
Abstract
Mutations in the TP53 tumour suppressor gene are very frequent in cancer, and attempts to restore the functionality of p53 in tumours as a therapeutic strategy began decades ago. However, very few of these drug development programmes have reached late-stage clinical trials, and no p53-based therapeutics have been approved in the USA or Europe so far. This is probably because, as a nuclear transcription factor, p53 does not possess typical drug target features and has therefore long been considered undruggable. Nevertheless, several promising approaches towards p53-based therapy have emerged in recent years, including improved versions of earlier strategies and novel approaches to make undruggable targets druggable. Small molecules that can either protect p53 from its negative regulators or restore the functionality of mutant p53 proteins are gaining interest, and drugs tailored to specific types of p53 mutants are emerging. In parallel, there is renewed interest in gene therapy strategies and p53-based immunotherapy approaches. However, major concerns still remain to be addressed. This Review re-evaluates the efforts made towards targeting p53-dysfunctional cancers, and discusses the challenges encountered during clinical development.
Similar content being viewed by others


Introduction
The p53 tumour suppressor protein acts as a major barrier against cancer initiation and progression. Biochemically, p53 functions primarily as a sequence-specific transcription factor, capable of binding to defined DNA sequences within the genome (known as p53 response elements or p53-binding sites) and activating the transcription of adjacent genes, as well as the transcription of more distant genes that are regulated by enhancers with p53-binding sites. In addition, p53 can also repress the transcription of a large subset of genes, usually by indirect mechanisms. In normal unstressed cells, p53 protein levels are kept low via constitutive proteasomal degradation, instructed by the E3 ubiquitin ligase MDM2, which is the major inhibitor of p53. Furthermore, the biochemical activity of p53 as a transcription factor is restrained also by the MDM4 protein (also known as MDMX), which thus serves as an additional physiological p53 inhibitor.
Mutations in the TP53 gene, which abrogate the tumour suppressor activities of its encoded protein, p53, are the most common single gene alterations in human cancers, and are recognized as driver events in many types of tumour. Consequently, attempts to restore the functionality of p53 in tumours as a therapeutic strategy, prompted by laboratory experiments that documented the ability of such restoration to trigger cancer cell death, began decades ago. However, these efforts have mostly met with limited success: very few of the p53 drug development initiatives have reached advanced clinical trial phases, and none so far has made it to FDA or EMA approval.
The main consequence of TP53 mutations in cancer is loss of tumour suppressor function, calling for therapeutic reactivation of the protein, whereas most cancer-relevant small-molecule drugs actually work by inhibiting excessive protein activity. Consequently, p53 has long been considered undruggable. A vivid illustration of the scope of this problem is provided by cancer gene panel tests, increasingly being used to guide treatment decisions1. TP53 is included in all such panels, and TP53 alterations are often top hits. Sadly, they are not in the ‘actionable’ column. Nevertheless, several promising approaches towards p53-based therapy have emerged in recent years, giving hope that TP53 alterations will finally relocate to the ‘actionable’ column. Some of those approaches are a continuation of earlier therapeutic attempts that have incorporated new knowledge and better understanding of mechanisms of action and modes of delivery. Other approaches take advantage of the progress made in drug design, including novel strategies for targeted degradation of any protein of choice. Notably, small molecules that can either protect p53 from its negative regulators or restore the functionality of mutant p53 (mutp53) proteins are increasingly gaining interest. Furthermore, efforts are now being made to develop drugs that target selectively one or a few specific mutants, with the expectation that such ‘personalized’ drugs will have fewer undesirable side effects.
