
Abstract
Aspirin and its active metabolite salicylate have emerged as promising agents for the chemoprevention of colorectal cancer (CRC). Moreover, aspirin suppresses the progression of established CRCs. However, the underlying molecular mechanisms are not completely understood. Here we found that salicylate induces the expression of the miR-34a and miR-34b/c genes, which encode tumor suppressive microRNAs, in a p53-independent manner. Salicylate activated AMPK, thereby activating NRF2, which directly induced miR-34a/b/c expression via ARE motifs. In addition, salicylate suppressed c-MYC, a known repressor of NRF2-mediated transactivation, via activating AMPK. The suppression of c-MYC by salicylate was necessary for NRF2-mediated activation of miR-34a/b/c. Inactivation of miR-34a/b/c largely abrogated the inhibitory effects of salicylate on migration, invasion and metastasis formation by CRC cells. In the future, aspirin and its derivates may be used therapeutically to activate miR-34a and miR-34b/c in tumors that have lost p53.

Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed malignancy with more than 1.9 million annual diagnoses worldwide and the second leading cause of cancer mortality, responsible for more than 900,000 deaths [1]. Colonoscopy-based screening strategies have demonstrated their potential for decreasing the incidence and mortality of CRC. Another strategy to reduce CRC occurrence is the use of chemo-preventive drugs. Among them, aspirin is perhaps the most promising substance [2].
Aspirin, also known as acetylsalicylic acid, is a nonsteroidal anti-inflammatory drug primarily known for its analgesic and antipyretic actions [3]. However, several decades of research also provided considerable evidence demonstrating its potential for the prevention of cancer, particularly CRC [2]. A pooled analysis of 10 case–control studies with more than 8000 CRC cases demonstrated that intake of aspirin was associated with a 29% reduction in the incidence of CRC (odds ratio (OR) = 0.71; 95% confidence interval (CI) 0.66–0.77) [4]. Another case-control meta-analysis of 17 studies with more than 12000 CRC cases showed that regular use of aspirin was associated with reduced risk for CRC (OR = 0.52, 95% CI 0.58–0.67). Furthermore, a meta-analysis of four randomized controlled trials with more than 14000 CRC patients found that aspirin treatment for 5 or more years at doses of 75–300 mg per day reduced the long-term risk of CRC by 24% (hazard ratio (HR) = 0.75; 95% CI 0.56–0.97) [5]. In 2015 the United States Preventive Services Task Force endorsed aspirin as the first pharmacological agent for cancer chemoprevention in a population not characterized as having a high risk of developing cancer [2]. In addition to cancer prevention, several studies have shown that aspirin use after CRC diagnosis was associated with reduced mortality [3]. A recent meta-analysis of 27 studies with more than 230,000 CRC patients showed that aspirin use after diagnosis was associated with an improvement in CRC-specific survival (HR = 0.74 (95% CI: 0.62–0.89) and overall survival (HR = 0.82 (95% CI: 0.74–0.90) [6]. CRC patients treated with a daily low-dose aspirin are less likely to develop advanced stage CRC, suggesting that aspirin affects the progression of established CRCs [7]. Following oral consumption, aspirin is absorbed mainly in the stomach and upper small intestine [8]. The half-life of aspirin is about 20 minutes as it undergoes hydrolysis to generate salicylic acid and salicylate [8, 9].
The molecular mechanisms by which aspirin decreases CRC initiation and progression are incompletely understood. Aspirin acts directly on CRC cells and indirectly via affecting the tumor microenvironment. Aspirin inhibits COX enzymes in epithelial and stromal cells, which reduces the synthesis of prostaglandins [10]. Aspirin also inhibits the Wnt/β-catenin pathway, which is the oncogenic driver in the majority of CRCs [11]. Activated Wnt signaling induces the expression of the oncogene c-MYC. Consistently, it has been shown that aspirin down-regulates c-MYC levels in numerous cancer entities [12, 13].
miR-34a and miR-34b/c represent p53-inducible genes, which encode microRNAs with tumor-suppressive properties [14, 15]. They are frequently down-regulated in human CRCs by methylation [16]. The loss of miR-34a promotes CRC development in mouse models of sporadic CRC (6xAOM treatment [17]), colitis-associated CRC (AOM/DSS treatment [18]), and inherited CRC (ApcMin/+ mice [19]). Therefore, down-regulation of miR-34a does not only commonly occur during intestinal carcinogenesis, but is also causally involved in CRC formation.
Here we hypothesized that miR-34a and miR-34b/c may represent mediators of the suppressive effects of aspirin on CRC cells. Indeed, we found, that salicylate, the main metabolite of aspirin, induces the expression of miR-34a and miR-34b/c in a p53-independent manner via activating of AMP-activated protein kinase (AMPK), which activates nuclear factor erythroid 2–related factor 2 (NRF2). Furthermore, we demonstrated that the suppression of c-MYC by salicylate is required for NRF2-mediated activation of miR-34a expression. The induction of miR-34a was necessary for multiple tumor suppressive effects of salicylate, including the repression of cell viability, migration, invasion, and metastasis, as well as for the induction of apoptosis and mesenchymal-to-epithelial transition (MET). In the future, this knowledge may be used to activate miR-34 function in tumors, which have lost p53 function.
Results
Salicylate inhibits cell viability and induces apoptosis in CRC cells
It has been previously shown that salicylate, which is the active metabolite of aspirin, represses the viability of CRC cells [20]. In order to systematically investigate whether p53 mediates the effect of salicylate on CRC cells, we treated HCT116 cells harboring wild-type p53 and isogenic HCT116 cells with homozygous deletion of p53 with increasing concentrations of salicylate for 48 h. p53-proficient and p53-deficient HCT116 cells displayed similar IC50 values (Fig. 1A). Based on the IC50 values and previous publications [21,22,23,24], in which CRC cells are treated with salicylate, a concentration of 5 mM was chosen for the subsequent experiments. Notably, this concentration of salicylate did not show obvious cytotoxicity in the non-transformed human colonic epithelial cells (HCEC-1CT) and human intestinal fibroblasts (CCD-18Co) (Fig. 1B), indicating that non-transformed epithelial cells and fibroblasts in the intestine may be resistant to the effects of salicylate. In contrast, salicylate strongly inhibited proliferation of p53-proficient and p53-deficient HCT116 CRC cells as determined by impedance measurements (Fig. 1C). The changes in cell numbers were confirmed at the final time point (Fig. 1D). Salicylate treatment resulted in an increase of cells in the G0/G1-phases and a decrease of cells in S- and G2/M-phases, irrespective of their p53 status (Fig. 1E and Supplementary Fig. S1A). The increase of cells in the sub-G1-phase indicated that salicylate induced apoptosis, which was more prominent in p53-deficient than in p53-proficient HCT116 cells as determined by flow-cytometric analysis of Annexin V/PI staining (Fig. 1F and Supplementary Fig. S1B). These results were confirmed by detection of cleaved-PARP protein levels (Fig. 1G). Similar effects were observed in p53-proficient or p53-deficient RKO CRC cell lines (Supplementary Fig. S1C). Altogether, these results demonstrate that salicylate represses proliferation and induces apoptosis in a p53-independent manner in CRC cells.

