
Abstract
Immunotherapy has rapidly evolved in the past decades in the battle against cancer. Chimeric antigen receptor (CAR)-engineered T cells have demonstrated significant success in certain hematologic malignancies, although they still face certain limitations, including high costs and toxic effects. Natural killer cells (NK cells), as a vital component of the immune system, serve as the “first responders” in the context of cancer development. In this literature review, we provide an updated understanding of NK cell development, functions, and their applications in disease therapy. Furthermore, we explore the rationale for utilizing engineered NK cell therapies, such as CAR-NK cells, and discuss the differences between CAR-T and CAR-NK cells. We also provide insights into the key elements and strategies involved in CAR design for engineered NK cells. In addition, we highlight the challenges currently encountered and discuss the future directions in NK cell research and utilization, including pre-clinical investigations and ongoing clinical trials. Based on the outstanding antitumor potential of NK cells, it is highly likely that they will lead to groundbreaking advancements in cancer treatment in the future.
Similar content being viewed by others
Facts
-
As a crucial immune cell type, natural killer (NK) cells function as the “first responders” in the context of cancer development.
-
With the rapid advancement of immunotherapy, particularly in cell therapy, research on NK cells has intensified due to their pivotal role in disease treatment.
-
An increasing number of NK cell-based therapeutics are under development, holding substantial promise for future breakthroughs in cancer treatment.
-
Discussing the biological background, cell sources, and the use of NK cells in disease therapy, as well as the primary advantages and disadvantages of engineered NK cells (e.g., chimeric antigen receptor [CAR]-NK cells), is a valuable avenue for future research in cell therapy.
Open questions
-
What is the current understanding of NK cell development, function, and their application in disease therapy?
-
How do NK cells exert their anticancer effects, and what are the associated mechanisms?
-
What is the rationale behind CAR cell therapy, and how do CAR-T and CAR-NK cells differ? What elements and strategies are relevant in the CAR design for engineered NK cells?
-
What are the existing challenges and development directions for this novel technology of CAR-NK cells? Furthermore, what is the current status of CAR cell therapy from laboratory research to clinical application (bench to bedside)?
Introduction
Cancer, a significant class of diseases, ranks among the leading causes of human mortality. This malignant disease now stands as the primary cause of death, second only to cardiovascular and cerebrovascular diseases [1]. In addition to traditional treatment methods such as surgery, chemotherapy, and radiotherapy [2], current cancer therapies have rapidly evolved, including targeted therapy and immunotherapy [3]. Targeted therapy is based on the advancements of cellular and molecular biology, and it seeks to inhibit cancer cells by identifying specific target molecules expressed by these cells. While notable progress has been made in enhancing therapeutic effects and reducing side effects, challenges such as drug resistance and off-target effects continue to persist in targeted therapies. On the other hand, immunotherapy has rapidly developed in recent years. It is primarily divided into two categories: immune checkpoint blockade therapy [4] and immune cell therapy [5]. These approaches have yielded promising clinical recoveries for certain patients, indicating a bright future for cancer treatment.
Natural killer (NK) cells, as a vital type of immune cell, possess dual functions involving cytotoxicity and immune regulation [6]. These cells originate from hematopoietic stem cells (HSCs) in the bone marrow and acquire the ability to differentiate between “friend” and “foe” through self-major histocompatibility complex (MHC) I molecule recognition [7]. NK cells exert broad-spectrum anticancer effects without the need for specificity and MHC restriction typically observed in cancer cell inhibition. They act as the “first responders” in the battle of the immune system against cancer development and viral infections. NK cells can spontaneously identify aberrant cells in the body, swiftly eliminating them via cytotoxic means while simultaneously generating various pro-inflammatory cytokines and chemokines. Furthermore, they have the capacity to activate other immune cells, initiating the adaptive immune response [8]. NK cells were initially discovered in the 1970s and primarily play a role in combating infected microorganisms and malignantly transformed allogeneic and autologous cells. As a distinct population of innate lymphocytes, NK cells inherently possess the ability to recognize and eliminate virus-infected and cancer cells. This recognition and elimination capacity rendered NK cells safe and effective as immunotherapeutic agents for patients with advanced leukemia nearly two decades ago [9]. In recent years, NK cells have received increasing attention as potential candidates for immunotherapy.
With the rapid development of immunotherapy, especially in the realm of cell therapy, research on NK cells has become increasingly intensive due to their pivotal role in disease treatment. In this paper, we aim to elucidate the biological background, cell sources, and the use of NK cells in disease therapy. We have also discussed the principal advantages and disadvantages of engineered NK cells, offering valuable insights into future avenues for NK cell therapy research.
NK cell biology
NK cell origination, development and classification
Peripheral blood mononuclear cells (PBMCs) stem from HSCs in the bone marrow, responsible for generating all the blood cells in the immune system through hematopoiesis [10]. HSCs possess the capacity to differentiate into two primary lineages: myeloid (including monocytes, macrophages, granulocytes, megakaryocytes, dendritic cells, and erythrocytes) and lymphoid (involving T cells, B cells, and NK cells). In healthy human bodies, approximately 70–90% of PBMCs are lymphocytes, and the majority of T cells remain in a naive state (mature but not stimulated by antigens). Only a small fraction of T cells become activated upon antigen recognition, initiating a cell-mediated immune response. Similarly, in normal human bodies, B cells predominantly exist in a naive or memory state, awaiting antigen stimulation and constituting approximately 5–15% of lymphocytes. Upon activation, B cells differentiate into plasma cells, which release substantial quantities of antibodies, triggering a humoral immune response. NK cells account for approximately 5–10% of the lymphocyte population and play a crucial role in the body’s innate immunity [11].
The development of NK cells primarily occurs from common lymphoid progenitors (CLPs) in the bone marrow, progressing through distinct stages, mainly including NK precursors (NKP), immature NK cells, and mature NK cells as illustrated in Fig. 1. In the course of normal hematopoiesis, one of the earliest markers of NK cells is IL2RB (CD122), which is expressed when CD34+ CLPs enter the NK cell fate lineage. Recent research has revealed the complexity of NK cell development. A “branched” model associated with NK cell development has been described, and the investigation into NK cell trafficking, tissue residence, and tissue-specific specialization is ongoing [12,13,14,15,16]. Subpopulations of NK cell precursors can be differentiated by their varied gene expression, including markers such as CD34, KIT, KLRB1, CD244, and IL-15R [17]. To identify immature NK cells, molecular markers like CD244, natural toxicity receptor (NCR)1, NCR2, NCR3, and KLRB1 have been reported. Subsequent differentiation to the mature stage is characterized by the expression of markers such as KLRD1, ITGB2, killer Ig-like receptors (KIRs), PRF1, IFNG, CD56, and CD16 [18]. The CD56bright subset is often considered an early stage of NK cell maturation [19]. Notably, CD56bright cells exhibit a higher proliferative capacity than CD56dim cells, likely due to their prominent expression of several key proteins, including CCR7, CSF2, CXCR3, IL2RB, KLRC1, and SELL [20]. This subpopulation is known for its elevated IFNG expression. Downregulation of CD56 is followed by CD16 expression, resulting in the CD56dim NK cell subset [21]. This subgroup is distinguished by its potent cytotoxic activity [22, 23], with KIRs being among the genes associated with this subgroup [24]. Additional genes linked to the CD56dim subset include CX3CR1 [25], KLGR1 [26], and PRF1 [12].

The maturation steps of NK cells in hematopoietic stem cell (HSC) differentiation and hematopoiesis process.
While the CD56bright cells make up a smaller portion, estimated at only 5–10% of the total NK cell population in peripheral blood (PB), the CD56dim NK cells constitute over 90%. However, CD56bright cells are more prevalent in specific tissues, including secondary lymphoid tissues [11, 21]. Recent findings have suggested that NK cell development can also take place in secondary lymphoid tissues such as lymph nodes and the spleen [27]. During this developmental phase, NK cells are trained to recognize MHC-I, thereby avoiding the targeting of healthy, normal cells.
