
Abstract
Breast cancer is one of the world’s leading causes of oncological disease-related death. It is characterized by a high degree of heterogeneity on the clinical, morphological, and molecular levels. Based on molecular profiling breast carcinomas are divided into several subtypes depending on the expression of a number of cell surface receptors, e.g., ER, PR, and HER2. The Her2-positive subtype occurs in ~10–15% of all cases of breast cancer, and is characterized by a worse prognosis of patient survival. This is due to a high and early relapse rate, as well as an increased level of metastases. Several FDA-approved drugs for the treatment of Her2-positive tumors have been developed, although eventually cancer cells develop drug resistance. These drugs target either the homo- or heterodimerization of Her2 receptors or the receptors’ RTK activity, both of them being critical for the proliferation of cancer cells. Notably, Her2-positive cancers also frequently harbor mutations in the TP53 tumor suppressor gene, which exacerbates the unfavorable prognosis. In this review, we describe the molecular mechanisms of RTK-specific drugs and discuss new perspectives of combinatorial treatment of Her2-positive cancers through inhibition of the mutant form of p53.
Similar content being viewed by others

Facts
-
Her2-positive tumors are characterized by a high level of Ki67 expression, a worse prognosis of patient survival due to a high and early relapse rate as well as an increased level of metastatic potential.
-
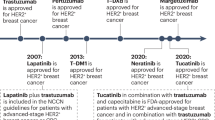
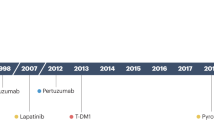
Only several drugs were officially approved by the U.S. Food and Drug Administration (FDA) for the treatment of Her2-positive breast cancer, known as trastuzumab, pertuzumab, trastuzumab emtansine (TDM-1), trastuzumab deruxtecan, lapatinib, neratinib, and pyrotinib.
-
Mutant p53 protein induces overexpression of HER2.
Open questions
-
Can we use inhibitors of mutp53 for future treatment Her2-positive breast cancer?
-
Is it important to detect the status of p53 in Her2-positive breast cancer?
-
What approaches should be used to suppress mutp53?
Breast cancer classification
Breast cancer, despite its heterogeneity, can be categorized into five subtypes: luminal type A (estrogen receptor (ER) and (or) PR positive, HER2 negative), luminal type B (ER and (or) progesterone receptor (PR) positive, human epidermal growth factor receptor 2 (HER2) positive), HER2 overexpressing (ER and PR negative, HER2 positive), triple negative or basal-like (ER, PR, and HER2 negative)1, and claudine-low breast cancer2,3 (Table 1). Based on immunohistochemical studies, pathologists subdivide the disease according to the expression status of receptors and growth factors, with particular attention being payed to the ER, PR, and Her22,4. Her2-positive subtype occurs in ~10–15% of all cases of breast cancer. Her2-positive tumors are usually characterized by a high level of Ki67 expression, a worse prognosis of patient survival due to a high and early relapse rate as well as increased level of metastatic potential2,5,6. Out of those, 30–40% of breast cancer patients with overexpressed Her2 also have high levels of ER, while the rest exhibit diminished expressions of ER and PR hormone receptors, making them extremely resistant to targeting with antihormone therapy.
Receptor tyrosine-protein kinase Her2 (erB-2)
Receptor tyrosine-protein kinase Her2 (ErbB2) is a member of the ERBB family of receptor tyrosine kinases (RTKs). The ErbB receptor family includes four members: ErbB1 (EGFR), ErbB2 (Her2), ErbB3, and ErbB4. ErbB receptors are localized on the cell membrane surface as inactive monomers. However, in the presence of ligands RTKs undergo homo- or heterodimerization, which changes their conformation and promotes autophosphorylation of their intracellular domains7. Ligand-induced conformational changes induce autophosphorylation on the tyrosine residue of the C-terminal domain, which serves as a platform for protein–protein interactions mediated by Src-domains resulting in the transfer of signaling through the PI3K/Akt pathways8. For example, Her2, besides homodimerization, can also form heterodimers not only with other members of the ErbB family, including EGFR4,9,10 but also with other RTKs, including insulin growth factor receptor (IGF1R) and c-Met. The latter event confers resistance to the receptor-specific inhibitory antibodies11. It is important to note that the extracellular domain of Her2 is always maintained in an open conformation ready to bind the activating ligands10,12,13. Notably, the specific ligand that can directly activate Her2 has not been identified so far10. Apparently, this promiscuity of Her2 in binding different partners and accepting various ligands allows tumor cells to actively proliferate.
