
Abstract
The long-sought discovery of HER2 as an actionable and highly sensitive therapeutic target was a major breakthrough for the treatment of highly aggressive HER2-positive breast cancer, leading to approval of the first HER2-targeted drug — the monoclonal antibody trastuzumab — almost 25 years ago. Since then, progress has been swift and the impressive clinical activity across multiple trials with monoclonal antibodies, tyrosine kinase inhibitors and antibody–drug conjugates that target HER2 has spawned extensive efforts to develop newer platforms and more targeted therapies. This Review discusses the current standards of care for HER2-positive breast cancer, mechanisms of resistance to HER2-targeted therapy and new therapeutic approaches and agents, including strategies to harness the immune system.
Similar content being viewed by others
Introduction
Innovations in pathology, molecular biology and drug development have enabled HER2-positive breast cancer (BC), a historically aggressive subtype, to become one with impressive outcomes. The field was energized in 1978 when the first tyrosine kinase, epidermal growth factor receptor (EGFR), was discovered followed by the identification of the neu or HER2 (also known as ERBB2) gene in 19841,2. The discovery that amplification or overexpression of HER2 was associated with extremely poor survival in BC ultimately led to the development of a monoclonal antibody (mAb) to HER2, trastuzumab3 (Box 1).
Until this point, triple-negative and HER2-overexpressing disease were widely regarded as the most aggressive BC histologies, with unfavourable prognoses. Advanced BC was considered incurable, and treatment was purely palliative. However, newer and novel therapeutic strategies have led to markedly improved survival outcomes. The dependence of the tumour on HER2, coupled with effective HER2-targeted drugs such as trastuzumab, pertuzumab and most recently, tucatinib and trastuzumab deruxtecan (T-DXd), have contributed to these survival improvements in patients with HER2-positive (HER2+) BC4. Currently, survival rates exceed 90% in HER2+ early breast cancer (EBC) treated with chemotherapy and dual antibody therapy5. More than half of patients with metastatic HER2+ disease are diagnosed de novo, further demonstrating that most patients presenting with early disease are cured6.
However, despite this success, metastatic HER2+ tumours inevitably develop resistance, leading to disease progression. As such, the goal of therapy in HER2+ BC is to expand the number of patients cured in the early setting and prevent recurrence. In those HER2+ cancers that do present with de novo stage IV disease or ultimately recur, development of novel therapies is needed as these tumours continue to be dependent on HER2 signalling. Therefore, extensive research is ongoing in the preclinical, translational and clinical arenas to develop original and more potent therapies for this exceptionally sensitive target, HER2.
Advances in targeting HER2 include further exploitation of antibody–drug conjugates (ADCs), altering the linkers, payload or antibody scaffold to optimize efficacy7,8. Another approach is the development of bispecific antibodies, which use binding of two different HER2 epitopes to maximize efficacy9. As immunotherapy has shown benefit in triple-negative breast cancer (TNBC), attempts to mobilize the immune system in HER2+ disease are also ongoing. Immunotherapy is being approached from various angles including administering checkpoint inhibitors, linking effector T cells to HER2 antibodies, cellular therapy and vaccines9,10,11,12.
This Review summarizes the successful therapeutic approaches approved for treatment of HER2+ BC, which are essential to understanding ensuing developments, exploring the potential areas of drug resistance that are the foundation for future drug development. A survey of the landscape of platforms being used to maximize HER2-targeted therapeutic efficacy are enumerated, which includes mAbs, tyrosine kinase inhibitors (TKIs), ADCs, bispecific antibodies, immune system targeting agents, cellular therapy and targeted protein degraders. As HER2 is such a sensitive target, continued investigation to advance therapeutic benefit will undoubtedly lead to improvements in survival.
Current standard of care for HER2+ BC
As the understanding of HER2 biology has evolved (Box 1), so has the development of agents that target HER2. HER receptors contain an extracellular ligand-binding domain, a transmembrane domain, and an intracellular tyrosine kinase domain. Ligand binding to the HER proteins results in homodimerization or heterodimerization of these receptors, leading to activation of downstream signalling pathways that promote cell division and growth and inhibit apoptosis13. There is no known ligand for HER2, but it is a preferred dimerization partner for the other HER proteins, especially HER3 (ref.14). HER2 overexpression or amplification leads to ligand-independent dimerization and abnormal signalling in addition to increased signalling through ligand-dependent heterodimerization13. The efficacy of HER2-targeted agents is most prominent in these ‘HER2-positive’ tumours.
The definition of HER2 positivity according to American Society of Clinical Oncology–College of American Pathologists (ASCO–CAP) guidelines, includes tumours that have 3+ positive staining by immunohistochemistry (IHC) in ≥10% of tumour cells, or HER2 gene amplification detected by fluorescence in situ hybridization (FISH)15,16 (Box 2). Recent research has identified a subset of patients with ‘HER2-low’ (HER2low) BC that is responsive to novel HER2-targeted ADCs17. HER2low is defined as HER2 IHC 1+ by itself or 2+ in the absence of HER2 gene amplification by ISH (in situ hybridization). The cut-off for the level of HER2 expression by IHC is only a crude estimation of those who may actually benefit from anti-HER2 therapies. With the introduction of the new HER2low definition, additional diagnostic tools may need to be considered.
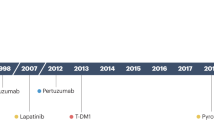
Since the initial approval of trastuzumab for HER2+ BC, multiple agents exhibiting various mechanisms of action and safety profiles have been approved for the treatment of early-stage and metastatic disease (Fig. 1 and Box 3). Below, agents that have been approved by regulatory agencies are briefly described and the advantages and limitations of each strategy are summarized.

Timeline of preclinical discovery milestones for HER2 biology and regulatory approval for anti-HER2 therapies. A, adjuvant setting; M, metastatic setting; N, neoadjuvant setting; +, approved in China only; *, M. Bishop and H. Varmus awarded Nobel Prize in 1989 for this discovery; **, S. Cohen and R. Levi-Montalcini awarded Nobel Prize in 1986 for discovery of growth factors and their receptors.
Monoclonal antibodies
Trastuzumab was the first humanized mAb developed to HER2 that achieved remarkable success in the treatment of HER2+ BC. Trastuzumab binds to the extracellular domain (ECD) of HER2, suppresses intracellular HER2 signalling pathways, inhibits cell cycle arrest and mediates antibody-dependent cell-mediated cytotoxicity (ADCC)18. Preclinical data demonstrating synergy between cytotoxic agents and trastuzumab paved the way for clinical trial designs with chemotherapy combinations for treatment of HER2-overexpressing metastatic breast cancer (MBC) and subsequently in the adjuvant setting (Supplementary Table 1)19,20,21,22,23,24,25,26. All these trials demonstrated favourable outcomes with trastuzumab and chemotherapy, leading to swift approvals from the regulatory agencies in the metastatic and curative settings (Fig. 1).
With these successes, trastuzumab became firmly established as the treatment of choice for patients with HER2+ BC, and HER2 drug development that would revolutionize the outcome for patients facing this disease commenced.
However, although trastuzumab improves responses and outcomes, a substantial number of patients develop therapeutic resistance and disease relapse27. Early studies showed that antibodies targeting multiple domains in HER2 can exert synergistic antitumour effects28. Consequently, a second humanized anti-HER2 mAb, pertuzumab, was developed. Unlike trastuzumab, which binds to ECD IV of HER2, pertuzumab binds to ECD II, preventing HER2 heterodimerization with HER1, HER3 and HER4, blocking downstream tumour signalling29. Trastuzumab is more effective at inhibiting cell growth in the absence of HER3 ligand30,31. These complementary mechanisms of action and the effect of the two agents on immune system-mediated antitumour activity via ADCC and/or complement-mediated cytotoxicity (CDC), suggested that combination therapy could be synergistic32,33,34.
Trials that combined these two mAbs with chemotherapy as treatment for HER2+ BC in the metastatic, adjuvant and neoadjuvant settings demonstrated better outcomes than trastuzumab and chemotherapy combinations, leading to FDA approval of pertuzumab in these settings5,35,36,37 (Supplementary Table 1). These trials changed the course of disease for patients with HER2+ BC and improved survival.
The efficacy of trastuzumab, in part, is dependent on ADCC mediated through its Fc domain. Patients whose immune effector cells (natural killer (NK) cells or dendritic cells (DCs)) can bind more tightly to the Fc domain have stronger responses to trastuzumab38,39. A novel anti-HER2 IgG1 mAb, margetuximab, has an engineered Fc domain to increase the affinity for the activating Fcγ receptor (CD16A) and to decrease the affinity for the inhibitory Fcγ receptor (CD32B) expressed on immune effector cells. The optimized Fc domain confers enhanced ADCC activity against HER2+ tumours and more potently stimulates ADCC than trastuzumab or pertuzumab in vitro40. The FDA granted regulatory approval for margetuximab plus chemotherapy for the treatment of HER2+ MBC on the basis of the results from the SOPHIA study, adding another drug to the HER2 armamentarium41 (Supplementary Table 1).
Trastuzumab biosimilars
Unlike novel synthetic HER2-targeting antibodies, trastuzumab biosimilars are biologic agents created from living cells and have similar pharmacokinetic and pharmacodynamic properties to the original product. They may have minor differences in clinically inactive components from the original biologic medication, but there are no clinically meaningful differences between the two with respect to safety, purity and potency (see Related links: https://www.fda.gov/media/82647/download). There are currently five trastuzumab biosimilars approved by the FDA for US markets, with additional agents in development (see Related links: https://www.fda.gov/drugs/biosimilars/biosimilar-productinformation). Trastuzumab-dkst (MYL1401O) was the first trastuzumab biosimilar to receive FDA approval (in 2017) for patients with HER2-overexpressing BC and gastrointestinal (GI) cancers. Subsequent approvals included trastuzumab-pkrb (CT-P6) for HER2+ BC (December 2018); trastuzumab-dttb (SB3) (January 2019), trastuzumab-qyyp (PF-05280014; March 2019) and trastuzumab-anns (ABP980) for HER2+ breast and GI tumours (June 2019). Although clinical guidelines support biosimilars, their adoption in clinical practice has been slow, hampered by physician and commercial payer awareness, perceptions and preferences.
Formulations of monoclonal antibodies
In an effort to conserve resources and reduce the burden of intravenous (i.v.) infusions to patients, a subcutaneous version of trastuzumab (trastuzumab–hyaluronidase-oysk) was developed and validated in clinical trials and approved in the EU in 2013 and in the USA in 201942. Pertuzumab–trastuzumab–hyaluronidase-zzxf, a subcutaneous formulation of a fixed dose formulation of trastuzumab and pertuzumab demonstrated safety and non-inferiority in pathological complete response (pCR) versus corresponding i.v. versions in patients with EBC and was granted FDA approval in the adjuvant and metastatic settings in 202043.
Tyrosine kinase inhibitors
TKIs are small molecules that target the intracellular catalytic kinase domain of HER2, competing with ATP, blocking phosphorylation and activating downstream signalling cascades. Lapatinib, an oral, 4-anilinoquinazoline TKI derivative, is a reversible inhibitor of EGFR (also known as HER1) and HER2, with activity in HER2-driven tumours that are insensitive to trastuzumab44. Lapatinib overcomes trastuzumab resistance mediated by insulin-like growth factor 1 receptor (IGF1R) upregulation by blocking the crosstalk between HER2 and IGF1R45. Cell lines and xenografts expressing the p95HER2 variant that lack the trastuzumab-binding ECD were susceptible to lapatinib, presumably because it targets the intracellular kinase domain of HER2 (ref.46). In vitro studies showed that pTEN-deficient, trastuzumab-resistant HER2+ BC cell lines remained sensitive to lapatinib47. These and other data supported the clinical development of lapatinib in trastuzumab-resistant BC48. Lapatinib in combination with capecitabine was superior to capecitabine alone in trastuzumab-treated HER2+ MBC, and the lapatinib–letrozole doublet was more efficacious than letrozole alone in hormone receptor-positive (HR+)/HER2+ MBC, leading to regulatory approval of these combinations and offering an alternative to HER2-targeting antibody combinations for these patients49,50,51 (Supplementary Table 1). Several trials were conducted with lapatinib in HER2+ EBC, the details of which are succinctly summarized in a recent review52.
A significant sanctuary for recurrent HER2+ disease is the central nervous system (CNS), and up to 50% of patients with HER2+ BC develop brain metastases53,54,55. Moreover, the blood–tumour barrier (BTB), which evolves from the blood–brain barrier (BBB), regulates drug distribution to brain metastases, posing a clinical challenge56. It has also been suggested that the large size of the HER2 antibodies (for example, trastuzumab) prevents penetration of the BBB and efficacy in the CNS. Lapatinib, by virtue of its small size and potent anti-HER2 activity, found a niche in this arena. Lapatinib monotherapy and combination therapy demonstrated lower rates of CNS progression and responses in the CNS in HER2+ MBC, including in patients with previously untreated brain metastases49,57,58. These data provided the impetus for further refinements in next-generation TKIs to tackle the growing problem of brain metastases associated with HER2+ MBC.
In contrast to lapatinib, neratinib (HKI-272) is an irreversible pan-HER TKI that targets EGFR, HER2 and HER4 (ref.59). Neratinib inhibits growth in trastuzumab-resistant cell lines and is synergistic with trastuzumab60,61. A unique feature of neratinib is its activity in cell lines with somatic HER2 mutations in the absence of HER2 amplification, suggesting that it can overcome possible resistance to other anti-HER2 therapies61. The co-occurrence of somatic HER2 mutations and HER2 amplification has also been observed, and a prevalence of 7.1% was reported in patients with HER2+ MBC, all of whom had poor response to dual anti-HER2 antibody therapy62. Durable tumour shrinkage was seen with neratinib, but not with lapatinib or trastuzumab, in animal models with concomitant HER2 mutation and amplification, and similar efficacy with neratinib was also evident in the clinical setting62.
Treatment with neratinib after standard adjuvant trastuzumab improved invasive disease-free survival (iDFS) rates in HER2+ BC, especially in patients with HR+ tumours63,64 (Supplementary Table 1). Similarly to lapatinib, the neratinib–capecitabine combination improved outcomes in HER2+ MBC, earning a nod from the FDA as third-line therapy65 (Supplementary Table 1). Given the rapid development of newer HER2-targeted therapies, including novel TKIs with less toxicity and higher efficacy, the role of the neratinib–capecitabine combination appears limited.
Pyrotinib, another oral, irreversible pan-HER TKI that targets EGFR, HER2 and HER4, has been approved by the Chinese regulatory authority in combination with capecitabine in HER2+ MBC treated with prior trastuzumab and taxane66 (Supplementary Table 1). Pyrotinib is similar to the other pan-HER2 TKIs, with diarrhoea as its most common toxicity66. Several trials with pyrotinib are ongoing in breast and other cancers, but it is unclear whether its use in HER2+ MBC will expand to other countries.
Tucatinib is a potent, oral, HER2-specific TKI with >1,000-fold greater potency for HER2 than EGFR67. Tucatinib demonstrated CNS penetration in intracranial xenograft models and was superior to lapatinib in preclinical studies68,69,70,71. The pivotal HER2CLIMB trial demonstrated superiority of the tucatinib plus capecitabine–trastuzumab combination in extending progression-free survival (PFS) and overall survival (OS) in HER2+ MBC previously treated with anti-HER2 therapies72,73 (Supplementary Table 1). The innovative design of the trial allowed patients with stable as well as active brain metastases, and HER2CLIMB is the first randomized trial to demonstrate clinically meaningful benefits in patients with HER2+ brain metastases, marking an important milestone in the treatment of HER2+ BC. Tucatinib was FDA approved in April 2020 for the treatment of HER2+ MBC, including patients with CNS metastases. Ongoing trials with tucatinib include a maintenance trial in first-line HER2+ MBC with the goal of delaying CNS progression (NCT05132582), an adjuvant study for patients with residual disease following neoadjuvant chemotherapy (NCT04457596) and the neoadjuvant I-SPY 2 trial (NCT01042379) (see Related links).
Antibody–drug conjugates
ADCs were designed to channel the cytotoxic effects of chemotherapy to specific tumour cells. ADCs contain a tumour-targeting antibody covalently bound to a cytotoxic drug (payload) via a synthetic linker74. The ADC is directed to cancer cells expressing the target (for example, HER2) on the cell surface, followed by internalization of the ADC and release of the cytotoxic payload, resulting in tumour cell death. Cleavable linkers in ADCs enable release of the cytotoxic payload from the target cell to the extracellular space, leading to destruction of surrounding cancer cells that may not have high target protein expression. This bystander effect further enhances the efficacy of ADCs against tumour cells75.