A Cell viability of HCT116 cells was determined by MTT assay after treatment with indicated concentrations of salicylate for 48 h. IC50 was determined using GraphPad Prism based on changes in viability. B Cell viability of non-transformed CCD-18-Co and HCEC-1CT cells was determined by MTT assay after treatment with 5 mM salicylate for 48 h. C Impedance of HCT116 cells treated with salicylate. D Determination of cell numbers at the final time point of the experiment indicated in C. E Cell cycle analysis using propidium iodide (PI) staining. F Analysis of apoptosis in salicylate-treated HCT116 cells by Annexin V FITC and propidium iodide staining. G Levels of cleaved PARP and PARP were determined by Western blot analysis in HCT116 cells after treatment with 5 mM salicylate for the indicated times. α-Tubulin served as a loading control. In panels A, B, and F (n = 3), and D (n = 4) mean values ± SD are shown. *p < 0.05, ****p < 0.0001, n.s not significant.
Salicylate inhibits migration, invasion and induces MET in CRC cells
Next, we evaluated the effects of salicylate on cell migration and invasion. Salicylate repressed cell migration and invasion in p53-proficient and p53-deficient HCT116 cells (Fig. 2A, B). Similar results were observed in p53-proficient and p53-deficient RKO CRC cell lines (Supplementary Fig. S2A, S2B). Salicylate treatment resulted in a switch from a mesenchymal to an epithelial morphology in p53-proficient and p53-deficient HCT116 cells (Fig. 2C), indicating a mesenchymal-to-epithelial transition (MET). Indeed, treatment of p53-proficient or p53-deficient HCT116 cells with salicylate resulted in the repression of the mesenchymal markers Vimentin (VIM) and SNAIL, while the expression of the epithelial marker E-cadherin/CDH1 was increased on mRNA and protein levels (Fig. 2D–H). Similar results were observed in p53-proficient or p53-deficient RKO CRC cell lines (Supplementary Fig. S2C–S2E). Taken together, these results show that salicylate suppresses migration and invasion, and induces MET in CRC cells in a p53-independent manner.

A Wound healing assay. The scratch width was determined 24 h after the indicated treatment (left panel). Results represent the mean (%) of wound closure (right panel). B Determination of invasion by a Modified Boyden chamber assay. After treatment for 48 h, cells that invaded through the matrigel were counted after crystal violet staining. C Phase-contrast images showing cell morphology of HCT116 cells treated with 5 mM salicylate for 48 h. Scale bar: 50 μm. qPCR analysis of CDH1 (D), VIM (E), and SNAIL (F) expression in HCT116 cells after the indicated treatments for 48 h. H Western blot analysis of protein expression after salicylate treatment for the indicated periods. In panels A–F (n = 3), mean values ± SD are shown. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Salicylate up-regulates miR-34a and miR-34b/c independent of p53
It has been previously shown that miR-34a and miR-34b/c suppress cell viability, migration and invasion, and induce apoptosis and MET [25]. Therefore, miR-34a/b/c represent candidate mediators of the effects of salicylate and we analyzed whether they are induced by salicylate. Indeed, treatment with salicylate induced the expression of pri-miR-34a, mature miR-34a, and pri-miR-34b/c in three CRC cell lines with wildtype p53 and also in three p53 mutant CRC cell lines (Fig. 3A–C). Furthermore, salicylate induced the expression of primary pri-miR-34a and pri-miR-34b/c in a dose- and time-dependent manner in HCT116 cells with deletion of p53 in a similar manner as in HCT116 wt p53 cells (Fig. 3D–F). Similar results were obtained in wt and KO p53 RKO cells (Supplementary Fig. S3A, B). The levels of mature miR-34a were also up-regulated by salicylate in p53-deficient HCT116 and RKO cells (Fig. 3G and Supplementary Fig. S3C). Therefore, salicylate induces miR-34a and miR-34b/c expression in CRC cell lines in a p53-independent manner.

qPCR analysis of pri-miR-34a (A), mature miR-34a (B), and pri-miR-34b/c (C) expression after treatment of the indicated cells with salicylate for 48 h. D qPCR analysis of pri-miR-34a expression after treatment with the indicated concentration of salicylate for 48 h. qPCR analysis of pri-miR-34a (E) and pri-miR-34b/c (F) expression after treatment with salicylate for the indicated periods. G qPCR analysis of mature miR-34a expression after treatment with salicylate for 48 h. In panels A–G (n = 3), mean values ± SD are shown. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s: not significant.
miR-34a and miR-34b/c mediate the effects of salicylate in CRC cells
To determine whether the induction of miR-34a and miR-34b/c expression mediates the effects of salicylate, we analyzed isogenic wt and miR-34a- or miR-34a/b/c-deficient HCT116 cells, that were recently generated using a CRISPR/CAS9 approach and characterized by us [26]. After treatment with salicylate for 48 h the viability of miR-34a- or miR-34a/b/c-deficient HCT116 cells was higher than that of miR-34-proficient HCT116 cells (Fig. 4A). Consistently, miR-34a- or miR-34a/b/c-deficiency resulted in an attenuated induction of apoptosis by salicylate in HCT116 cells (Fig. 4B and Supplementary Fig. S4A). Furthermore, inhibition of migration and invasion by salicylate was significantly lower in miR-34a and miR-34a/b/c-deficient cells when compared to miR-34-proficient HCT116 cells (Fig. 4C, D, Supplementary Fig. S4B, S4C). Finally, the deletion of miR-34a or miR-34a/b/c partly abolished salicylate-induced MET, as salicylate treatment resulted in attenuated induction of CDH1 and decreased suppression of VIM and SNAIL in miR-34a−/− and miR-34a/b/c−/− HCT116 cells (Fig. 4E–H). Taken together, our results demonstrate that miR-34a and miR-34b/c mediate the effects of salicylate on cell viability, apoptosis, migration, invasion, and MET to a large extent.

A Treatment of the indicated CRC cell lines with increasing concentrations of salicylate for 48 h. IC50 was determined by MTT assay. B After treatment with salicylate for the indicated periods apoptosis was determined by quantification of Annexin V-FITC and PI staining. C Migration was assessed by wound-healing assay 24 h after salicylate treatment. D Invasion was analyzed using a modified Boyden chamber assay 48 h after salicylate treatment. qPCR analysis of CDH1 (E), VIM (F), and SNAIL (G) expression in HCT116 cells after the indicated treatments for 48 h. H Western blot analysis of E-cadherin and SNAIL protein levels after salicylate treatment for the indicated periods. In panels A–G (n = 3), mean values ± SD are shown.