In parallel, there is renewed interest in gene therapy strategies, which were extensively championed in the 1990s. Having gone into disfavour in the early years of the twenty-first century, gene therapy strategies are gradually making a comeback, including in the p53 arena. Likewise, approaches that fall within the broad realm of cancer immunotherapy are also seeing a renaissance. Needless to say, immunotherapy has revolutionized the treatment of several previously highly deadly cancers, generating great excitement and great hopes. This has also kindled new interest in recruiting the immune system to recognize and attack cancer cells that harbour TP53 mutations, with an additional twist: a growing number of studies indicate that loss of p53 function in cancer cells exerts cell non-autonomous effects on the tumour immune microenvironment (TIME), enabling the cancer cell to better evade immune attack. Hence, restoration of p53 functionality in such cancer cells might be expected to sensitize them to regimens such as immune checkpoint inhibition, raising growing interest in exploring relevant drug combinations.
Although p53-based therapy has already been addressed in several excellent reviews over the past decade2,3,4,5, the introduction of novel approaches and the growing number of ongoing clinical trials call for constant re-evaluation of current knowledge.
This Review attempts to reassess the efforts made towards targeting p53-dysfunctional cancers using small molecules that restore or enhance wild-type p53 (wtp53) activity in cancer cells, as well as p53-based immunotherapy strategies and p53-based gene therapy. Challenges and concerns that hamper the introduction of such treatments into the clinic are also discussed.
p53, an appealing target in cancer
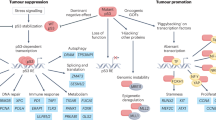
Extensive knowledge has been gained about the functions of the p53 protein and its relationship to cancer since its first description in 1979 (for a recent review see ref.6). Yet, new functions keep emerging. Consensually, p53 is a powerful tumour suppressor that inhibits tumour growth in multiple ways. As a transcription factor, p53 orchestrates the expression of target genes that can promote cell cycle arrest, apoptosis, DNA repair and more7,8,9,10. Furthermore, p53 can exert antiproliferative effects also by transcription-independent mechanisms10,11. In fact, p53 has been reported to affect almost all cellular compartments and organelles, including mitochondria, lysosomes, endoplasmic reticulum and more12,13,14,15 (Fig. 1). Importantly, these activities of p53 are compromised when TP53 is mutated. Unlike what is observed with other tumour suppressor genes, cancer-associated TP53 mutations are predominantly missense mutations, causing single amino acid substitutions16,17,18,19. Often, the resultant mutp53 accumulates abundantly in the cancer cells, to the extent that intense p53 staining in a tumour section is regarded as a good proxy for the presence of a missense mutation. Although many hundreds of different p53 mutations have been recorded, some of them are particularly common and are therefore referred to as hotspot mutations16,17,18,19. Typically, most cancer-associated mutations fall into one of two categories: DNA contact mutants, in which p53 residues responsible for sequence-specific binding to DNA are altered but the overall structure of the protein is only mildly affected, and conformational or structural mutants, in which the protein becomes extensively misfolded16,17,18,19. There is growing evidence that, in addition to the loss of the cancer-inhibitory activities of wtp53, some p53 mutants can also acquire new gain-of-function (GOF) activities that further promote cancer. Similar to wtp53, mutp53 can impinge on a wide variety of cellular processes16,17,18,19 (Fig. 1). Notably, although having lost the ability to interact directly with p53-binding sites within the DNA, mutp53 can still piggyback on other transcription factors to drive tumour-promoting gene transcription20,21. In addition to their cell-autonomous activities, both wtp53 and mutp53 can also modulate the tumour microenvironment (TME), rendering it more cancer inhibitory or cancer supportive, respectively22,23 (Fig. 1). For example, p53 can suppress tumour progression by controlling the composition of exosome-carried microRNAs and the pattern of secreted cytokines, thereby maintaining the differentiation state of tumour-associated nerves and inhibiting neutrophil infiltration, respectively22,24. Conversely, mutp53 can support tumour progression by modulating exosome content in a manner that leads to reprogramming of macrophages to an M2 state, thereby generating a more favourable TME25. For a recent review on cell non-autonomous effects of wtp53 and mutp53 see ref.26.