NK cell membrane protein characteristics and biology functions
NK cells eliminate infected and cancerous cells upon activation of activating receptors, such as the NCRs, which include NKp30, NKp44, and NKp46 [28,29,30]. As mentioned earlier, CD56dim NK cells primarily display robust cytotoxic activity, whereas CD56bright NK cells primarily secrete cytokines and exhibit lower cytotoxic activity. The regulation of NK cell cytotoxicity involves a complex interplay of activation and inhibition signals. The killing efficiency of NK cells on target cells relies on the delicate balance between inhibitory and activation signals, closely tied to cell membrane receptors and proteins (Fig. 2). Activated NK cells employ various mechanisms for target cell elimination (Fig. 3), including ① release of perforin and granzyme, leading to target cell lysis or apoptosis; ② promotion of FasL expression, inducing cell apoptosis; ③ release of tumor necrosis factor (TNF)-α, interferon (IFN)-γ, granulocyte macrophage colony-stimulating factor (GM-CSF), and chemokines (such as CCL1, CCL2, CCL3, CCL4, CCL5, and CXCL8) to recruit and activate other effector immune cells; ④ antibody-dependent cell-mediated cytotoxicity (ADCC). NK cells express various membrane proteins, as depicted in Fig. 2, to modulate inhibitory and activation signals [16]; however, they lack T-cell receptors (TCRs) and B-cell receptors, among which CD56 [31] (an adhesion molecule mediating homotypic adhesion) and CD16 [32] (a low-affinity Fc receptor FcγRIII, contributing to NK cell-mediated ADCC) serve as primary surface markers. Unlike T cells, NK cells do not express antigen-specific recognition receptors. The surface receptors of NK cells are broadly categorized into activating and inhibitory receptors, both capable of recognizing classical or non-classical human MHC-class I molecules on normal cells. The interaction between MHC-I molecules and inhibitory receptors signals convey a “Do not eat me” message to NK cells, enabling cell evasion from NK cell-mediated killing [33].

NK cells primarily express different cell surface proteins exhibiting various effects.

NK cells exhibit broad-spectrum anticancer effects via various mechanisms, including antibody-dependent cell-mediated cytotoxicity (ADCC) via CD16 binding, secretion of cytokines (IFN-γ, TNFα), release of cytolytic granules (e.g., granzyme B), and expression of death ligands. The balance of NK cell activity is regulated by activating and inhibitory receptors.
NK cell engineering
Numerous studies suggest the presence of potential neoantigens, such as HSP70, expressed exclusively on tumor cells and absent in normal cells, can activate NK cells. This discovery opens a promising avenue in target-driven cell-based immunotherapies, as the HSP70 protein becomes a viable target for NK cells [34,35,36]. Currently, several approaches exist in NK cell engineering (Fig. 4). A prominent method involves chimeric antigen receptors (CARs), receptor proteins that confer immune cells with the ability to target specific antigenic proteins. CAR-T cell therapy has achieved significant success in hematological malignancies, including acute lymphoblastic leukemia [37, 38] and diffuse large B-cell lymphoma [39]. Various CAR-T therapies have gained approval [40,41,42]. While CAR-T cell therapy stands as a groundbreaking biotechnological advancement in cancer treatment, it faces challenges such as side effects, toxicity [43], T cell exhaustion [44], and limited efficacy against solid tumors [45]. Presently, new cell therapies such as CAR-NK, CAR-NKT, CAR-macrophage (CAR-M), CAR-Treg, CAR-γδT, with CAR technology at their core, have emerged, especially CAR-NK showing promising prospects in tumor immunotherapy [46].

1) CAR-NK cells: NK cells that are modified to express extracellular receptors (such as scFvs, nanobodies, peptides, and ligands) for HLA-independent antigen recognition. 2) TCR NK cells: NK cells that are modified to express TCR complexes for MHC-I presented cancer antigens. 3) Multi-specific Ab-based NK cell therapy: NK cells that utilize mono-antibodies, bi-specific antibodies, or tri-specific antibodies to recognize cancer antigens (including but not limited to bi-specific NK-cell-engager, BiKE).
NK cell sources
There are four primary sources of NK cells in NK cell engineering (e.g., CAR-NK), depicted in Fig. 5: Peripheral blood (PB), induced pluripotent stem cells (iPSCs), mesenchymal stem cells (e.g., umbilical cord blood), and NK cell lines (such as NK-92, etc.). As previously mentioned, NK cells, as a subset of lymphocytes, possess the ability to recognize and eliminate tumor cells without prior sensitization, antibody involvement, and are not restricted by MHC. Commonly utilized markers for NK cells in experiments include CD16 and CD56. The characteristics and limitations of NK cells from different sources in clinical treatment are detailed below.

There are four primary sources of NK cells including peripheral blood (PB), induced pluripotent stem cells (iPSC), mesenchymal stem cells (e.g., umbilical cord blood), and NK cell lines (e.g., NK-92), especially in CAR-NK cell therapy.
Peripheral blood (PB-NK)
NK cells derived from the patient’s own body or a healthy donor. Due to disease and treatment limitations, the function of the patient’s own NK cells may be compromised. Allogeneic NK cells are clinically favored [47], but careful T cell removal is essential to mitigate graft-versus-host disease (GVHD) [48]. PB-NK cells are mature and do not necessitate induction of differentiation, but gene transduction efficiency is relatively low. Prolonged in vitro expansion may result in shortened telomeres and reduced cytotoxicity. Cryopreservation diminishes the viability and toxicity of PB-NK cells. NK cell therapy typically involves reinfusion of 106–108 cells per kilogram of body weight [49, 50], yet the proportion of NK cells in PB is low [51], posing challenges for large-scale in vitro culture. However, there are advantages; for instance, these cells are mature and bypass the need for an extended differentiation period.
Umbilical cord blood (UCB-NK)
These cells exhibit high proliferation efficiency, allowing real-time selection of human leukocyte antigen (HLA)-mismatched products by establishing an NK cell bank [52]. Cord blood serves as a promising source for NK cells in clinical applications, with two main strategies: direct use of NK cells in cord blood or differentiation of HSCs in cord blood into NK cells. It is crucial to note that mesenchymal cells in cord blood are not utilized for these purposes. Cord blood remains a relatively stable source of NK cells. Due to their high proliferative capacity, only 10% of a cord blood unit is required to generate a nearly pure cell pool of over 109 NK cells within 2 weeks, typically suitable for one treatment cycle [53, 54]. However, UCB-NK cells are not fully differentiated, exhibiting relatively low expression of NK receptors and limited cell inhibition ability, with a potential risk of tumorigenesis in allogeneic transplantation [55]. Nevertheless, they demonstrate a robust bone marrow homing ability [56]. Moreover, the higher proportion of hemoglobin and red blood cells in cord blood can impact the isolation and culture of PBMCs.
Stem cell-derived NK cells
NK cells are commonly induced from human embryonic stem cells (hESCs), HSCs or iPSCs [57], with an expansion period extending beyond 3–5 weeks. This extended period helps mitigate the heterogeneity of NK cells between the recipient and the donor. However, NK cells induced by iPSCs present potential risks of malignant transformation and tumorigenesis in vivo, along with the possibility of triggering unexpected immune responses due to their potential immunogenicity. iPSCs efficiently clone, expand, and differentiate in vitro, producing a substantial quantity of uniform NK cell products [58]. Nevertheless, iPSC-derived NK cells often express low levels of endogenous CD16, a drawback that can be addressed through genetic engineering [59, 60]. Moreover, iPSCs may retain DNA methylation signatures consistent with their somatic tissue origin, contributing to “epigenetic memory,” which could influence the development of specific cell lineages distinct from the donor cells. Systemic administration of cytokines in a clinical setting is highly undesirable due to its expense and potential dangers. Additionally, iPSC-derived cells carry the risk of malignant transformation and potential immunogenicity, leading to the destruction of ES cells and even adverse immune responses such as cytokine release storms [61].
NK cell line
Various NK cell lines, including NK-92, HANK-1, KHYG-1, NK-YS, NKG, NK101, NK3.3, YTS, and NKL, have been constructed to date, serving as excellent cell models for studying NK cell biology and associated applications. Among these, the NK-92 cell line is the only one applied in clinical trials, demonstrating a relatively satisfactory response outcome with controllable adverse effects. NK-92 cells lack the CD16 receptor-mediated ADCC effect, but this can be addressed through modification [62]. These cells are easily genetically manipulated, allowing effective introduction of exogenous genes through electroporation without the need for viral vectors [63]. Because NK-92 is a tumor-derived aneuploid immortalized cell line [64], it requires irradiation before use to inhibit in vivo proliferation [65], negatively impacting long-term persistence and overall therapeutic potential.
CAR-NK cells
CAR-NK cells typically share the similar CAR structures as CAR-T cells. NK cells enhance their cytotoxic capacity and cytokine production through co-stimulatory molecules like NKG2D and CD244, providing probably stronger tumor-specific targeting and cytotoxicity than CAR-T cells [66]. CAR-NK cell therapy is a potential alternative to CAR-T therapy due to several unique features. Firstly, allogeneic NK cells are generally safe for adoptive cell therapy (ACT) as they do not typically mediate GVHD [66]. Moreover, NK cells only secrete small amounts of IFN-γ and GM-CSF [67], without producing IL-1 and IL-6 that initiate cytokine release syndrome (CRS) [68]. Secondly, in addition to inhibiting cancer cells through single-chain antibody recognition of tumor surface antigens, NK cells can also recognize various ligands through multiple receptors such as NCRs (NKp46, NKp44, and NKp30), NKG2D, and DNAM-1 (CD226) [69,70,71]. Lastly, NK cells are abundant in clinical samples [13] and can be generated from various sources, including PB [72], UCB [54], hESCs, iPSCs [57], and even NK-92 cell lines [73] as mentioned above.
