whether such effect is dependent on NAT10 acetylase activity to p65 or other targets merits further investigation. Indeed, we failed to detect interaction between p65 and NAT10, suggesting a less possibility of NAT10 directly acetylating p65. We assume an existence of some indirect mechanisms, for instances, through SIRT1 and SIRT7, two NAD-dependent deacetylases which are reported to form complex with NAT10 [10, 13], by which NAT10 might affect the activity of nuclear p65. As previously reported, SIRT1 S-nitrosylation-dependent acetylation of p65 contributes to muscle wasting in burn wound repair; and SIRT7 regulating p65 is involved in inflammation process, and TGF-β signaling transduction in the tissue repair process [41, 42].
Due to embryo lethality by Nat10 total KO in mice, we used the heterozygotes for most of the in vivo phenotypical analyses. While NAT10 haploinsufficiency does remarkably delay wound skin repair, which, to some extent, demonstrates our views, we realize that keratinocyte-specific KO of Nat10 mice would be helpful to exclude the unexpected effects and further consolidate the conclusion. Moreover, the seemingly mild phenotype that Nat10+/− mice only showed delayed speed rate of wound healing at day 5 and 7 and finally completed wound repair at day 10, could be also explained by half loss of NAT10. Supporting this notion, abrogation of NAT10 in HaCaT keratinocyte cells in vitro elicits great inhibition of keratinocyte migration without apparent effect on cell viability and proliferation. We also identify that NAT10 has higher expression level in skin keratinocytes by analysis of single cell RNA sequencing data profiles from skin tissue. The results confirm the roles of NAT10 in wound healing via regulating keratinocyte migration. Nevertheless, in future study, generating Nat10 conditional KO mice to evaluate the keratinocyte specific function is warranted.
In summary, our study demonstrates that NAT10 ensures efficient skin wound repair by potentiating NF-κB/p65-IL-6/8-STAT3 signaling which promotes keratinocyte migration. Our findings highlight NAT10 as a potential target for the treatment of skin wound dysfunction and related diseases.
Materials and methods
Animals
Nat10+/− C57BL6 mice were generated by using the standard CRISPR-Cas9 methodology, purchased from Cyagen Company (Suzhou, China). The Nat10+/− mice were backcrossed for at least three generations to separate potential off-target deletions. To create skin wound healing, the fur was removed from the back of the mice and full-thickness excision wounds using a 6-mm biopsy punch were made on the dorsal skin. The general pictures of wounds were taken by Canon camera (Oita, Japan) and skin wound tissues were harvested at days 0, 1, 5, 9 post-punching. The wound area was calculated using ImageJ Software (NIH, Bethesda, MD), and the wound healing speed was measured as a ratio of the wound area at each time normalized by the area at day 0. To perform histological analysis, those collected tissues at 5 days post-punching were fixed and then stained with Hematoxylin-Eosin (H&E staining). Relevant images were captured by microscope (Olympus, IXplore, Japan) for further analysis. Above mentioned data collection were performed by researchers who were blinded to the group information. Each genotype of mice (at least 6 mice) was randomly grouped and the mice initially punched with irregular skin wounds would be excluded for further study. All mice were maintained under specific pathogen-free conditions and the procedures were performed in accordance with protocols approved by the Committee for Experimental Animal Research of Shenzhen University.
Cell culture
HaCaT (immortalized human keratinocyte cell line) cells were purchased from Kepu Company (Shenzhen, China) and cultured in Dulbecco’s modified Eagle’s medium (DMEM, high glucose, Corning) supplemented with 10% Fetal Bovine Serum (FBS, Gibco) and penicillin-streptomycin antibiotics (Gibco) in a 5% CO2 incubator at 37 °C. Confluent Cells were passed by trypsin with 0.25% EDTA (Gibco) with a ratio of 1:4. Other reagents used for additional treatments were: Recombinant Human IL-6 (rhIL6) (Peprotech, 200-06), Recombinant Human TNF-α (rhTNF-α) (Peprotech, 300-01 A), Leptomycin B (Selleck, S7580), LB 1-00 (Selleck, S7537) and MG132 (Beyotime, S1748). For analysis from cell culture, we plated cells in a random distribution and randomly assigned them to experimental groups.
Cell migration assays
For scratching-wound healing assay, HaCaT cells were plated on 6-well culture plates until they reached 80–90% confluence, and then cells were scratched using a 100-μl pipette tip after they were starved in serum-free medium for 24 h. Next, cells were washed three times with PBS and cultured in fresh DMEM with 2% FBS. The scratched areas were recorded after scratching at the different time point using the ZEISS ZEN microscopy (ZEISS, Germany). The percentage of unhealed area at each time point was calculated by normalizing with the wound area at zero by using ImageJ software. For trans-well assay, a suitable number of cells (5*104 cells/ml) were seeded in the upper chamber (Corning, China) and the lower chamber was incubated by DMEM with 2% FBS. Migrated cells were fixed, stained by DAPI (Beyotime, China) and migrated cell number was counted after pictures were taken.