Her2-specific therapies
Her2, being the most important biomarker for several types of cancer, regulates such important cellular processes as proliferation, angiogenesis, cell adhesion and mobility, and also takes part in organogenesis and development14. Overexpression of Her2 or gene amplification occurs in 20–30% of cases of breast cancer and correlates with a poor prognosis of survival in patients. Anti-Her2 therapy is currently approved for Her2 overexpression in breast, stomach, and gastroesophageal cancers. However, the overexpression of Her2 has also been shown for other tumors found in places such as the bladder, cervix, colon, endometrium, glioblastoma, head and neck, liver, lungs, ovary, pancreas, and salivary ducts15. Her2-directed therapies include specific monoclonal antibodies that prevent the dimerization of Her2, such as trastuzumab and pertuzumab15,16,17,18, and/or small molecule inhibitors of its RTK activity to block the signaling cues, including lapatinib, neratinib, and pyrotinib19.
Monoclonal anti-Her2 antibodies
Trastuzumab (Herceptin), the first monoclonal anti-Her2 antibody, which was developed in 1990, inhibits Her2 signaling through several mechanisms: inhibition of dimerization; internalization and/or degradation of the receptor; inhibition of the PI3K-AKT signaling pathway; and antibody-dependent cell cytotoxicity (ADCC). Trastuzumab has been a standard treatment for both metastatic, and earlier stages of, Her2-positive breast cancer for more than 20 years20,21,22. Trastuzumab binds to the extracellular domain of Her223. It is considered that the extracellular domain of the Her family of receptors consists of four subdomains (I–IV). Structural studies show that extracellular domains can be in two forms: tethered form through interaction II and IV subdomains and constitutively extended form through interactions between I and III subdomains24. It was shown that trastuzumab targets the IV subdomain of Her225 (Fig. 1). Heterodimerization of Her2 with other RTKs blocks the access of trastuzumab to the IV subdomain domain thereby making the cells resistant to the therapy26.

Antibody-based therapeutics pertuzamab and trastuzamab interact with the extracellular domain of Her2. Tyrosin kinasse inhibitors (lapatinib, neratinib, and pyrotinib) interact with the intracellular domain of Her2.
Pertuzumab is a recombinant monoclonal antibody against Her2, the binding site of which is different from the site of trastuzumab. Pertuzumab binds to the dimerization domain (subdomain II of extracellular domain) of Her2, which leads to the inhibition of the heterodimerization of Her2 with Her3 (Fig. 1)27,28,29,30. Proliferation is suppressed by inactivating several downstream signaling pathways, including PI3K/AKT/mTOR and the mitogen-activated protein kinase pathway (RAS/RAF/MEK/ERK)31. In addition, to trastuzumab, pertuzumab triggers the ADCC reaction32. Although pertuzumab monotherapy has shown only moderate efficacy against Her2, a synergistic effect is observed when combined with trastuzumab33.
Tyrosine kinase inhibitors
A wide range of tyrosine kinase inhibitors (TKI) have also shown a good level of efficacy in women with Her2-positive breast cancer. TKI interact with the intracellular catalytic kinase domain of Her2 (Fig. 1).
Lapatinib (Tykerb/Tyverb) acts as a double reversible inhibitor (TKI) of EGFR (Her1) and Her2, achieving a high overall inhibitory effect34. Lapatinib binds to the ATP-binding pocket of these receptors and hence prevents their self-phosphorylation, thereby blocking the activation of MAPK/Erk1/2 and PI3K/AKT downstream pathways35,36. Lapatinib has been shown to enhance Trastuzumab-dependent cellular cytotoxicity against breast tumor cells in in vitro studies37. Later studies have shown that lapatinib is metabolized by the P450 cytochrome system through 3A4 isozyme, resulting in the only metabolite activity against EGFR without targeting Her238.
Neratinib (Nerlynx), an irreversible inhibitor of Her1, Her2, and Her4, which improves patient survival by 2-year when given to women with Her2-positive breast cancer after chemotherapy and adjuvant trastuzumab therapy39. Neratinib covalently binds to the ATP-binding site thus blocking the RTK kinase activity40.
Pyrotinib (Irene) is a novel irreversible anti-Her2 therapeutic drug that inhibits Her1, Her2, and Her4 by covalently binding the ATP-binding pocket of intracellular kinase regions41. A recent study of patients with HER2-positive relapsed or metastatic breast cancer showed that pyrotinib increased progression-free survival and objective response rates over lapatinib42.
Antibody–drug conjugates
Trastuzumab emtansinem (adotrastuzumab) is an antibody–drug conjugate consisting of the monoclonal antibody trastuzumab bound to the cytotoxic agent mertansine (DM1), which is a tubulin inhibitor. This conjugate was designed to overcome trastuzumab resistance, which is developed in most Her2-positive patients and uses trastuzumab to deliver cytotoxic agent to Her2 overexpression cells. Adotrastuzumab is approved as the only treatment for Her2-positive metastatic breast cancer in patients who have already received trastuzumab and taxane individually or in combination. The approval is based on the results of a study, which compared adotrastuzumab with lapatinib and capecitabine. The study showed significantly longer survival without disease progression and overall survival with less toxicity than lapatinib plus capecitabine43.