Similar to CAR-T cells, the functional CAR molecule expressed on NK cells comprises three components: an extracellular domain, a transmembrane region, and an intracellular signaling domain (Fig. 6A). The extracellular domain includes a signal peptide and a single-chain antibody fragment (scFv) responsible for recognizing the antigen. A hinge region connects this structure to the transmembrane region, which, in turn, links to the intracellular domain containing the activation signal. The commonly utilized transmembrane segment for CAR-NK is adapted from CD3ζ, CD8, or CD28, with T cell-specific CD8 and CD28 being the most frequently employed [74]. The intracellular segment is pivotal for cell activation post-reception of the target antigen signal and constitutes a linear structure of co-stimulatory molecules and signaling domains recruited downstream of signal transduction. Successful CAR design is achieved via a combination of meticulous design and functional testing. The evolution of CAR generations includes the first generation containing only CD3ζ [75], the second and third generations adding one or two co-stimulatory domains, respectively, based on the first generation [76], the fourth generation incorporating a cytokine secretion segment based on the third generation [77], and the fifth generation introducing a special binding motif (Fig. 6A). Logic-gated control of CAR cells has also been developed to achieve precision therapy and avert potential toxicity (Fig. 6B). Currently, the second-generation CAR structures CD28-CD3ζ and 41BB-CD3ζ are most commonly used in the field of CAR-NK, whereas in the third generation, CD28-41BB-CD3ζ is also frequently employed [74, 78]. A detailed description of each CAR element is discussed below.

A The first generation of CAR contained only CD3ζ. The second and third generations added one or two co-stimulatory domains, respectively, building on the first generation. The fourth generation introduced a cytokine secretion segment based on the third generation, and the fifth generation incorporated special binding motifs. B Logic-gated control of CAR cells was developed for precision therapy and to avoid potential toxicity, including “AND,” “AND NOT,” and “OR” in three logic-gated control manners.
Vector backbone and promoter
The vector backbone incorporates all elements necessary for CAR expression, including a promoter, polyA signal, and transcriptional regulatory fragments. The choice of promoter directly impacts the expression of the transgene. Current reports on CAR-NK cells reveal the use of various promoters to drive CAR expression, whether derived from cell lines [79] or primary NK cells [80]. In primary CAR-NK and CAR-NK cell lines, viral promoters (such as MPSV and MMLV) are more commonly utilized than constitutively active promoters (such as EF1α and PGK) [81].
Signal peptide
Signal peptides exhibit substantial heterogeneity, leading to varying levels of protein production efficiency. For CAR-NK and CAR-T cells, comparative studies identifying the optimal signal peptide are lacking. Currently, CD8a-SP is the most commonly used signal peptide sequence for NK cells, and immunoglobulin heavy or light chain signal peptides are reported for NK cell lines [82].
Single-chain antibody fragment (scFv)
The scFv serves as the tumor antigen-binding domain of CAR [83], determining the specificity and function of CAR-NK cells. As single-chain antibodies deviate from the natural form of antibodies, the order of the heavy and light chains is artificially determined [84]. For CAR-NK designs, the VH-VL direction is preferred over the VL-VH direction [85,86,87]. Fujiwara et al. revealed that the order of the heavy and light chains does not affect the expression of CARs on T cells [88].
Furthermore, cells can be equipped with multiple scFvs, thereby expanding the antigen recognition capacity of CAR effector cells. Several options exist: the CAR can be transduced with a two-element vector, inducing the expression of two CAR constructs; or two scFvs can be fused in one construct, creating a “single-handle” CAR with tandem scFvs [44, 89]. While these technologies have been utilized to produce CAR-T cells [90], their application in CAR-NK cells is not well-documented. In most current clinical CAR-T cell trials, single-chain antibodies derived from mouse antibodies are commonly used, increasing the risk of GVHD in anti-mouse IgG cells. This risk can be mitigated through humanization or screening of fully human antibodies [91]. Unfortunately, even humanized scFvs may induce host anti-idiotypic immune responses due to the chimeric nature of CAR receptors [92]. However, in the limited number of CAR-NK clinical trials to date, no major adverse effects associated with anti-CAR immune responses have been identified [66]. Moreover, several other forms like nanobody were also explored to serve as the tumor antigen-binding domain.
Linking region
The linking region between the heavy and light chains contributes to stabilizing the conformation of the single-chain antibody. A too-short linking region may lead to multimer formation, whereas a too-long linking region can cause hydrolysis or reduce the association between VH and VL domains [93]. For CAR-NK cells, the GGGGS pentapeptide is widely used in multimers, typically in 3 repeats. Another linker designed to enhance proteolytic stability is the Whitlow “218” linker (GSTSGSGKPGSGEGSTKG) [88, 94].
Hinge region
The hinge region, the extracellular domain of the CAR connecting the single-chain antibody unit and transmembrane domain, maintains the stability required for robust CAR expression and activity in effector cells. Most CAR-NK constructions use derivatives of CD8α or CD28 extracellular domains or IgG-based hinge regions. The type and length of the hinge region significantly affect the functional activity of CAR [95]. Although most information comes from CAR-T, the direct transformation into CAR-NK remains unproven. A direct comparison between CD28 and CD8α hinge regions revealed that CD28 is more likely to promote CAR molecule dimerization, resulting in a stronger activation stimulus [93]. While beneficial, this can also lead to more serious adverse effects. IgG-based hinge regions, made up of the Fc portion of IgG1 or the CH2/CH3 domains of the Fc portion, offer flexibility in structure. The length of the hinge region can be adjusted to adapt to antigen recognition; however studies have revealed that a shorter spacer region results in higher cytokine production, faster CAR cell proliferation, and improved persistence and antitumor effects in vivo [95].
Transmembrane domain
The transmembrane domain connects the CAR extracellular domain and the intracellular activation signal domain. The most commonly used transmembrane domains for CAR-NK originate from CD3ζ, CD8, and CD28. The choice of transmembrane domain influences the activation extent of the CAR construct in cellular functions. Transmembrane domains from molecules typically expressed on NK cells, such as DNAM-1, 2B4, and NKG2D, lead to increased CD107a degranulation and higher cytotoxicity. Thus, the specific source of the transmembrane domain determines the activity of CAR-NK [96]. An important aspect of the transmembrane domain is that it should follow the natural orientation (order of N-terminal to C-terminal) of transmembrane proteins on NK cells. At present, CD8α- and CD28-modified transmembrane regions are most common in primary CAR-NK cells, whereas CD28 is the preferred transmembrane region for CAR-NK cell lines [93].
Activation signal
The number of intracellular activation signals in a CAR determines its “generation”. First-generation CAR-NK cells, akin to CAR-T cells, contain only the CD3ζ signal. Second-generation and third-generation CAR-NKs carry one and two additional co-stimulatory signals, respectively, typically derived from the CD28 family (CD28 and ICOS), the TNF receptor family (4-1BB, OX40, and CD27), or the signaling lymphocytic activation molecule-related receptor family (2B4) [97]. The published CAR-NK clinical trial used a second-generation CAR-NK construct that improved activity by incorporating IL-15 expression. Most current CAR structures depend on the CD3ζ chain signaling domain, and robust activation signals are crucial for eliciting potent antitumor responses but may also result in rapid effector cell exhaustion. Combinations of co-stimulatory domains can be employed to calibrate desired immune cell responses. CD28-based CARs exhibit a faster effector profile than 4-1BB-based CARs, inducing higher levels of IFN-γ, granzyme B, and TNF-α. However, this strong co-stimulatory signal also results in activation-induced cell death (AICD). Conversely, 4-1BB-CD3ζ signaling preferentially induces memory-related genes and sustained antitumor activity [98, 99]. This difference may be attributed to the amelioration of T cell exhaustion induced by the 4-1BB domain in contrast to the CD28 domain [44]. CD3ζ was universally used as the primary activation domain in studies of CAR-NK cell lines and primary CAR-NK cells, with approximately half carrying an additional activation domain, generally with the addition of 4-1BB or CD28. For third-generation constructs, the combination CD28/4-1BB/CD3ζ is most commonly employed. Intracellular signaling domains, such as CD28, 4-1BB, and OX40, often function to trigger immune cell activation and inhibition [100]. A recent report using iPSC-derived CAR-NK therapy identified the crucial role of the NKG2D transmembrane domain and emphasized the critical role of the 2B4 co-stimulation domain [96]. Different researchers also performed similar studies using 2B4 to highlight the importance of the activation signal in immune cell therapy [101, 102]. At present, four generations of CAR structures have been developed and are available for CAR-NK research.