a | Wild-type p53 (wtp53) acts predominantly as a transcription factor to restrict cancer cell proliferation and survival. Many non-transcriptional effects are also involved. Wtp53 can promote mitochondrially induced apoptosis by interacting with multi-domain members of the apoptosis regulator BCL-2 family (such as BCL-2 and BCL-X), unleashing the activity of pro-apoptotic BH3-only proteins such as BAK and BAX. Wtp53 can also increase Ca2+ load upon stress, resulting in induction of apoptosis. Additionally, wtp53 is an important regulator of autophagy. b | Mutant p53 (mutp53) can modulate transcription by piggybacking on other transcription factors (TFX) and can also promote cancer by non-transcriptional mechanisms. Mutp53 can inhibit peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α), a master regulator of mitochondrial biogenesis and oxidative phosphorylation. Moreover, in contrast to wtp53, mutp53 inhibits endoplasmic reticulum (ER) stress-induced apoptosis by modulating the unfolded protein response (UPR), which increases cell survival upon ER stress. Mutp53 also induces the transcription of many genes that encode proteasome subunits. This transcriptional activation, mediated by the binding of mutp53 to the transcription factor nuclear factor erythroid 2-related factor 2 (NRF2), results in elevated proteasome activity and enhanced degradation of tumour suppressor proteins. Both wtp53 and mutp53 impinge on multiple cellular organelles and compartments, and can also elicit non cell-autonomous effects through secretion of various molecules, both soluble and those carried by exosome or extracellular vesicle. The presence of wtp53 inhibits the expression of Golgi scaffolding proteins, thus inhibiting secretory vesicle biogenesis in the Golgi apparatus, while mutp53 can modulate Golgi apparatus function by inducing microRNA miR-30d expression through interacting with the hypoxia responsive factor HIF1α. Consequently, miR-30d modulates tubulo-vesiculation of the Golgi apparatus, promoting vesicular trafficking and secretion. RE, response element.
The multitude of deleterious effects exerted by wtp53 on cancer cells and on the TME, such as induction of cell death or replicative senescence and promotion of a tumour-restrictive immune TME, probably explains why TP53 is so frequently mutated in human cancer25,27 (Fig. 1); it also makes restoration of p53 activity in cancer cells very promising, as demonstrated experimentally in various genetically modified mouse models28,29,30,31,32 and as discussed in the following sections. Although many obstacles still remain to be overcome, our understanding and capabilities to target p53 are gradually improving, raising hopes for eventual impact on the lives of many patients.
Small molecules
The pursuit of small molecules capable of activating p53 signalling started several decades ago and has steadily intensified since then (Fig. 2). Importantly, tumours with different TP53 status call for very different small-molecule strategies. Thus, for tumours that harbour TP53 missense mutations, small-molecule drug development has mainly focused on compounds that restore wild-type-like conformation and activity to the mutp53 proteins. Conversely, for cancers that maintain wtp53, the main approach has been to identify small molecules that liberate p53 from inhibition by its negative regulators, most notably MDM2, thereby unleashing full p53 activity.

Clinical trials with p53-targeted therapies initiated after 1 January 2000 were stratified by year blocks and category. Gene therapy clinical trials were popular before the year 2000 (12 clinical trials initiated), but their number declined sharply soon thereafter, owing to mounting concerns about the safety of this strategy. These numbers increased again in the course of 2011–2015, mostly reflecting the clinical trials driven by Shenzhen SiBiono GeneTech and several trials of SGT-53 (SynerGene Therapeutics). Immune-based clinical trials targeting p53 were rather uncommon before 2000 (two clinical trials). With the introduction of new anticancer vaccination approaches, the number of relevant p53-based clinical trials has increased. Presently, most p53-based immunotherapy clinical trials use a combination of immune checkpoint inhibition and a p53-activating agent (either gene therapy or small molecules). It is expected that, owing to the growing interest in bispecific antibodies and T cell receptor (TCR)-like antibodies (see Fig. 4), p53-centric clinical trials that use these strategies will become more popular in the coming years. Visibly, the biggest increase in p53-based clinical trials in the past decade involved small-molecule drugs. This may be attributed, at least in part, to the emergence of new screening methods and improved compound libraries, along with better understanding of the deregulation of p53 in cancer.