Salicylate inhibits lung metastases formation through induction of miR-34a
Next, we aimed to determine the effect of salicylate on metastases formation of CRC cells in mice. For this we utilized SW620 CRC cells stably expressing luciferase (SW620-Luc2). We have previously shown that mature miR-34b and miR-34c are expressed at very low levels in SW620-Luc2 cells when compared to mature miR-34a [27]. Therefore, we focused our analyses on the inhibition of miR-34a. First, we confirmed that salicylate repressed migration and invasion in SW620-Luc2 cells (Fig. 5A, B, Supplementary Fig. S5A, S5B). Notably, this effect was abolished by miR-34a-specific antagomirs. Next, SW620-Luc2 cells were treated with salicylate or/and miR-34a-specific antagomirs for 48 h. After washing cells with HBSS for 3 times and counting the number of living cells (using trypan blue), 4 ×106 cells were injected into the lateral tail vein of NOD/SCID mice. Longitudinal, non-invasive imaging revealed that treatment of SW620-Luc2 cells with salicylate completely abolished lung metastasis formation within 5 weeks after injection (Fig. 5C, D). However, concomitant treatment with miR-34a-specific antagomirs partially restored metastasis formation after salicylate treatment (Fig. 5C, D). Five weeks after injection, excised lungs showed no macroscopic metastasis in mice injected with salicylate-treated SW620-Luc2 cells (Fig. 5E). Hematoxylin and eosin (H&E) staining confirmed the absence of metastatic nodules in mice injected with salicylate-treated cells (Fig. 5E, F). However, SW620-Luc2 cells concomitantly treated with miR-34a-antagonist and salicylate developed lung metastases (Fig. 5E, F). Therefore, salicylate suppresses metastases formation by inducing miR-34a in xenografted CRC cells.

Analysis of migration (A) and invasion (B) after transfection of SW620-Luc2 cells with miR-34a antagomirs and/or treatment with salicylate for 72 h. C–F SW620-Luc2 cells were treated for 48 h as indicated and then injected into the tail vein of NOD/SCID mice. Representative images of luciferase signals (C) and quantification of total photon flux (D) at indicated time points after xenografting. E left: representative lungs 5 weeks after tail vein injection. Arrows indicate metastatic tumor nodules. right: representative images of H&E-stained resected lungs. Scale bar: 500 μm; 50 μm (insert). F Quantification of metastatic nodules in the lungs of the indicated mice. In panels A, B, D, and F (n = 3), mean values ± SD are shown. *p < 0.05, **p < 0.01, ***p < 0.001.
The induction of miR-34a/b/c by salicylate is mediated by NRF2
We recently showed that curcumin induces miR-34a and miR-34b/c expression via the activation of NRF2 independent of p53 [27]. Here, salicylate also induced the expression of NRF2 and NQO1, which is a conserved NRF2 target gene and can serve to monitor the activity of the NRF2 pathway (Fig. 6A–C). Furthermore, NRF2 protein accumulated and translocated to the nucleus after treatment with salicylate, confirming that salicylate activates NRF2 (Fig. 6D, E). We have previously identified three NRF2 binding sites (TGAG/CnnnGC), so-called AREs (Antioxidant Response Elements) in the miR-34a promoter and one in the miR-34b/c promoter (Fig. 6F) [27]. A qChIP assay confirmed enhanced NRF2 occupancy at these four ARE sites within the miR-34a and miR-34b/c promoters after salicylate treatment for 48 h (Fig. 6G). Ectopic NRF2 expression increased pri-miR-34a and pri-miR-34b/c expression, as well as mature miR-34a and NQO1 mRNA expression, in p53-deficient HCT116 cells (Fig. 6H). Conversely, silencing of NRF2 by a pool of 4 different siRNAs prevented the activation of pri-miR-34a and pri-miR-34b/c after treatment with salicylate (Fig. 6I, J). The inhibitory effect of salicylate on migration, invasion, and EMT was also partly prevented after silencing NRF2 (Supplementary Fig. S6A–S6B, Fig. 6K). We have recently reported, that curcumin activates NRF2 via generating reactive oxygen species/ROS in CRC cells [27]. However, ROS levels were not increased after treatment with salicylate (Fig. 6L). Moreover, addition of the ROS-inhibitor N-acetyl-cystein/NAC did not prevent the induction of pri-miR-34a and pri-miR-34b/c expression by salicylate in p53-proficient or p53-deficient HCT116 cells (Supplementary Fig. S6C, D). In summary, these results show that salicylate induces miR-34a and miR-34b/c expression by activating NRF2 via a ROS-independent mechanism.

mRNA (A, B) and protein (C) levels of NRF2 and NQO1 were determined by qPCR and Western blot analysis, respectively. D Immunofluorescence was used to detect the location of NRF2 protein after the indicated treatments for 48 h. Nuclear DNA was detected by DAPI. The scale bar represents 20 µm. E Western blot analysis of NRF2 protein levels in cytoplasmic and nuclear cellular fractions after salicylate treatment. F Map of human miR-34a and miR-34b/c genomic regions with NRF2 binding sites A–D and respective sequences depicted. G qChIP analysis of NRF2 occupancy at the human miR-34a and miR-34b/c genomic regions in HCT116 p53−/− cells 48 h after the indicated treatments. NQO1 and 16q22 served as positive and negative controls, respectively. H qPCR analysis of indicated mRNAs in p53-deficient HCT116 cells after transfection with empty pcDNA3.1 (NC) or NRF2 pcDNA3.1 vectors for 72 h. qPCR analysis of pri-miR-34a (I) and pri-miR-34b/c (J) expression after indicated treatment and/or transfection with the indicated siRNA pools for 24 h in p53-deficient HCT116 cells. K Western blot analysis of NQO1 and SNAIL protein expression after treatment and/or transfection with siRNA pools for 24 h in p53-deficient HCT116 cells. L Analysis of ROS formation in HCT116 cells treated as indicated, left panel: representative fluorescence intensity pictures. Scale bar: 100 µm. right panel: quantification of fluorescence intensity. In panels A, B, G–J, and L (n = 3), mean values ± SD are shown. *p < 0.05, **p < 0.01, ***p < 0.001, n.s not significant.