CAR transfection or transduction vector
With the advancement of gene modification technology, various methods have been employed to generate CAR-NK cells. The two primary methods include viral transduction [103] (using lentiviruses or retroviruses) and transfection of naked plasmid DNA [87], transposase DNA-mediated integration [104], and mRNA electroporation [96]. Lentiviruses can efficiently transduce both periodic and non-cyclical cells and have been widely utilized in gene therapy [105]. They have been successfully used as vectors in studies on primary CAR-NK cells and CAR-NK cell lines. Both second-generation and third-generation lentiviruses have been used in preclinical studies to generate CAR-expressing NK cell lines and primary CAR-NK cells. Retroviral vectors are also commonly used for NK cells [54, 66]. Retroviruses have been used as gene therapy vectors for decades, including CAR-NK cell lines and primary NK cells [106]. In a recent phase I clinical trial, retroviral-transduced CD19 CAR-NK cells were used to treat CD19+ non-Hodgkin’s lymphoma and chronic lymphocytic leukemia. The study results stated that 73% of patients responded to the treatment, with 7 of 8 patients achieving a complete response. Responses were rapid, occurring within 30 days of CAR-NK administration at all dose levels. After the 1-year follow-up, expanded CAR-NK cells remained detectable [66]. Following infusion, CAR-NK DNA copy numbers remained stable in PB for up to 1 year, indicating, for the first time, that retroviral-transduced CAR-NK cells can exhibit long-term in vivo survival. Different retroviruses types have been used to generate CAR-NK cells. The RD114 retrovirus was reported to be more efficient at transducing primary NK cells than the γ retrovirus and lentivirus [107]. Although long-term stable CAR expression in NK cells can be achieved using various retroviruses, the safety of retroviral systems remains a concern, especially when compared with the safer lentiviruses. CAR-encoding mRNA electroporation is a rapid, efficient, but short-lived method. To date, mRNA electroporation has been used in CAR-NK cell lines and primary CAR-NK cell studies. Generally, expanded or activated NK cells exhibit much higher mRNA transfection efficiency than freshly isolated NK cells [108]. Because mRNA synthesis is a good manufacturing practice (GMP)-compliant manner and electroporation can be conducted in a clean room, it is feasible to generate GMP-compliant CAR-NK cells through mRNA electroporation. However, the primary disadvantage of this approach is the short window for CAR expression: after electroporation, CAR-NK cells should be infused back into the patient within 7 days. The Sleeping Beauty transposon system has also been developed. Transposon-based systems offer important advantages over conventional methods, such as the efficient introduction of CAR transgenes at predetermined locations. Transposons are primarily introduced into NK cells via electroporation and then integrated into the host genome by transposonases [50]. Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 is another powerful genetic modification technology that introduces the Cas9 protein into NK cells along with gRNA. This technique was initially used to disrupt the CD38 gene in primary NK cells [109], aiming to prevent NK cell cannibalism in combination with daratumumab (anti-CD38) because CD38 is expressed in NK cells, multiple myeloma [110], and acute myeloid leukemia (AML) cells [111]. CRISPR/Cas9 has also been recently used to introduce some other new genes [112]. Overall, using CRISPR/Cas9 strategy is a promising strategy to precisely delete, repair, or introduce specific genes, facilitating the generation of potent antitumor NK cells.
While we have established the superiority of a specific domain among multiple candidates based on findings from T cells expressing the CAR applied into NK cells, it is crucial to note that the superiority of CAR domains is complex and depends on factors such as the target and the interaction with other domains. Further exploration is needed to determine if the results and experiences obtained from T cells are applicable to NK cells and to anticipate optimization. Moreover, the expansion of NK cells may result in an adverse phenomenon of “suicide” or “fratricide” as mentioned above, where cells recognize receptors or ligands on the surface of other similar cells and trigger cytotoxic activity against them. The Fas/FasL axis is among the most relevant mechanisms. FasL-mediated cytotoxicity plays a crucial role in NK cell function, triggering caspase-dependent apoptosis when it binds to the receptor Fas in the target cell. Fas can also serve as a steady-state mechanism for inhibiting NK cell activity expressed by NK cells, known as AICD. NKG2D is another receptor that may lead to self-killing among NK cells, which is a natural receptor primarily comprising NK, CD8+T, and γδ T cell expression, displaying recognition of various stress-induced ligands. Cannibalism may also occur in CAR-NK cells due to CAR ligand/antigen recognition if certain target antigens are also expressed on these effector cells [113]. Furthermore, trogocytosis is a common phenomenon that often takes place during NK cell-mediated cancer inhibition. Trogocytosis ultimately leads to the transfer of antigens to NK cells, mediating the inhibition of NK cells by other NK cells. The knock out of target antigens in effector cells can overcome fratricide, but this method is unsuitable for antigens transferred to effector cells during trogocytosis. Low affinity of CAR to antigen or optimized CAR signaling transduction may serve as alternative approaches [114].
Popular targets of CAR-NK research
Solid tumor targets
In several cancer types, programmed cell death ligand 1 (PD-L1) is upregulated in the tumor microenvironment (TME) and in immunosuppressive cells [115]. Preclinical tests have revealed that PD-L1-targeted CAR-NK cells exhibit specific antitumor effects against several in vitro tumor cell lines, and exhibit robust in vivo antitumor effects against triple-negative breast cancer [116], bladder cancer [117], and lung cancer [118]. Human epidermal growth factor receptor 2 (HER2)/erythroblastic oncogene B 2 (ERBB2) is often overexpressed in breast, gastric, esophageal, ovarian, and endometrial cancers [119]. HER2 is associated with poor survival and also expressed in most glioblastomas [120]. Extensive studies have been conducted on the application of CAR constructs targeting HER2 [121]. NKG2D is an activating NK cell receptor modulating the anticancer cytotoxic potential of NK cells by interacting with its tumor-associated overexpressed ligands [122]. NKG2D ligands include MICA, MICB, and ULBPs (ULBP1, ULBP2, ULBP3, ULBP4, ULBP5, ULBP6) [123, 124]. Altogether, the targets in solid tumor (pre-) clinical therapy are relatively fewer than hematologic malignancies, but display potentials that CAR-NK cells congregate in these tumors [50].
Hematologic malignancy targets
Clinical studies have demonstrated the significant efficacy of anti-CD19 CAR-T cell therapy in tumor immunotherapy [66]. However, CAR-T cell therapy is constrained by various adverse effects and manufacturing challenges. CAR-NK cells present themselves as an alternative therapeutic strategy for hematological malignancies, focusing on the currently popular targets (Fig. 7). Multiple myeloma (MM) is a hematologic malignancy, and numerous CAR-T and CAR-NK cell therapies are being currently developed [125], with B-cell maturation antigen being the most popular cell therapy target [126]. Furthermore, CD38 and CD138 are also common targets for MM treatment with CAR constructs [127]. CD19, CD20, and CD22 are commonly employed targets for CAR-T cell therapy in B-cell lymphoma and leukemia. Two major challenges in constructing CARs against AML include shared antigen expression and heterogeneity with hematopoietic progenitor cells. Shared antigenic expression of CD123 and CD33 can result in on-target nontumor toxicity [128, 129]. In several cases, targeting various AML-associated antigens (such as FLT3 [130], CD123 [131], CD33 [132], CLL-1 [133], and GRP78 [134]) using multiple CARs may be necessary because certain tumor-associated antigens (TAAs) may not be expressed on all leukemia cells.

The currently popular targets in hematologic malignancy cell therapy are listed in the categories.
Challenges of NK cell therapy
At present, CAR-NK cells demonstrate obvious advantages compared with CAR-T cells, as summarized in previous literature [135,136,137] (Table 1). Registered clinical trials using these cells have seen a rapid increase, as shown in Table 2. Unlike CAR-T cell clinical applications, NK cells exhibit relatively acceptable toxicity adverse effects, avoiding effects such as cytokine release syndrome. Several clinical trial evidences support this, for instance, Tang et al. reported the first-in-man CD33+ CAR-NK-92 cell clinical trial, testing safety in patients with AML experiencing relapsed and refractory conditions. The dose of 5 × 109 cells in each patient led to no evident adverse effects [138]. Additionally, Liu et al. reported phase I and II trial outcomes using CD19 CAR-NK cells, revealing a rapid response in patients with CD19+ cancer with relapsed or refractory conditions, without apparent associations between effectors and adverse effects, including neurotoxicity, cytokine release syndrome, or GVHD [66]. However, challenges persist in this type of immunotherapy, summarized as follows:
Low persistence
A major drawback is the lack of in vivo persistence of infused cells in the absence of cytokine support, limiting the effectiveness of NK cell immunotherapy. While exogenous cytokines have been reported to increase proliferation and persistence of adoptive NK cells [139], they can also lead to undesired adverse effects, including the expansion of suppressive immune subsets, such as Tregs [140]. Rejection of allogeneic NK cells by host T cells is also a critical consideration in cell therapy using allogeneic NK cells. Further exploration of the role of transmembrane-bound IL-15 in promoting NK cell persistence is warranted.