Targeting TP53 missense mutant tumours
As mentioned earlier, TP53 missense mutations are exceptionally common in human cancer, representing about 70% of all TP53 alterations. The resulting structural or conformational mutants have been viewed as promising targets for small molecules that can restore the proper folding and functionality of misfolded mutp53. The first compound to possess mutp53-reactivating capability was described in 1999 (ref.33). This drug, CP31398, identified by Pfizer through a synthetic compound library screen, restored the transcriptional activity of p53 and reduced tumour growth in vivo. Mechanistically it was suggested that CP31398, which possesses Michael acceptor characteristics, stabilizes the wild-type conformation of p53 and prevents its degradation through inhibition of its ubiquitylation34. However, later work revealed more complex effects, including nonspecific toxicity caused by intercalation of the drug into the DNA35 and p53-independent upregulation of the pro-apoptotic protein BAX35. Consequently, CP31398 was not taken into clinical development. Nonetheless, this prototype drug marked the beginning of the era of small molecules that target mutp53.
In 2002, a chemical drug screen identified that PRIMA-1 (p53 reactivation with induction of massive apoptosis 1), a compound that restored wtp53 function upon binding to mutp53, triggered apoptosis in vitro in Saos-2 cells transduced to express the R273H mutant protein (p53(R273H)), and suppressed tumour formation in vivo by these cells36. Pharmacologically, PRIMA-1 is actually a prodrug; its degradation product, MQ, reacts covalently with thiol groups of cysteine residues in the core domain of mutp53, restoring wtp53 conformation37. In addition, MQ can shift the cellular redox balance by binding directly to cysteine in glutathione and by inhibiting thioredoxin reductase, thereby promoting cancer cell death by disrupting a crucial homeostatic mechanism required to maintain cell viability38. Furthermore, PRIMA-1 also promotes apoptosis by inducing BAX-dependent cytochrome c release from the mitochondria and thus driving caspase activation39.
Over the years, many additional mutp53-reactivating drugs have been described. Examples include MIRA-1, which contains a maleimide group that can participate in a nucleophilic addition reaction, and STIMA-1, a 2-styrylquinazolin-4(3H)-one-related derivative40,41. Like PRIMA-1, both MIRA-1 and STIMA-1 also possess Michael acceptor activity, and can potentially modify cysteines in the p53 protein to stabilize the wild-type conformation and prevent mutp53 unfolding. Although both compounds demonstrated p53-dependent effects in vitro and in vivo, neither entered clinical trials, owing to solubility issues41 (STIMA-1) and toxicity to normal cells42 (MIRA-1) (Fig. 3). In a different approach, a panel of mutp53-reactivating small peptides (pCAPs) were developed using phage display selection, and showed wtp53-like effects in vitro and in vivo when applied to cancer cells harbouring TP53 missense mutations41. Mechanistic studies suggested that pCAPs bind preferentially to the wtp53 conformation; when a mutp53 molecule assumes transiently a wild-type-like conformation, the peptide binds and stabilizes it, gradually shifting the dynamic equilibrium of the p53 population in that direction43.