Plasmid transfection and RNA interference (RNAi)
Gene expression by RNAi was performed by specific siRNA oligos (GenePharma, China) which was transfected by using Lipofectamine 3000 (Thermo, USA) according to the manufacturer’s instructions. The two sequences of NAT10 siRNAs were listed as follows: 5'-AUGGAACACUGAACAUAAATT-3' and 5'-GGCCAAAGCUGUCUUGAAATT-3'. Knockdown efficiency was validated by immunoblotting or real-time PCR after 48–72 h of transfection as indicated in figure legends, and all the NAT10 knockdown studies were performed within 72 h after transfection.
Total RNA extraction and quantitative qPCR analysis
Total RNA was isolated in Trizol reagent (Takara, Japan) according to a standard method. Total mRNA was reversely transcripted into cDNA by 5 × Primescript RT Master Mix (Takara, Japan). Quantitative real-time PCR was performed with 2 × SYBR Green Mix (Takara, Japan) in Bio-Rad detection system. Primer sequences used in this research are provided in Supplementary Table 1 (Table S1).
Protein extraction and immunoblotting
Protein samples derived from mouse skins and cells were extracted by RIPA lysis buffer containing phosphatases and proteases inhibitor cocktails (Roche, USA). Protein concentration was determined by BCA protein assay kit (Pierce, USA). For immunoblotting analysis, protein samples were subjected with SDS-polyacrylamide gel electrophoresis and proteins were transferred to PVDF membranes (Millipore). After being blocked, and incubated with primary antibodies and indicated HRP-coupled secondary antibody, membranes were visualized with ECL and images were captured using the Bio-Rad system. Band intensities were detected, normalized, and quantified with the Image Lab 5.0 software (Bio-Rad). The following antibodies were used in this research: NAT10 (Abcam, ab194297, 1:1000 dilution), STAT3 (CST, 30835, 1:1000 dilution), p-STAT3 (Abcam, ab76315, 1:1000 dilution), p-p65 (CST, 3033, 1:1000 dilution), p65 (CST, 8242, 1:1000 dilution), p-FAK(CST, 8556, 1:1000 dilution), FAK (Abcam, ab40794, 1:1000 dilution), Lamin B1 (Abcam, ab133741, 1:1000 dilution), poly-Ubiquitin (CST, 3936, 1:1000 dilution), Fibronectin(CST, 26836, 1:1000 dilution), α-SMA(Proteintech, 14395-1-AP, 1:1000 dilution), α-Tubulin (Beyotime, AT819, 1:5000 dilution), and GAPDH (Beyotime, AG019, 1:5000 dilution).
Immunoprecipitation (IP)
Cell samples were prepared in IP lysis buffer (150 mM NaCl, 25 mM Tris-HCl (pH 7.9), 5 mM MgCl2, 10% glycerol, 0.2 mM EDTA, 0.1% NP-40) with protease inhibitors (Roche Complete). Cleared cell lysates were then incubated with indicated antibodies and its normal IgGs, and 15 μl Protein A/G agarose beads (Invitrogen, USA) at 4 °C for 4 h. The immunoprecipitates were washed, eluted in Laemmli loading buffer, and analyzed by immunoblotting.
Statistical analysis
Sample size was chosen according to generally accepted number in the field and no particular statistical methods were used to predetermine it. Some of the data collection and analyzes were not performed blind due to obvious differences between groups but at least two observers performed the experiments and independently analyzed the data. The data were presented as means ± s.e.m. or means ± s.d. with indicated statements in the manuscript. Statistical analyses were performed by SPSS 22.0 (IBM Corp, Armonk, NY) or GraphPad Prism 8 (GraphPad Software, Inc, La Jolla, CA). Statistical differences were determined by Student’s t test. P < 0.05 was considered with statistical significance. All experiments were repeated at least three times and all sample sizes were shown in the figures legends.
Data availability
All data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
Harris-Tryon TA, Grice EA. Microbiota and maintenance of skin barrier function. Science. 2022;376:940–5.
Sonnemann KJ, Bement WM. Wound repair: toward understanding and integration of single-cell and multicellular wound responses. Annu Rev Cell Dev Biol. 2011;27:237–63.
Jones RE, Foster DS, Longaker MT. Management of chronic wounds-2018. JAMA. 2018;320:1481–2.
Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–46.
Rodrigues M, Kosaric N, Bonham CA, Gurtner GC. Wound healing: a cellular perspective. Physiol Rev. 2019;99:665–706.