Transport to the desired tumor site
The efficient homing of NK cells to tumor sites has been debated, as rapid homing to the tumor bed is critical for adoptive cell therapy efficacy. This process is regulated via complex interactions between NK cells and chemokines released by tumor cells [141]. Various engineering approaches have been explored to enhance NK cell migration to tumor sites. For instance, NK cells have been subjected to electroporation with mRNA encoding the chemokine receptor CCR7 to enhance migration to lymph nodes expressing the chemokine CCL19 [20, 142]. To improve the success rate of NK cell immunotherapy in patients with solid tumors, mouse models have been used to explore novel techniques promoting NK cell translocation to tumor sites [143]; however, the effectiveness of these approaches requires further verification in clinical trials.
Immunosuppressive tumor microenvironment (TME)
The TME, encompassing immunosuppressive molecules, immunosuppressive cells, and an unfavorable environment hindering immune cell function, poses a major obstacle for CAR-NK cell therapy. Immunoregulatory factors such as transforming growth factor (TGF)-β and others present in the TME can impair NK cell activity [144]. Researchers are investigating the development of CAR-NK cells that counteract some of these immunosuppressive effects, such as knocking out associated genes of NK cells using CRISPR/Cas9 technology [145]. Another strategy to overcome NK cell depletion [146] in the TME is to eliminate checkpoint components using genome editing to improve their function.
Low transduction efficiency of lentivirus
Lentivirus-based transduction systems represent one of the most commonly used methods for intracellular gene modification and delivery. However, the natural resistance of NK cells to lentivirus poses a challenge to efficient transduction. Various chemicals, such as protamine sulfate, are employed to enhance viral transduction [147].
Altogether, while CAR-T cell immunotherapy provides a promising approach to treating certain cancers, there are still several limitations: 1) high costs leading to unavailability; 2) long production cycles resulting in patients being unable to afford waiting; 3) poor cell quality of patient samples potentially leading to production failure; 4) CRS and neurotoxicity contributing to high treatment risks. Therefore, the general CAR holds great promise. NK cells, with their unique biological characteristics, demonstrate distinct advantages as potential “off-the-shelf” universal CAR-NK cells. As a promising alternative, different sources of NK cells (including UCB, PB, cellular lines, and iPSCs) could be utilized. Additionally, the allogeneic context without obvious toxic adverse effects presents a significant advantage, even though most CAR-NK cells are still in preclinical or early clinical trial stages. However, the short persistence of NK cells after infusion in vivo remains a major setback. Optimization and standardization of cell expansion and target gene transfection also need further definition, considering the differences between T cells and NK cells. Lastly, akin to CAR-T cells, the lack of tumor-specific targets poses a significant challenge for CAR-NK cell applications in various cancer treatments, including hematologic and solid tumors, necessitating further development for precision medicine.
Development directions of NK cell therapy
Recognition of novel target antigens
As mentioned above, identifying highly consistently expressed target tumor antigens is a critical step in CAR design. Most TAAs are also expressed by some healthy cells, potentially causing a “targeting nontumor” effect [148]. Furthermore, the expression of these TAAs can vary greatly among single-cell clones of the same tumor. To address this issue, bispecific CARs have been designed to target multiple antigens simultaneously. This can be achieved by injecting different CAR-NK cells targeting distinct antigens simultaneously or designing one CAR to recognize multiple antigens through “tandem CARs,” wherein two combined elements are attached to individual molecules to enhance the immune synapse [149]. Additionally, multiple CARs can be simultaneously produced on the same immune cell using a vector.
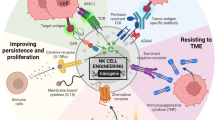
Improving NK cell activity
Various immune checkpoints, such as PD-L1, regulate and suppress NK cell activity. For instance, a new NK-92 cell line designed with a CAR targeting PD-L1, known as PD-L1-targeting haNK, demonstrated specific antitumor effects against several tumors in preclinical data [150]. Another strategy to improve the activity of CAR-NK cells involves regulating tumor metabolism, an area that has not received sufficient attention. Under hypoxic conditions, adenosine is produced via ATP metabolism by CD39 and CD73, contributing to immune evasion, preventing NK cell trafficking to tumor sites, and inhibiting NK cell maturation. NKG2D-engineered CAR-NK cells exhibited efficacy in treating lung cancer following anti-CD73 antibody inhibition [151]. Therefore, immune checkpoint regulation remains a crucial consideration in cell-based immunotherapy.
Overcoming the immunosuppressive TME
Tumors harbor various immunosuppressive factors, including TGF-β, IL-10, and PD-1. Several strategies are utilized to mitigate their inhibitory effects. Combining TGF-β kinase inhibitors with NK cells has been observed to restore NK cell cytotoxicity and preserve NKG2D and CD16 expression [152]. Additionally, hybrid CARs incorporating extracellular TGF-β receptor domains have proven successful in improving the antitumor potential of NK-92 cells [153]. Furthermore, the concurrent use of immune checkpoint blockade inhibitors presents a promising avenue.
Improving security
Enhancing the safety of CAR-NK cell-based therapy may involve modifying the CAR structure by incorporating suicide genes [154]. Developing bispecific CAR molecules to better target tumor-specific antigens is another crucial approach. CAR-NK cells exhibit the unique ability to target tumors in both a CAR-dependent and CAR-independent manner. This ability can be harnessed to achieve enhanced tumor inhibition by developing nonsignaling CARs. These nonsignaling CARs lack direct killing signals but can augment the specific killing of NK cells by promoting residence and adhesion to target cells [136]. Another intriguing strategy involves designing CAR-NKs capable of modulating the TME. These highly specialized CAR-NK cells express several foreign genes that can modulate the local TME to prevent any harmful effects.
Improving accessibility
Addressing the accessibility of CAR-NK cells in solid tumors necessitates various approaches, including topical, intraperitoneal, and focused ultrasound-guided drug delivery. For instance, pleural injections proved highly effective in an orthotopic model mimicking human pleural malignancies, demonstrating an even longer duration of function compared to intravenous injections [155]. The topical administration of CAR immune cells may also help reduce treatment doses.
Prospective future
NK cells stand as a unique cohort of antitumor effector cells, wielding functions such as MHC-independent cytotoxicity, cytokine production, and immune memory. These attributes position them as pivotal contributors to both innate and adaptive immune response systems. The field of CAR-NK cell therapy holds promise in clinical research, demonstrating commendable safety and preliminary efficacy in certain patients with cancer. In comparison to CAR-T cells, CAR-NK cells boast distinct advantages, yet they grapple with challenges. Enhancing cell proliferation, facilitating more efficient activation of cytotoxicity, and ultimately optimizing NK cell reconstitution are concerns. Consequently, advancements in large-scale preparation methods, cryopreservation measures, and efficacy are imperative. Addressing the short duration of in vivo persistence and exhaustion remains an unresolved frontier. Overall, CAR-NK is poised to evolve into a versatile cell product, holding greater advantages in single-drug or combined transplantation, monoclonal antibody applications, and other treatments. With the formidable antitumor lineage of NK cells as a foundation, overcoming these challenges is likely to usher in groundbreaking developments in tumor treatment. The rapid evolution of NK cell-based immunotherapy (Fig. 8), reflected in the expanding cancer cell therapy pipelines [156, 157], proves that CAR-NK modifications will pave the way for new breakthroughs. In the near future, the maturation of CAR-NK cell therapy technology promises uplifting news for a broader spectrum of patients with cancer, propelling humanity closer to conquering the challenges of refractory and recurrent cancer treatments.

Clinical pipeline changes and the year-on-year growth rate of cell therapy in the past 3 years. NK natural killer, TCR T cell receptor, TIL tumor-infiltrating lymphocyte.
Data availability
The relevant information is available from the corresponding author upon reasonable request.
References
GlobalSurg Collaborative and National Institute for Health Research Global Health Research Unit on Global Surgery. Global variation in postoperative mortality and complications after cancer surgery: a multicentre, prospective cohort study in 82 countries. Lancet. 2021;397:387–97.
Albano D, Benenati M, Bruno A, Bruno F, Calandri M, Caruso D, et al. Imaging side effects and complications of chemotherapy and radiation therapy: a pictorial review from head to toe. Insights Imaging. 2021;12:76.
Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer. 2017;17:286–301.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61.