Salicylate activates NRF2 via AMPK in CRC cells
Next, we studied alternative modes of NRF2 activation, that would explain how salicylate induces miR-34 expression. Previously, AMPK was reported to phosphorylate and activate NRF2 [28]. Interestingly, salicylate directly binds to AMPK in the cleft between the β-CBM and the α-KD N-lobe, which causes both allosteric activation and protection against Thr172 dephosphorylation of AMPK [29]. Here, salicylate activated AMPK, which was largely prevented by the AMPK inhibitor dorsomorphin (Fig. 7A). Furthermore, the induction of NRF2 and NQO1, which is encoded by a NRF2 target gene, by salicylate was largely prevented by dorsomorphin (Fig. 7A). Finally, the induction of pri-miR-34a by salicylate was completely abolished by dorsomorphin (Fig. 7B). Also, suppression of AMPKα1 by siRNA pools attenuated the induction of NRF2, NQO1, NQO1 mRNA, and pri-miR-34a by salicylate (Fig. 7C–E). Furthermore, suppression of AMPKβ1 by siRNA pools also attenuated the induction of NRF2, NQO1 and pri-miR-34a by salicylate (Fig. 7F–H). Similar results were observed in RKO cells (Supplementary Fig. S7A–H). Taken together, these results demonstrate that salicylate activates NRF2 and induces miR-34a via activating AMPK in CRC cells.

A Western blot analysis of indicated proteins in p53-deficient HCT116 cells treated with salicylate and/or 10 μm dorsomorphin for 24 h. β-actin served as a loading control. B qPCR analysis of pri-miR-34a expression after treatment with salicylate and/or 10 μm dorsomorphin for 24 h. C Western blot analysis of indicated proteins 48 h after transfection with AMPK α1 siRNA or control siRNA pools with or without salicylate for the last 24 h. β-actin served as a loading control. qPCR analysis of NQO1 (D) and pri-miR-34a (E) expression 48 h after transfection with AMPK α1-specific siRNA pools or control siRNA pools with or without salicylate for the last 24 h. F Western blot analysis of indicated proteins after transfection with AMPK β1-specific siRNA or control siRNA pools for 48 h with or without salicylate for 24 h. β-actin served as a loading control. qPCR analysis of NQO1 (G) and pri-miR-34a (H) expression after indicated treatments with siRNA-pools and/or salicylate for 24 h. In panels B, D, E, G, and H (n = 3), mean values ± SD are shown. ***p < 0.001, n.s not significant.
c-MYC abrogates NRF2 activation and miR-34a/b/c induction by salicylate
It has been shown that salicylate suppresses c-MYC expression at both transcriptional and post-transcriptional levels [12]. Furthermore, c-MYC was shown to interact with NRF2, thereby preventing the binding of NRF2 to ARE elements and induction of NRF2 target genes [30]. Therefore, we asked whether a down-regulation of c-MYC is involved in the regulation of NRF2 activity by salicylate. First, we analyzed 13 public Gene Expression Omnibus (GEO) datasets for the effects of ectopic expression or knockdown/knockout of c-MYC in cell lines or mice on NQO1 expression. Notably, the NRF2 target gene NQO1 was repressed after ectopic expression of c-MYC and up-regulated after repression of c-MYC in the majority of these GEO studies (Fig. 8A). Treatment with salicylate repressed c-MYC at the mRNA and protein levels in a p53-independent manner (Fig. 8B, C). The suppression of c-MYC by salicylate was completely abolished by the AMPK inhibitor dorsomorphin or AMPKα1/AMPKβ1-specific siRNA pools (Fig. 8D–F and Supplementary Fig. S8A–C), demonstrating that salicylate suppresses the expression of c-MYC or the levels of c-MYC via AMPK. The induction of NRF2, NQO1, pri-miR-34a and pri-miR-34b/c by salicylate was largely abolished after DOX-mediated activation of a conditional c-MYC allele in DLD-1/pRTR-c-MYC-VSV cells (Fig. 8G–I and Supplementary Fig. S8D, E). Conversely, suppression of c-MYC by specific siRNA pools resulted in elevated expression of NQO1 protein and pri-miR-34a (Fig. 8J, K). In summary, these results imply that the suppression of c-MYC is necessary for the activation of NRF2 and induction of miR-34a and miR-34b/c by salicylate.

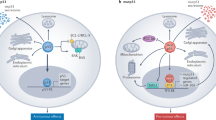
A Fold changes in NQO1 expression in GEO datasets representing studies of ectopic expression or silencing of c-MYC in the indicated cell lines. qPCR and Western blot analysis of c-MYC mRNA (B) and protein (C) levels after treatment with salicylate for the indicated periods. D Western blot analysis of c-MYC protein levels in p53-deficient HCT116 cells treated with salicylate and/or 10 μM dorsomorphin for 24 h. β-actin served as a loading control. Western blot analysis of indicated proteins after transfection with AMPK α1 (E) or AMPK β1 (F) siRNA pools or control siRNA pool for 48 h with or without salicylate for 24 h. β-actin served as a loading control. G Western blot analysis of c-MYC, NRF2, and NQO1 protein levels in DLD-1/pRTR-c-MYC-VSV cells after treatment with salicylate or/and DOX (c-MYC on) for 48 h. qPCR analyses of pri-miR-34a (H) and pri-miR-34b/c (I) expression in DLD-1/pRTR-c-MYC-VSV cells treated with salicylate or/and DOX (c-MYC on) for 48 h. J Western blot analysis of c-MYC, NRF2, and NQO1 protein levels in DLD-1 cells after transfection c-MYC siRNA pool or control siRNA pool for 48 h. K qPCR analysis of pri-miR-34a expression in DLD-1 cells after transfection c-MYC siRNA pool or control siRNA pool for 48 h. L Schematic model of the findings obtained in this study. In panels B, H, I, and K (n = 3) mean values ± SD are shown. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
Here we characterized the activation of an AMPK-NRF2-miR-34a/b/c axis by salicylate as a novel mechanism of salicylate-mediated suppression of CRC (see Fig. 8L). Our results suggest that this mechanism plays a central role in mediating the tumor suppressive effects of salicylate since the deletion or suppression of miR-34a/b/c prevented the salicylate-mediated suppression of CRC cell viability, migration, invasion, and metastasis. We recently showed that curcumin induces miR-34a/b/c expression via the activation of NRF2 [27]. NRF2 is a transcription factor that regulates the cellular response to stress [31]. Under normal conditions, NRF2 is rapidly degraded and sequestered in the cytoplasm by a KEAP1-CUL3-RBX1 complex [32]. In response to stress NRF2 is released from the complex and translocates to the nucleus, where it binds to ARE motifs and induces the expression of corresponding genes [33]. The activation of NRF2 by curcumin was mediated by the induction of reactive oxygen species (ROS). Here we found that salicylate also activates NRF2. However, salicylate achieves this by activating AMPK in the absence of any detectable generation of ROS. Since ROS may cause DNA damage, which may lead to cell death or induce mutations, not only in cancer cells, but also in normal cells, using salicylate as a chemo-preventive drug may have less side effects than curcumin.