Spallotta F, Cencioni C, Straino S, Sbardella G, Castellano S, Capogrossi MC, et al. Enhancement of lysine acetylation accelerates wound repair. Commun Integr Biol. 2013;6:e25466.
Sleiman S, Dragon F. Recent advances on the structure and function of RNA Acetyltransferase Kre33/NAT10. Cells 2019;8:1035.
Liu HY, Liu YY, Yang F, Zhang L, Zhang FL, Hu X, et al. Acetylation of MORC2 by NAT10 regulates cell-cycle checkpoint control and resistance to DNA-damaging chemotherapy and radiotherapy in breast cancer. Nucleic Acids Res. 2020;48:3638–56.
Liu X, Tan Y, Zhang C, Zhang Y, Zhang L, Ren P, et al. NAT10 regulates p53 activation through acetylating p53 at K120 and ubiquitinating Mdm2. EMBO Rep. 2016;17:349–66.
Liu X, Cai S, Zhang C, Liu Z, Luo J, Xing B, et al. Deacetylation of NAT10 by Sirt1 promotes the transition from rRNA biogenesis to autophagy upon energy stress. Nucleic Acids Res. 2018;46:9601–16.
Balmus G, Larrieu D, Barros AC, Collins C, Abrudan M, Demir M, et al. Targeting of NAT10 enhances healthspan in a mouse model of human accelerated aging syndrome. Nat Commun. 2018;9:1700.
Arango D, Sturgill D, Alhusaini N, Dillman AA, Sweet TJ, Hanson G, et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell 2018;175:1872–86.
Xu C, Zhang J, Zhang J, Liu B. SIRT7 is a deacetylase of N4-acetylcytidine on ribosomal RNA. Genome Instab Dis. 2021;2:253-60. https://doi.org/10.1007/s42764-021-00046-x.
Yang W, Li HY, Wu YF, Mi RJ, Liu WZ, Shen X, et al. ac4C acetylation of RUNX2 catalyzed by NAT10 spurs osteogenesis of BMSCs and prevents ovariectomy-induced bone loss. Mol Ther Nucleic Acids. 2021;26:135–47.
Zhang Y, Jing Y, Wang Y, Tang J, Zhu X, Jin WL, et al. NAT10 promotes gastric cancer metastasis via N4-acetylated COL5A1. Signal Transduct Target Ther. 2021;6:173.
Li D, Wang A, Liu X, Meisgen F, Grünler J, Botusan IR, et al. MicroRNA-132 enhances transition from inflammation to proliferation during wound healing. J Clin Invest. 2015;125:3008–26.
Serezani APM, Bozdogan G, Sehra S, Walsh D, Krishnamurthy P, Sierra Potchanant EA, et al. IL-4 impairs wound healing potential in the skin by repressing fibronectin expression. J Allergy Clin Immunol. 2017;139:142–51.
Dey A, Varelas X, Guan KL. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov. 2020;19:480–94.
Wilson VG. Growth and differentiation of HaCaT keratinocytes. Methods Mol Biol. 2014;1195:33–41.
Hodde JP, Johnson CE. Extracellular matrix as a strategy for treating chronic wounds. Am J Clin Dermatol. 2007;8:61–6.
Johnson BZ, Stevenson AW, Prêle CM, Fear MW, Wood FM. The role of IL-6 in skin fibrosis and cutaneous wound healing. Biomedicines 2020;8:101.
Hsu PC, Chen YH, Cheng CF, Kuo CY, Sytwu HK. Interleukin-6 and Interleukin-8 regulate STAT3 activation migration/invasion and EMT in chrysophanol-treated oral cancer cell lines. Life. 2021;11:423.
Wang W, Tang YA, Xiao Q, Lee WC, Cheng B, Niu Z, et al. Stromal induction of BRD4 phosphorylation results in chromatin remodeling and bet inhibitor resistance in colorectal cancer. Nat Commun. 2021b;12:4441.
Georganas C, Liu H, Perlman H, Hoffmann A, Thimmapaya B, Pope RM. Regulation of IL-6 and IL-8 expression in rheumatoid arthritis synovial fibroblasts: the dominant role for NF-kappa B but not C/EBP beta or c-Jun. J Immunol. 2000;165:7199–206.
Lim R, Barker G, Lappas M. SIRT6 is decreased with preterm labor and regulates key terminal effector pathways of human labor in fetal membranes. Biol Reprod. 2013;88:17.
Na J, Shin JY, Jeong H, Lee JY, Kim BJ, Kim WS, et al. JMJD3 and NF-kappaB-dependent activation of Notch1 gene is required for keratinocyte migration during skin wound healing. Sci Rep. 2017;7:6494.