Trastuzumab deruxtecan (Enhertu) is a novel antibody–drug conjugate recently approved by FDA for the treatment of metastatic Her2-positive breast cancer. This drug comprises of a monoclonal Her2-targeting antibody (trastuzumab) and a topoisomerase I inhibitor (deruxtecan) that showed very promising antitumor effects in preclinical xenograft models and clinical trials. This drug was designed to reach the cytotoxicity effect in tumor cells expressing Her244.
However, none of these drugs can deliver a complete cure to patients, and eventually drug resistance develops in ~80% of HER2 + metastatic breast cancer patients45.
Mechanisms of drug resistance
HER2-targeted therapies (trastuzumab, pertuzumab, TDM1, and lapatinib) are efficient at attenuating primary tumor progression and cancer relapse, but on the other hand, they apply a high selection pressure on the tumor to favor the clones with mutations in Her2 that become resistant to the therapy. For example, mutations in HER2 could perturb the antibody recognition or physical interaction between the drug and receptor. The T798M mutation in HER2 showed increased autophosphorylation activity and preference for heterodimerization with EGFR in human breast cancer cells. Interestingly, the treatment of Her2 T798M-expressing cells with an EGFR-specific antibody, cetuximab, reverted the trastuzumab resistance46. Similar to EGFR, which develops drug resistance to gefitinib by acquiring secondary mutations, Her2 also follows the same route. Specifically, in response to neratininb, mutant HER2 L869R was reported to acquire a secondary mutation at HER2T798I, reducing the binding between RTK and neratinib47.
Another mechanism of drug resistance to anti-RTK therapies in breast cancer is executed via the activation of other RTKs. For example, IGF1R is overexpressed in HER2+ breast cancer, and forms a heteromeric complex with HER2 and HER3 to activate the PI3K signaling pathway, conferring trastuzumab resistance to breast cancer patients48. Simultaneous treatment with anti-HER2 (trastuzumb) and anti-IGF1R mAbs (figitumumab) reportedly produced synergetic effects in breast cancer cells. Another RTK, c-Met, is frequently overexpressed in HER2+ breast cancer patients and similar to IGF1R contributes to trastuzumab resistance. Pharmacological inhibition of c-Met sensitizes the cancer cells to trastuzumab treatment49.
The p53 family of tumor suppressors in breast cancer
Tp53 is one of the most important tumor suppressors in mammals. Being a transcriptional factor, p53 plays a key role in the maintenance of genome integrity by regulating such important processes as cell cycle progression, apoptosis, senescence, DNA repair, and cell metabolism in response to various forms of cell stress. Perhaps not surprisingly, the TP53 gene is the most commonly mutated gene in various tumors including breast cancer50,51,52,53. Importantly, about 30% of all breast cancer cases are characterized by mutations in the TP53 gene, while for HER2-positive subtypes, the proportion of TP53 mutations reaches 70%54,55.
The vast majority of p53 mutations occurs within the DNA-binding domain, which resides between amino acids 120 and 300. Only one-tenth of them leads to loss of p53 functions and the rest are missense mutations that result in the expression of a defective protein56. However, there are a few hotspot mutations, e.g., in the positions R248, R273, R175, and G245, that are enriched in breast cancer cells and are associated with an increased tumor growth rate, chemo- and radioresistance, and an unfavorable prognosis57,58. These mutations not only exhibit dominant-negative activity over wild-type p53 in case of heterozygosity but also confer new oncogenic properties to the protein, i.e., providing it with “gain of function” (GOF) features50,59,60.
Mutant p53 alters the gene expression program in various ways. One way that p53mut can affect transcription is through preventing p53 homologs, p63, and p73, from activating their target pro-apoptotic genes61,62. On the other hand, GOF p53 can act as a cofactor for other transcription factors such as NF-Y and ETS2, and promote cancer progression50,63,64.