AMPK is a sensor of cellular energy status and a regulator of energy balance [34]. AMPK is composed of a catalytic α subunit, as well as the regulatory β and γ subunits [35]. It has been shown that salicylate binds to AMPK between the α and β subunits [36], which causes a conformational change that promotes the phosphorylation of Thr172 by upstream kinases while inhibiting dephosphorylation by phosphatases [35]. The activation of AMPK by salicylate was abolished by the loss or mutation of the AMPK β1 subunit [37]. Our results show that both AMPK α1 and AMPK β1 subunits are important for the induction of miR-34a/b/c by salicylate, since the suppression of either subunit prevented the activation of NRF2 and the induction of miR-34a/b/c.
It has previously been shown that salicylate suppresses the expression of c-MYC [12]. However, the mechanism remained unknown. Here, the repression of c-MYC by salicylate was p53-independent. The inhibition of AMPK by siRNA or chemical inhibitors abolished the repression of c-MYC by salicylate, suggesting that salicylate suppresses the expression of c-MYC via AMPK. Interestingly, another inducer of AMPK, Metformin, also down-regulates c-MYC expression and inhibits CRC cell proliferation [38]. Moreover, the AMPK activator AICAR suppresses c-MYC via SIRT1 and GSK3β [39]. Finally, another AMPK activator, 4-O-Methly-ascochlorin, suppresses c-MYC activity via mTOR [40]. Therefore, the suppression of c-MYC by AMPK probably involves multiple mechanisms. In addition, salicylate suppresses WNT signaling at multiple levels, i.e. by inhibition of COX-2 and by activation of protein phosphatase 2A, which promotes β-catenin degradation [41]. Since c-MYC is a target gene of the WNT/TCF4 pathway [42], the suppression of c-MYC by salicylate might also be mediated by inhibition of WNT signaling.
Interestingly, a single nucleotide polymorphism (SNP) rs6983267 located on chromosome 8q24 that regulates the expression of c-MYC has been associated with the chemo-preventive effect of aspirin [43]. The protective effect of aspirin was confined to individuals with the T allele of the SNP, which was associated with reduced expression of c-MYC. The SNP affects a binding site for the WNT-regulated transcription factor TCF4, with the T allele showing weaker binding [44, 45]. The allele frequency of the T allele is high (49% among Europeans) and thus the majority of the population may benefit from the chemo-preventive use of aspirin [41].
We showed that the ectopic expression of c-MYC prevents the activation of NRF2 and the induction of miR-34a/b/c by salicylate. It has been shown that c-MYC interacts with NRF2, which prevents the binding of NRF2 to the ARE elements and the induction of NRF2 target genes [30, 46]. Therefore, the binding of c-MYC to NRF2 might sequester NRF2 from the miR-34a/b/c promoters and thereby prevents the induction of miR-34a/b/c expression by NRF2. We showed that salicylate on one hand activates NRF2 and on the other hand suppresses the expression of c-MYC. Our results suggest that both mechanisms contribute to the salicylate-mediated induction of miR-34a/b/c.
Aspirin was shown to be effective in cancer prevention when given for extended periods [47,48,49,50]. Dominick J et al. [51]. determined that the Cmax is 25–53 μg /ml (160–332 μM) after intake of 650 mg aspirin as tablets. Since we intended to analyze the acute effects of salicylate, the main metabolite of aspirin, on CRC cells and characterize the mediators of these effects in periods covering a few days at most, we chose to apply a higher concentration (5 mM) of salicylate based on our IC50 value determination. In addition, a concentration of 5 mM was used in previous studies, which examined the effect of salicylate on CRC cells [21,22,23,24]. Presumably, lower doses of aspirin given over extended periods for cancer prevention exert the same or similar effects, which we observed here for 5 mM salicylate. In the future, in vivo analyses to confirm this assumption are warranted.
We challenge the notion that aspirin prevents cancer through a single, dominant pathway (the inhibition of COX enzymes [10]) and propose an integrative multi-pathway model for its mode of action, with miR-34a and miR-34b/c representing important effectors. As of today, more than 200 experimentally validated miR-34a/b/c target mRNAs have been identified [52]. In the future it will be important to identify the miR-34 targets that mediate the tumor suppressive effects of aspirin.
Materials and methods
Cell culture and treatments
The CRC cell lines (HCT116, HCT116 miR-34a−/−, HCT116 miR-34a/b/c−/−, RKO, SW48, SW480, DLD-1, SW620-Luc2, DLD1-pRTR-c-MYC-VSV) and Human colon fibroblast (CCD18-co) were cultured in McCoy’s 5A medium (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS) (Invitrogen) containing 100 units/ml penicillin and 0.1 mg/ml streptomycin at 20% O2, 5% CO2, and 37 °C. p53-deficient and p53-proficient HCT116 and RKO cells were a gift from Bert Vogelstein (Johns Hopkins Medical School, Baltimore, Maryland, USA) [53]. The isogenic wt and miR-34a- or miR-34a/b/c-deficient HCT116 cells were generated in our lab using a CRISPR/CAS9 approach [26]. HCEC-1CT immortalized human colonic epithelial cells (Evercyte GmbH) were maintained in DMEM medium supplemented with 2% FBS, 1 × N2 supplement (containing insulin, apolipoprotein, and sodium selenite; Thermo Fisher Scientific), 20 ng/ml epidermal growth factor (EGF; AF-100-15, PeproTech, part of Thermo Fisher Scientific), 1 μg/ml hydrocortisone (Sigma, Merck) and 100 units/ml penicillin and 0.1 mg/ml streptomycin (Gibco, Thermo Fisher Scientific). Sodium salicylate (Lot # SLCC 7389, Sigma-Aldrich) was dissolved in distilled H2O to a 600 mM stock solution and applied at 5 mM working concentration. Dorsomorphin (Compound C, ab120843, Abcam) was dissolved in DMSO with a concentration of 10 mM. siGENOME Human NRF2 SMARTPOOL (Horizon Discovery, USA, #M-003755-02-0010), Human PRKAA1 siRNA SMARTPOOL (Horizon Discovery, USA, #M-005027-02-0010), Human PRKAB1 siRNA SMARTPOOL (Horizon Discovery, USA, # M-007675-00-0010), control siRNA pool (Horizon Discovery, USA), miRNA antagonist (hsa-miR-34a-5p inhibitor, Invitrogen, MH11030), and corresponding negative controls were transfected at a concentration of 10 nM using HiPerfect transfection reagent (Qiagen, Hilden, Germany).