a | In p53-wild-type (wtp53) tumours, small-molecule drug development is mainly focused on inhibiting or degrading negative regulators of p53, including MDM2, MDM4 and human papillomavirus (HPV) E6. Such inhibition increases p53 protein abundance and wtp53 activity in the cancer cells, promoting the expression of p53 target genes. ALRN-6924 is a stapled peptide that blocks both MDM2–p53 and MDM4–p53 interactions. p53-activating proteolysis targeting chimeras (PROTACs) work by targeting MDM2 for ubiquitylation by particular E3 ligases, resulting in MDM2 degradation. b | In tumours expressing missense mutant p53 (mutp53) proteins, drug development aims to restore wtp53 conformation and/or inhibit gain-of-function activities of mutp53 such as inhibition of p73. From left to right: (1) small molecules such as RETRA (reactivation of transcriptional reporter activity) or NSC59984, which inhibit the interaction of mutp53 with p73, unleash p73 and enable it to enter the nucleus and transactivate target genes that partly overlap with p53 target genes. (2) Some small molecules (arsenic trioxide (ATO), ZMC1) act predominantly on structural p53 mutants (such as p53(R175H)) to restore wtp53 conformation and induce p53 target gene expression. (3) The p53(Y220C) mutant has an accessible crevice near the site of mutation, which can be targeted by small molecules to thermodynamically stabilize the mutant protein and shift it towards a wild-type-like state. (4) Many compounds (such as APR-246 and pCAPs) target a broad spectrum of p53 mutants to restore a wtp53-like structure, thus enabling p53 target gene activation (for a more detailed mechanistic description see ref.195). (5) Some small molecules — such as ReACp53 or ADH-6 — act by inhibiting mutp53 aggregation, restoring wtp53-like structure and activating p53 target genes. c | Other small molecules inhibit the recognition of premature termination codons (PTCs), enabling translational readthrough and synthesis of full-length p53 protein in cells that harbour truncating TP53 mutations. The overarching goal of all these drugs is to restore the expression of wtp53 target genes as a means to induce cancer cell death or replicative senescence, thereby curtailing tumour growth. RITA, reactivation of p53 and induction of tumour cell apoptosis.
Still, only a minority of the reported mutp53-reactivating small molecules have made it into clinical trials (Table 1). The first to reach a clinical trial was PRIMA-1 MET, a methylated derivative of PRIMA-1, also known as APR-246. In in vitro and preclinical studies, APR-246 demonstrated better activity than PRIMA-1, with increased apoptotic effects in acute myeloid leukaemia (AML) cell lines and primary cells from patients44. Likewise, APR-246 promoted apoptosis in cell lines derived from small-cell lung carcinoma (SCLC), and delayed tumour growth in mice injected with such cells45. Interestingly, PRIMA-1 MET did not exert apoptotic effects in cells carrying the p53(Y220C) hotspot mutation46, suggesting that not all p53 mutants are sensitive to it. Besides its effects when administered as monotherapy, APR-246 also increased cancer cell death when combined with chemotherapy both in vitro and in lung and ovarian cancer mouse xenograft models47,48,49. Importantly, the synergistic effect was more robust in primary ovarian cancer cells from patients bearing TP53 missense mutations than in those carrying wtp53 or TP53 nonsense mutations49.
Clinical trials with APR-246 are currently ongoing. Two phase I/II trials demonstrated substantial effects when APR-246 was combined with azacitidine, the first drug approved by the FDA for treatment of myelodysplastic syndrome (MDS), in patients with MDS or AML carrying p53 mutations50,51. The combination was well tolerated and displayed a similar safety profile to that reported for each monotherapy alone. Some of the patients harbouring TP53 truncating mutations also gained clinical benefit from APR-246 treatment, suggesting that p53-independent effects of APR-246 also contribute to clinical outcome50. In January 2020, the FDA granted breakthrough designation to APR-246 for MDS treatment, and a phase III multicentre randomized trial for the combined treatment was initiated (NCT03745716). However, although the results of this trial revealed a complete remission rate of 33% in the combined treatment group compared with only 22% in the azacitidine-alone group, statistical significance was not achieved. Recently, encouraging results were announced for a phase II trial evaluating APR-246 plus azacitidine as a post-transplant management therapy in patients with MDS and AML with mutations in TP53. Compared with previous trials, which attained around 30% relapse-free survival (RFS) 1 year after transplant, with median overall survival (OS) of 5–8 months after transplantation, combined treatment with APR-246 and azacitidine achieved 58% RFS with median OS of 19.3 months52. More trials with APR-246 as monotherapy or in combination, as well as with the orally administered next generation compound APR-548 (NCT04638309), are ongoing or planned, targeting both haematological cancers and solid tumours.