Importantly, recent studies in mice have shown that GOF p53 (R172H, which is homologous to the human R175H mutant form) facilitates p53LOH after DNA damage65. This means that genotoxic therapy administered to breast cancer patients with such mutations in the TP53 gene will eventually result in the ablation of the wild-type copy of TP53 and worsen the survival prognosis. Furthermore, the p53 GOF forms were shown to be more stable on the protein level in cancer cells compared to their normal counterparts66,67. The main mechanism responsible for the degradation of both p53wt and p53mut is through the ubiquitin-dependent proteasome system67,68. MDM2 is the main p53-specific E3 ubiquitin ligase that targets the p53 protein for proteasomal degradation69,70. Being a direct transcription target of p53, Mdm2 expression is attenuated in the presence of mutant p53 and hence the latter is accumulated in the cell71. Furthermore, upon DNA damage, p53 undergoes multiple post-translational modifications, which compete with MDM2-mediated ubiquitination, thereby indirectly stabilizing the p53 protein72,73,74. On a related note, mutant p53 is constitutively phosphorylated on Ser15 by ATM in the human breast carcinoma cell lines MDA-MB231 and MDA-MB46875,76,77. Moreover, chaperones HSP70 and HSP90 and co-chaperones BAG contribute to the maintenance of mutant forms of p53 in cancer cells via their interactions with mutp53 and hence preventing its proteasomal degradation78,79. Importantly, in response to DNA damage proteasomes themselves undergo post-translational modifications that affect their activity and composition80,81,82. Taken together, post-translational modifications play a very important role in the regulation of p53. Future experiments should clarify how various covalent modifications affect the interactome of mutant forms of p53 thereby affecting their functions.
Since GOF mutant forms of p53 are more stable in cancer cells, it is not surprising that strong p53 signals obtained by immunohistochemistry are associated with more aggressive forms of cancer and worse survival prognosis for patients.
Analyzing the association of p53 and Her2 in breast cancer
In breast cancer patients, the majority of Her2-positive cases harbor p53 mutations, which correlate with a poor prognosis. Also, women patients with p53 germline mutations (Li–Fraumeni syndrome) have been shown to have a significantly higher probability (83%) of developing breast cancer with Her2 overexpression83. Analysis of these patients showed that tumors with a higher Ki67 proliferation index were associated with greater Her2 overexpression, larger size, and greater lymph node damage compared to other types, and are considered more aggressive84.
A relationship between p53 mutations and the early onset of Her2-positive breast cancer has been demonstrated85. These data indicate an indirect association of p53 and Her2 expression. In the study of Ferrari et al. the authors divided 64 Her2-positive breast tumors into four subgroups (A, B, C, and D) with subgroups C and D overexpressing Her2. They found that primarily these C and D subgroups were enriched with p53 mutations86.
To obtain more insights into the relation between Her2 expression and the p53 mutation status in breast cancer patients we performed an analysis of the breast cancer dataset (GSE22358) from the public GEO database. ERBB2 (Her2) gene expression levels from breast cancer patients were analyzed using a Wilcoxon–Mann–Whitney statistical test to evaluate Her2 gene expression differences in tumor tissues with a different p53 status (Fig. 2). In Her2-positive tumors, Her2 mRNA levels are significantly higher in samples with mutant p53 (p value = 0.046) comparing to samples with wild-type p53. On the other hand, in Her2-negative tumors, the Her2 mRNA level is significantly lower in samples with mutant p53 (p value = 0.0024) compared to samples with wild-type p53. Collectively, our data indicate at an association between the presence of mutant p53, but not wild type, and Her2 expression.
In the mouse model, mutp53 R172H was shown to interact with oncogenic Her2 signaling in the development of breast cancer. The authors found that mutp53 enhanced Her2/EGFR signaling, thereby facilitating the proliferation of breast cancer cells and increasing the population of breast cancer stem cells87. Mechanistically, mutp53 can increase the level of Her2 twofold: first, mutp53 can enhance the transcriptional activity of HSF1 (heat shock transcription factor 1), whose target (among several others) is chaperon Hsp90, which in turn, stabilizes Her2 and mutp53 itself on the protein level88. Importantly, double Her2/EGFR inhibitor, lapatinib, inhibits the Her2–HSF1–mutp53 interaction, which leads to the destabilization of mutp53 protein in cancer cells89.
The second mechanism of Her2 regulation by mutp53 was shown by Roman-Rosales et al.90 The authors showed that the transfection of mutant forms of the p53 protein (R248Q and R273C) in cell lines led to an increase of Her2 expression. Conversely, suppression of mutp53 reduced the expression of Her2 on the level of transcription90. Thus, the status of p53 affects the expression of Her2 in opposite ways, either promoting the proliferation or apoptosis of cancer cells91.

Her2-negative tumors with mutp53 (beige) were compared to Her2-negative tumors with wild-type p53 (blue). Her2-positive tumors with mutp53 (beige) were compared to Her2-positive tumors with wild-type p53 (blue).