Ado-trastuzumab emtansine (T-DM1), the first anti-HER2 ADC to be developed, is composed of trastuzumab connected via a stable linker to DM1, a maytansine derivative with a drug-to antibody ratio (DAR) of ~3.5. T-DM1 caused mitotic disruption and apoptosis in HER2-overexpressing cell lines regardless of their sensitivity to trastuzumab and lapatinib76,77. T-DM1 prolonged PFS and OS in patients with HER2+ MBC compared with current standard of care in large, randomized trials, validating HER2 overexpression as a target in trastuzumab-resistant BC and showcasing the efficacy of ADCs in this setting. These results led to FDA approval of T-DM1 for HER2+ MBC (Fig. 1 and Supplementary Table 1). Although the attempt to replace trastuzumab with T-DM1 as part of the dual anti-HER2 (neo)adjuvant regimens was not successful, T-DM1 reduced the risk of recurrence in patients with HER2+ EBC without upfront pCR after neoadjuvant chemotherapy78,79,80 (Fig. 1 and Supplementary Table 1). T-DM1 also demonstrated activity in a subset of patients with HER2+ MBC and brain metastases in the KAMILLA study81.
T-DXd is a HER2 ADC comprising a humanized HER2 antibody with the same sequence as trastuzumab conjugated to deruxtecan (DXd); T-DXd has a DAR of 8 and exhibits the enhanced features of DXd. The novel DXd ADC technology consists of a cleavable tetrapeptide-based linker, a self-immolative amino methylene spacer and a novel topoisomerase inhibitor payload derivative of exatecan (DS-8951)82. The linker of DXd is selectively cleaved by cathepsins, which are upregulated in tumours, releasing the payload preferentially inside cancer cells. This feature, coupled with the short half-life of DXd in vivo, limits systemic exposure of the cytotoxic agent, with the goal of reducing toxicity. The high membrane permeability of DXd enables local bystander effects, leading to the death of tumour cells in the tumour microenvironment (TME)82.
Single-agent T-DXd demonstrated impressive antitumour activity in refractory HER2+ MBC and an unprecedented improvement in PFS when compared head to head with T-DM1 in second-line HER2+ MBC leading to its FDA approval in these patient populations83,84,85,86 (Fig. 1 and Supplementary Table 1). T-DXd has also shown encouraging activity in BC brain metastases, in DESTINY-Breast01 (ref.87). DEBBRAH is evaluating T-DXd in patients with HER2+ or HER2low MBC with brain metastases and/or leptomeningeal metastases; preliminary data are promising88.
A unique feature of T-DXd is its ability to target HER2low MBC as evidenced by activity in this subset of patients in a phase I trial83. This remarkable efficacy appears multifactorial based on enhanced features of T-DXd compared with T-DM1, the ability to deliver a higher dose and the bystander effect tackling intratumour HER2 heterogeneity. T-DXd demonstrated statistically significant and clinically meaningful improvements in PFS and OS versus treatment of physician’s choice (TPC) chemotherapy in the phase III DESTINY-Breast04 trial in HER2low MBC17. These results are likely to redefine nomenclature around HER2 expression and what is considered actionable in terms of HER2 expression. The FDA recently approved the use of T-DXd for patients with HER2low MBC on the basis of data from the DESTINY-Breast04 study (see Related links: https://www.fda.gov/news-events/press-announcements/fda-approves-first-targeted-therapy-her2-low-breast-cancer).
Recent results from the DAISY trial showed that T-DXd antitumour activity was associated with level of HER2 expression in HER2+ MBC89. Interestingly, a high percentage of HER2 IHC 0 cells in the tumour and their spatial distribution relative to HER2-overexpressing cells were associated with a decreased response to T-DXd. This highlights the need to develop more sensitive methods for detection of HER2 expression and novel technologies to assess heterogeneous HER2 expression within the tumour.
Mechanisms of HER2-targeted resistance
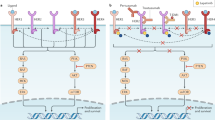
Resistance to anti-HER2 therapies may occur via multiple mechanisms, some of which appear to be shared between different agents. A common reason for trastuzumab treatment failure is incomplete inhibition of the HER family of receptors, which can be overcome by dual HER2-targeted therapy or ADCs with potent payloads that have activity even with lower HER2 expression. Effective inhibition of HER2 may also be thwarted by the emergence of HER2-activating mutations. Other resistance mechanisms include generation of p95HER2, a truncated form of HER2 that lacks the ECD that is recognized by anti-HER2 antibodies and Δ16HER2, a splice variant lacking the ECD encoded by exon 16, which leads to stabilization of homodimers and constitutive activation of downstream signalling. Figure 2 depicts selected mechanisms of HER2-targeted resistance.

a, Mutations and/or alterations in the HER family of receptors that lead to activation of downstream signalling pathways. (1) Mutations in HER2 leading to P13K–AKT and RAS–MAPK pathway activation. (2) Co-occuring mutations in HER2 and HER3 leading to PI3K–AKT pathway activation. b, Loss of HER2 extracellular domain in cells overexpressing p95HER2 receptor. Masking of the trastuzumab-binding site on HER2 owing to overexpression of mucin 4 (MUC4) and CD44–polymeric hyaluronan complex. (3) p95HER2 overexpression. (4) MUC4 overexpression and CD44–polymeric hyaluronan complex. c, Activation of compensatory pathways. (5) Mutations in HER2 promote MEK–ERK signalling, which activates CDK2 kinase. (6) PIK3CA mutations lead to P13K–AKT pathway activation. (7) Cyclin D1 gene overexpression leads to resistance to anti-HER2 therapies. d, Heterogeneous expression of the HER2 receptor in tumours leads to decreased sensitivity to HER2-targeted therapies that are dependent on overexpression of HER2. ER, oestrogen receptor.
HER family alterations
HER2 mutations can drive BC growth even in the absence of HER2 overexpression or amplification. The frequency of HER2 mutations in BC is ~3%. HER2(L755S) is the most common alteration associated with lapatinib resistance in MBC treated with prior trastuzumab90,91. This activating HER2 mutation has also conferred resistance to dual blockade by trastuzumab and pertuzumab and reduced sensitivity to T-DM1. Second-generation TKIs (for example, afatinib and neratinib) can overcome treatment resistance in BC models, suggesting that they may be therapeutic alternatives for treatment of patients with HER2+ tumours harbouring the HER2L755S mutation92. Neratinib was shown to be active against HER2 mutant/HER2-non-amplified MBC in the SUMMIT basket trial93.
HER2 mutations frequently co-occur with HER3 mutations, and cancers with both of these mutations respond poorly to neratinib. The HER3E928G kinase domain mutation has been shown to enhance the affinity of HER2/HER3 and reduce binding of HER2 to neratinib94. Co-expression of HER2 and HER3 mutations leads to enhanced downstream PI3K–AKT pathways and resistance to neratinib. Thus, combined anti-HER2 and PI3K inhibition with a PI3Kα inhibitor such as alpelisib may be a promising strategy to overcome HER2 resistance due to co-occurring HER2 and HER3 mutations.
The generation of tucatinib-resistant BC cell lines revealed significant phosphorylation of HER2 receptors and reactivation of downstream signalling pathways, unlike the partial reactivation of HER signalling seen in lapatinib- and neratinib-resistant models95. Acquired resistance to tucatinib was dependent on amplified EGFR signalling and could be overcome by a combination of gefitinib and tucatinib or pan-HER TKIs, such as neratinib, pyrotinib or poziotinib. Using organoids derived from xenograft tumours of HER2+ BC resistant models, neratinib-resistant models were shown to be cross-resistant to other single-agent HER2 TKIs, but the presence of co-occurring HER2 and PIK3CA mutations suggested susceptibility to combination with a PI3K or AKT inhibitor96.
Loss or masking of HER2 epitope
The presence of p95HER2 has been associated with poor outcomes in patients with HER2+ EBC, and patients with MBC overexpressing p95HER2 had lower response rates to trastuzumab than those expressing full-length HER2 (ref.97). Chemotherapy sensitized p95HER2/611CTF xenografts to trastuzumab, presumably via HER2 stabilization induced by chemotherapy98. Lapatinib inhibited p95HER2 phosphorylation in cell lines, reducing downstream activation of AKT and MAPK and inhibiting cell growth46. Retrospective analyses from clinical trials with lapatinib noted that the presence of p95HER2 had no influence on lapatinib efficacy99. Similarly, in the CHER-LOB neoadjuvant study, which randomized patients to receive trastuzumab, lapatinib or their combination, p95HER2 expression was not predictive of outcome nor did it predict for sensitivity to either anti-HER2 agent100. Therefore, the role of p95HER2 as a biomarker of resistance or sensitivity remains to be confirmed.
Overexpression of mucin 4 (MUC4) and the CD44–hyaluronan polymer complex interferes with trastuzumab binding by masking the HER2 epitope and activating HER2. Increased MUC4 expression in oestrogen receptor-positive (ER+)/HER2+ tumours leads to reduced trastuzumab-binding sites101. Soluble TNF upregulated MUC4 expression, resulting in trastuzumab resistance, and combining a soluble TNF inhibitor with trastuzumab prevented tumour growth in preclinical models102. Recent data also show that MUC4 expression results in an immunosuppressive TME in HER2+ BC and emphasize the role of tumour-infiltrating macrophages in mounting an antitumour response103. INB03, a second-generation TNF inhibitor that increases antitumour macrophage phagocytosis and increases lymphocyte function in the TME, is being considered for evaluation in clinical trials.
Activation of compensatory pathways
The activation of compensatory signalling pathways to overcome effects of trastuzumab treatment has been a subject of extensive exploration. Nearly 25–50% of BCs harbour PIK3CA mutations, with enrichment in HR+ (~35%) and HER2+ (~25%) subtypes. PIK3CA mutations are associated with reduced pCR rates to neoadjuvant anti-HER2 therapies and decreased efficacy with trastuzumab or pertuzumab in HER2+ MBC104,105. Loss of PTEN (a key tumour suppressor), which leads to hyperactivation of the PI3K pathway, has also been observed in trastuzumab-resistant tumours106,107. Development of PI3K and mTOR inhibitors offered hope of a therapeutic option, but early trials with buparlisib (a pan-PI3K inhibitor) in the neoadjuvant setting did not yield the desired outcomes108. The BOLERO-1 and BOLERO-3 trials evaluated the addition of the mTOR inhibitor everolimus to trastuzumab plus chemotherapy in first-line and trastuzumab-resistant HER2+ MBC settings, respectively. Biomarker analysis from the pooled study populations indicated PI3K–AKT–mTOR pathway aberrations in approximately 40% of tumours, with everolimus treatment leading to a consistent benefit in these patients versus patients with tumours not exhibiting the aberrations109. Although these results suggested proof of concept, the triplet combinations led to increased toxicity. The advent of isoform-specific PI3K inhibitors such as alpelisib has led to a maintenance study of triplet alpelisib-trastuzumab-pertuzumab in patients with PIK3CA-mutant HER2+ MBC (NCT04208178; see Related links).
Bidirectional crosstalk exists between the HER2 and ER pathways, and preclinical studies have demonstrated a role for ER signalling in promoting resistance to anti-HER2 therapies110,111. Clinical evidence also shows that ER pathway activation offers an escape from HER2 inhibition, and concomitant inhibition of both ER and HER2 signalling may be necessary as demonstrated in trials of ER+/HER2+ BC51,112,113,114. The cyclin D1–CDK4/6 axis also has a role in resistance to anti-HER2 therapies115. CDK4/6 inhibitors appear to be synergistic with trastuzumab and/or lapatinib in inhibiting growth of HER2+ cell lines116,117. This was evidenced in the clinical setting in which dual inhibition of CDK4/6 and HER2 led to improved outcomes in monarcHER and PATRICIA trials in heavily pretreated HR+/HER2+ MBC118,119. The PATINA trial is evaluating whether addition of palbociclib to front-line trastuzumab, pertuzumab and taxane plus endocrine therapy (ET) in HR+/HER2+ MBC will delay the onset of therapeutic resistance (NCT02947685; see Related links). Preclinical and clinical data corroborate cyclin D1-mediated resistance to HER2-targeted therapies, and CDK4/6 inhibitors can overcome this resistance. Cyclin D1 overexpression correlated with lower pCR rates in patients receiving neoadjuvant trastuzumab plus chemotherapy117. In the Na-PHER2 study, trastuzumab and pertuzumab given with fulvestrant and palbociclib neoadjuvantly to patients with ER+/HER2+ BC, led to a significant Ki67 reduction after 2 weeks of therapy, clinical complete response (CR) in 50% of patients and a 27% pCR rate (breast and axilla)120. These data suggest that a combination blocking HER2 and ER and CDK4/6–cyclin D1 activation offers a chemotherapy-free alternative for treatment of HR+/HER2+ BC. An ongoing trial is evaluating trastuzumab plus palbociclib with or without letrozole in HER2+ and ER+/− MBC (NCT02448420; see Related links).
Genomic sequencing analysis of 733 HER2-amplified primary and metastatic breast tumours revealed significant enrichment of mutations that activate RAS–MAPK signalling in advanced tumours treated with prior anti-HER2 therapies121. These mutations, including NF1 and HER2 activating mutations, contribute to resistance to tucatinib and neratinib. The resistant tumours were highly sensitive to MEK–ERK inhibition, with susceptibility due to MEK-dependent activation of CDK2 kinase, thus offering the possibility of overcoming HER2 resistance with MEK–ERK inhibitors.
HER2 heterogeneity
HER2 heterogeneity — the variable expression of HER2 across the tumour — is another potential source of resistance to HER2-targeted therapies. HER2 heterogeneity, defined as HER2 positivity by FISH in 5–50% of tumour cells, or an area of tumour that tested HER2-negative (HER2−) in multiple core biopsies, was found in 10% of patients in a phase II trial of neoadjuvant T-DM1 plus pertuzumab122. A significant association was found between HER2 heterogeneity and lack of pCR following dual HER2-targeted therapy; none of the patients with HER2 heterogeneity achieved a pCR, whereas 55% of patients not classified as HER2 heterogeneous had a pCR. T-DXd demonstrated significant activity in HER2low MBC, significantly improving PFS and OS, and this attribute may also enable T-DXd to overcome resistance due to heterogeneous expression of HER2, which could potentially become even more important in the early setting for patients with tumour heterogeneity17.
Host and tumour immunity
ADCC is a key mechanism mediating the antitumour activity of trastuzumab, and it can be hampered by an immunosuppressive TME. NK cells have an important role in antitumour immunity, and their activity is regulated by careful modulation of inhibitory and activating receptor signalling123. Tumour cells expressing high levels of HLA class I molecules can inhibit NK cells through the engagement of killer cell immunoglobulin-like receptors (KIRs). HLA-G was shown to desensitize BC cells to trastuzumab by binding to the NK cell receptor KIR2DL4, and blocking this HLA-G–KIR2DL4 signalling made HER2+ BC susceptible to trastuzumab treatment in vivo123. Moreover, trastuzumab increased the production of TGFβ and interferon-γ (IFN-γ) by BC cells and NK cells, respectively. TGFβ induced PD1 expression on NK cells, and PD1 blockade significantly increased cytotoxicity of NK cells. Accordingly, combined blockade of HLA-G and PDL1/PD1 may be necessary for effective treatment of trastuzumab-resistant BC.
Trastuzumab can also engage Fcγ receptors on macrophages to promote antibody-dependent cellular phagocytosis (ADCP), which contributes to its antitumour efficacy. Magrolimab, a humanized mAb that targets CD47 was studied to combat trastuzumab resistance by activating ADCP124. CD47 is a protein that acts as a ‘don’t eat me’ signal via its interaction with signal regulatory protein-α (SIRPα) on macrophages to inhibit phagocytosis. CD47 has been shown to be upregulated in HER2+ BC125. The combination of magrolimab and trastuzumab was found to eliminate HER2+ BC cells with increased efficacy due to enhancement of ADCP by macrophages, even in HER2+ BC resistant to ADCC124. This offers a novel therapeutic approach to treat trastuzumab-sensitive or trastuzumab-resistant HER2+ BC, provided the trastuzumab-binding epitope on HER2 is accessible.
Other potential mechanisms of resistance
Several other mechanisms of anti-HER2 therapy resistance have recently been elucidated. A study modelling resistance of HER2+ PIK3CA-mutant BC using two patient-derived xenografts, one resistant to paclitaxel and T-DM1 and the other insensitive to T-DM1 and pertuzumab, demonstrated that alveolar epithelial and fibroblastic reticular as well as lymphatic vessel endothelial hyaluronan receptor 1-positive (Lyve1+) macrophages may be putative drivers of therapeutic resistance126. These intriguing findings require further studies comparing data from transcriptome and exome profiling from trials with anti-HER2 therapies. Preclinical studies hint that abnormal transit of T-DM1 through the endosomal maturation pathway may be responsible for resistance to T-DM1 treatment, but this has not been validated or studied in clinical trials127.