MTT assay
Cells were seeded at a density of 3000 cells per well in 96-well plates and treated with salicylate at different concentrations for 48 h. Then, 10 μl of MTT solution was added to each well and incubated for 4 h. This assay is based on reducing MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) by viable cells to form a purple formazan product. Cells were treated with DMSO to release the formazan and the absorbance was measured at 570 nm using a Varioscan system (Thermo Fisher).
Assessment of proliferation by real-time impedance
Cell proliferation was analyzed using the xCELLigence system. The cells were seeded at a density of 3 × 103 cells in 100 μL per well of E-Plates®. After 24 h, cells were exposed to 5 mM salicylate. The xCELLigence system was used to monitor the cells for an additional 116 h. The system automatically calculates a parameter called “Cell Index” based on the measured impedance. Real-time monitoring of electrical impedance, represented as the cell index, provided continuous information about cell behavior and growth, which was further confirmed by manual cell counting at the end of the experiment.
Cell cycle analysis by flow cytometry
HCT116 cell lines were seeded into 6-well plates at the density of 2 × 105 cells/ml. The supernatant with detached cells was transferred into 15 ml tubes. Cells were harvested by adding trypsin and neutralized with 1 ml of fresh medium. The cell suspension was transferred to a 15 ml tube, centrifuged at 1200 rpm for 4 min, and the supernatant was discarded. The cells were washed with 3 ml of 1% BSA in PBS. 100 μl of Click-iT® fixative was added. The cells were incubated for 15 min at room temperature, protected from light. Next, cells were washed with 3 ml of 1% BSA in PBS and pelleted to remove the supernatant. The cells were resuspended in 100 μl of 1X Click-iT® saponin-based permeabilization solution and mixed well. Cells were incubated for 15 min, followed by the addition of 0.5 ml of Click-iT® reaction cocktail to each tube and incubated for 30 min at room temperature, protected from light. Cells were resuspended in Propidium Iodide (PI) staining solution and analyzed by flow cytometry using a CFlow6 device (Accuri, Ann Arbor, MI).
RNA isolation and quantitative real-time polymerase chain reaction (qPCR)
Total cellular RNA was isolated and purified from CRC cell lines according to the manufacturer’s instructions (High Pure RNA Isolation Kit; Roche). 1 µg of RNA was reverse transcribed using a Verso cDNA Synthesis Kit from Thermo Fisher Scientific (Waltham, MA, USA). qPCR was conducted using Fast SYBR Green Master Mix from Applied Biosystems on a LightCycler 480 system from Roche. The data obtained from qPCR analysis was normalized to the expression of the housekeeping genes GAPDH or β-actin. The analysis was performed using the ΔΔCt method. The primer sequences used in the qPCR assay are provided in Supplementary Table S1.
Detection of apoptosis by flow cytometry
HCT116 cells were seeded into 6 well plates at a cell density of 2 × 105 cells per well. After 24 h cells were treated with 5 mM of salicylate for indicated times. Cells were harvested, washed with HBSS, and resuspended in 1 X binding buffer. FITC Annexin V and PI were added and incubated for 15 min at room temperature in the dark. After addition of 1x binding buffer, cells were analyzed by flow cytometry using a CFlow6 device (Accuri, Ann Arbor, MI).
Immunofluorescence and confocal laser-scanning microscopy
The cells were first cultured on glass cover slides and then fixed in 4% paraformaldehyde/PBS for 15 min, permeabilized with 0.2% Triton X-100 for 5 min and blocked in 1% BSA/PBS for 1 h at room temperature. Next, the cells were incubated with a primary antibody against NRF2 (D1Z9C, #12721, Cell Signaling Technology) for 1 h at room temperature, washed three times with PBS-Tween, and then incubated with a Cy3-labeled secondary antibody (ab6939, Abcam). The chromatin was stained with DAPI (Roth). Finally, confocal laser scanning microscopy (CLSM) was used to acquire images of the cells. For imaging a LSM700 microscope with a Plan Apochromat 20×/0.8 M27 objective and ZEN 2009 software (Zeiss) was used.
Western blot analysis
Cells were washed by HBSS and lysed in RIPA lysis buffer (50 mM Tris/HCl, pH 8.0, 250 mM NaCl, 1% NP40, 0.5% [w/v] sodium deoxycholate, 0.1% SDS, complete mini protease inhibitors [Roche, Basel, Switzerland], PhosSTOP Phosphatase Inhibitor Cocktail Tablets [Roche]). Lysates were sonicated for 5 s and centrifuged at 13,000 rpm for 20 min at 4 °C. Supernatants containing proteins were transferred to new tubes and quantified by Pierce™ BCA Protein Assay Kit (Termo Fisher Scientifc). 40 μg of protein from each sample was loaded and separated on 10% SDS-acrylamide gels. Gel electrophoresis and transfer to Immobilon PVDF membranes (Millipore, Burlington, MA, USA) were performed according to manufacturer’s instructions (BioRad Laboratories, Hercules, CA). Membranes were blocked with non-fat dry milk. For immuno-detection, the membrane was incubated with the primary antibodies listed in Supplementary Table S3. The signal from the HRP-conjugated secondary antibody was generated by enhanced chemi-luminescence (Millipore) and detected using a LI-COR Odyssey Fc imaging system (LI-COR, Lincoln, NE). Antibodies used in the Western blot are provided in Supplementary Table S3.
Wound healing assay
A wound healing assay was used to assess migration of cells. 70 μl cell suspension at a density of 1 × 106 cells/ml was seeded into a 2-well culture-Insert (80241; IBIDI, Martinsried, Germany). After attaching, mitomycin C (10 ng/ml) was added 2 h before removal of the insert. After 0 and 24 h the scratched area was documented using an Axiovert Observer Z.1 microscope with an AxioCam MRm camera and Axiovision software (Axiovs 40 Version 4.8.0.0, Zeiss, Oberhochen, Germany). The cell-free area was determined using ImageJ software.
Modified Boyden-chamber assay
For determination of invasion, 60 µl of matrigel in serum-free medium (BD Biosciences, East Rutherford, NJ) at a concentration of 300 µg/ml was added to the chamber 4 h before adding cells. Then 5 × 104 cells were seeded in the upper chamber (membranes with 8.0 µm pore size, Corning, NY). Salicylate was added to the top chamber at a final concentration of 5 mM. Then 500 µl of 10% FBS medium was added to the lower chamber. After 48 h the invaded cells located on the lower surface of the cell culture inserts were fixed, stained with crystal violet, and counted. Fold changes in invasive cells were calculated by normalizing them to the corresponding control group.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) analysis was performed using HCT116 p53−/− cells. The cells were treated with salicylate for 48 h before cross-linking. ChIP experiments were performed using the iDeal ChIP-qPCR kit from Diagenode, Belgium, following the manufacturer’s instructions. Sequences of qChIP primers and antibodies are provided in Supplementary Tables S2 and S3.