Perspective therapeutic approaches to treat mutant p53- and Her2-positive cancers
The wealth of accumulated experimental data strongly suggests that combination therapeutic approaches are more efficient in targeting solid tumors compared to monotherapy. Following these logics, it would be reasonable to assume that different combinations of Her2-specific drugs combined with anti-mutp53 therapy would give the best result. Unfortunately, not many chemicals that would specifically target mutant p53 are available today. In this respect, high expectations are associated with a new experimental drug, APR-246, which reverts the mutant conformation of p53 to its wild-type form92. It would be interesting to see whether the combination of APR-246 and Her2 inhibitors produce a synergistic effect on killing the breast cancer cells.
Besides targeting mutp53 directly, there are several alternative approaches. For example, it has been shown that the mutant forms of the members of the p53 family increase the immunogenicity of cancer cells93,94. Bioinformatic analysis revealed that almost all analyzed immune signatures showed significantly higher enrichment levels in TP53-mutated breast cancer cells compared to wild-type TP53 cells95. Taken these results together, it seems as a viable approach to test immune therapy on Her2-positive/mutp53 either with specific CAR-Ts or using anti-PDL1/CTLA4-therapy.
One of the most common forms of mutp53 expressed in Her2-positive breast cancers is GOF p53 R248Q p53. Since this protein assumes mutant conformation, its stability is critically dependent on the presence of Hsp90 chaperon. Accordingly, Schulz-Heddergott et al. have shown that Hsp90 inhibition reduced the level of mutp53 R248Q and inhibited colon cancer progression. In this respect, it would be interesting to see whether the same approach, when combined with Her2 inhibitors, inhibits the growth of breast cancer cells96. Supporting this finding is the recent report demonstrating that Hsp90 inhibitor 17-AAG eliminates R248Q by stimulating macroautophagy under normal growth conditions96.
Work by the group of Del Sal97 and by others98 highlighted that statins, a class of MVA pathway inhibitors and a very common drug used in the clinic for treatment of cardiovascular diseases, elicit mutp53 destabilization and reducing cancer cell proliferation. Future experiments will assess the validity of our hypotheses (Fig. 3).

Mutp53 can influence transcription level of Her2, and as a result stabilize Her2. Mutp53 physically interacts with and enhances the transcriptional activity of HSF1, which is the main transcriptional regulator of Hsp90. Hsp90 stabilizes Her2 and mutp53 itself. Moreover, Her2 inhibitor lapatinib inhibits the ternary Her2–HSF1–mutp53 interaction, which leads to the destabilization of the mutp53 protein in cancer cells.
References
Eliyatkın, N., Yalçın, E., Zengel, B., Aktaş, S. & Vardar, E. Molecular classification of breast carcinoma: from traditional, old-fashioned way to a new age, and a new way. J. Breast Health 11, 59–66 (2015).
Perou, C. M. et al. Molecular portraits of human breast tumours. Nature 406, 747–752 (2000).
Butti, R. et al. Receptor tyrosine kinases (RTKs) in breast cancer: signaling, therapeutic implications and challenges. Mol. Cancer 17, 34 (2018).
Malhotra, G. K., Zhao, X., Band, H. & Band, V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol. Ther. 10, 955–960 (2010).
Perou, C. M. Molecular stratification of triple-negative breast cancers. Oncologist 15, 39–48 (2010).
Lund, M. J. et al. Age/race differences in HER2 testing and in incidence rates for breast cancer triple subtypes: a population-based study and first report. Cancer 116, 2549–2559 (2010).
Yarden, Y. & Pines, G. The ERBB network: at last, cancer therapy meets systems biology. Nat. Rev. Cancer 12, 553–563 (2012).
Roskoski, R. Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharm. Res 79, 34–74 (2014).
Weigelt, B., Geyer, F. C. & Reis-Filho, J. S. Histological types of breast cancer: how special are they? Mol. Oncol. 4, 192–208 (2010).
Graus-Porta, D., Beerli, R. R., Daly, J. M. & Hynes, N. E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 16, 1647–1655 (1997).
Kennedy, S. P. et al. Targeting promiscuous heterodimerization overcomes innate resistance to ERBB2 dimerization inhibitors in breast cancer. Breast Cancer Res. 21, 43 (2019).
Cho, H. S. et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756–760 (2003).
Garrett, T. P. et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell 11, 495–505 (2003).
Carney, W. P. HER2 status is an important biomarker in guiding personalized HER2 therapy. Personalized Med. 2, 317–324 (2005).
Yan, M., Parker, B. A., Schwab, R. & Kurzrock, R. HER2 aberrations in cancer: implications for therapy. Cancer Treat. Rev. 40, 770–780 (2014).
Dean-Colomb, W. & Esteva, F. J. Her2-positive breast cancer: herceptin and beyond. Eur. J. Cancer 44, 2806–2812 (2008).
Zhang, J. L. et al. Effects of Herceptin on circulating tumor cells in HER2 positive early breast cancer. Genet Mol. Res. 14, 2099–2103 (2015).