Small molecules that disrupt the interaction between mutp53 and the p53 family member p73 and unleash its tumour suppressor activity have also been described53,54. p73 shares many transcriptional targets with p53, but its transcriptional activities are suppressed when it is sequestered by mutp53 (ref.55). Molecules that release p73 from the hold of mutp53 enable it to elicit wtp53-like effects, including growth inhibition and cancer cell death53,54. One example is RETRA (reactivation of transcriptional reporter activity), identified in a chemical library screen and shown to promote apoptosis in vitro and tumour regression in vivo53. Another small molecule, NSC59984, also disrupts mutp53–p73 complexes54. Interestingly, NSC59984 has recently been demonstrated to induce mutp53 degradation in colorectal cancer cell lines and mouse xenografts, resulting in cancer cell death56. Mechanistically, NSC59984 was found to promote ERK and MDM2 phosphorylation in the presence of reactive oxygen species (ROS), leading to MDM2–mutp53 binding, mutp53 ubiquitylation and subsequent proteasomal degradation56 (Fig. 3).
Missense mutp53 proteins can form molecular aggregates, and might promote GOF effects by sequestering the p53 family member proteins p73 and p63 (refs.57,58). This propelled a rational design study that resulted in ReACp53, a peptide that inhibits mutp53 aggregation58. ReACp53 restores the wild-type conformation and nuclear localization of p53, enabling p53-dependent gene transcription and promoting cancer cell apoptosis and cell cycle arrest in vitro, and tumour suppression in vivo in an ovarian cancer model58. Subsequently, by screening an oligopyridylamide library previously shown to inhibit amyloid formation associated with Alzheimer disease, the tripyridylamide ADH-6 was identified as an inhibitor of mutp53 aggregation that enhances cell death and inhibits tumour growth, with high selectivity for mutp53-expressing cancer cells, no toxicity to healthy tissues, p53-null cells or cells with wtp53, and greater efficacy than ReACp53 (ref.59). However, none of those drugs has yet made it into clinical trials.
Whereas the above compounds are relatively broad spectrum, each targeting diverse p53 missense mutants, approaches aimed at targeting specifically a single mutant or a distinct group of similar mutants have also been pursued. High-resolution crystal structure analysis of the p53(Y220C) mutant protein revealed an accessible crevice near the site of mutation60. Using a structure-based in silico screen, the small molecule PhiKan083 was found to bind this crevice and thermodynamically stabilize p53(Y220C), shifting it towards a wtp53-like state61. Subsequent screening of a synthesized fragment library yielded PK7088, which, like PhiKan083, binds the p53(Y220C) crevice and stabilizes its correct folding. PK7088 induced apoptosis and decreased the viability of gastric cancer and hepatoblastoma cells harbouring the Y220C mutation46, and cooperated with the wtp53-activating drug nutlin-3 (see below) to transactivate p53 target genes. These effects were specific for cancer cells harbouring the Y220C mutation. A similar approach has been adopted by PMV Pharmaceuticals, which generated the p53(Y220C)-specific small molecule PC14586. PC14586, bioavailable orally, is now in phase I/II clinical trial (NCT04585750), and encouraging initial results were presented at the recent ASCO annual meeting62. Overall, the success of p53(Y220C)-specific drugs such as PC14586 is encouraging. However, the Y220C mutation is not very abundant, and its unique structure that enabled the development of such drugs is not shared with other p53 mutants. Applying similar structure-based approaches to other p53 mutants may therefore be much more challenging.