Metastases formation in a xenograft mouse model
SW620 cells stably expressing Luc2 were described previously [54]. After the treatments Luciferase-tagged cells were washed with HBSS for 3 times and the number of living cells was determined after staining with trypan blue. 4 ×106 living cells were injected into the lateral tail vein of NOD/SCID mice using 25-gauge needles. Starting from the second week after injection, the mice were injected intraperitoneally (i.p.) with D-luciferin at a dose of 150 mg/kg. The mice were then subjected to weekly bioluminescent imaging using the IVIS Illumina System from Caliper Life Sciences. The imaging was performed for 5 min. Five weeks after the initial tail vein injection, the mice were sacrificed, and their lung tissues were examined for the presence of metastases using hematoxylin and eosin (H&E) staining. All animal experiments and analyses were conducted in compliance with the regulations and guidelines set by the Government of Upper Bavaria, Germany (Approval number 55.2-2532.vet_02-18-57).
DCFDA/H2DCFDA - cellular ROS assay
Reactive oxygen species (ROS) were analyzed using DCFDA/H2DCFDA staining (Cellular ROS Assay Kit (ab113851, Abcam). HCT116 cells were seeded in 96-well plates at a density of 104 cells per well. 24 h after seeding, cells were treated with or without salicylate for 48 h. As a positive control, tBHP (tert-Butyl hydroperoxide) was added to the HCT116 cells for 4 h before staining. After the indicated treatment period, the medium was removed, and 100 µl of 1X buffer was added to each well. Subsequently, the cells were stained with DCFDA solution (100 µl/well) and incubated for 45 min at 37 °C in the dark. After the staining period, the DCFDA solution was removed, and the plates were immediately analyzed using a fluorescence plate reader with excitation/emission wavelengths set at 485/535 nm, which corresponds to the excitation and emission wavelengths of the DCFDA dye. To analyze the images obtained from the plate reader Image J software was used.
Statistical analysis
The GraphPad Prism software was used for statistical analyses. The statistical differences between two groups were calculated using a Student’s t test (two-tailed; unpaired). When more than 2 groups were compared, a one-way analysis of variance (ANOVA) combined with a Tukey multiple comparison post-test was used. Asterisks generally indicate: *p < 0.05, **p < 0.01 and ***p < 0.001, n.s. not significant.
Data availability
All data, analytic methods, and study materials will be made available to other researchers upon reasonable request.
References
GLOBOCAN Global Cancer Observatory (GCO): Cancer Today. Available online: https://gco.iarc.fr/today/#. Accessed 15 August 2021.
Drew DA, Cao Y, Chan AT. Aspirin and colorectal cancer: the promise of precision chemoprevention. Nat Rev Cancer. 2016;16:173–86.
Grancher A, Michel P, Di Fiore F, Sefrioui D. Colorectal cancer chemoprevention: is aspirin still in the game? Cancer Biol Ther. 2022;23:446–61.
Nan H, Hutter CM, Lin Y, Jacobs EJ, Ulrich CM, White E, et al. Association of aspirin and NSAID use with risk of colorectal cancer according to genetic variants. JAMA. 2015;313:1133–42.
Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376:1741–50.
Madge JC, Stallmach A, Kleebusch L, Schlattmann P. Meta-analysis of aspirin-guided therapy of colorectal cancer. J Cancer Res Clin Oncol. 2022;148:1407–17.
Garcia Rodriguez LA, Soriano-Gabarro M, Bromley S, Lanas A, Cea Soriano L. New use of low-dose aspirin and risk of colorectal cancer by stage at diagnosis: a nested case-control study in UK general practice. BMC Cancer. 2017;17:637.
Needs CJ, Brooks PM. Clinical pharmacokinetics of the salicylates. Clin Pharmacokinet. 1985;10:164–77.
Dovizio M, Tacconelli S, Sostres C, Ricciotti E, Patrignani P. Mechanistic and pharmacological issues of aspirin as an anticancer agent. Pharmaceuticals. 2012;5:1346–71.
Sankaranarayanan R, Kumar DR, Altinoz MA, Bhat GJ. Mechanisms of colorectal cancer prevention by aspirin-a literature review and perspective on the role of cox-dependent and -independent pathways. Int J Mol Sci. 2020;21:9018.
Bos CL, Kodach LL, van den Brink GR, Diks SH, van Santen MM, Richel DJ, et al. Effect of aspirin on the Wnt/beta-catenin pathway is mediated via protein phosphatase 2A. Oncogene. 2006;25:6447–56.
Ai G, Dachineni R, Muley P, Tummala H, Bhat GJ. Aspirin and salicylic acid decrease c-Myc expression in cancer cells: a potential role in chemoprevention. Tumour Biol. 2016;37:1727–38.
Law BK, Waltner-Law ME, Entingh AJ, Chytil A, Aakre ME, Norgaard P, et al. Salicylate-induced growth arrest is associated with inhibition of p70s6k and down-regulation of c-myc, cyclin D1, cyclin A, and proliferating cell nuclear antigen. J Biol Chem. 2000;275:38261–7.
Hermeking H. p53 enters the microRNA world. Cancer Cell. 2007;12:414–8.
Rokavec M, Li H, Jiang L, Hermeking H. The p53/miR-34 axis in development and disease. J Mol Cell Biol. 2014;6:214–30.
Shi X, Kaller M, Rokavec M, Kirchner T, Horst D, Hermeking H. Characterization of a p53/miR-34a/CSF1R/STAT3 feedback loop in colorectal cancer. Cell Mol Gastroenterol Hepatol. 2020;10:391–418.
Oner MG, Rokavec M, Kaller M, Bouznad N, Horst D, Kirchner T, et al. Combined inactivation of TP53 and MIR34A promotes colorectal cancer development and progression in mice via increasing levels of IL6R and PAI1. Gastroenterology. 2018;155:1868–82.
Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, Lodygin D, et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest. 2014;124:1853–67.
Jiang L, Hermeking H. miR-34a and miR-34b/c suppress intestinal tumorigenesis. Cancer Res. 2017;77:2746–58.
Pathi S, Jutooru I, Chadalapaka G, Nair V, Lee SO, Safe S. Aspirin inhibits colon cancer cell and tumor growth and downregulates specificity protein (Sp) transcription factors. PLoS One. 2012;7:e48208.
Din FV, Stark LA, Dunlop MG. Aspirin-induced nuclear translocation of NFkappaB and apoptosis in colorectal cancer is independent of p53 status and DNA mismatch repair proficiency. Br J Cancer. 2005;92:1137–43.
Fernandez HR, Linden SK. The aspirin metabolite salicylate inhibits lysine acetyltransferases and MUC1 induced epithelial to mesenchymal transition. Sci Rep. 2017;7:5626.