Yaal-Hahoshen, N. & Safra, T. Herceptin (trastuzumab): adjuvant and neoadjuvant trials. Isr. Med. Assoc. J. 8, 416–421 (2006).
Xuhong, J. C., Qi, X. W., Zhang, Y. & Jiang, J. Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer. Am. J. Cancer Res. 9, 2103–2119 (2019).
Piccart-Gebhart, M. J. et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 353, 1659–1672 (2005).
Romond, E. H. et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 353, 1673–1684 (2005).
Slamon, D. et al. Adjuvant trastuzumab in HER2-positive breast cancer. N. Engl. J. Med. 365, 1273–1283 (2011).
Gemmete, J. J. & Mukherji, S. K. Trastuzumab (herceptin). Ajnr. Am. J. Neuroradiol. 32, 1373–1374 (2011).
Adams, C. W. et al. Humanization of a recombinant monoclonal antibody to produce a therapeutic HER dimerization inhibitor, pertuzumab. Cancer Immunol. Immunother. 55, 717–727 (2006).
Nahta, R. Molecular mechanisms of trastuzumab-based treatment in HER2-overexpressing breast cancer. ISRN Oncol. 2012, 428062 (2012).
Luque-Cabal, M., García-Teijido, P., Fernández-Pérez, Y., Sánchez-Lorenzo, L. & Palacio-Vázquez, I. Mechanisms behind the resistance to trastuzumab in HER2-amplified breast cancer and strategies to overcome it. Clin. Med. Insights Oncol. 10, 21–30 (2016).
Mann, M. et al. Targeting cyclooxygenase 2 and HER-2/neu pathways inhibits colorectal carcinoma growth. Gastroenterology 120, 1713–1719 (2001).
Mendoza, N., Phillips, G. L., Silva, J., Schwall, R. & Wickramasinghe, D. Inhibition of ligand-mediated HER2 activation in androgen-independent prostate cancer. Cancer Res. 62, 5485–5488 (2002).
Mullen, P., Cameron, D. A., Hasmann, M., Smyth, J. F. & Langdon, S. P. Sensitivity to pertuzumab (2C4) in ovarian cancer models: cross-talk with estrogen receptor signaling. Mol. Cancer Ther. 6, 93–100 (2007).
Sakai, K. et al. Pertuzumab, a novel HER dimerization inhibitor, inhibits the growth of human lung cancer cells mediated by the HER3 signaling pathway. Cancer Sci. 98, 1498–1503 (2007).
Capelan, M. et al. Pertuzumab: new hope for patients with HER2-positive breast cancer. Ann. Oncol. 24, 273–282 (2013).
von Minckwitz, G. et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N. Engl. J. Med. 377, 122–131 (2017).
Bachelot, T. et al. Preliminary safety and efficacy of first-line pertuzumab combined with trastuzumab and taxane therapy for HER2-positive locally recurrent or metastatic breast cancer (PERUSE). Ann. Oncol. 30, 766–773 (2019).
Blandino, G. & Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 37, 30 (2018).
Loibl, S. et al. PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab. Ann. Oncol. 27, 1519–1525 (2016).
Nishimura, R. et al. Role of HER2-related biomarkers (HER2, p95HER2, HER3, PTEN, and PIK3CA) in the efficacy of lapatinib plus capecitabine in HER2-positive advanced breast cancer refractory to trastuzumab. Oncology 93, 51–61 (2017).
Oakman, C., Pestrin, M., Zafarana, E., Cantisani, E. & Di Leo, A. Role of lapatinib in the first-line treatment of patients with metastatic breast cancer. Cancer Manag. Res. 2, 13–25 (2010).
Zhang, D. et al. Activity of lapatinib is independent of EGFR expression level in HER2-overexpressing breast cancer cells. Mol. Cancer Ther. 7, 1846–1850 (2008).
Chan, A. et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 17, 367–377 (2016).
Feldinger, K. & Kong, A. Profile of neratinib and its potential in the treatment of breast cancer. Breast Cancer 7, 147–162 (2015).
Zhang, K. et al. CDK4/6 inhibitor palbociclib enhances the effect of pyrotinib in HER2-positive breast cancer. Cancer Lett. 447, 130–140 (2019).
Ma, F. et al. Pyrotinib or lapatinib combined with capecitabine in HER2-positive metastatic breast cancer with prior taxanes, anthracyclines, and/or trastuzumab: a randomized, phase II study. J. Clin. Oncol. 37, 2610–2619 (2019).
Verma, S. et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med 367, 1783–1791 (2012).
Modi, S. et al. Antitumor activity and safety of trastuzumab deruxtecan in patients with HER2-low-expressing advanced breast cancer: results from a phase Ib study. J. Clin. Oncol. JCO1902318, https://doi.org/10.1200/JCO.19.02318 (2020).