Wang H, Lu H, Wu Y. Knockdown of Dual Oxidase 1 (DUOX1) promotes wound healing by regulating Reactive Oxygen Species (ROS) by activation of Nuclear Kactor kappa B (NF-kappaB) signaling. Med Sci Monit. 2021a;27:e926492.
Pereira SG, Oakley F. Nuclear factor-kappaB1: regulation and function. Int J Biochem Cell Biol. 2008;40:1425–30.
Gough P, Myles IA. Tumor necrosis factor receptors: pleiotropic signaling complexes and their differential effects. Front Immunol. 2020;11:585880.
Shen Q, Zheng X, McNutt MA, Guang L, Sun Y, Wang J, et al. NAT10, a nucleolar protein, localizes to the midbody and regulates cytokinesis and acetylation of microtubules. Exp Cell Res. 2009;315:1653–67.
Hsieh CY, Hsu MJ, Hsiao G, Wang YH, Huang CW, Chen SW, et al. Andrographolide enhances nuclear factor-kappaB subunit p65 Ser536 dephosphorylation through activation of protein phosphatase 2A in vascular smooth muscle cells. J Biol Chem. 2021;286:5942–55.
Yang J, Fan GH, Wadzinski BE, Sakurai H, Richmond A. Protein phosphatase 2A interacts with and directly dephosphorylates RelA. J Biol Chem. 2001;276:47828–33.
Zerfaoui M, Errami Y, Naura AS, Suzuki Y, Kim H, Ju J, et al. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation. J Immunol. 2001;185:1894–902.
Hamed M, Caspar B, Port SA, Kehlenbach RH. A nuclear export sequence promotes CRM1-dependent targeting of the nucleoporin Nup214 to the nuclear pore complex. J Cell Sci. 2021;134:jcs258095.
Saccani S, Marazzi I, Beg AA, Natoli G. Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor kappaB response. J Exp Med. 2004;200:107–13.
Tanaka T, Shibazaki A, Ono R, Kaisho T. HSP70 mediates degradation of the p65 subunit of nuclear factor kappaB to inhibit inflammatory signaling. Sci Signal. 2014;77:ra119.
Cabanel M, da Costa TP, El-Cheikh MC, Carneiro K. The epigenome as a putative target for skin repair: the HDAC inhibitor trichostatin a modulates myeloid progenitor plasticity and behavior and improves wound healing. J Transl Med. 2019;17:247.
Liu B, Ghosh S, Yang X, Zheng H, Liu X, Wang Z, et al. Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab. 2012;16:738–50.
Wang Y, Zhao X, Shi D, Chen P, Yu Y, Yang L, et al. Overexpression of SIRT1 promotes high glucose-attenuated corneal epithelial wound healing via p53 regulation of the IGFBP3/IGF-1R/AKT pathway. Investig Ophthalmol Vis Sci. 2013;54:3806–14.
Tanaka T, Grusby MJ, Kaisho T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. 2007;8:584–91.
Araki S, Izumiya Y, Rokutanda T, Ianni A, Hanatani S, Kimura Y, et al. Sirt7 contributes to myocardial tissue repair by maintaining transforming growth factor-beta signaling pathway. Circulation. 2015;132:1081–93.
Tang X, Shi L, Xie N, Liu Z, Qian M, Meng F, et al. SIRT7 antagonizes TGF-beta signaling and inhibits breast cancer metastasis. Nat Commun. 2017;8:318.
Funding
This work was supported by the National Natural Science Foundation of China (grant nos. 91849208, 91749114, 32090030, 81972602, 81702909, and 81974480), the National Key R&D Program of China (grant no. 2017YFA0503900), the Natural Science Foundation of Guangdong Province (grant no. 2019A1515010472), Shenzhen Municipal Commission of Science and Technology Innovation (grant nos. ZDSYS20190902093401689, KQJSCX20180328093403969, and JCYJ20210324120213038), The Fundamental Research Funds for the Central Universities (521119200099), Natural Science Foundation of Hunan Province (grant nos. 2021JJ31098, 2023JJ20021), China Postdoctoral Science Foundation funded project (grant nos. 2021T140752, 2021M693560).
Author information
Authors and Affiliations
Contributions
BL and XT supervised the work. BL, XT and BW conceived the ideas of this project and wrote the manuscript. BW performed the majority of the experiments. YW, YL, JZ, CX, LY, JZ, GL, ZL, MW, and JL analyzed and discussed the results.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, B., Zhang, J., Li, G. et al. N-acetyltransferase 10 promotes cutaneous wound repair via the NF-κB-IL-6 axis. Cell Death Discov. 9, 324 (2023). https://doi.org/10.1038/s41420-023-01628-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-023-01628-2