Goel A, Chang DK, Ricciardiello L, Gasche C, Boland CR. A novel mechanism for aspirin-mediated growth inhibition of human colon cancer cells. Clin Cancer Res. 2003;9:383–90.
Williams JL, Borgo S, Hasan I, Castillo E, Traganos F, Rigas B. Nitric oxide-releasing nonsteroidal anti-inflammatory drugs (NSAIDs) alter the kinetics of human colon cancer cell lines more effectively than traditional NSAIDs: implications for colon cancer chemoprevention. Cancer Res. 2001;61:3285–9.
Siemens H, Jackstadt R, Hunten S, Kaller M, Menssen A, Gotz U, et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle. 2011;10:4256–71.
Huang Z, Kaller M, Hermeking H. CRISPR/Cas9-mediated inactivation of miR-34a and miR-34b/c in HCT116 colorectal cancer cells: comprehensive characterization after exposure to 5-FU reveals EMT and autophagy as key processes regulated by miR-34. Cell Death Differ. 2023;30:2017–34.
Liu C, Rokavec M, Huang Z, Hermeking H. Curcumin activates a ROS/KEAP1/NRF2/miR-34a/b/c cascade to suppress colorectal cancer metastasis. Cell Death Differ. 2023;30:1771–85.
Joo MS, Kim WD, Lee KY, Kim JH, Koo JH, Kim SG. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol Cell Biol. 2016;36:1931–42.
Hardie DG, Schaffer BE, Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016;26:190–201.
Davies KJA, Forman HJ. Does Bach1 & c-Myc dependent redox dysregulation of Nrf2 & adaptive homeostasis decrease cancer risk in ageing? Free Radic Biol Med. 2019;134:708–14.
Petsouki E, Cabrera SNS, Heiss EH. AMPK and NRF2: interactive players in the same team for cellular homeostasis? Free Radic Biol Med. 2022;190:75–93.
Yu C, Xiao JH. The Keap1-Nrf2 system: a mediator between oxidative stress and aging. Oxid Med Cell Longev. 2021;2021:6635460.
Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–91.
Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19:121–35.
Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62.
Calabrese MF, Rajamohan F, Harris MS, Caspers NL, Magyar R, Withka JM, et al. Structural basis for AMPK activation: natural and synthetic ligands regulate kinase activity from opposite poles by different molecular mechanisms. Structure. 2014;22:1161–72.
Hardie DG, Ross FA, Hawley SA. AMP-activated protein kinase: a target for drugs both ancient and modern. Chem Biol. 2012;19:1222–36.
Mogavero A, Maiorana MV, Zanutto S, Varinelli L, Bozzi F, Belfiore A, et al. Metformin transiently inhibits colorectal cancer cell proliferation as a result of either AMPK activation or increased ROS production. Sci Rep. 2017;7:15992.
Gollavilli PN, Kanugula AK, Koyyada R, Karnewar S, Neeli PK, Kotamraju S. AMPK inhibits MTDH expression via GSK3beta and SIRT1 activation: potential role in triple negative breast cancer cell proliferation. FEBS J. 2015;282:3971–85.
Shin JM, Jeong YJ, Cho HJ, Magae J, Bae YS, Chang YC. Suppression of c-Myc induces apoptosis via an AMPK/mTOR-dependent pathway by 4-O-methyl-ascochlorin in leukemia cells. Apoptosis. 2016;21:657–68.
Gala MK, Chan AT. Molecular pathways: aspirin and Wnt signaling-a molecularly targeted approach to cancer prevention and treatment. Clin Cancer Res. 2015;21:1543–8.
He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–12.
Nan H, Morikawa T, Suuriniemi M, Imamura Y, Werner L, Kuchiba A, et al. Aspirin use, 8q24 single nucleotide polymorphism rs6983267, and colorectal cancer according to CTNNB1 alterations. J Natl Cancer Inst. 2013;105:1852–61.
Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, Doddapaneni H, et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet. 2009;41:882–4.
Tuupanen S, Turunen M, Lehtonen R, Hallikas O, Vanharanta S, Kivioja T, et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat Genet. 2009;41:885–90.
Levy S, Forman HJ. C-Myc is a Nrf2-interacting protein that negatively regulates phase II genes through their electrophile responsive elements. IUBMB Life. 2010;62:237–46.
Burn J, Bishop DT, Mecklin JP, Macrae F, Moslein G, Olschwang S, et al. Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N Engl J Med. 2008;359:2567–78.
Burn J, Gerdes AM, Macrae F, Mecklin JP, Moeslein G, Olschwang S, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. 2011;378:2081–7.
Flossmann E, Rothwell PM, British Doctors Aspirin T, the UKTIAAT. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet. 2007;369:1603–13.
Logan RF, Grainge MJ, Shepherd VC, Armitage NC, Muir KR, uk CAPTG. Aspirin and folic acid for the prevention of recurrent colorectal adenomas. Gastroenterology. 2008;134:29–38.
Angiolillo DJ, Bhatt DL, Lanza F, Cryer B, Dong JF, Jeske W, et al. Pharmacokinetic/pharmacodynamic assessment of a novel, pharmaceutical lipid-aspirin complex: results of a randomized, crossover, bioequivalence study. J Thromb Thrombolysis. 2019;48:554–62.
Rokavec M, Huang Z, Hermeking H. Meta-analysis of miR-34 target mRNAs using an integrative online application. Comput Struct Biotechnol J. 2023;21:267–74.
Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104:263–9.
Hahn S, Jackstadt R, Siemens H, Hunten S, Hermeking H. SNAIL and miR-34a feed-forward regulation of ZNF281/ZBP99 promotes epithelial-mesenchymal transition. EMBO J. 2013;32:3079–95.
Acknowledgements
We thank Bert Vogelstein for p53-deficient and p53-proficient HCT116 and RKO cells, Jinjiang Chou for DLD-1/pRTR-c-MYC cells and Uschi Götz for technical assistance. CL and ZH are recipients of Chinese Scholarship Council fellowships. HH received grants from the Rudolf Bartling-Stiftung.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
HH conceived, planned and supervised the project; HH, CL, and MR designed experiments; CL performed experiments and analyzed results; MR performed and analyzed mouse experiments. ZH generated miR-34a/b/c KO cells. HH, CL and MR wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
Animal experimentations and analyses were approved by the Government of Upper Bavaria, Germany (55.2-2532.vet_02-18-57).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Gerry Melino
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, C., Rokavec, M., Huang, Z. et al. Salicylate induces AMPK and inhibits c-MYC to activate a NRF2/ARE/miR-34a/b/c cascade resulting in suppression of colorectal cancer metastasis. Cell Death Dis 14, 707 (2023). https://doi.org/10.1038/s41419-023-06226-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-023-06226-9