Three novel markers, RAC1, CDK12 and VTCN1, have been found to correlate with response to lapatinib, neratinib and tucatinib in a study that compared the TKI anti-proliferative effects using a 115-cancer cell line panel to identify novel markers of TKI response and/or resistance markers128. Prior data have implicated these genes in resistance to anti-HER2 therapies or immunotherapy129,130,131. Hence, combinations of HER2 TKIs and CDK12 and RAC1 inhibitors may offer a therapeutic strategy in high CDK12- or RAC1-expressing HER2+ BC.
Next-generation therapies for HER2+ BC
The ever-changing face of cancer and its ability to evade existing therapies have underscored the need for continued development of therapeutics based on existing and/or novel platforms and for uncovering new vulnerabilities in resistant tumours. Antibodies targeting alternative HER2 domains or other HER family members have been explored with the goal of achieving a more complete blockade of HER2 and dampening the effects on downstream signalling pathways. Novel ADCs carrying different payloads to avoid cross-resistance to existing therapies or new linkers to offset off-target toxicity are also being actively pursued.
Monoclonal antibodies
Disruption of HER2–HER3 dimerization is important for HER2-driven signalling and is targeted effectively by pertuzumab132. The positive results from pertuzumab trials supported a strategy of targeting HER3, which has a crucial role in HER2-mediated tumorigenesis. HER3 is unique compared with other HER family members as it is defined by the absence of a functional kinase domain and thus any catalytic activity. HER3 is the preferred dimerization partner for HER2, and HER2–HER3 dimerization leads to oncogenic activation of the PI3K signalling pathway, mediating resistance to HER2-targeted therapy133. Several HER3-targeted antibodies have been evaluated in the past decade, mainly targeting the ECD of HER3 (for example, seribantumab and patritumab) and some with modifications to improve ADCC (for example, lumretuzumab, TrasGex) or trap HER3 in an inactive conformation (elgemtumab). Although these drugs have shown some promising preliminary activity, most are no longer in clinical development for HER2+ BC given the high bar of efficacy set by standard-of-care anti-HER2 therapies133,134,135,136,137,138,139. Now, there is greater focus on novel ADCs that target HER2 or HER3. Additionally, bispecific antibodies that target multiple epitopes of HER2, or HER2 and HER3 together in one molecule, are under clinical investigation and are discussed in the next section.
Antibody–drug conjugates
ADCs have successfully combined the antitumoural features of cytotoxics and HER2 antibodies into a single pharmacological entity that has greater efficacy than the sum of its parts7,8. In addition to the ability of ADCs to hone in on the cells expressing the target protein and cause tumour cell lysis, the membrane-permeable payload can diffuse into the surrounding tumour milieu, inducing the bystander effect. This feature enables activity against tumours with low or heterogeneous target expression, thus expanding the pool of susceptible tumour cells. Furthermore, the ADCC of the Fc fragment of the antibody may also contribute to antitumour efficacy. HER2 ADCs can also retain trastuzumab-mediated activity such as inhibiting the HER2 dimerization and suppression of downstream signalling. There are encouraging data against HER2+ brain metastases with these HER2 ADCs81,87,140. Coupled with their demonstrated activity in HER2low MBC, further evaluation of these ADCs is warranted in this high-risk patient population17.
Given the success of T-DM1 and T-DXd, there are more than a dozen HER2-targeted ADCs now in clinical development, with the aim of improving therapeutic index and efficacy. These ADCs differ from the approved agents in cytotoxic payload, DAR, linker or the HER2 epitope targeted. The development of linkers for ADCs has been a very important area of investigation and has been recently reviewed141. Several ADCs currently in development for HER2+ BC are listed in Table 1, and some of these are discussed in further detail below.
Trastuzumab duocarmycin (SYD985)
Trastuzumab duocarmycin (SYD985) is a HER2-targeted ADC based on trastuzumab with a cleavable linker duocarmycin (vc-seco-DUBA) payload. The novel payload is an active toxin (DUBA) that alkylates DNA, causing DNA damage in both dividing and non-dividing cells. The protease-cleavable linker, and subsequent release of the payload into the TME by diffusion, promotes a bystander effect that allows activity in tumour cells with low HER2 expression142. In the phase III TULIP study of SYD985 versus physician’s choice of chemotherapy plus anti-HER2 therapy in patients with HER2+ MBC who had received two or more lines of MBC therapy, SYD985 was associated with a significant improvement in PFS143,144. Ocular toxicity was the most common adverse event (AE) reported, and as for other HER2 ADCs, interstitial lung disease (ILD)/pneumonitis was also observed in a small percentage of patients. A biologics licence application for SYD985 has been recently submitted to the FDA (see Related links: https://go.nature.com/3VZlL4a), and it remains to be seen how it may integrate into the standard of care with T-DXd and tucatinib.
ARX788
ARX788 is a next-generation HER2 ADC created using site-specific oxime conjugation technology and a non-cleavable linker designed for homogeneity and chemical stability145. It also employs a highly hydrophilic payload (AS269, synthetic dolastatin) with limited cell permeability, unlike other ADCs that use highly permeable payloads to elicit a bystander killing effect. Preclinical data with ARX788 demonstrated activity in HER2+ tumours, T-DM1-resistant BCs, and HER2low tumours145. Promising efficacy data and low systemic toxicity (due to the stable linker) were reported from a phase I trial, and a phase II randomized trial is ongoing (NCT04829604; see Related links)146.
Disitamab vedotin (RC48-ADC)
Disitamab vedotin (RC48-ADC) comprises the humanized anti-HER2 antibody hertuzumab coupled via a cleavable linker to the cytotoxic agent monomethyl auristatin E (MMAE). Disitamab targets different epitopes of the HER2 receptor and has better molecular affinity for HER2 than trastuzumab147. The linker in disitamab vedotin uses random coupling of cysteine, which is more homogeneous than lysine. Furthermore, this agent has better endocytosis, which is independent of V-ATPase activity, and has no lysosomal resistance147. Preclinical data demonstrated good activity in HER2-overexpressing cancer cells and a robust bystander effect targeting neighbouring tumour cells147. Multiple clinical trials evaluating disitamab vedotin are ongoing in solid tumours including MBC and gastric cancer. Disitamab vedotin has already received regulatory approval in China for treatment of HER2+ gastric cancer and urothelial cancer147,148.
Zanidatamab zovodotin (ZW49)
Zanidatamab zovodotin (ZW49) is a bispecific HER2-targeted ADC combining the unique design of zanidatamab (ZW25) (binds ECDs II and IV of HER2) with a cytotoxic and cleavable linker. The biparatopic antibody of ZW49 demonstrated lysosomal trafficking and superior internalization relative to a HER2-targeted monospecific ADC149. Preclinical data indicated efficacy in HER2low- and HER2high-expressing models and also in a brain metastasis model149. ZW49 is being evaluated in a phase I trial in patients with metastatic HER2-expressing cancers that have progressed following standard therapies, including HER2-targeted agents (NCT03821233; see Related links).
ALTA-ADC
In an effort to improve the potency of HER2-targeting ADCs, a pertuzumab-based ADC with lower affinity for HER2 at acidic endosomal pH was developed150. Engineering the ADC to confer endosomal dissociation from its target is expected to enable payload entry into lysosomes and recycling of unbound target. This engineered HER2 ADC variant (referred to as an ALTA-ADC) demonstrated increased lysosomal delivery and cytotoxicity even on tumour cells with intermediate levels of HER2 expression, and higher efficacy in xenograft models in mice compared with T-DM1. Furthermore, the ability of the ALTA-ADCs to achieve a therapeutic effect at lower doses may help to overcome the dose-limiting toxicities for other tumour targets.
Targeted thorium-227 conjugates
Targeted α-therapy aims to deliver α-particle-emitting radionuclides selectively to cancer cells in the TME. These α-particles are highly cytotoxic, inducing difficult-to-repair, clustered double strand DNA breaks, leading to cell death151. Targeted thorium-227 conjugates (TTCs) have been generated using efficient chelators that connect the α-particle-emitting radionuclide to an antibody to a target expressed on tumours151. Exposure of cancer cells to TTCs releases markers of danger-associated molecular patterns (DAMPs), which are upregulated by dying cells to alert the immune system and to initiate immunogenic cell death152. The synthetic lethal effect of the combination of TTCs with other DNA-damaging agents was demonstrated using colorectal cancer xenografts153. Combination of a HER2 TTC with olaparib resulted in complete growth inhibition in a human DLD-1 BRCA2–/– xenograft. A first-in-human (FIH) study has been initiated with a HER2 TTC in patients with metastatic breast or gastric cancer (NCT04147819; see Related links).
Tyrosine kinase inhibitors
Targeting the intracellular kinase domain of HER2 using small-molecule inhibitors continues to be investigated with next-generation TKIs. This class of compounds is attractive owing to their unique ability to cross the BBB and BTB although some compounds pose a challenge owing to promiscuous activity and EGFR side effects of rash and diarrhoea. Selected new TKIs in development are listed in Table 2.
DZD1516 was designed as an oral, reversible and selective HER2 kinase that has full BBB penetration. It demonstrated tumour regressions in xenograft mouse models including subcutaneous, brain metastasis and leptomeningeal metastasis models. Phase I pharmacokinetic data supported once-daily dosing. DZD1516 was also well tolerated, with most AEs being grade 1 events154. Interestingly, diarrhoea was not noted as an AE in this study, distinguishing it from most of the other HER2 TKIs studied.
Oncogenic mutations in HER2 have been identified in multiple solid tumours including BC, and most of these occur at allosteric sites outside the ATP-binding site of HER2. BDTX-189 is an oral, ATP-competitive, irreversible, small-molecule inhibitor of EGFR/HER2 alterations and HER2 wild type, designed to spare EGFR wild type to minimize toxicity155. BDTX-189 demonstrated potent, sustained inactivation of multiple allosteric ERBB mutants in vivo. A phase I trial in advanced solid tumours harbouring specific allosteric HER2 or HER3 mutations or other EGFR/HER2 alterations is ongoing. Early data show activity with BDTX-189 in HER2-amplified tumours and in patients with non-small-cell lung cancer (NSCLC) with EGFR and HER2 exon 20 insertions155.
In general, it appears that the focus of newer HER2 TKIs has shifted from HER2+ BC to solid tumours harbouring HER2 point mutations or alterations given that the frequency of these alterations varies from 1% to 2% in BC and up to 5–10% in other cancers (for example, stomach and bladder cancers)156. Preliminary activity of poziotinib in HER2 exon 20 mutant NSCLC has been reported, and there is an ongoing basket trial with neratinib in solid tumours with HER2 mutations (NCT01953926; see Related links)157.
Bispecific antibodies
Advances in antibody biology and engineering have led to the development of bispecific antibodies that contain two binding sites directed against two separate antigens or conversely, can target two separate epitopes on the same antigen. Great diversity is possible with this format as bispecifics can also target an antigen with one binding site, and the other site can be an immune target that could elicit a synergistic effect. Key examples of bispecific antibodies currently in development are discussed below.
Zanidatamab (ZW25)
Zanidatamab (ZW25) is a humanized, bispecific, IgG1 antibody directed against the ECD IV and the dimerization domain (ECD II) of HER2, the same domains as are targeted by trastuzumab and pertuzumab, respectively. Unlike trastuzumab, where each receptor can be bound by only one mAb, zanidatamab promotes receptor clustering whereby each HER2 receptor can be targeted by two zanidatamab antibodies. Hence, treatment with zanidatamab leads to enhanced HER2 internalization, downregulation and potent effector-function-mediated cytotoxicity158. Zanidatamab showed promising antitumour activity as monotherapy or in combination with chemotherapy in patients with advanced HER2-expressing cancers that had progressed on anti-HER2 therapies159,160. Zanidatamab in combination with palbociclib and fulvestrant is currently under evaluation in HR+/HER2+ MBC (NCT04224272; see Related links), and zanidatamab in combination with ALX148 (a CD47 blocker) is being investigated in HER2high and HER2low BC (NCT05027139; see Related links). A neoadjuvant pilot trial with single-agent zanidatamab is also being planned in HER2+ EBC (NCT05035836; see Related links).
Zenocutuzumab (MCLA-128)
Another bispecific, humanized IgG1 antibody that is under investigation is zenocutuzumab (MCLA-128), which acts via two independent mechanisms of action: inhibition of HER2–HER3 signalling and elimination of tumour cells via ADCC. MCLA-128 functions via a ‘dock and block’ mechanism whereby one arm of the antibody binds HER2 domain I and optimally positions the anti-HER3 arm to block the ligand–HER3 receptor interaction, preventing HER2–HER3 dimerization and activation of downstream signalling161. Zenocutuzumab in combination with trastuzumab and vinorelbine demonstrated a 35% clinical benefit rate at 6 months in patients with HER2+ MBC who had progressed on prior anti-HER2 therapy including T-DM1 (ref.162). Further development of zenocutuzumab in HER2+ BC is uncertain; however, its ability to bind HER2 and block NRG1 fusion protein binding and subsequent HER2–HER3 dimerization is being actively explored in NRG1 fusion-positive cancers (NCT02912949; see Related links).
KN026
KN026 is a bispecific antibody that targets two distinct epitopes on HER2 (domains II and IV) leading to dual HER2 signal blockade, presumably by causing HER2 to aggregate on the cell surface and endocytose. Results of a FIH trial of KN026 in heavily pretreated patients with HER2+ MBC showed a 28% objective response rate (ORR) and a median PFS of 6.8 months163. Translational research suggested that patients with co-amplification of CDK12 and HER2 had better responses to KN026 than patients without the co-amplification. On the basis of these results, additional trials are ongoing or planned with KN026 in HER2+ BC (NCT04521179, NCT04881929, NCT04778982; see Related links).
Targeted protein degraders
Targeted protein degradation is being explored as an alternative strategy in cancer, whereby the natural protein degradation system is co-opted for therapeutic purposes. Recently developed novel molecules called proteolysis-targeting chimeras (PROTACs) are heterobifunctional molecules with two ligands joined by a linker. One ligand binds to the ‘protein of interest’ (POI) and the second ligand binds to an E3 ubiquitin ligase. Simultaneous binding of the PROTAC to the POI and ligase induces ubiquitylation of the POI and its degradation by the ubiquitin–proteasome system, followed by regeneration of the PROTAC to tackle another copy of the target164. PROTACs are unique because they exhibit a catalyst-type mechanism of action, unlike classic inhibitors which have a one-to-one relationship with the target protein. Two PROTACs, one targeting ER (ARV-471) and the other targeting the androgen receptor (AR) (ARV-110) have demonstrated clinical efficacy; however, these are not tissue specific because they use E3 ligases that have a broad expression profile. In an effort to optimize the therapeutic window and potentially minimize side effects of broad-spectrum PROTACs, an antibody–PROTAC conjugate that specifically targets HER2-expressing cells was developed165. This trastuzumab–PROTAC conjugate cages E3 ligase-directed degrader activity with an antibody linker that can be hydrolysed after antibody–PROTAC internalization, releasing the active PROTAC that induces catalytic protein degradation. Studies of a trastuzumab–BRD4 degrader conjugate demonstrated that it selectively targets BRD4 for degradation only in HER2-overexpressing BC cell lines, but not in HER2-negative cell lines165. This novel antibody–PROTAC strategy combines the catalytic potency of PROTACs with the tissue specificity of ADCs, enabling the development of new molecules that can target degradation of specific molecules in selected tissues.
An emerging technology aims to selectively degrade HER2-expressing cells by coupling targeted protein degraders to a HER2-specific antibody, generating an antibody neodegrader conjugate (AnDC). Conjugating the protein degrader to the HER2 antibody directs the degrader specifically to the cytosol of the target cells. ORM-5029 is designed to deliver catalytic GSPT1 protein degrader (SMol006) to HER2-expressing tumours via antibody targeting (pertuzumab). Once the antibody and degrader enter the HER2+ tumour cell by endocytosis, the antibody is degraded in the lysosome, releasing the free degrader, which binds to GSPT1 in the cytosol. The natural protein degradation system of the cell is then harnessed to destroy GSPT1, leading to cancer cell death. ORM-5029 displayed in vitro and in vivo efficacy that was comparable to that of other GSPT1 degraders and approved ADCs166.
Harnessing the immune system
In addition to directly targeting the oncogenic driver HER2 and inhibiting its function, exploiting the innate and adaptive immune system to tackle proliferating cancer cells is an area of promise and active investigation in HER2+ BC. Some of these strategies are outlined below and in Table 3 and Fig. 3.