Raffin C, Vo LT, Bluestone JA. T(reg) cell-based therapies: challenges and perspectives. Nat Rev Immunol. 2020;20:158–72.
Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immunol. 2013;132:536–44.
Kärre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319:675–8.
Malmberg KJ, Carlsten M, Björklund A, Sohlberg E, Bryceson YT, Ljunggren HG. Natural killer cell-mediated immunosurveillance of human cancer. Semin Immunol. 2017;31:20–9.
Mace EM. Human natural killer cells: form, function, and development. J Allergy Clin Immunol. 2023;151:371–85.
Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–44.
Dogra P, Rancan C, Ma W, Toth M, Senda T, Carpenter DJ, et al. Tissue determinants of human NK cell development, function, and residence. Cell. 2020;180:749–763.e13.
Shannon MJ, Mace EM. Natural killer cell integrins and their functions in tissue residency. Front Immunol. 2021;12:647358.
Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov. 2020;19:200–18.
Abel AM, Yang C, Thakar MS, Malarkannan S. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. 2018;9:1869.
Cichocki F, Grzywacz B, Miller JS. Human NK cell development: one road or many? Front Immunol. 2019;10:2078.
Quatrini L, Della Chiesa M, Sivori S, Mingari MC, Pende D, Moretta L. Human NK cells, their receptors and function. Eur J Immunol. 2021;51:1566–79.
Rosmaraki EE, Douagi I, Roth C, Colucci F, Cumano A, Di Santo JP. Identification of committed NK cell progenitors in adult murine bone marrow. Eur J Immunol. 2001;31:1900–9.
Freud AG, Caligiuri MA. Human natural killer cell development. Immunol Rev. 2006;214:56–72.
Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood. 2001;97:3146–51.
Somanchi SS, Somanchi A, Cooper LJ, Lee DA. Engineering lymph node homing of ex vivo-expanded human natural killer cells via trogocytosis of the chemokine receptor CCR7. Blood. 2012;119:5164–72.
Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–40.
Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandström N, et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J Exp Med. 2019;216:2113–27.
Björkström NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood. 2010;116:3853–64.
Pende D, Falco M, Vitale M, Cantoni C, Vitale C, Munari E, et al. Killer Ig-Like Receptors (KIRs): their role in nk cell modulation and developments leading to their clinical exploitation. Front Immunol. 2019;10:1179.
Sciumè G, De Angelis G, Benigni G, Ponzetta A, Morrone S, Santoni A, et al. CX3CR1 expression defines 2 KLRG1+ mouse NK-cell subsets with distinct functional properties and positioning in the bone marrow. Blood. 2011;117:4467–75.
Ponzetta A, Sciumè G, Benigni G, Antonangeli F, Morrone S, Santoni A, et al. CX3CR1 regulates the maintenance of KLRG1+ NK cells into the bone marrow by promoting their entry into circulation. J Immunol. 2013;191:5684–94.
Ferlazzo G, Thomas D, Lin SL, Goodman K, Morandi B, Muller WA, et al. The abundant NK cells in human secondary lymphoid tissues require activation to express killer cell Ig-like receptors and become cytolytic. J Immunol. 2004;172:1455–62.
Luczo JM, Ronzulli SL, Tompkins SM. Influenza A virus hemagglutinin and other pathogen glycoprotein interactions with nk cell natural cytotoxicity receptors NKp46, NKp44, and NKp30. Viruses. 2021;13:156.
Hecht ML, Rosental B, Horlacher T, Hershkovitz O, De Paz JL, Noti C, et al. Natural cytotoxicity receptors NKp30, NKp44 and NKp46 bind to different heparan sulfate heparin sequences. J Proteome Res. 2008;8:712–20.
Horton NC, Mathew PA. NKp44 and natural cytotoxicity receptors as damage-associated molecular pattern recognition receptors. Front Immunol. 2015;6:31.
Poznanski SM, Ashkar AA. Shining light on the significance of NK cell CD56 brightness. Cell Mol Immunol. 2018;15:1071–3.
Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood. 2013;121:3599–608.
He Y, Tian Z. NK cell education via nonclassical MHC and non-MHC ligands. Cell Mol Immunol. 2017;14:321–30.
Gross C, Holler E, Stangl S, Dickinson A, Pockley AG, Asea AA, et al. An Hsp70 peptide initiates NK cell killing of leukemic blasts after stem cell transplantation. Leuk Res. 2008;32:527–34.
Bashiri Dezfouli A, Yazdi M, Benmebarek M-R, Schwab M, Michaelides S, Miccichè A, et al. CAR T cells targeting membrane-bound Hsp70 on tumor cells mimic Hsp70-Primed NK cells. Front Immunol. 2022;13:883694.
Sharifzad F, Mardpour S, Mardpour S, Fakharian E, Taghikhani A, Sharifzad A, et al. HSP70/IL-2 treated NK cells effectively cross the blood brain barrier and target tumor cells in a rat model of induced glioblastoma multiforme (GBM). Int J Mol Sci. 2020;21:2263.
Gu R, Liu F, Zou D, Xu Y, Lu Y, Liu B, et al. Efficacy and safety of CD19 CAR T constructed with a new anti-CD19 chimeric antigen receptor in relapsed or refractory acute lymphoblastic leukemia. J Hematol Oncol. 2020;13:122.
Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic Leukemia. N. Engl J Med. 2018;378:449–59.
Kamdar M, Solomon SR, Arnason J, Johnston PB, Glass B, Bachanova V, et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): results from an interim analysis of an open-label, randomised, phase 3 trial. Lancet. 2022;399:2294–308.
Jaklevic MC. CAR-T therapy is approved for non-hodgkin lymphoma. Jama. 2021;325:1032.
Mullard A. FDA approves fourth CAR-T cell therapy. Nat Rev Drug Discov. 2021;20:166.
First-Ever CAR T-cell Therapy Approved in U.S. Cancer Discov. 2017;7:OF1.
Nastoupil LJ, Jain MD, Feng L, Spiegel JY, Ghobadi A, Lin Y, et al. Standard-of-care axicabtagene ciloleucel for relapsed or refractory large B-Cell Lymphoma: Results from the US Lymphoma CAR T Consortium. J Clin Oncol. 2020;38:3119–28.
Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21:581–90.
Zhang BL, Qin DY, Mo ZM, Li Y, Wei W, Wang YS, et al. Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors. Sci China Life Sci. 2016;59:340–8.
Pan K, Farrukh H, Chittepu V, Xu H, Pan CX, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. 2022;41:119.
Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 2014;123:3855–63.
Simonetta F, Alvarez M, Negrin RS. Natural killer cells in graft-versus-host-disease after allogeneic hematopoietic Cell transplantation. Front Immunol. 2017;8:465.
Heipertz EL, Zynda ER, Stav-Noraas TE, Hungler AD, Boucher SE, Kaur N, et al. Current perspectives on “off-the-shelf” allogeneic NK and CAR-NK cell therapies. Front Immunol. 2021;12:732135.
Lamers-Kok N, Panella D, Georgoudaki AM, Liu H, Özkazanc D, Kučerová L, et al. Natural killer cells in clinical development as non-engineered, engineered, and combination therapies. J Hematol Oncol. 2022;15:164.
Yang Y, Badeti S, Tseng HC, Ma MT, Liu T, Jiang JG, et al. Superior expansion and cytotoxicity of human primary NK and CAR-NK cells from various sources via enriched metabolic pathways. Mol Ther Methods Clin Dev. 2020;18:428–45.
Laughlin MJ, Eapen M, Rubinstein P, Wagner JE, Zhang MJ, Champlin RE, et al. Outcomes after transplantation of cord blood or bone marrow from unrelated donors in adults with leukemia. N. Engl J Med. 2004;351:2265–75.
Kotylo PK, Baenzinger JC, Yoder MC, Engle WA, Bolinger CD. Rapid analysis of lymphocyte subsets in cord blood. Am J Clin Pathol. 1990;93:263–6.
Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–31.
Sarvaria A, Jawdat D, Madrigal JA, Saudemont A. Umbilical cord blood natural killer cells, their characteristics, and potential clinical applications. Front Immunol. 2017;8:329.
Dolstra H, Roeven MWH, Spanholtz J, Hangalapura BN, Tordoir M, Maas F, et al. Successful transfer of umbilical cord blood CD34(+) hematopoietic stem and progenitor-derived NK cells in older acute myeloid leukemia patients. Clin Cancer Res. 2017;23:4107–18.
Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ, et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med. 2013;2:274–83.
Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell. 2020;27:224–237.e6.
Cichocki F, Goodridge JP, Bjordahl R, Mahmood S, Davis ZB, Gaidarova S, et al. Dual antigen-targeted off-the-shelf NK cells show durable response and prevent antigen escape in lymphoma and leukemia. Blood. 2022;140:2451–62.