Geyer, C. E. et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med 355, 2733–2743 (2006).
Rexer, B. N. et al. Human breast cancer cells harboring a gatekeeper T798M mutation in HER2 overexpress EGFR ligands and are sensitive to dual inhibition of EGFR and HER2. Clin. Cancer Res. 19, 5390–5401 (2013).
Hanker, A. B. et al. An acquired HER2(T798I) gatekeeper mutation induces resistance to neratinib in a patient with HER2 mutant-driven. Breast Cancer Cancer Discov. 7, 575–585 (2017).
Rexer, B. N. & Arteaga, C. L. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit. Rev. Oncog. 17, 1–16 (2012).
Shattuck, D. L., Miller, J. K., Carraway, K. L. 3rd & Sweeney, C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 68, 1471–1477 (2008).
Muller, P. A. & Vousden, K. H. p53 mutations in cancer. Nat. Cell Biol. 15, 2–8 (2013).
Coles, C. et al. p53 mutations in breast cancer. Cancer Res. 52, 5291–5298 (1992).
Duffy, M. J., Synnott, N. C. & Crown, J. Mutant p53 in breast cancer: potential as a therapeutic target and biomarker. Breast Cancer Res. Treat. 170, 213–219 (2018).
Fedorova, O. et al. Orphan receptor NR4A3 is a novel target of p53 that contributes to apoptosis. Oncogene 38, 2108–2122 (2019).
Nik-Zainal, S. et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54 (2016).
Silwal-Pandit, L. et al. TP53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin. Cancer Res. 20, 3569–3580 (2014).
Baugh, E. H., Ke, H., Levine, A. J., Bonneau, R. A. & Chan, C. S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 25, 154–160 (2018).
Walerych, D., Napoli, M., Collavin, L. & Del Sal, G. The rebel angel: mutant p53 as the driving oncogene in breast cancer. Carcinogenesis 33, 2007–2017 (2012).
Thor, A. D. et al. Accumulation of p53 tumor suppressor gene protein: an independent marker of prognosis in breast cancers. JNCI 84, 845–855 (1992).
Harvey, M. et al. A mutant p53 transgene accelerates tumour development in heterozygous but not nullizygous p53–deficient mice. Nat. Genet. 9, 305–311 (1995).
Blagosklonny, M. V. p53 from complexity to simplicity: mutant p53 stabilization, gain-of-function, and dominant-negative effect. FASEB J. 14, 1901–1907 (2000).
Stindt, M. H. et al. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene 34, 4300–4310 (2015).
Gaiddon, C., Lokshin, M., Ahn, J., Zhang, T. & Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 21, 1874–1887 (2001).
Di Agostino, S. et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 10, 191–202 (2006).
Sampath, J. et al. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 276, 39359–39367 (2001).
Ghaleb, A., Yallowitz, A. & Marchenko, N. Irradiation induces p53 loss of heterozygosity in breast cancer expressing mutant p53. Commun. Biol. 2, 436 (2019).
Midgley, C. A. & Lane, D. P. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene 15, 1179–1189 (1997).
Frum, R. A. & Grossman, S. R. in Mutant p53 and MDM2 in Cancer 187–197 (Springer, 2014).
Lukashchuk, N. & Vousden, K. H. Ubiquitination and degradation of mutant p53. Mol. Cell. Biol. 27, 8284–8295 (2007).
Karni-Schmidt, O., Lokshin, M. & Prives, C. The roles of MDM2 and MDMX in cancer. Annu. Rev. Pathol. 11, 617–644 (2016).
Sane, S. & Rezvani, K. Essential roles of E3 ubiquitin ligases in p53 regulation. Int. J. Mol. Sci. 18, https://doi.org/10.3390/ijms18020442 (2017).
Lezina, L. et al. KMT Set7/9 affects genotoxic stress response via the Mdm2 axis. Oncotarget 6, 25843–25855 (2015).
Morgunkova, A. & Barlev, N. A. Lysine methylation goes global. Cell Cycle 5, 1308–1312 (2006).
Li, J. & Kurokawa, M. Regulation of MDM2 stability after DNA damage. J. Cell Physiol. 230, 2318–2327 (2015).
Landré, V. et al. Regulation of transcriptional activators by DNA-binding domain ubiquitination. Cell Death Differ. 24, 903–916 (2017).
Jin, S. et al. Gadd45a contributes to p53 stabilization in response to DNA damage. Oncogene 22, 8536–8540 (2003).
Lavin, M. A. & Gueven, N. The complexity of p53 stabilization and activation. Cell Death Differ. 13, 941–950 (2006).