a, Immune-stimulating antibody conjugate (ISAC). (1) ISAC binds to cognate tumour-associated antigen (TAA). (2) Fc receptor-dependent phagocytosis of the tumour cell by myeloid antigen-presenting cell (APC). (3) Toll-like receptor (TLR)-mediated activation of the myeloid cells leads to chemokine and/or cytokine secretion and enhanced antigen presentation. (4) T cell priming by expression of major histocompatibility complex (MHC)–tumour peptide on myeloid cells and expansion of activated T cells. (5) Chemokines attract immune effector cells. Increased myeloid APC phagocytosis. Migration of activated T cells to the tumour and killing of tumour cells. b, Chimeric antigen receptor–macrophage (CAR-M). (1) Targeting of CAR-M to tumour cell expressing the antigen leads to its activation. (2) Phagocytosis of the tumour cell by CAR-M. (3) CAR-M activates the tumour microenvironment (TME) and primes T cells. (4) Primed T cells induce antitumour immune response. c, Radiation plus checkpoint inhibitor. (1) Exposure to radiation results in release of chemokines from tumour cells. (2) Migration of CXC chemokine receptor type 6 (CXCR6)-expressing T cells attracted by chemokines to tumour. (3) Addition of anti-CTLA4 antibody helps to neutralize the CTLA4-mediated inhibition of T cell activation. Activated T cells cause tumour cell lysis. d, (1) HER2-engineered toxin body (ETB) binds to HER2 receptor, followed by (2) forced internalization, (3) intracellular self-routing and (4) ribosome inactivation.
Combinations with checkpoint inhibitors
Immune checkpoint inhibitors (CPIs) have transformed the treatment landscape of solid tumours (for example, melanoma and lung cancer) by inducing the immune system to attack cancer cells, resulting in durable tumour regression and prolonged survival. However, success with these agents in BC has been limited compared with other tumour histologies. There have been no trials in metastatic disease showing benefit outside of the 30–40% of metastatic TNBCs that express PDL1 in the first-line setting. CPI benefits in neoadjuvant TNBC have been broader, irrespective of PDL1 status. Disappointingly, the efficacy of CPI in HER2+ BC has been modest despite high levels of PDL1 expression. Levels of tumour-infiltrating lymphocytes (TILs) in primary HER2+ breast tumours are on par with that in TNBC, indicating a potential for leveraging the immune system11. Furthermore, the immune-mediated ADCC mechanism of trastuzumab and pertuzumab suggests that combination immunotherapies may be effective167,168. Combinations of HER2-targeted agents with CPI have demonstrated preliminary antitumour activity in phase I trials, and T-DXd is being investigated in combination with pembrolizumab (NCT04042701; see Related links)169,170. In the KATE2 randomized phase II trial of T-DM1 with atezolizumab or placebo in previously treated HER2+ MBC, the addition of atezolizumab did not improve PFS relative to T-DM1 alone171. However, this was a later-line setting, and we have learned from TNBC that both setting and line may have profound implications for the activity of immunotherapy in BC. On the basis of the trend to improvement in the PDL1+ subset in KATE2, the phase III KATE3 trial is enrolling patients with PDL1+HER2+ MBC to receive T-DM1 plus atezolizumab or placebo (NCT04740918; see Related links). Further investigations of anti-HER2 therapies in combination with CPI in earlier lines of treatment are ongoing (NCT03199885, NCT04538742; see Related links). The underlying complexity of tumour–immune system interactions may limit the efficacy of targeting a single immune checkpoint in isolation. Hence, combination strategies that simultaneously hinder multiple checkpoints or those that invoke lasting immunological memory may be more effective and may also counter the development of resistance.
Radiation therapy (RT), a common modality for BC treatment, has an immunostimulatory effect by altering the TME, exposing tumour antigen and inducing anti-inflammatory responses172. Hence, addition of RT to CPI may enhance responses (Fig. 3). Results from a trial of whole-brain radiation therapy (WBRT) and concurrent CTLA4-mediated immune modulation with tremelimumab plus or minus trastuzumab in patients with BC with brain metastases, demonstrated a 12-week non-CNS disease control rate (primary end point) of 10% in patients with HER2− MBC and 33% in patients with HER2+ MBC173. Tremelimumab plus durvalumab (anti-PD1 antibody) with brain RT has also been evaluated in MBC including in HER2+ disease (NCT02563925; see Related links). Most ongoing trials exploring RT plus CPI are restricted to patients with HER2− or TNBC disease, although there is one trial that aims to combine pembrolizumab with single-fraction radiation boost to enhance efficacy in operable BC including HER2+ disease (NCT04454528; see Related links).
Bispecific engagers
The poor activity of immunotherapy in BC is attributed to the immunosuppressive TME after chemotherapy, potentially due to loss of the MHC class I molecules on metastatic tumours leading to reduced or delayed recovery of the T cell repertoire clones that target BC-specific antigens174. Novel bispecific antibody (BsAb) formats can overcome this barrier by simultaneously binding a tumour-specific antigen and an immune cell to cause tumour cell death. HER2 is a common antigen targeted by these BsAbs. Bispecific T cell engagers (BiTEs) redirect T cells to target HER2-expressing tumour cells using a BsAb directed against CD3 and HER2 (Fig. 4). The advantage of the BiTE approach is that T cell activation is independent of antigen specificity, and a large fraction of the T cells are activated. Bispecific killer cell engagers (BiKEs) bind to CD16 on natural killer (NK)/monocytic cells and HER2 on tumour cells to eradicate HER2-expressing cancer cells.

a, Structure of HER2–CD3 bispecific antibody with ‘knobs-in-hole’ technology. b, Mechanism of action of a HER2–CD3 bispecific antibody. Step 1: binding of HER2–CD3 bispecific antibody to HER2 on tumour cell and CD3 on T cell. Step 2: activation of T cell causing release of cytokines (TNF and interferon-γ (IFNγ)). Step 3: tumour cell lysis.
An early candidate, 2B1, was a murine-derived HER2 with a Fcγ bispecific antibody that activated NK cells against HER2-expressing tumour cells. Although 2B1 did not demonstrate antitumour activity in a phase I trial, there was evidence of immune activation in 10 of 20 patients175. These findings contributed to a better understanding of the mechanism by which antibody-induced ADCC may exert antitumour activity.
Innovations led to the ‘knobs-in-hole’ technology, which involves replacing a smaller amino acid with a larger amino acid (T336Y) in the CH3 region of an antibody chain to form a ‘knobs’ structure and at the same time substituting a larger amino acid in the other chain with a smaller amino acid to form a ‘hole’ structure (Y407T). This technology, which enables Fc heterodimerization, results in a BsAb that is more stable, has favourable pharmacokinetics and can be produced with high quality and reproducibility176. M-802 and runimotamab (BTRC4017A) are examples of bispecifics that use this technology that are under evaluation in clinical trials (Table 3 and Fig. 4; NCT04501770, NCT03448042; see Related links). An alternative approach is to use antibodies directed against specific receptors expressed on T cells or NK cells (for example, CD137, NKG2A) in combination with HER2-targeted therapies to enhance antitumour activity. Monalizumab is an antibody that blocks the interaction between the inhibitory checkpoint receptor NKG2A expressed on NK cells and CD8+ T cells and HLA-E, which is overexpressed on malignant tumours177. Blocking this inhibitory interaction between NKG2A and HLA-E overcomes tumour resistance to NK cells. Since monalizumab can act simultaneously on both NK cells and T cells as well as tumour-infiltrating immune cells, it can enhance the cytotoxic potential of therapeutic antibodies such as trastuzumab and as such, is being investigated in the phase II MIMOSA trial in patients with HER2+ MBC (NCT04307329; see Related links). CD137 is a costimulatory immune receptor expressed on activated T and B cells and NK cells and a potential target for cancer immunotherapy as it is expressed on TILs. Upon activation, it promotes increased proliferation, cytokine production and cell lysis by T and NK cells178,179.
Other bispecific antibodies
A different approach combining anti-HER2 and PDL1 antibodies into one BsAb (IBI315) was employed to redirect anti-PDL1 response to HER2-expressing tumour cells. Results of a phase I dose-escalation trial in patients with advanced solid tumours treated with various dose levels of IBI315 showed a 20% ORR (see Innovent press release in Related links: https://go.nature.com/3f91toe). Theoretically, this may be a very clean way to use immunotherapy: effectively, targeting just the HER2-expressing cancer cells without as many off-target effects.
PRS-343 (cinrebafusp alfa) (Table 3) is a novel bispecific fusion protein that combines a 4-1BB (CD137)-targeting anticalin protein and a modified version of trastuzumab. The HER2-targeting moiety localizes the drug to the HER2-expressing cells in the TME and facilitates 4-1BB cross-linking, which ameliorates T cell exhaustion and enables T cell expansion and eventual tumour regression. In a phase I study in patients with HER2-expressing solid tumours, PRS-343 demonstrated activity as monotherapy and in combination with atezolizumab180.
Another BsAb innovation is the generation of patient-derived activated T cells (ATCs) armed with an anti-CD3–anti-HER2 bispecific antibody (HER2Bi), meaning these T cells are coated with the BsAb. Essentially, polyclonal T cell populations expanded and activated ex-vivo can be armed with a HER2–CD3 bispecific antibody to specifically recognize and kill HER2-expressing tumour cells in vivo. Arming ATCs with HER2Bi redirects the non-MHC-restricted cytotoxicity of ATCs to a HER2-specific target. A phase I trial that evaluated HER2Bi–aATCs administered with IL-2 and granulocyte–macrophage colony-stimulating factor (GM-CSF) in 23 patients with MBC demonstrated preliminary efficacy, and no dose-limiting toxicities (DLTs) were observed181.
Immune-stimulating antibody conjugates
Immune-stimulating antibody conjugates (ISACs) covalently attach immune stimulants to tumour-specific antibodies such as trastuzumab and can trigger target tumour-dependent activation of the innate and adaptive immune systems to eradicate tumours182 (Fig. 3).
Toll-like receptors (TLRs) are important components that initiate innate immunity. These receptors also bridge innate and adaptive immunity183. BDC-1001 is an example of an ISAC that consists of a trastuzumab biosimilar conjugated to a TLR7/8 agonist with a non-cleavable linker. BDC-1001 employs a three-pronged approach — direct antitumour effects mediated by trastuzumab, localized phagocytosis and elimination of HER2-expressing tumour cells by the immune-stimulating TLR7/8 molecules that activate the myeloid antigen-presenting cells (APCs), that is, macrophages and DCs. This results in cytotoxicity and subsequent processing and presentation of neoantigens that stimulates T cell-mediated durable immunity. The advantage with this construct is that TLR7/8 activation occurs only after internalization into the APCs, thus mitigating the risk of nonspecific immune activation. In essence, these ISACs convert the TME from ‘cold’ to ‘hot’ by localized stimulation of the immune system. Additional agents conjugating trastuzumab or biosimilar to TLR7/8 that are in development are listed in Table 3.
Engineered toxin bodies
Engineered toxin bodies (ETBs) are novel immunotoxins that combine the specificity of antibodies with the potent mechanism of bacterial toxin cell destruction. MT-5111 (Table 3), an example of a de-immunized ETB fused to a HER2 antibody, has a novel mechanism of action that induces direct cell-kill via enzymatic and permanent ribosome destruction, thus potentially bypassing resistance mechanisms that exist for HER2 TKIs, ADCs or antibody modalities (Fig. 3). Furthermore, MT-5111 binds to an epitope on HER2 that is distinct from that of trastuzumab and pertuzumab, enabling combination with existing HER2-targeting agents184. Preliminary results of a phase I trial of MT-5111 in HER2-expressing solid tumours showed stable disease as best response, no DLTs and maximum tolerated dose not yet reached185. An expansion cohort for patients with MBC is ongoing.
CAR-M and CAR-NK therapies
Chimeric antigen receptors (CARs), engineered molecules that combine the specificity of antibodies with the downstream signalling of T cells, represent a key class of cellular immunotherapies. Although FDA-approved CAR-T therapies are available for haematological malignancies, their application to solid tumours has been hindered by the need for active trafficking and penetration of T cells into the TME. As such, alternative immune cells such as human macrophages have been genetically engineered with CARs (CAR-M) to redirect their phagocytic activity against solid tumours186 (Fig. 3). A single infusion of these CAR-Ms was able to shrink tumours and improve OS in solid tumour xenograft mouse models10. Furthermore, the CAR-Ms converted bystander immunosuppressive M2 macrophages to pro-inflammatory M1 macrophages, upregulated the APC machinery and reprogrammed the TME by activating immature DCs and recruiting activated CD8+ T cells to the tumour sites, thus amplifying the antitumour response.
CT-0508 is a cell product comprising autologous peripheral blood monocyte-derived macrophages that are transduced with an adenoviral vector containing an anti-HER2 CAR-M and locked into a pro-inflammatory M1 phenotype. This agent is being investigated in an ongoing FIH trial in patients with HER2-overexpressing solid tumours, and early data demonstrate good tolerability187. Correlative analyses demonstrated evidence of broad reprogramming of the TME as well as intratumoural T cell expansion and activation with altered peripheral T cell repertoire.
CAR-engineered NK cells are superior to CAR-T cells; they appear to be safer because they do not induce cytokine release syndrome (CRS) and they engage multiple mechanisms to promote cytotoxicity188. They also lend themselves to ‘off-the shelf’ manufacturing, that is, they can be generated from peripheral blood cells or from stem cell sources or NK-92 cell lines instead of using a patient’s own immune cells188. The CAR-NK cells represent receptors against tumour-associated antigens (TAAs) and redirect the effector NK cells to target specific tumours. CAT-179 is an off-the-shelf CAR-NK cell therapy in development, with an optimized CAR that targets HER2, an IL-15 cytokine that enables enhanced and sustained NK cell activity and a TME switch to counteract the immunosuppressive TGFβ signal in the TME.
Cancer vaccines targeting HER2
Cancer vaccines are designed to kindle the patient’s own immune system to identify and kill tumour cells by stimulating CD8+ and CD4+ T cell responses to tumour-specific antigens189. Several platforms have been used to develop cancer vaccines, including HER2-specific vaccines. These range from simple peptide vaccines to the more complex autologous or allogenic cell-based vaccines summarized in Table 4.
A key consideration regarding cancer vaccines is the treatment setting. It is well recognized that disease burden and the immunosuppressive TME in metastatic disease may limit T cell activity. Vaccines may be more successful in a setting of very low disease burden, such as post-operative EBC as well as the pre-malignant ductal carcinoma in situ (DCIS) setting12,190. Furthermore, combinations with CPIs or chemotherapy and targeted agents may enhance vaccine efficacy in the (neo)adjuvant setting and potentially help to overcome the suppressive TME in the advanced disease setting191.
Peptide vaccines
These may be the most common vaccine design and have several advantages over other vaccine types: production is very easy, more cost-effective and easier to administer with relatively few side effects. Peptide vaccines using various domains of the HER2 protein have been evaluated in patients with BC. E75 is a nine-amino-acid immunogenic peptide derived from the extracellular domain of HER2 and is expressed in 60–75% of the population. Nelipepimut-S/NeuVax is an MHC class I vaccine that consists of E75 combined with an immunoadjuvant GM-CSF192. A disadvantage is that by being more specific, their T cell immunological responses may be more limited in terms of cell type and may not stimulate T helper cells and B cells sufficiently.
Protein-based vaccines
These offer an advantage over peptide vaccines as they contain both HLA class I and II epitopes and hence are not subject to HLA restrictions. Moreover, they can significantly activate T cells, leading to an enhanced immune response and better T cell activation. Although this approach has not been explored extensively, early trials using a fragment from the intracellular domain (ICD) of HER2 was shown to be well tolerated, and patients developed HER2 ICD-specific T cell immunity193. A phase I trial evaluating dHER2 vaccine, a recombinant protein comprising the ECD and a fragment of the ICD combined with the adjuvant AS15, combined with lapatinib in 12 patients with trastuzumab-refractory HER2+ MBC demonstrated anti-HER2 antibody induction in all patients. HER2-specific T cells were detected in one patient, and importantly, there were no signals of cardiotoxicity194.
Cell-based vaccines
These can be derived from the patient’s own tumour cells to generate autologous vaccines or alternatively, allogeneic tumour cell lines can be used to develop cell-based vaccines. Allogeneic vaccines offer an alternative because they can be obtained from established cancer cell lines that express specific TAAs. Initial data with an allogeneic HER2–GM-CSF vaccine given with cyclophosphamide and weekly trastuzumab appear promising, warranting further studies195 (Table 4). One disadvantage of autologous vaccines is variability in the vaccine and a lengthy manufacturing process.
Dendritic cells
DCs are potent modulators of the immune response and are specialized APCs that can stimulate naive T lymphocytes while generating memory T lymphocytes. The DC vaccine platform allows for antigen processing by the patient’s own immune system, eliciting immunological responses against multiple epitopes on the target, versus a single epitope approach with an antibody or a small-molecule inhibitor. Preliminary studies using DC vaccines have been conducted in patients with invasive BC and even DCIS; the latter hinting at the potential for possible development of a vaccine for prevention of BC196,197,198,199. Although DC vaccines are a promising individualized strategy, the vaccine manufacturing process is complex and will need to be streamlined to make it more accessible. Exploration of allogeneic or artificial APCs should be pursued to overcome this limitation.