p53(R175H) belongs to the class of p53 mutants in which the conformation of the DNA binding domain is severely distorted, and hence these are referred to as structural or conformational mutants. Specifically, the p53(R175H) mutation results in impaired zinc binding, causing misfolding and inactivation of the p53 protein. Using computational analysis of the NCI60 anticancer drug screen, NSC319726 (also known as ZMC1), a metal ion chelator with high affinity for zinc63, was identified as a p53(R175H)-targeting drug. NSC319726 promoted p53-dependent apoptosis in vitro and tumour regression in vivo, in a manner highly specific to p53(R175H). Non-transformed cells and cancer cells harbouring wtp53 were not affected, while cells expressing other p53 mutants such as p53(R273H) and p53(R248Q), which abolish the ability of p53 to engage in sequence-specific DNA binding without causing a gross distortion of the overall structure of the protein (DNA contact mutants), were only mildly affected63. Furthermore, NSC319726 inhibited the growth of tumours in mice transplanted with xenografts from human cancer cells harbouring the p53(R175H) mutation, as well as in knock-in mice carrying the p53(R172H) mutation (corresponding to the human p53(R175H)). ZMC2 and ZMC3, belonging to the same family of thiosemicarbazones, were also found to promote a wild-type-like conformation of p53(R175H) in vitro64. COTI-2, a third-generation thiosemicarbazone identified in a combined computational platform65, showed preferential selectivity for p53-mutated cancer cell lines, but also had some activity in p53-wild-type cells. Indeed, a subsequent study in head and neck squamous cell carcinoma (HNSCC) cells, in vitro and in an orthotopic mouse model, confirmed that COTI-2 has both p53-dependent and p53-independent effects66. Interestingly, triggering of cell death by COTI-2 was proposed to be due to induction of DNA damage and replication stress and activation of p53 target genes by the p53 family member p63 (ref.66). With the exception of COTI-2, which has reached a phase I trial67, all other R175H-reactivating thiosemicarbazones have so far not gone into clinical trials.
Recently, through in silico analysis, Chen et al.68 discovered that arsenic trioxide (ATO; Trisenox), an FDA-approved drug for the treatment of acute promyelocytic leukaemia, can rescue the functionality of structural p53 mutants, with only a limited effect on DNA contact mutants68. Intriguingly, although ATO restored the proper folding of a wide range of p53 mutants, only a subset of those regained wtp53-like transcriptional activity68. A phase I trial, treating patients with TP53-mutated AML and MDS with a combination of decitabine and ATO, is currently ongoing (NCT03855371). The ‘rediscovery’ of ATO as a mutp53 reactivator raises the intriguing possibility that additional FDA-approved small-molecule compounds, including some that are used to treat conditions other than cancer, may also possess unappreciated mutp53-reactivating capabilities.
Targeting wtp53 tumours
MDM2 and MDM4 inhibitors
In tumours that retain wtp53 expression, the p53-targeted therapy approach pursued most extensively is inhibition of p53 degradation. The best studied mechanism of p53 degradation involves ubiquitylation of p53 by the E3 ubiquitin ligase MDM2, leading to proteasomal degradation of p53. Importantly, MDM2 amplification is observed in many cancer types69,70, typically in tumours that retain wtp53. MDM2-mediated ubiquitylation and degradation relies on its direct binding to p53, prompting the search for small molecules that inhibit MDM2–p53 binding as a means to stabilize p53 and enable it to regain potency.