Allen AG, Khan SQ, Margulies CM, Viswanathan R, Lele S, Blaha L, et al. A highly efficient transgene knock-in technology in clinically relevant cell types. Nat Biotechnol. 2023; Online ahead of print.
Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature. 2017;545:229–33.
Huang RS, Shih HA, Lai MC, Chang YJ, Lin S. Enhanced NK-92 cytotoxicity by CRISPR genome engineering Using Cas9 ribonucleoproteins. Front Immunol. 2020;11:1008.
Ingegnere T, Mariotti FR, Pelosi A, Quintarelli C, De Angelis B, Tumino N, et al. Human CAR NK cells: a new non-viral method allowing high efficient transfection and strong tumor cell killing. Front Immunol. 2019;10:957.
Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994;8:652–8.
Schönfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. 2015;23:330–8.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl J Med. 2020;382:545–53.
Lopes N, Vivier E, Narni-Mancinelli E. Natural killer cells and type 1 innate lymphoid cells in cancer. Semin Immunol. 2023;66:101709.
Zhang C, Hu Y, Xiao W, Tian Z. Chimeric antigen receptor- and natural killer cell receptor-engineered innate killer cells in cancer immunotherapy. Cell Mol Immunol. 2021;18:2083–100.
Hu Z, Xu X, Wei H. The adverse impact of tumor microenvironment on NK-Cell. Front Immunol. 2021;12:633361.
Whalen KA, Rakhra K, Mehta NK, Steinle A, Michaelson JS, Baeuerle PA. Engaging natural killer cells for cancer therapy via NKG2D, CD16A and other receptors. MAbs. 2023;15:2208697.
Barrow AD, Martin CJ, Colonna M. The natural cytotoxicity receptors in health and disease. Front Immunol. 2019;10:909.
Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, et al. Cytokine activation induces human memory-like NK cells. Blood. 2012;120:4751–60.
Pinz KG, Yakaboski E, Jares A, Liu H, Firor AE, Chen KH, et al. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. Oncotarget. 2017;8:112783–96.
Drent E, Poels R, Ruiter R, van de Donk N, Zweegman S, Yuan H, et al. Combined CD28 and 4-1BB costimulation potentiates affinity-tuned chimeric antigen receptor-engineered T cells. Clin Cancer Res. 2019;25:4014–25.
Kruschinski A, Moosmann A, Poschke I, Norell H, Chmielewski M, Seliger B, et al. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc Natl Acad Sci USA. 2008;105:17481–6.
Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106:376–83.
Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–98.
Ramos CA, Heslop HE, Brenner MK. CAR-T cell therapy for lymphoma. Annu Rev Med. 2016;67:165–83.
Kulemzin SV, Matvienko DA, Sabirov AH, Sokratyan AM, Chernikova DS, Belovezhets TN, et al. Design and analysis of stably integrated reporters for inducible transgene expression in human T cells and CAR NK-cell lines. BMC Med Genom. 2019;12:44.
Song DG, Ye Q, Carpenito C, Poussin M, Wang LP, Ji C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. 2011;71:4617–27.
Jones S, Peng PD, Yang S, Hsu C, Cohen CJ, Zhao Y, et al. Lentiviral vector design for optimal T cell receptor gene expression in the transduction of peripheral blood lymphocytes and tumor-infiltrating lymphocytes. Hum Gene Ther. 2009;20:630–40.
Li L, Liu LN, Feller S, Allen C, Shivakumar R, Fratantoni J, et al. Expression of chimeric antigen receptors in natural killer cells with a regulatory-compliant non-viral method. Cancer Gene Ther. 2010;17:147–54.
Sadelain M, Rivière I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45.
Burns WR, Zhao Y, Frankel TL, Hinrichs CS, Zheng Z, Xu H, et al. A high molecular weight melanoma-associated antigen-specific chimeric antigen receptor redirects lymphocytes to target human melanomas. Cancer Res. 2010;70:3027–33.
Eitler J, Wotschel N, Miller N, Boissel L, Klingemann HG, Wels W, et al. Inability of granule polarization by NK cells defines tumor resistance and can be overcome by CAR or ADCC mediated targeting. J Immunother Cancer. 2021;9:e001334.
Batchu RB, Gruzdyn OV, Tavva PS, Kolli BK, Dachepalli R, Weaver DW, et al. Engraftment of mesothelin chimeric antigen receptor using a hybrid Sleeping Beauty/minicircle vector into NK-92MI cells for treatment of pancreatic cancer. Surgery. 2019;166:503–8.
Kim KS, Han JH, Park JH, Kim HK, Choi SH, Kim GR, et al. Multifunctional nanoparticles for genetic engineering and bioimaging of natural killer (NK) cell therapeutics. Biomaterials. 2019;221:119418.
Fujiwara K, Masutani M, Tachibana M, Okada N. Impact of scFv structure in chimeric antigen receptor on receptor expression efficiency and antigen recognition properties. Biochem Biophys Res Commun. 2020;527:350–7.
Cronk RJ, Zurko J, Shah NN. Bispecific chimeric antigen receptor T cell therapy for B cell malignancies and multiple myeloma. Cancers (Basel). 2020;12:2523.
Smith EL, Harrington K, Staehr M, Masakayan R, Jones J, Long TJ, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med. 2019;11:eaau7746.
Stoiber S, Cadilha BL, Benmebarek MR, Lesch S, Endres S, Kobold S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells. 2019;8:472.
Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5:22.
Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJW, Sievers SA, et al. Function of novel Anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther. 2017;25:2452–65.
Whitlow M, Bell BA, Feng SL, Filpula D, Hardman KD, Hubert SL, et al. An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng. 1993;6:989–95.
Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3:125–35.
Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23:181–192.e5.
MacKay M, Afshinnekoo E, Rub J, Hassan C, Khunte M, Baskaran N, et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat Biotechnol. 2020;38:233–44.
Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol. 2009;183:5563–74.
Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. 2018;11:eaat6753.
Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 2007;18:712–25.
Xu Y, Liu Q, Zhong M, Wang Z, Chen Z, Zhang Y, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol Oncol. 2019;12:49.
Huang Y, Zeng J, Liu T, Xu Q, Song X, Zeng J. DNAM1 and 2B4 costimulatory domains enhance the cytotoxicity of anti-gpc3 chimeric antigen receptor-modified natural killer cells against hepatocellular cancer cells in vitro. Cancer Manag Res. 2020;12:3247–55.
Tseng H-C, Xiong W, Badeti S, Yang Y, Ma M, Liu T, et al. Efficacy of anti-CD147 chimeric antigen receptors targeting hepatocellular carcinoma. Nat Commun. 2020;11:4810.
You F, Wang Y, Jiang L, Zhu X, Chen D, Yuan L, et al. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am J Cancer Res. 2019;9:64–78.
June CH, Blazar BR, Riley JL. Engineering lymphocyte subsets: tools, trials and tribulations. Nat Rev Immunol. 2009;9:704–16.
Suerth JD, Labenski V, Schambach A. Alpharetroviral vectors: from a cancer-causing agent to a useful tool for human gene therapy. Viruses. 2014;6:4811–38.
Suerth JD, Morgan MA, Kloess S, Heckl D, Neudörfl C, Falk CS, et al. Efficient generation of gene-modified human natural killer cells via alpharetroviral vectors. J Mol Med (Berl). 2016;94:83–93.
Shimasaki N, Fujisaki H, Cho D, Masselli M, Lockey T, Eldridge P, et al. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy. 2012;14:830–40.
Clara JA, Levy ER, Reger R, Barisic S, Chen L, Cherkasova E, et al. High-affinity CD16 integration into a CRISPR/Cas9-edited CD38 locus augments CD38-directed antitumor activity of primary human natural killer cells. J Immunother Cancer. 2022;10:e003804.
Naeimi Kararoudi M, Nagai Y, Elmas E, de Souza Fernandes Pereira M, Ali SA, Imus PH, et al. CD38 deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood. 2020;136:2416–27.
Gurney M, Stikvoort A, Nolan E, Kirkham-McCarthy L, Khoruzhenko S, Shivakumar R, et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica. 2022;107:437–45.
Afolabi LO, Adeshakin AO, Sani MM, Bi J, Wan X. Genetic reprogramming for NK cell cancer immunotherapy with CRISPR/Cas9. Immunology. 2019;158:63–9.
Valeri A, García-Ortiz A, Castellano E, Córdoba L, Maroto-Martín E, Encinas J, et al. Overcoming tumor resistance mechanisms in CAR-NK cell therapy. Front Immunol. 2022;13:953849.
Kilgour MK, Bastin DJ, Lee S-H, Ardolino M, McComb S, Visram A. Advancements in CAR-NK therapy: lessons to be learned from CAR-T therapy. Front Immunol. 2023;14:1166038.
Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, et al. The mechanism of Anti-PD-L1 antibody efficacy against PD-L1-negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discov. 2019;9:1422–37.
Klopotowska M, Bajor M, Graczyk-Jarzynka A, Kraft A, Pilch Z, Zhylko A, et al. PRDX-1 supports the survival and antitumor activity of primary and CAR-modified NK cells under oxidative stress. Cancer Immunol Res. 2022;10:228–44.
Ranti D, Bieber C, Wang YS, Sfakianos JP, Horowitz A. Natural killer cells: unlocking new treatments for bladder cancer. Trends Cancer. 2022;8:698–710.
Menon T, Gopal S, Rastogi Verma S. Targeted therapies in non-small cell lung cancer and the potential role of AI interventions in cancer treatment. Biotechnol Appl Biochem. 2023;70:344–56.
Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–12.
Strecker MI, Wlotzka K, Strassheimer F, Roller B, Ludmirski G, König S, et al. AAV-mediated gene transfer of a checkpoint inhibitor in combination with HER2-targeted CAR-NK cells as experimental therapy for glioblastoma. Oncoimmunology. 2022;11:2127508.
Xia W, Chen J, Hou W, Chen J, Xiong Y, Li H, et al. Engineering a HER2-CAR-NK cell secreting soluble programmed cell death protein with superior antitumor efficacy. Int J Mol Sci. 2023;24:6843.
Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013;31:413–41.
Zhong J, Yang X, Chen J, He K, Gao X, Wu X, et al. Circular EZH2-encoded EZH2-92aa mediates immune evasion in glioblastoma via inhibition of surface NKG2D ligands. Nat Commun. 2022;13:4795.
Daher M, Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr Opin Immunol. 2018;51:146–53.
Huang R, Wen Q, Zhang X. CAR-NK cell therapy for hematological malignancies: recent updates from ASH 2022. J Hematol Oncol. 2023;16:35.
Roex G, Campillo-Davo D, Flumens D, Shaw PAG, Krekelbergh L, De Reu H, et al. Two for one: targeting BCMA and CD19 in B-cell malignancies with off-the-shelf dual-CAR NK-92 cells. J Transl Med. 2022;20:124.
Reiser J, Chan SR, Mathavan K, Sillitti D, Mottershead C, Mattson B, et al. FT555: Off-the-Shelf CAR-NK Cell Therapy Co-Targeting GPRC5D and CD38 for the Treatment of Multiple Myeloma. Blood. 2022;140:4560–1.
Klöß S, Oberschmidt O, Morgan M, Dahlke J, Arseniev L, Huppert V, et al. Optimization of human NK cell manufacturing: fully automated separation, improved Ex Vivo expansion Using IL-21 with autologous feeder cells, and generation of Anti-CD123-CAR-expressing effector cells. Hum Gene Ther. 2017;28:897–913.
De Propris MS, Raponi S, Diverio D, Milani ML, Meloni G, Falini B, et al. High CD33 expression levels in acute myeloid leukemia cells carrying the nucleophosmin (NPM1) mutation. Haematologica. 2011;96:1548–51.
Li KX, Wu HY, Pan WY, Guo MQ, Qiu DZ, He YJ, et al. A novel approach for relapsed/refractory FLT3(mut+) acute myeloid leukaemia: synergistic effect of the combination of bispecific FLT3scFv/NKG2D-CAR T cells and gilteritinib. Mol Cancer. 2022;21:66.
Caruso S, De Angelis B, Del Bufalo F, Ciccone R, Donsante S, Volpe G, et al. Safe and effective off-the-shelf immunotherapy based on CAR.CD123-NK cells for the treatment of acute myeloid leukaemia. J Hematol Oncol. 2022;15:163.
Nixdorf D, Sponheimer M, Berghammer D, Engert F, Bader U, Philipp N, et al. Adapter CAR T cells to counteract T-cell exhaustion and enable flexible targeting in AML. Leukemia. 2023;37:1298–310.
Jin X, Zhang M, Sun R, Lyu H, Xiao X, Zhang X, et al. First-in-human phase I study of CLL-1 CAR-T cells in adults with relapsed/refractory acute myeloid leukemia. J Hematol Oncol. 2022;15:88.
Hebbar N, Epperly R, Vaidya A, Thanekar U, Moore SE, Umeda M, et al. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat Commun. 2022;13:587.
Zhao Y, Zhou X. Engineering chimeric antigen receptor-natural killer cells for cancer immunotherapy. Immunotherapy. 2020;12:653–64.
Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine. 2020;59:102975.
Pan K, Farrukh H, Chittepu VCSR, Xu H, Pan CX, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. 2022;41:119.
Tang X, Yang L, Zheng L, Ansel PN, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;6:1083–9.
Stravokefalou V, Stellas D, Karaliota S, Nagy BA, Valentin A, Bergamaschi C, et al. Heterodimeric IL-15 (hetIL-15) reduces circulating tumor cells and metastasis formation improving chemotherapy and surgery in 4T1 mouse model of TNBC. Front Immunol. 2022;13:1014802.
Pedroza-Pacheco I, Madrigal A, Saudemont A. Interaction between natural killer cells and regulatory T cells: perspectives for immunotherapy. Cell Mol Immunol. 2013;10:222–9.
Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res. 2011;17:678–89.
Carlsten M, Levy E, Karambelkar A, Li L, Reger R, Berg M, et al. Efficient mRNA-Based Genetic Engineering of Human NK Cells with High-Affinity CD16 and CCR7 Augments Rituximab-Induced ADCC against Lymphoma and Targets NK Cell Migration toward the Lymph Node-Associated Chemokine CCL19. Front Immunol. 2016;7:105.
Ng YY, Tay JCK, Wang S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol Ther Oncolytics. 2020;16:75–85.
Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16:7–19.
Morgan MA, Büning H, Sauer M, Schambach A. Use of cell and genome modification technologies to generate improved “Off-the-Shelf” CAR T and CAR NK cells. Front Immunol. 2020;11:1965.
Sun H, Sun C. The rise of NK cell checkpoints as promising therapeutic targets in cancer immunotherapy. Front Immunol. 2019;10:2354.
Yang YW, Hsieh YC. Protamine sulfate enhances the transduction efficiency of recombinant adeno-associated virus-mediated gene delivery. Pharm Res. 2001;18:922–7.
Han X, Wang Y, Wei J, Han W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J Hematol Oncol. 2019;12:128.
Di S, Li Z. Treatment of solid tumors with chimeric antigen receptor-engineered T cells: current status and future prospects. Sci China Life Sci. 2016;59:360–9.
Fabian KP, Padget MR, Donahue RN, Solocinski K, Robbins Y, Allen CT, et al. PD-L1 targeting high-affinity NK (t-haNK) cells induce direct antitumor effects and target suppressive MDSC populations. J Immunother Cancer. 2020;8:e000450.
Wang J, Lupo KB, Chambers AM, Matosevic S. Purinergic targeting enhances immunotherapy of CD73(+) solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J Immunother Cancer. 2018;6:136.
Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, et al. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS One. 2018;13:e0191358.
Nayyar G, Chu Y, Cairo MS. Overcoming resistance to natural killer cell based immunotherapies for solid tumors. Front Oncol. 2019;9:51.
Minagawa K, Jamil MO, Al-Obaidi M, Pereboeva L, Salzman D, Erba HP, et al. In vitro pre-clinical validation of suicide gene modified Anti-CD33 redirected chimeric antigen receptor T-Cells for acute myeloid leukemia. PLoS One. 2016;11:e0166891.
Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6:261ra151.
Upadhaya S, Yu JX, Shah M, Correa D, Partridge T, Campbell J. The clinical pipeline for cancer cell therapies. Nat Rev Drug Discov. 2021;20:503–4.
Saez-Ibanez AR, Upadhaya S, Partridge T, Shah M, Correa D, Campbell J. Landscape of cancer cell therapies: trends and real-world data. Nat Rev Drug Discov. 2022;21:631–2.
Funding
This research was supported by Yunnan Fundamental Research Projects, China (Grant no. 202201AT070198) and the National Natural Science Foundation of China (Grant no. 82260038).
Author information
Authors and Affiliations
Contributions
BZ, NY and TR conceived and contributed to the writing of the manuscript. MY, WZ, NL, DW, LJ, NX contributed to the writing of the manuscript. BZ, NY and TR revised and wrote the manuscript. All authors reviewed the manuscript. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Yufang Shi
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, B., Yang, M., Zhang, W. et al. Chimeric antigen receptor-based natural killer cell immunotherapy in cancer: from bench to bedside. Cell Death Dis 15, 50 (2024). https://doi.org/10.1038/s41419-024-06438-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-024-06438-7