Recombinant DNA- or virus-based vaccines
These can be used to deliver tumour-specific antigens and generate antigen-specific cellular and humoral immune responses. DNA-based vaccines are an easy and practical approach given their simplicity, safety and cost effectiveness. Multiple preclinical studies have attested to the efficacy of this strategy. Clinical trials with plasmid-based vaccines are underway in patients with HER2+ EBC with the goal of evaluating the immunogenicity of these vaccines (Table 4). The natural immunogenic nature of viruses is an inherent advantage. Unlike DNA-based vaccines, TAAs are presented along with viral antigens, potentially leading to enhanced and long-lasting immune T cell responses200. Preliminary results from a phase I trial with a novel alphaviral vector encoding a self-amplifying replicon RNA encoding HER2 segment (VRP-HER2) have been encouraging201. Subsequent preclinical studies have shown that only vaccines targeting true oncogenic drivers of HER2 elicited significant antitumour responses, and long-term tumour control was observed only in combination with a CPI. This has led to a phase II study evaluating a combination of VRP-HER2 with pembrolizumab (Table 4; NCT03632941; see Related links).
Despite being inherently well tolerated and inducing immunogenicity and long-term survival in select patients, cancer vaccines have not demonstrated efficacy in randomized phase II or III trials202,203. Novel delivery vehicles such as lipid nanoparticles, virus-like particles, polymeric and non-degradable particles are being explored for cancer vaccines191,204. Previous iterations of vaccines have focused on using TAAs over neoantigens; the latter arise from mutations in the tumour cells owing to their inherent genetic instability, whereas the former are non-mutated antigens, which may explain the lacklustre efficacy observed with vaccine therapies. Neoantigens are able to induce T cell responses comparable to that found in antiviral T cells. New bioinformatic tools for neoantigen prediction have been developed with promising results, which may spur further development of personalized, neoantigen-based therapeutic cancer vaccines including for BC205,206. The success of the coronavirus disease 2019 (COVID-19) mRNA vaccines has breathed new life into exploring this technology for cancer, and trials with mRNA-based vaccines against solid tumours are in development.
Conclusions and future directions
The remarkable journey of discovery of HER2 as an oncogene, biomarker and target for treatment of a very aggressive form of BC has led to an unprecedented improvement in survival. This success results from the exquisite response of the HER2 receptor to HER2-targeted therapy, which is maintained even after multiple lines of treatment. The huge interest in further development and drug discovery for this focused group of BCs is evidenced by the 1,922 clinical trials for HER2+ BC currently listed in Clinical Trials.gov (see Related links: https://clinicaltrials.gov/ct2/results?cond=HER2-positive+Breast+Cancer&term=&cntry=&state=&city=&dist=). This success and continued enthusiasm truly represent an enormous effort and cooperation between many basic and clinical scientists to improve the lives of patients. The bridging of drug development from academia to industry and vice versa is a crucial element of the process.
Several pathways have been successfully targeted, and there are various mechanisms of resistance that can be explored to potentially cure HER2+ MBC. The recent update of the hallmarks of cancer adding ‘enabling characteristics’ serves to remind us of the constant discoveries in the area of cancer research and the dynamics of thought regarding the carcinogenic process207. There are many fascinating fields of research that are being explored, such as harnessing the microbiome and continued exploration of the role of the gut in immunotherapy as one example208. In addition, many tools have been discovered and flourished to enhance drug development, such as artificial intelligence, CRISPR–Cas9, single-cell sequencing, spatial transcriptomics, spatial proteomics and theranostics; the list keeps growing209,210,211,212,213,214,215. Recent drug constructs such as the ADCs and upcoming bispecifics will continue to improve our ability to safely target HER2+ cells, while limiting AEs for our patients. Building on the past accomplishments in treatment with HER2-targeted therapy, the investigation of these newer concepts and use of these methods will ultimately lead to continued advances.
References
Carpenter, G., King, L. Jr & Cohen, S. Epidermal growth factor stimulates phosphorylation in membrane preparations in vitro. Nature 276, 409–410 (1978).
Schechter, A. L. et al. The neu oncogene: an erb-B-related gene encoding a 185,000-Mr tumour antigen. Nature 312, 513–516 (1984).
Slamon, D. J. et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182 (1987). This work demonstrated that HER2 amplification is a prognostic factor and predictive of outcomes in breast cancer.
Giordano, S. H. et al. Systemic therapy for advanced human epidermal growth factor receptor 2-positive breast cancer: ASCO guideline update. J. Clin. Oncol. 40, 2612–2635 (2022).
von Minckwitz, G. et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N. Engl. J. Med. 377, 122–131 (2017).
Tripathy, D. et al. De novo versus recurrent HER2-positive metastatic breast cancer: patient characteristics, treatment, and survival from the SystHERs registry. Oncologist 25, e214–e222 (2020).
Kostova, V., Désos, P., Starck, J.-B. & Kotschy, A. The chemistry behind ADCs. Pharmaceuticals 14, 442 (2021).
Fu, Z., Li, S., Han, S., Shi, C. & Zhang, Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal. Transduct. Target. Ther. 71, 93 (2022).
Roussos Torres, E. T. & Emens, L. A. Emerging combination immunotherapy strategies for breast cancer: dual immune checkpoint modulation, antibody-drug conjugates and bispecific antibodies. Breast Cancer Res. Treat. 191, 291–302 (2022).
Klichinsky, M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 38, 947–953 (2020).
Savas, P. et al. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat. Rev. Clin. Oncol. 13, 228–241 (2016).
Disis, M. L. & Cecil, D. L. Breast cancer vaccines for treatment and prevention. Breast Cancer Res. Treat. 191, 481–489 (2022).
Tebbutt, N., Pedersen, M. W. & Johns, T. G. Targeting the ERBB family in cancer: couples therapy. Nat. Rev. Cancer 13, 663e73 (2013).
Baselga, J. & Swain, S. M. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 9, 463–475 (2009).
Wolff, A. C. et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J. Clin. Oncol. 31, 3997–4013 (2013).
Wolff, A. C. et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline focused update. J. Clin. Oncol. 36, 2105–2122 (2018). Important guidelines for interpretation of HER2 testing results from IHC and FISH.
Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med. 387, 9–20 (2022). A phase III trial that demonstrated the efficacy of the anti-HER2 ADC, trastuzumab deruxtecan, in HER2-low metastatic breast cancer.
Baselga, J. Treatment of HER2-overexpressing breast cancer. Ann. Oncol. 21, vii36–vii40 (2010).
Baselga, J., Norton, L., Albanell, J., Kim, Y. M. & Mendelsohn, J. Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res. 58, 2825–2831 (1998).
Pegram, M. et al. Inhibitory effects of combinations of HER-2/neu antibody and chemotherapeutic agents used for treatment of human breast cancers. Oncogene 18, 2241–2251 (1999).
Pietras, R. J. et al. Antibody to HER-2/neu receptor blocks DNA repair after cisplatin in human breast and ovarian cancer cells. Oncogene 9, 1829–1838 (1994).
Pietras, R. J., Pegram, M. D., Finn, R. S., Maneval, D. A. & Slamon, D. J. Remission of human breast cancer xenografts on therapy with humanized monoclonal antibody to HER-2 receptor and DNA-reactive drugs. Oncogene 17, 2235–2249 (1998).
Piccart-Gebhart, M. J. et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 353, 1659–1672 (2005).
Romond, E. H. et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 353, 1673–1684 (2005).
Slamon, D. et al. Adjuvant trastuzumab in HER2-positive breast cancer. N. Engl. J. Med. 365, 1273–1283 (2011).
Moja, L. et al. Trastuzumab containing regimens for early breast cancer. Cochrane Database Syst. Rev. 2012, CD006243 (2012). A review of ~12,000 patients with breast cancer enrolled in randomized controlled trials evaluating trastuzumab alone or in combination with chemotherapy versus no treatment or standard chemotherapy alone as adjuvant therapy for breast cancer.
Nahta, R. & Esteva, F. J. Herceptin: mechanisms of action and resistance. Cancer Lett. 232, 123–138 (2006).
Drebin, J. A., Link, V. C. & Greene, M. I. Monoclonal antibodies reactive with distinct domains of the neu oncogene-encoded p185 molecule exert synergistic anti-tumor effects in vivo. Oncogene 2, 273–277 (1988).
Ishii, K., Morii, N. & Yamashiro, H. Pertuzumab in the treatment of HER2-positive breast cancer: 671 an evidence-based review of its safety, efficacy, and place in therapy. Core Evid. 14, 51–70 (2019).
Agus, D. B. et al. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2, 127–137 (2002).
Lee-Hoeflich, S. T. et al. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 68, 5878–5887 (2008).
Nahta, R., Hung, M. C. & Esteva, F. J. The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells. Cancer Res. 64, 2343–2346 (2004).
Scheuer, W. et al. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res. 69, 9330–9336 (2009).
Mamidi, S., Cinci, M., Hasmann, M., Fehring, V. & Kirschfink, M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol. Oncol. 7, 580–594 (2013).
Swain, S. M. et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 372, 724–734 (2015). The first randomized phase III trial to demonstrate superiority of dual HER2 therapy of pertuzumab and trastuzumab in HER2+ metastatic breast cancer.
Gianni, L. et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 13, 25–32 (2012).
Schneeweiss, A. et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Ann. Oncol. 24, 2278–2284 (2013).
Musolino, A. et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu positive metastatic breast cancer. J. Clin. Oncol. 26, 1789–1796 (2008).
Nordstrom, J. L. et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcγ receptor binding properties. Breast Cancer Res. 13, R123 (2011).
Liu, L., Yang, Y. & Burns, R. Margetuximab mediates greater Fc-dependent anti-tumor activities than trastuzumab or pertuzumab in vitro. Cancer Res. 79 (Suppl. 13), Abstr. 1538 (2019).
Rugo, H. S. et al. Efficacy of margetuximab vs trastuzumab in patients with pretreated ERBB2-positive advanced breast cancer: a phase 3 randomized clinical trial. JAMA Oncol. 7, 573–584 (2021).
Jackisch, C. et al. Subcutaneous vs intravenous trastuzumab for patients with ERBB2-positive early breast cancer: final analysis of the HannaH phase 3 randomized clinical trial. JAMA Oncol. 5, e190339 (2019).
Tan, A. R. et al. Fixed-dose combination of pertuzumab and trastuzumab for subcutaneous injection plus chemotherapy in HER2-positive early breast cancer (FeDeriCa): a randomised, open-label, multicentre, non-inferiority, phase 3 study. Lancet Oncol. 22, 85–97 (2021).
Konecny, G. E. et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2- overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 66, 1630–1639 (2006).
Nahta, R., Yuan, L. X., Du, Y. & Esteva, F. J. Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling. Mol. Cancer Ther. 6, 667–674 (2007).
Scaltriti, M. et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl Cancer Inst. 99, 628–638 (2007).
Xia, W. et al. Lapatinib antitumor activity is not dependent upon phosphatase and tensin homologue deleted on chromosome 10 in ErbB2-overexpressing breast cancers. Cancer Res. 67, 1170–1175 (2007).
O’Brien, N. A. et al. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol. Cancer Ther. 9, 1489–1502 (2010).
Geyer, C. E. et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 355, 2733–2743 (2006). The first trial to demonstrate efficacy of a HER2 TKI + chemotherapy in trastuzumab-resistant HER2+ MBC.
Cameron, D. et al. A phase III randomized comparison of lapatinib plus capecitabine versus capecitabine alone in women with advanced breast cancer that has progressed on trastuzumab: updated efficacy and biomarker analyses. Breast Cancer Res. Treat. 112, 533–543 (2008).
Johnston, S. et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J. Clin. Oncol. 27, 5538–5546 (2009).
Schlam, I. & Swain, S. M. HER2-positive breast cancer and tyrosine kinase inhibitors: the time is now. NPJ Breast Cancer 7, 56 (2021).
Lin, N. U. & Winer, E. P. Brain metastases: the HER2 paradigm. Clin. Cancer Res. 13, 1648–1655 (2007). A review that highlights the increase in brain metastases in patients with HER2+ MBC, the probable causes of this increase, and the development of novel treatments.
Pestalozzi, B. C. et al. Identifying breast cancer patients at risk for central nervous system (CNS) metastases in trials of the International Breast Cancer Study Group (IBCSG). Ann. Oncol. 17, 935–944 (2006).
Clayton, A. J. et al. Incidence of cerebral metastases in patients treated with trastuzumab for metastatic breast cancer. Br. J. Cancer 91, 639–643 (2004).
Steeg, P. S. The blood–tumour barrier in cancer biology and therapy. Nat. Rev. Clin. Oncol. 18, 696–714 (2022).
Lin, N. U. et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin. Cancer Res. 15, 1452–1459 (2009). The first prospective multicentre study for patients with HER2+ MBC and brain metastases that demonstrated the modest activity of lapatinib in this group of patients.
Bachelot, T. et al. Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): a single-group phase 2 study. Lancet Oncol. 14, 64–71 (2013).
Collins, D. M. et al. Preclinical characteristics of the irreversible pan-HER kinase inhibitor neratinib compared with lapatinib: implications for the treatment of HER2-positive and HER2-mutated breast cancer. Cancers 11, 737 (2019).
Rabindran, S. K. et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 64, 3958–3965 (2004).
Canonici, A. et al. Neratinib overcomes trastuzumab resistance in HER2 amplified breast cancer. Oncotarget 4, 1592–1605 (2013).
Cocco, E. et al. Neratinib is effective in breast tumors bearing both amplification and mutation of ERBB2 (HER2). Sci. Signal. 11, eaat9773 (2018).
Chan, A. et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 17, 367–377 (2016).
Chan, A. et al. Final efficacy results of neratinib in HER2-positive hormone receptor-positive early-stage breast cancer from the phase III ExteNET trial. Clin. Breast Cancer 21, 80–91.e7 (2021).
Saura, C. et al. Neratinib plus capecitabine versus lapatinib plus capecitabine in HER2-positive metastatic breast cancer previously treated with ≥2 HER2-directed regimens: phase III NALA Trial. J. Clin. Oncol. 38, 3138–3149 (2020).
Xu, B. et al. Pyrotinib plus capecitabine versus lapatinib plus capecitabine for the treatment of HER2-positive metastatic breast cancer (PHOEBE): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 22, 351–360 (2021).
Kulukian, A. et al. Preclinical activity of HER2-selective tyrosine kinase inhibitor tucatinib as a single agent or in combination with trastuzumab or docetaxel in solid tumor models. Mol. Cancer Ther. 19, 976–987 (2020).
Dinkel, V. et al. ARRY-380, a potent, small molecule inhibitor of ErbB2, increases survival in intracranial ErbB2+ xenograft models in mice. Cancer Res. 72, 852 (2012).
Olson, D. J. et al. Preclinical characterization of tucatinib in HER2-amplified xenograft and CNS implanted tumors. Cancer Res. 80 (Suppl. 16), Abstr. 1962 (2020).
Stringer-Reasor, E. M. et al. Pharmacokinetic (PK) analyses in CSF and plasma from TBCRC049, an ongoing trial to assess the safety and efficacy of the combination of tucatinib, trastuzumab and capecitabine for the treatment of leptomeningeal metastasis (LM) in HER2 positive breast cancer. J. Clin. Oncol. 39, 1044 (2021).
O’Brien, N. A. et al. The small molecule inhibitor of HER2, tucatinib, has potent and highly selective activity in preclinical modes of HER2-driven cancer. Cancer Res. 79 (Suppl. 4), Abstr. P6-17-11 (2019).
Murthy, R. K. et al. Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. N. Engl. J. Med. 382, 597–609 (2020). The first randomized trial in HER2+ MBC that enrolled patients with active brain metastases and demonstrated improved outcomes with tucatinib + trastuzumab + capecitabine in this patient population.
Hamilton, E. et al. Tucatinib vs placebo in combination with trastuzumab and capecitabine for patients with locally advanced unresectable or HER2-positive metastatic breast cancer (HER2CLIMB): Outcomes by hormone receptor status. Cancer Res. 81 (Suppl. 4), Abstr. PD3-08 (2021).
Beck, A., Goetsch, L., Dumontet, C. & Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug. Discov. 16, 315–337 (2017).
Staudacher, A. H. & Brown, M. P. Antibody drug conjugates and bystander killing: Is antigen-dependent internalisation required? Br. J. Cancer 117, 1736–1742 (2017).
Lewis Phillips, G. D. et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 68, 9280–9290 (2008).
Junttila, T. T., Li, G., Parsons, K., Phillips, G. L. & Sliwkowski, M. X. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res. Treat. 128, 347–356 (2011).
von Minckwitz, G. et al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N. Engl. J. Med. 380, 617–628 (2019).
Hurvitz, S. A. et al. Neoadjuvant trastuzumab, pertuzumab, and chemotherapy versus trastuzumab emtansine plus pertuzumab in patients with HER2-positive breast cancer (KRISTINE): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 19, 115–126 (2018).
Krop, I. E. et al. Trastuzumab emtansine plus pertuzumab versus taxane plus trastuzumab plus pertuzumab after anthracycline for high-risk human epidermal growth factor receptor 2-positive early breast cancer: the phase III KAITLIN study. J. Clin. Oncol. 40, 438–448 (2022).