Li, D. et al. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 9, 577–588 (2011).
Li, D., Marchenko, N. D. & Moll, U. M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 18, 1904–1913 (2011).
King, F. W., Wawrzynow, A., Höhfeld, J. & Zylicz, M. Co‐chaperones Bag‐1, Hop and Hsp40 regulate Hsc70 and Hsp90 interactions with wild‐type or mutant p53. EMBO J. 20, 6297–6305 (2001).
Fedorova, O. A. et al. Proteomic analysis of the 20S proteasome (PSMA3)-interacting proteins reveals a functional link between the proteasome and mRNA metabolism. Biochem. Biophys. Res. Commun. 416, 258–265 (2011).
Kulichkova, V. A. et al. 26S proteasome exhibits endoribonuclease activity controlled by extra-cellular stimuli. Cell Cycle 9, 840–849 (2010).
Moiseeva, T. N., Bottrill, A., Melino, G. & Barlev, N. A. DNA damage-induced ubiquitylation of proteasome controls its proteolytic activity. Oncotarget 4, 1338–1348 (2013).
Wilson, J. R. et al. A novel HER2-positive breast cancer phenotype arising from germline TP53 mutations. J. Med Genet 47, 771–774 (2010).
Shokouh, T. Z., Ezatollah, A. & Barand, P. Interrelationships between Ki67, HER2/neu, p53, ER, and PR status and their associations with tumor grade and lymph node involvement in breast carcinoma subtypes: retrospective-observational analytical study. Medicine 94, e1359 (2015).
Melhem-Bertrandt, A. et al. Early onset HER2-positive breast cancer is associated with germline TP53 mutations. Cancer 118, 908–913 (2012).
Ferrari, A. et al. A whole-genome sequence and transcriptome perspective on HER2-positive breast cancers. Nat. Commun. 7, 12222 (2016).
Yallowitz, A. R. et al. Mutant p53 amplifies epidermal growth factor receptor family signaling to promote mammary tumorigenesis. Mol. Cancer Res. 13, 743–754 (2015).
Li, D., Yallowitz, A., Ozog, L. & Marchenko, N. A gain-of-function mutant p53-HSF1 feed forward circuit governs adaptation of cancer cells to proteotoxic stress. Cell Death Dis. 5, e1194–e1194 (2014).
Li, D. & Marchenko, N. D. ErbB2 inhibition by lapatinib promotes degradation of mutant p53 protein in cancer cells. Oncotarget 8, 5823–5833 (2017).
Roman-Rosales, A. A., Garcia-Villa, E., Herrera, L. A., Gariglio, P. & Diaz-Chavez, J. Mutant p53 gain of function induces HER2 over-expression in cancer cells. BMC Cancer 18, 709 (2018).
Casalini, P., Botta, L. & Menard, S. Role of p53 in HER2-induced proliferation or apoptosis. J. Biol. Chem. 276, 12449–12453 (2001).
Synnott, N. C., Murray, A. M., O’Donovan, N., Duffy, M. J. & Crown, J. Combined treatment using the anti-p53 drug, APR-246 and eribulin: Synergistic growth inhibition in p53-mutated breast cancer cells. J. Clin. Oncol. 35, e14098–e14098 (2017).
Low, L., Goh, A., Koh, J., Lim, S. & Wang, C. I. Targeting mutant p53-expressing tumours with a T cell receptor-like antibody specific for a wild-type antigen. Nat. Commun. 10, 5382 (2019).
Qu, J. et al. Mutant p63 affects epidermal cell identity through rewiring the enhancer landscape. Cell Rep. 25, 3490–3503.e3494 (2018).
Liu, Z. et al. TP53 mutations promote immunogenic activity in breast cancer. J. Oncol. 2019, 5952836 (2019).
Schulz-Heddergott, R. et al. Therapeutic ablation of gain-of-function mutant p53 in colorectal cancer inhibits Stat3-mediated tumor growth and invasion. Cancer Cell 34, 298–314.e297 (2018).
Ingallina, E. et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat. Cell Biol. 20, 28–35 (2018).
Parrales, A. et al. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 18, 1233–1243 (2016).
Acknowledgements
O.F., A.K., A.P., Y.G. wrote the article, designed pictures and table and acknowledged the support from RSF grant 18–75–10076. A.D., O.S., N.B. executed general leadership, wrote the section on perspective therapeutic approaches to treat Her2-positive breast cancer, and acknowledged the support from RFBR grant 18-29-09144.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Rufini
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fedorova, O., Daks, A., Shuvalov, O. et al. Attenuation of p53 mutant as an approach for treatment Her2-positive cancer. Cell Death Discov. 6, 100 (2020). https://doi.org/10.1038/s41420-020-00337-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-020-00337-4