Montemurro, F. et al. Trastuzumab emtansine (T-DM1) in patients with HER2-positive metastatic breast cancer and brain metastases: exploratory final analysis of cohort 1 from KAMILLA, a single-arm phase IIIb clinical trial. Ann. Oncol. 31, 1350–1358 (2020).
Yver, A., Agatsuma, T. & Soria, J. C. The art of innovation: clinical development of trastuzumab deruxtecan and redefining how antibody-drug conjugates target HER2-positive cancers. Ann. Oncol. 31, 430–434 (2020).
Tamura, K. et al. Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive breast cancer previously treated with trastuzumab emtansine: a dose-expansion, phase 1 study. Lancet Oncol. 20, 816–826 (2019).
Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N. Engl. J. Med. 382, 610–621 (2020).
Cortés, J. et al. Trastuzumab deruxtecan versus trastuzumab emtansine for breast cancer. N. Engl. J. Med. 386, 1143–1154 (2022).
Swain, S. M. et al. Multidisciplinary clinical guidance on trastuzumab deruxtecan (T-DXd)–related interstitial lung disease/pneumonitis — focus on proactive monitoring, diagnosis, and management. Cancer Treat. Rev. 106, 102378 (2022).
Jerusalem, G. H. M. et al. Trastuzumab deruxtecan (T-DXd) in patients with HER2+ metastatic breast cancer with brain metastases: a subgroup analysis of the DESTINY-Breast01 trial. J. Clin. Oncol. 39, 526 (2021).
Pérez-García, J. M. et al. Trastuzumab deruxtecan in patients with central nervous system involvement from HER2-positive breast cancer: the DEBBRAH trial. Neuro-oncol. https://doi.org/10.1093/neuonc/noac144 (2022).
Mosele, M. F. et al. Unraveling the mechanism of action and resistance to trastuzumab deruxtecan (T-DXd): biomarker analyses from patients from DAISY trial. Ann. Oncol. 33, S123 (2022).
Kancha, R. K. et al. Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS ONE 6, e26760 (2011).
Wagle, N. et al. Whole exome sequencing (WES) of HER2+ metastatic breast cancer (MBC) from patients with or without prior trastuzumab (T): a correlative analysis of TBCRC003. Cancer Res. 75 (Suppl. 9), Abstr. PD3-5 (2015).
Xu, X. et al. HER2 reactivation through acquisition of the HER2 L755S mutation as a mechanism of acquired resistance to HER2-targeted therapy in HER2(+) breast cancer. Clin. Cancer Res. 23, 5123–5134 (2017).
Hyman, D. M. et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 554, 189–194 (2018).Results from the SUMMIT basket trial which demonstrated the clinical actionability of HER2 and HER3 mutations.
Hanker, A. B. et al. Co-occurring gain-of-function mutations in HER2 and HER3 modulate HER2/HER3 activation, oncogenesis, and HER2 inhibitor sensitivity. Cancer Cell 39, 1099–1114.e8 (2021).
Veeraraghavan J. et al. Acquired resistance to tucatinib is associated with EGFR amplification in HER2+ breast cancer (BC) models and can be overcome by a more complete blockade of HER receptor layer. Cancer Res. 82 (Suppl. 4), Abstr. PD8-06 (2022).
Bose, S. et al. Resistance to next generation tyrosine kinase inhibitors (TKIs) in HER2-positive breast cancer (BC): role of HER and PIK3CA mutations and development of new treatment strategies and study models. Cancer Res. 82 (Suppl. 4), Abstr. P4-01-01 (2022).
Derakhshani, A. et al. Overcoming trastuzumab resistance in HER2-positive breast cancer using combination therapy. J. Cell Physiol. 235, 3142–3156 (2020).
de Melo Gagliato, D., Jardim, D. L. F., Marchesi, M. S. P. & Hortobagyi, G. N. Mechanisms of resistance and sensitivity to anti-HER2 therapies in HER2+ breast cancer. Oncotarget 7, 64431–64446 (2016).
Scaltriti, M. et al. Clinical benefit of lapatinib-based therapy in patients with human epidermal growth factor receptor 2–positive breast tumors coexpressing the truncated p95HER2 receptor. Clin. Cancer Res. 16, 2688–2695 (2010).
Guarneri, V. et al. Prospective biomarker analysis of the randomized CHER-LOB study evaluating the dual anti-HER2 treatment with trastuzumab and lapatinib plus chemotherapy as neoadjuvant therapy for HER2-positive breast cancer. Oncologist 20, 1001–1010 (2015).
Miranda, F., Prazeres, H., Mendes, F., Martins, D. & Schmitt, F. Resistance to endocrine therapy in HR+ and/or HER2+ breast cancer: the most promising predictive biomarkers. Mol. Biol. Rep. 49, 717–733 (2022).
Schillaci, R. et al. Neutralizing soluble tumor necrosis factor alpha overcomes trastuzumab-resistant breast cancer immune evasion by downregulating mucin 4, improving NK cell function and decreasing myeloid-derived suppressor cells in tumor microenvironment. Cancer Res. 79 (Suppl. 4), Abstr. P6-20-14 (2019).
Schillaci, R. et al. Mucin 4 expression in high risk breast cancer: predicting and overcoming resistance to immunotherapy. Cancer Res. 82 (Suppl. 4), Abstr. P5-13-32 (2022).
Loibl, S. et al. PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab. Ann. Oncol. 27, 1519–1525 (2016).
Baselga, J. et al. Biomarker analyses in CLEOPATRA: a phase III, placebo-controlled study of pertuzumab in human epidermal growth factor receptor 2-positive, first-line metastatic breast cancer. J. Clin. Oncol. 32, 3753–3761 (2014).
Chandarlapaty, S. et al. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin. Cancer Res 18, 6784–6791 (2012).
Rimawi, M. F., De Angelis, C. & Schiff, R. Resistance to anti-HER2 therapies in breast cancer. Am. Soc. Clin. Oncol. Ed. Book 35, e157–e164 (2015).
Loibl, S. et al. Neoadjuvant buparlisib plus trastuzumab and paclitaxel for women with HER2+ primary breast cancer: a randomised, double-blind, placebo-controlled phase II trial (NeoPHOEBE). Eur. J. Cancer 85, 133–145 (2017).
André, F. et al. Molecular alterations and everolimus efficacy in human epidermal growth factor receptor 2-overexpressing metastatic breast cancers: combined exploratory biomarker analysis from BOLERO-1 and BOLERO-3. J. Clin. Oncol. 34, 2115–2124 (2016).
Xia, W. et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc. Natl Acad. Sci. USA 103, 7795–7800 (2006).
Wang, Y. C. et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers-role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 13, R121 (2011).
Vaz-Luis, I., Winer, E. P. & Lin, N. U. Human epidermal growth factor receptor-2-positive breast cancer: does estrogen receptor status define two distinct subtypes? Ann. Oncol. 24, 283–291 (2013).
Kaufman, B. et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: results from the randomized phase III TAnDEM study. J. Clin. Oncol. 27, 5529–5537 (2009).
Arpino, G. et al. Final analysis of PERTAIN: a randomized, two-arm, open-label, multicenter phase II trial assessing the efficacy and safety of first-line pertuzumab given in combination with trastuzumab plus an aromatase inhibitor in patients with HER2-positive and hormone receptor-positive metastatic or locally advanced breast cancer. Cancer Res. 81 (Suppl. 4), Abstr. PD3-02 (2021).
Vernieri, C. et al. Resistance mechanisms to anti-HER2 therapies in HER2-positive breast cancer: current knowledge, new research directions and therapeutic perspectives. Crit. Rev. Oncol. Hematol. 139, 53–66 (2019).
Finn, R. S. et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 11, R77 (2009).
Goel, S. et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell 29, 255–269 (2016).
Tolaney, S. M. et al. Abemaciclib plus trastuzumab with or without fulvestrant versus trastuzumab plus standard-of-care chemotherapy in women with hormone receptor-positive, HER2-positive advanced breast cancer (monarcHER): a randomised, open-label, phase 2 trial. Lancet Oncol. 21, 763–775 (2020).
Ciruelos, E. et al. Palbociclib and trastuzumab in HER2-positive advanced breast cancer: results from the phase II SOLTI-1303 PATRICIA trial. Clin. Cancer Res. 26, 5820–5829 (2020).
Gianni, L. et al. Neoadjuvant treatment with trastuzumab and pertuzumab plus palbociclib and fulvestrant in HER2-positive, ER-positive breast cancer (NA-PHER2): an exploratory, open-label, phase 2 study. Lancet Oncol. 19, 249–256 (2018).
Smith, A. E. et al. HER2+ breast cancers evade anti-HER2 therapy via a switch in driver pathway. Nat. Commun. 12, 6667 (2021).
Metzger Filho, O. et al. Impact of HER2 heterogeneity on treatment response of early-stage HER2-positive breast cancer: phase II neoadjuvant clinical trial of T-DM1 combined with pertuzumab. Cancer Discov. 11, 2474–2487 (2021). A prospective trial evaluating heterogenous HER2 expression in tumours, and its impact on response to anti-HER2 therapy.
Zheng, G. et al. Interaction between HLA-G and NK cell receptor KIR2DL4 orchestrates HER2-positive breast cancer resistance to trastuzumab. Signal. Transduct. Target. Ther. 6, 236 (2021).
Upton, R. et al. Combining CD47 blockade with trastuzumab eliminates HER2-positive breast cancer cells and overcomes trastuzumab tolerance. Proc. Natl Acad. Sci. USA 118, e2026849118 (2021).
Betancur, P. A. et al. A CD47-associated super-enhancer links pro-inflammatory signalling to CD47 upregulation in breast cancer. Nat. Commun. 8, 14802 (2017).
Janiszewska, M. et al. The impact of tumor epithelial and microenvironmental heterogeneity on treatment responses in HER2+ breast cancer. JCI Insight 6, e147617 (2021).
Hunter, F. W. et al. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 122, 603–612 (2020).
Conlon, N. T. et al. Comparative analysis of drug response and gene profiling of HER2-targeted tyrosine kinase inhibitors. Br. J. Cancer 124, 1249–1259 (2021).
Kim, J. Y. et al. Immune signature of metastatic breast cancer: Identifying predictive markers of immunotherapy response. Oncotarget 8, 47400–47411 (2017).
Choi, H. et al. CDK 12 drives breast tumor initiation and trastuzumab resistance via WNT and IRS 1‐ErbB‐PI 3K signaling. EMBO Rep. 20, e48058 (2019).
Zhao, Y., Wang, Z., Jiang, Y. & Yang, C. Inactivation of Rac1 reduces trastuzumab resistance in PTEN deficient and insulin-like growth factor I receptor overexpressing human breast cancer SKBR3 cells. Cancer Lett. 313, 54–63 (2011).
Holbro, T. et al. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl Acad. Sci. USA 100, 8933–8938 (2003).
Reynolds, K. L. A phase I open-label dose-escalation study of the anti-HER3 monoclonal antibody LJM716 in patients with advanced squamous cell carcinoma of the esophagus or head and neck and HER2-overexpressing breast or gastric cancer. BMC Cancer 17, 646 (2017).
Saeki, T. & Mukai, H. Phase I study of HER3 targeted antibody patritumab in combination with trastuzumab and paclitaxel in patients with HER2-overexpressing metastatic breast cancer (MBC). J. Clin. Oncol. 33, 584 (2015).
Helsten, T. L. et al. Evaluation of patritumab/paclitaxel/trastuzumab over standard paclitaxel/trastuzumab in early stage, high-risk HER2 positive breast cancer: results from the neoadjuvant I-SPY 2 trial. Cancer Res. 80 (Suppl. 4) Abstr. P3-11-02 (2022).
Meulendijks, D. et al. First-in-human phase I study of lumretuzumab, a glycoengineered humanized anti-HER3 monoclonal antibody, in patients with metastatic or advanced HER3-positive solid tumors. Clin. Cancer Res. 22, 877–885 (2016).
Schneeweiss, A. et al. Phase Ib study evaluating safety and clinical activity of the anti-HER3 antibody lumretuzumab combined with the anti-HER2 antibody pertuzumab and paclitaxel in HER3-positive, HER2-low metastatic breast cancer. Invest. New Drugs 36, 848–859 (2018).
Im, S.-A. et al. A phase 1 dose-escalation study of anti-HER3 monoclonal antibody LJM716 in combination with trastuzumab in patients with HER2-overexpressing metastatic breast or gastric cancer. J. Clin. Oncol. 32, 2519 (2014).
Fielder, W. et al. Phase I study of TrasGEX, a glycooptimised anti-HER2 monoclonal antibody, in patients with HER2positive solid tumours. ESMO Open 3, e000381 (2018).
Bartsch, R. et al. Trastuzumab-deruxtecan (T-DXd) in HER2-positive breast cancer patients (pts) with active brain metastases: primary outcome analysis from the TUXEDO-1 trial. Ann. Oncol. 33, S198 (2022).
Su, Z. et al. Antibody-drug conjugates: recent advances in linker chemistry. Acta Pharmaceutica Sin. B 11, 3889–3907 (2021).
van der Lee, M. M. et al. The preclinical profile of the duocarmycin-based HER2-targeting ADC SYD985 predicts for clinical benefit in low HER2-expressing breast cancers. Mol. Cancer Ther. 14, 692–703 (2015).
Banerji, U. et al. Trastuzumab duocarmazine in locally advanced and metastatic solid tumours and HER2-expressing breast cancer: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 20, 1124–1135 (2019).
Saura Manich, C. et al. Primary outcome of the phase III SYD985.002/TULIP trial comparing [vic-]trastuzumab duocarmazine to physician’s choice treatment in patients with pre-treated HER2-positive locally advanced or metastatic breast cancer. Ann. Oncol. 32, S1283–S1346 (2021).
Skidmore, L. et al. ARX788, a site-specific anti-HER2 antibody-drug conjugate, demonstrates potent and selective activity in HER2-low and T-DM1-resistant breast and gastric cancers. Mol. Cancer Ther. 19, 1833–1843 (2020).
Zhang, J. et al. Phase I trial of a novel anti-HER2 antibody-drug conjugate, ARX788, for the treatment of HER2-positive metastatic breast cancer. Clin. Cancer Res. 28, 4212–4221 (2022).
Shi, F. et al. Disitamab vedotin: a novel antibody-drug conjugates for cancer therapy. Drug. Deliv. 29, 1335–1344 (2022).
Deeks, E. D. Disitamab vedotin: first approval. Drugs 81, 1929–1935 (2021).
Hamblett, K. J. et al. ZW49, a HER2-targeted biparatopic antibody drug conjugate for the treatment of HER2-expressing cancers. Cancer Res. 79 (Suppl. 4), Abstr. P6-17-13 (2019).
Kang, J. C. et al. Engineering a HER2-specific antibody–drug conjugate to increase lysosomal delivery and therapeutic efficacy. Nat. Biotechnol. 37, 523–526 (2019).
Hagemann, U. B. et al. Advances in precision oncology: targeted thorium-227 conjugates as a new modality in targeted alpha therapy. Cancer Biother. Radiopharm. 35, 497–510 (2020).
Hagemann, U. B. et al. Mesothelin-targeted thorium-227 conjugate (MSLN-TTC): preclinical evaluation of a new targeted alpha therapy for mesothelin-positive cancers. Clin. Cancer Res. 25, 4723–4734 (2019).
Wickstroem, K. et al. Preclinical combination studies of an FGFR2 targeted thorium-227 conjugate and the ATR inhibitor BAY 1895344. Int. J. Radiat. Oncol. Biol. Phys. 105, 410–422 (2019).
Zhang, J. et al. Preclinical and early clinical safety and pharmacokinetics data of DZD1516, an BBB-penetrant selective HER2 inhibitor for the treatment of HER2 positive metastatic breast cancer. Cancer Res. 82 (Suppl. 4), Abstr. P2-13-43 (2022).
Schram, A. M. et al. Safety and preliminary efficacy from the phase 1 portion of MasterKey-01: a first-in-human dose-escalation study to determine the recommended phase 2 dose (RP2D), pharmacokinetics (PK) and preliminary antitumor activity of BDTX-189, an inhibitor of allosteric ErbB mutations, in patients (pts) with advanced solid malignancies. J. Clin. Oncol. 39, 3086 (2021).
Connell, C. M. & Doherty, G. J. Activating HER2 mutations as emerging targets in multiple solid cancers. ESMO Open 2, e000279 (2017).
Elamin, Y. Y. et al. Poziotinib for patients with HER2 exon 20 mutant non–small-cell lung cancer: results from a phase II trial. J. Clin. Oncol. 40, 702–709 (2022).
Weisser, N. E. et al. The bispecific antibody zanidatamab’s (ZW25’s) unique mechanisms of action and durable anti-tumor activity in HER2-expressing cancers. Cancer Res. 81, 1005 (2021).
Meric-Bernstam, F. et al. Safety, anti-tumor activity, and biomarker results of the HER2-targeted bispecific antibody ZW25 in HER2-expressing solid tumors. Ann. Oncol. 30, V159–v193 (2019).
Bedard, P. L. et al. Zanidatamab (ZW25), a HER2-targeted bispecific antibody, in combination with chemotherapy (chemo) for HER2-positive breast cancer (BC): results from a phase 1 study. Cancer Res. 82 (Suppl. 4), Abstr. P2-13-07 (2022).
Geuijen, C. A. et al. Unbiased combinatorial screening identifies a bispecific IgG1 that potently inhibits HER3 signaling via HER2-guided ligand blockade. Cancer Cell 33, 922–936 (2018).
Hamilton, E. P. et al. Clinical activity of MCLA-128 (zenocutuzumab), trastuzumab, and vinorelbine in HER2 amplified metastatic breast cancer (MBC) patients (pts) who had progressed on anti-HER2 ADCs. J. Clin. Oncol. 38, 3093 (2020).
Zhang, J. et al. First-in-human HER2-targeted bispecific antibody KN026 for the treatment of patients with HER2-positive metastatic breast cancer: results from a phase I study. Clin. Cancer Res. 28, 618–628 (2022).
Békés, M., Langley, D. R. & Crews, C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug. Discov. 21, 181–200 (2022).
Maneiro, M. et al. Antibody–PROTAC conjugates enable HER2-dependent targeted protein degradation of BRD4. ACS Chem. Biol. 15, 1306–1312 (2020).
Palacino, J. et al. ORM-5029: A first-in-class targeted protein degradation therapy using antibody neodegrader conjugate (AnDC) for HER2-expressing breast cancer. Cancer Res. 82 (Suppl. 4), Abstr. 3933 (2022).
Stagg, J. et al. Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc. Natl Acad. Sci. USA 108, 7142–7247 (2011).
Varchetta, S. et al. Elements related to heterogeneity of antibody-dependent cell cytotoxicity in patients under trastuzumab therapy for primary operable breast cancer. Cancer Res. 67, 11991–11999 (2007).
Loi, S. et al. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): a single-arm, multicentre, phase 1b-2 trial. Lancet Oncol. 20, 371–382 (2019).
Hamilton, E. et al. Trastuzumab deruxtecan (T-DXd; DS-8201) with nivolumab in patients with HER2-expressing, advanced breast cancer: a 2-part, phase 1b, multicenter, open-label study. Cancer Res. 81 (Suppl. 4), PD3-07 (2021).
Emens, L. A. et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): a phase 2, multicentre, randomized, double-blind trial. Lancet Oncol. 21, 1283–1295 (2020).
Nguyen, A. T., Shiao, S. L. & McArthur, H. L. Advances in combining radiation and immunotherapy in breast cancer. Clin. Breast Cancer 21, 143–152 (2021).
Page, D. B. et al. Brain radiotherapy, tremelimumab-mediated CTLA-4-directed blockade +/− trastuzumab in patients with breast cancer brain metastases. NPJ Breast Cancer 8, 50 (2022).
Messaoudene, M. et al. T-cell bispecific antibodies in node-positive breast cancer: novel therapeutic avenue for MHC class I loss variants. Ann. Oncol. 30, 934–944 (2019).
Borghaei, H. et al. Induction of adaptive anti-HER2/neu immune responses in a phase 1B/2 trial of 2B1 bispecific murine monoclonal antibody in metastatic breast cancer (E3194): a trial coordinated by the eastern cooperative oncology group. J. Immunother. 30, 455–467 (2007).
Yu, S. et al. A novel asymmetrical anti-HER2/CD3 bispecific antibody exhibits potent cytotoxicity for HER2-positive tumor cells. J. Exp. Clin. Cancer Res. 38, 355 (2019).
Mandó, P., Rivero, S. G., Rizzo, M. M., Pinkasz, M. & Levy, E. M. Targeting ADCC: a different approach to HER2 breast cancer in the immunotherapy era. Breast 60, 15–25 (2021). A review summarizing the immunological mechanisms of antibody-mediated cellular cytotoxicity induced by anti-HER2 therapies, and novel strategies in drug development in this area.
Melero, I. et al. Amplification of tumor immunity by gene transfer of the co-stimulatory 4-1BB ligand: synergy with the CD28 co-stimulatory pathway. Eur. J. Immunol. 28, 1116–1121 (1998).
Melero, I., Murillo, O., Dubrot, J., Hervas-Stubbs, S. & Perez-Gracia, J. L. Multi-layered action mechanisms of CD137 (4-1BB)-targeted immunotherapies. Trends Pharmacol. Sci. 29, 383–390 (2008).
Ku, G. et al. A phase I dose escalation study of PRS-343, a HER2/4-1BB bispecific molecule, in patients with HER2-positive malignancies. Ann. Oncol. 31, S462–S463 (2020).
Lum, L. G. et al. Targeted T-cell therapy in stage IV breast cancer: a phase I clinical trial. Clin. Cancer Res. 21, 2305–2314 (2015).
Le Blanc, H. et al. Systemically administered HER2-targeted ISACs provoke a rapid, local response that engages the innate and adaptive arms of the immune system to eradicate tumors in preclinical models. J. Immunother. Cancer 8 (Suppl. 3), Abstr. 605 (2020).
Fitzgerald, K. A. & Kagan, J. C. Toll-like receptors and the control of immunity. Cell 180, 1044–1066 (2020).
Wainberg, Z. A. et al. Phase 1 study of the novel immunotoxin MT-5111 in patients with HER-2+ tumors. Cancer Res. 81, CT130 (2021).
Van Tine, B. et al. Interim results of a phase 1 study of the novel immunotoxin MT-5111 in patients with HER2+ tumors. Cancer Res. 8 (Suppl. 4), Abstr. P2-13-45 (2022).
Sloas, C., Gill, S. & Klichinsky, M. Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Front. Immunol. 12, 783305 (2021).
Reiss, K. et al. A phase 1 first in human study of adenovirally transduced anti-HER2 CAR macrophages in subjects with HER2 overexpressing solid tumors: preliminary safety, pharmacokinetics, and TME reprogramming data. J. Immunother. Cancer 9 (Suppl. 2), Abstr. 951 (2021).
Marofi, F. et al. CAR-NK cell: a new paradigm in tumor immunotherapy. Front. Oncol. 11, 673276 (2021).
Solinas, C., Aiello, M., Migliori, E., Willard-Gallo, K. & Emens, L. A. Breast cancer vaccines: heeding the lessons of the past to guide a path forward. Cancer Treat. Rev. 84, 101947 (2020). A comprehensive review of breast cancer vaccines.
Sharma, A. et al. HER-2 pulsed dendritic cell vaccine can eliminate HER-2 expression and impact ductal carcinoma in situ. Cancer 118, 4354–4362 (2012).
Antonarelli, G. et al. Therapeutic cancer vaccines revamping: technology advancements and pitfalls. Ann. Oncol. 32, 1537–1551 (2021).
Clifton, G. T., Peoples, G. E. & Mittendorf, E. A. The development and use of the E75 (HER2 369–377) peptide vaccine. Future Oncol. 12, 1321–1329 (2016).
Disis, M. L. et al. Effect of dose on immune response in patients vaccinated with an her-2/neu intracellular domain protein–based vaccine. J. Clin. Oncol. 22, 1916–1925 (2004).
Hamilton, E. et al. Phase 1 clinical trial of HER2-specific immunotherapy with concomitant HER2 kinase inhibition [corrected]. J. Transl. Med. 10, 28 (2012).
Chen, G. et al. A feasibility study of cyclophosphamide, trastuzumab, and an allogeneic GM-CSF-secreting breast tumor vaccine for HER2+ metastatic breast cancer. Cancer Immunol. Res. 2, 949–961 (2014).
Morse, M. A. et al. Long term disease-free survival and T cell and antibody responses in women with high-risk Her2+ breast cancer following vaccination against Her2. J. Transl. Med. 5, 42 (2007).
Maeng, H. M. et al. Phase I clinical trial of an autologous dendritic cell vaccine against HER2 shows safety and preliminary clinical efficacy. Front. Oncol. 11, 789078 (2021).
Lowenfeld, L. et al. Dendritic cell vaccination enhances immune responses and induces regression of HER2 pos DCIS independent of route: results of randomized selection design trial. Clin. Cancer Res. 23, 2961–2971 (2017).
Burke, E. E., Kodumudi, K., Ramamoorthi, G. & Czerniecki, B. J. Vaccine therapies for breast cancer. Surg. Oncol. Clin. N. Am. 28, 353–367 (2019).
Shanmugaraj, B. et al. Bacterial and viral vectors as vaccine delivery vehicles for breast cancer therapy. Life Sci. 250, 117550 (2020).
Crosby, E. J. et al. Vaccine-induced memory CD8+ T cells provide clinical benefit in HER2 expressing breast cancer: a mouse to human translational study. Clin. Cancer Res. 25, 2725–2736 (2019).
Mittendorf, E. A. et al. Primary analysis of a prospective, randomized, single-blinded phase II trial evaluating the HER2 peptide AE37 vaccine in breast cancer patients to prevent recurrence. Ann. Oncol. 27, 1241–1248 (2016).
Mittendorf, E. A. et al. Efficacy and safety analysis of nelipepimut-S vaccine to prevent breast cancer recurrence: a randomized, multicenter, phase III clinical trial. Clin. Cancer Res. 25, 4248–4254 (2019).
Caldeira, J. C. et al. Virus-like particles as an immunogenic platform for cancer vaccines. Viruses 12, 488 (2020).
Dafni, U. et al. Efficacy of cancer vaccines in selected gynaecological breast and ovarian cancers: a 20-year systematic review and meta-analysis. Eur. J. Cancer 142, 63–82 (2021).
Blass, E. & Ott, P. A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 18, 215–229 (2021).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022). An updated review discussing novel facets of cancer and proposing emerging hallmarks of cancer and enabling characteristics.
Park, E. M. et al. Targeting the gut and tumor microbiota in cancer. Nat. Med. 28, 690–703 (2022).
Liang, G. et al. The emerging roles of artificial intelligence in cancer drug development and precision therapy. Biomed. Pharmacother. 128, 110255 (2020).
Jinek, M. et al. Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Ran, F. A. et al. Genome engineering using the CRISPR–Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Tang, X. et al. The single-cell sequencing: new developments and medical applications. Cell Biosci. 9, 53 (2019).
Andersson, A. et al. Spatial deconvolution of HER2-positive breast cancer delineates tumor-associated cell type interactions. Nat. Commun. 12, 6012 (2021).
McNamara, K. L. et al. Spatial proteomic characterization of HER2-positive breast tumors through neoadjuvant therapy predicts response. Nat. Cancer 2, 400–413 (2021).
Vahidfar, N. et al. Theranostic advances in breast cancer in nuclear medicine. Int. J. Mol. Sci. 22, 4597 (2021).
Xu, Y. et al. Phase I study of the recombinant humanized anti-HER2 monoclonal antibody–MMAE conjugate RC48-ADC in patients with HER2-positive advanced solid tumors. Gastric Cancer 24, 913–925 (2021).
Hu, X. et al. Phase I study of A166 in patients with HER2-expressing locally advanced or metastatic solid tumors. J. Clin. Oncol. 39, 1024 (2021).
Park, Y. H. et al. First-in-human phase I study of ALT-P7, a HER2-targeting antibody-drug conjugate in patients with HER2-positive advanced breast cancer. J. Clin. Oncol. 38, 3551 (2020).
Hurvitz, S. et al. Safety and unique pharmacokinetic profile of ARX788, a site-specific ADC, in heavily pretreated patients with HER2-overexpresing solid tumors: Results from two phase 1 clinical trials. J. Clin. Oncol. 39, 1038 (2021).
Zhang, X. et al. Novel development strategies and challenges for anti-Her2 antibody-drug conjugates. Antib. Ther. 5, 18–29 (2022).
Fasching, P. A. Invited discussant LBA1. ESMO Breast Cancer Congress (4 May 2022); https://oncologypro.esmo.org/meeting-resources/esmo-breast-cancer-congress/invited-discussant-lba1
Jhaveri, K. et al. Preliminary results from a phase 1 study using the bispecific, human epidermal growth factor 2 (HER2)-targeting antibody-drug conjugate (ADC) zanidatamab zovodotin (ZW49) in solid cancers. Ann. Oncol. 33 (Suppl. 7), S197–S224 (2022).
Macpherson, I. R. et al. A phase I/II study of epertinib plus trastuzumab with or without chemotherapy in patients with HER2-positive metastatic breast cancer. Breast Cancer Res. 22, 1 (2019).
Brufsky, A. et al. A phase 2 study of poziotinib in patients with HER2-positive metastatic breast cancer heavily pre-treated with HER2-targeted therapy. Cancer Res. 81 (Suppl. 4), Abstr. PD1-07 (2021).
Lopez-Albaitero, A. et al. Overcoming resistance to HER2-targeted therapy with a novel HER2/CD3 bispecific antibody. Oncoimmunology 6, e1267891 (2017).
Ruiz, I. R. et al. p95HER2-T cell bispecific antibody for breast cancer treatment. Sci. Transl. Med. 10, eaat1445 (2018).
Oberg, H. H. et al. Tribody [(HER2) 2 xCD16] is more effective than trastuzumab in enhancing γδ T cell and natural killer cell cytotoxicity against HER2-expressing cancer cells. Front. Immunol. 9, 814 (2018).
Mittal, D. et al. CD96 is an immune checkpoint that regulates CD8+ T-cell antitumor function. Cancer Immunol. Res. 7, 559–571 (2019).
Sha, W. et al. SAR443216, a novel trispecific T cell engager with potent T cell-dependent cytotoxicity for HER2-low tumors. Cancer Res. 81 (Suppl. 13), Abstr. 1825 (2021).
Sharma, M. R. et al. Preliminary results from a phase I/II study of BDC-1001, a novel HER2 targeting TLR7/8 immune-stimulating antibody conjugate (ISAC), in patients (pts) with advanced HER2-expressing solid tumors. Ann. Oncol. 32, S1453–S1454 (2021).
Janku, F. et al. 378 A first in-human, multicenter, open-label, dose-finding phase 1 study of the immune stimulator antibody conjugate NJH395 in patients with nonbreast HER2+ advanced malignancies. J. Immunother. Cancer 8 (Suppl. 3), A230 (2020).
Klemper, S. J. et al. 209P Interim results of a phase I/Ib study of SBT6050 monotherapy and pembrolizumab combination in patients with advanced HER2-expressing or amplified solid tumors. Ann. Oncol. 32 (Suppl. 5), S450 (2021).
Mittendorf, E. A. et al. Primary analysis of a prospective, randomized, single-blinded phase II trial evaluating the HER2 peptide GP2 vaccine in breast cancer patients to prevent recurrence. Oncotarget 7, 66192–66201 (2016).
Bekaii-Saab, T. et al. Phase I immunotherapy trial with two chimeric HER-2 B-cell peptide vaccines emulsified in montanide ISA 720VG and nor-MDP adjuvant in patients with advanced solid tumors. Clin. Cancer Res. 25, 3495–3507 (2019).
Emens, L. A. et al. Timed sequential treatment with cyclophosphamide, doxorubicin, and an allogeneic granulocyte-macrophage colony-stimulating factor-secreting breast tumor vaccine: a chemotherapy dose-ranging factorial study of safety and immune activation. J. Clin. Oncol. 27, 5911–5918 (2009).
Disis, M. L. et al. A phase I trial of the safety and immunogenicity of a DNA-based vaccine encoding the HER2/neu (HER2) intracellular domain in subjects with HER2+ breast cancer. J. Clin. Oncol. 32, 616 (2014).
Cohen, S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J. Biol. Chem. 237, 1555–1562 (1962).
Cohen, S. The stimulation of epidermal proliferation by a specific protein (EGF). Dev. Biol. 12, 394–407 (1965).
Engelbreth-Holm, J. & Meyer, A. R. Variations in the percentage of takes in 3 strains of chicken leukosis. Acta Pathol. Microbiol. Scand. 12, 366–377 (1935).
Yamamoto, T. et al. The erbB gene of avian erythroblastosis virus is a member of the src gene family. Cell 35, 71–78 (1983).
Downward, J. et al. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature 307, 521–527 (1984).
Shih, C., Shilo, B. Z., Goldfarb, M. P., Dannenberg, A. & Weinberg, R. A. Passage of phenotypes of chemically transformed cells via transfection of DNA and chromatin. Proc. Natl Acad. Sci. USA 76, 5714–5718 (1979).
Shih, C., Padhy, L. C., Murray, M. & Weinberg, R. A. Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature 290, 261–264 (1981).
Padhy, L. C., Shih, C., Dowing, D., Finkelstein, R. & Weinberg, R. A. Identification of a phosphoprotein specifically induced by the transforming DNA of rat neuroblastomas. Cell 28, 865–871 (1982).
King, C. R., Kraus, M. H. & Aaronson, S. A. Amplification of a novel v-erbB-related gene in a human mammary carcinoma. Science 229, 974–976 (1985).
Ullrich, A. et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 309, 418–425 (1984).
Coussens, L. et al. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 230, 1132–1139 (1985).
Kraus, M. H., Issing, W., Miki, T., Popescu, N. C. & Aaronson, S. A. Isolation and characterization of ERBB3, a third member of the ERBB/epidermal growth factor receptor family: evidence for overexpression in a subset of human mammary tumors. Proc. Natl Acad. Sci. USA 86, 9193–9197 (1989).
Plowman, G. D. et al. Ligand-specific activation of HER4/p180erbB4, a fourth member of the epidermal growth factor receptor family. Proc. Natl Acad. Sci. USA 90, 1746–1750 (1993).
Drebin, J. A., Stern, D. F., Link, V. C., Weinberg, R. A. & Greene, M. I. Monoclonal antibodies identify a cell-surface antigen associated with an activated cellular oncogene. Nature 312, 545–548 (1984).
Drebin, J. A., Link, V. C., Stern, D. F., Weinberg, R. A. & Greene, M. I. Down-modulation of an oncogene protein product and reversion of the transformed phenotype by monoclonal antibodies. Cell 41, 697–706 (1985).
Drebin, J. A., Link, V. C., Weinberg, R. A. & Greene, M. I. Inhibition of tumor growth by a monoclonal antibody reactive with an oncogene-encoded tumor antigen. Proc. Natl Acad. Sci. USA 83, 9129–9133 (1986).
Di Fiore, P. P. et al. erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science 237, 178–182 (1987).
Hudziak, R. M., Schlessinger, J. & Ullrich, A. Increased expression of the putative growth factor receptor p185HER2 causes transformation and tumorigenesis of NIH 3T3 cells. Proc. Natl Acad. Sci. USA 84, 7159–7163 (1989).
Slamon, D. J. et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244, 707–712 (1989).
Fendly, B. M. et al. Characterization of murine monoclonal antibodies reactive to either the human epidermal growth factor receptor or HER2/neu gene product. Cancer Res. 50, 1550–1558 (1990).
Carter, P. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl Acad. Sci. USA 89, 4285–4289 (1992).
Slamon, D. J. et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 344, 783–792 (2001). The first publication to demonstrate the efficacy of trastuzumab + chemotherapy in HER2-overexpressing metastatic breast cancer.
Baselga, J. Phase I and II clinical trials of trastuzumab. Ann. Oncol. 12, S49–S55 (2001).
Payne, S. J., Bowen, R. L., Jones, J. L. & Wells, C. A. Predictive markers in breast cancer–the present. Histopathology 52, 82–90 (2008).
Chia, S. et al. Human epidermal growth factor receptor 2 overexpression as a prognostic factor in a large tissue microarray series of node-negative breast cancers. J. Clin. Oncol. 26, 5697–5704 (2008).
Fritzsche, F. R. et al. Tissue pretreatment with formic acid might lower HercepTest scores in breast cancer. Diagn. Mol. Pathol. 15, 237–242 (2006).
Siddiqui, S. & Rimm, D. L. Pre-analytic variables and phospho-specific antibodies: the Achilles heel of immunohistochemistry. Breast Cancer Res. 12, 113 (2010).
Press, M. F. et al. Diagnostic evaluation of HER-2 as a molecular target: an assessment of accuracy and reproducibility of laboratory testing in large, prospective, randomized clinical trials. Clin. Cancer Res. 11, 6598–6607 (2005).
Schnitt, S. J. & Jacobs, T. W. Current status of HER2 testing: caught between a rock and a hard place. Am. J. Clin. Pathol. 116, 806–810 (2001).
Choritz, H., Busche, G. & Kreipe, H. Quality assessment of HER2 testing by monitoring of positivity rates. Virchows Arch. 459, 283–289 (2011).
Yeh, I. T. et al. Clinical validation of an array CGH test for HER2 status in breast cancer reveals that polysomy 17 is a rare event. Mod. Pathol. 22, 1169–1175 (2009).
Tse, C. H. et al. Determining true HER2 gene status in breast cancers with polysomy by using alternative chromosome 17 reference genes: implications for anti-HER2 targeted therapy. J. Clin. Oncol. 29, 4168–4174 (2011).
Ballinger, T. J., Sanders, M. E. & Abramson, V. G. Current HER2 testing recommendations and clinical relevance as a predictor of response to targeted therapy. Clin. Breast Cancer 15, 171–180 (2015).
Onsum, M. D. et al. Single-cell quantitative HER2 measurement identifies heterogeneity and distinct subgroups within traditionally defined HER2-positive patients. Am. J. Pathol. 183, 1446–1460 (2013).
Gu, J., Tang, Z., Chen, H., Sfamenos, S. & Geiersbach, K. B. HER2 FISH for breast cancer: advances in quantitative image analysis and automation. OBM Genetics 4, 109 (2020).
Pizzamiglio, S. et al. What if the future of HER2-positive breast cancer patients was written in miRNAs? An exploratory analysis from NeoALTTO study. Cancer Med. 11, 332–339 (2022).
Moutafi, M. et al. Quantitative measurement of HER2 expression to subclassify ERBB2 unamplified breast cancer. Lab. Invest. 102, 1101–1108 (2022).
Prat, A. et al. Development and validation of the new HER2DX assay for predicting pathological response and survival outcome in early-stage HER2-positive breast cancer. EBioMedicine 75, 103801 (2022).
Triulzi, T. et al. The TRAR gene classifier to predict response to neoadjuvant therapy in HER2-positive and ER-positive breast cancer patients: an explorative analysis from the NeoSphere trial. Mol. Oncol. 16, 2355–2366 (2022).
FDA Approves Fam-Trastuzumab Deruxtecan-Nxki for HER2-Low Breast Cancer (US Food and Drug Administration, 2022); https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-fam-trastuzumab-deruxtecan-nxki-her2-low-breast-cancer
Fernandez, A. I. et al. Examination of low ERBB2 protein expression in breast cancer tissue. JAMA Oncol. 8, 1–4 (2022).
Slamon, D. J. et al. Ten year follow-up of BCIRG-006 comparing doxorubicin plus cyclophosphamide followed by docetaxel (AC→T) with doxorubicin plus cyclophosphamide followed by docetaxel and trastuzumab (AC→TH) with docetaxel, carboplatin and trastuzumab (TCH) in HER2+ early breast cancer. Cancer Res. 76, S5–S04 (2016).
Swain, S. M. et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): end-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 21, 519–530 (2020).
Piccart, M. et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer in the APHINITY trial: 6 years’ follow-up. J. Clin. Oncol. 39, 1448–1457 (2021).
Ewer, M. S. & Ewer, S. M. Trastuzumab cardiotoxicity after anthracycline exposure constitutes a complex and clinically important entity. JACC Heartfail. 7, 805–807 (2019).
Cameron, D. et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: final analysis of the HERceptin Adjuvant (HERA) trial. Lancet 389, 1195–1205 (2017).
Armenian, S. H. et al. Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 35, 893–911 (2017).
Curigliano, G. et al. Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann. Oncol. 31, 171–190 (2020).
Barcenas, C. H. et al. Improved tolerability of neratinib in patients with HER2-positive early-stage breast cancer: the CONTROL trial. Ann. Oncol. 31, 1223–1230 (2020).
Diéras, V. et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 18, 732–742 (2017).
Krop, I. E. et al. Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): final overall survival results from a randomised open-label phase 3 trial. Lancet Oncol. 18, 743–754 (2017).
Uppal, H. et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin. Cancer Res. 21, 123–133 (2015).
Powell, C. A. et al. Pooled analysis of drug-related interstitial lung disease and/or pneumonitis in nine trastuzumab deruxtecan monotherapy studies. ESMO Open 7, 100554 (2022).
Hamilton, E. P. et al. Trastuzumab deruxtecan (T-DXd) versus trastuzumab emtansine (T-DM1) in patients (pts) with HER2-positive (HER2+) unresectable and/or metastatic breast cancer (mBC): safety follow-up of the randomized, phase 3 study DESTINY-Breast03. J. Clin. Oncol. 40 (Suppl. 16), Abstr. 1000 (2022).
Acknowledgements
The authors thank C. Gilmore, of Phillips Gilmore Oncology Communications, Inc., for editorial assistance with manuscript preparation. Financial support was provided by Breast Cancer Research Foundation.
Author information
Authors and Affiliations
Contributions
All authors contributed to the design, research and writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.S. reports no conflict of interest. E.H.’s institution has received research funding from the following: AbbVie, Acerta Pharma, Accutar Biotechnology, ADC Therapeutics, AKESOBIO Australia, Amgen, Aravive, ArQule, Artios, AtlasMedx, Bliss BioPharmaceuticals, Cascadian Therapeutics, Clovis, Compugen, Cullen-Florentine, Curis, Dana Farber Cancer Institute, Duality Biologics, eFFECTOR Therapeutics, Ellipses Pharma, Elucida Oncology, EMD Serono, Fochon, FujiFilm, G1 Therapeutics, H3 Biomedicine, Harpoon, Hutchinson MediPharma, Immunogen, Immunomedics, Incyte, Infinity Pharmaceuticals, InventisBio, Jacobio, Karyopharm, Leap Therapeutics, Lycera, Mabspace Biosciences, Macrogenics, MedImmune, Merus, Millennium, Molecular Templates, Myraid Genetic Laboratories, Nucana, Olema, OncoMed, Onconova Therapeutics, ORIC Pharmaceuticals, Orinove, PharmaMar, Pieris Pharmaceuticals, Pionyr Immunotherapeutics, Plexxikon, Radius Health, Regeneron, Repertoire Immune Medicine, Rgenix, Sermonix Pharmaceuticals, Shattuck Labs, StemCentRx, Sutro, Syndax, Syros, Taiho, TapImmune, Tesaro, Tolmar, Torque Therapeutics, Treadwell Therapeutics, Verastem, Vincerx Pharma, Zenith Epigenetics and Zymeworks. E.H.’s institution has received consulting fees from the following: Arcus, Eisai, Greenwich Lifesciences, H3 Biomedicine, iTeos, Janssen, Loxo, Orum Therapeutics, Propella Therapeutics and Puma Biotechnology; and her institution has received research funding and consulting fees from the following: Arvinas, Black Diamond, Boehringer Ingelheim, CytomX, Dantari, Deciphera, Lilly, Merck, Mersana, Novartis, Pfizer, Relay Therapeutics, Roche/Genentech, SeaGen, Silverback. S.M.S. reports grants or contracts from Genentech/Roche, Kailos, Genetics, BCRF; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from Genentech/Roche, Daiichi Sankyo; support for attending meetings and/or travel from Genentech/Roche (travel 11/2019), Daiichi Sankyo (travel 9/2022) and Sanofi (travel 9/2022); participation on a Data Safety Monitoring Board for AstraZeneca; participation in an advisory board for AstraZeneca, Daiichi Sankyo, Exact Sciences, Biotheranostics, Natera, Merck, Silverback Therapeutics, Athenex, Lilly, Aventis; and participation in a Scientific Advisory Board for Inivata. S.M.S. reports leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: NSABP Vice Chairman; CCF, ASCO Director; and third party writing support from Genentech/Roche and AstraZeneca.
Peer review
Peer review information
Nature Reviews Drug Discovery thanks Jamuna Veeraraghavan, who co-reviewed with Rachel Schiff, Alvin Wong and the other, anonymous reviewer for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Bishop, J.M. Retroviruses and oncogenes II. Nobel lecture (1989): https://www.nobelprize.org/prizes/medicine/1989/press-release/.
Byondis B. V. FDA Accepts Byondis’ Biologics License Application for [Vic-] Trastuzumab Duocarmazine (SYD985) in HER2-Positive Metastatic Breast Cancer. Cision PR Newswire: https://go.nature.com/3VZlL4a (2022).
Cohen, S. Epidermal growth factor. Nobel lecture (1986): https://www.nobelprize.org/prizes/medicine/1986/cohen/lecture/.
Innovent Biologics. Innovent Releases Preliminary Results of the Phase Ia Dose-Escalation study of IBI315 (Anti-Her2/PD-1 Bispecific Antibody) in Patients with Advanced Solid Tumors at CSCO Annual Meeting 2021. Cision PR Newswire: https://go.nature.com/3f91toe (2021).
Levi-Montalcini, R. The nerve growth factor: thirty-five years later. Nobel lecture (1986): https://www.nobelprize.org/prizes/medicine/1986/levi-montalcini/lecture/.
US Food and Drug Administration: https://www.fda.gov/media/82647/download (2015).
US Food and Drug Administration: https://www.fda.gov/drugs/biosimilars/biosimilar-productinformation (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT05132582 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04457596 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT01042379 (2022).
US Food and Drug Administration: https://www.fda.gov/news-events/press-announcements/fda-approves-first-targeted-therapy-her2-low-breast-cancer.
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04208178 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT02947685 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT02448420 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04829604 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT03821233 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT01953926 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04224272 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT05027139 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT05035836 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT02912949 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04521179 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04881929 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04778982 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04042701 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04740918 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT03199885 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04538742 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT02563925 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04454528 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04501770 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT03448042 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04307329 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT03632941 (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/results?cond=HER2-positive+Breast+Cancer&term=&cntry=&state=&city=&dist (2022).
US National Library of Medicine. ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT04147819?term=NCT04147819&draw=2&rank=1 (2022).
Varmus, H.E. Retroviruses and oncogenes I. Nobel lecture (1989): https://www.nobelprize.org/prizes/medicine/1989/varmus/lecture/.
Supplementary information
Glossary
- Antibody dependent cell-mediated cytotoxicity
-
(ADCC). An immune mechanism through which Fc receptor-bearing cytotoxic effector cells can recognize and kill antibody-coated target cells that express tumour- or pathogen-derived antigens on their surface.
- Antibody-dependent cellular phagocytosis
-
(ADCP). An immunological mechanism of elimination whereby tumour cells are targeted with monoclonal antibodies to promote their clearance from the body by phagocytic immune cells such as macrophages.
- Biosimilar
-
A biologic product that is highly similar to and has no clinically meaningful differences from an existing FDA-approved reference product
- Complement-mediated cytotoxicity
-
(CDC). The mechanism by which antibody-coated target cells recruit and activate components of the complement cascade, leading to the formation of a membrane attack complex (MAC) on the cell surface and subsequent cell lysis.
- De novo stage IV disease
-
Cancer that is already metastatic or stage IV at the time of diagnosis
- Epitope
-
A portion of a protein or antigen that is recognized by the immune system (specifically by antibodies).
- Fc domain
-
The fragment crystallizable or the constant fragment region of the antibody, which is the tail region of the antibody that interacts with the cell surface receptors (Fc receptors) and some proteins of the complement system. This property enables antibodies to activate the immune system.
- Natural killer (NK) cells
-
A type of immune cell that has granules (small particles) with enzymes that can kill tumour cells or cells infected with a virus. A NK cell is a type of white blood cell.
- Proteolysis-targeting chimeras
-
(PROTACs). Protein degraders that use the cell’s own waste disposal machinery to eliminate a target protein. PROTACs can simultaneously bind to a target protein and an E3 ligase protein, which then ubiquitylates the target, marking it for proteasomal degradation.
- T cells
-
A type of white blood cell. T cells or T lymphocytes are part of the immune system and develop from stem cells in the bone marrow. They help protect the body from infection and may help fight cancer.
- Theranostics
-
A combination of the terms therapeutics and diagnostics. It is used to describe the combination of using one radioactive drug to identify (diagnose) and a second radioactive drug to deliver therapy to treat the main tumour and any metastatic tumours.
- Tumour-infiltrating lymphocytes
-
(TILs). A type of immune cell that has moved from the blood into a tumour. TILs can recognize and kill cancer cells.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Swain, S.M., Shastry, M. & Hamilton, E. Targeting HER2-positive breast cancer: advances and future directions. Nat Rev Drug Discov 22, 101–126 (2023). https://doi.org/10.1038/s41573-022-00579-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-022-00579-0
This article is cited by
-
Comprehensive review of CRISPR-based gene editing: mechanisms, challenges, and applications in cancer therapy
Molecular Cancer (2024)
-
Combined inhibition of HER2 and VEGFR synergistically improves therapeutic efficacy via PI3K-AKT pathway in advanced ovarian cancer
Journal of Experimental & Clinical Cancer Research (2024)
-
Folic acid-functionalized PEGylated niosomes co-encapsulated cisplatin and doxoribicin exhibit enhanced anticancer efficacy
Cancer Nanotechnology (2024)
-
Combined therapy of dabrafenib and an anti-HER2 antibody–drug conjugate for advanced BRAF-mutant melanoma
Cellular & Molecular Biology Letters (2024)
-
CDK7 in breast cancer: mechanisms of action and therapeutic potential
Cell Communication and Signaling (2024)