The first such inhibitors were the nutlins, cis-imidazolines identified by Vassilev et al.71 through a synthetic chemical library screen. Nutlins elicited p53 activation in wtp53 cancer cells, with no effect in mutp53 cells71. The nutlin derivative RG7112 (ref.72) was the first MDM2 inhibitor tested in clinical trials73. In patients with refractory relapsed AML and chronic myeloid leukaemia (CML), RG7112 triggered wtp53 activation, including p53 protein stabilization, and elevated expression of many p53 target genes, such as CDKN1A (encoding cyclin-dependent kinase inhibitor 1, also known as p21) and BBC3 (encoding Bcl-2-binding component 3, also known as PUMA)74. Reassuringly, anti-leukaemia activity was observed in many of the patients. Unexpectedly, clinical activity was seen also in a few patients devoid of TP53 mutations, suggesting that RG7112 may also possess p53-independent activities such as inhibition of angiogenesis through hypoxia-inducible factor 1α (HIF1α) suppression75. Yet, high doses of RG7112 were needed to achieve efficacy, causing undesirable adverse events such as gastrointestinal intolerance72 and suppression of thrombopoiesis76. Likewise, in patients with liposarcoma, high dose of RG7112 was associated with thrombocytopenia and neutropenia73. Conceivably, progenitor cells in the bone marrow and the gastrointestinal tract may be particularly sensitive to excess p53 activation76. This may be because such cells express relatively abundant TP53 mRNA, normally coupled with rapid turnover of the p53 protein77. Additionally, RG7112 was found to cause thrombocytopenia by impairing the ability of megakaryocytes to give rise to platelets76.
RG7112 was later replaced by a third-generation derivative, idasanutlin (RG7388). Several clinical trials are currently testing the safety and efficacy of idasanutlin in various types of cancer78 and the jury is still out (Table 1). However, a phase III clinical trial (MIRROS) of idasanutlin together with cytarabine in patients with relapsed or refractory AML79 did not meet its primary point of superiority over cytarabine plus placebo. Specifically, despite an improved overall response rate, adding idasanutlin to cytarabine did not improve OS or complete response rates in patients with relapsed or refractory AML80. Similarly, idasanutlin showed encouraging clinical effects in a phase I clinical trial in patients with polycythaemia vera81, but haematological and low-grade gastrointestinal toxicity led to frequent discontinuation of the drug in a subsequent phase II trial82. Thus, although nutlin derivatives have a strong scientific foundation and showed promising anticancer effects in preclinical studies and in early-phase human clinical trials, the fact that wtp53 is present in all normal tissues and its excessive activity is not well tolerated by at least some essential cell types still poses a major challenge to their successful clinical application.
Besides nutlin derivatives, numerous other molecules that interfere with MDM2–p53 binding have been, or are being, developed and tested. For example, APG-115, an orally bioavailable potent MDM2 inhibitor, demonstrated robust antitumour effects in preclinical models of AML83 and sensitized gastric cancer xenografts to radiotherapy84. APG-115 is presently being evaluated in several clinical trials (such as NCT02935907, NCT03611868, NCT04785196, NCT03781986), as monotherapy or in combination with chemotherapy or immune checkpoint inhibition (Table 1). AMG 232 is another orally bioavailable MDM2 inhibitor, shown to promote wtp53 functionality and tumour regression in osteosarcoma cells85. AMG 232 was reported to have superior activity in head-to-head comparison with other MDM2 inhibitors, including idasanutlin86. AMG 232 in combination with cytotoxic chemotherapy showed improved antitumour efficacy compared with AMG 232 or chemotherapy alone86. More than ten clinical trials with AMG 232 (subsequently renamed KRT-232) have been initiated in recent years, including a phase III trial for myelofibrosis after failure of JNK inhibitors87. In this trial, the impressive effects of AMG 232 led to an FDA fast-track designation, and earlier phase trials in other cancers88. Additional MDM2 inhibitors, including siremadlin and milademetan, are also in clinical trials (NCT03634228, NCT04116541). Interestingly, both drugs demonstrated better efficacy in preclinical and phase I clinical trials when administered in high intermittent doses, compared with continuous administration for 2 weeks89,90.
Although MDM2 inhibition is an appealing strategy, it still remains to be seen whether new MDM2 inhibitors will be less harmful to normal tissues. p53 is expressed in practically all normal tissues, particularly in their proliferative compartments. As such, it is not a cancer-specific target. Therefore, striving to produce an MDM2 inhibitor that has absolutely no adverse side effects is probably not a realistic goal, unless one can come up with combinations that involve lower, well-tolerated amounts of an MDM2 inhibitor together with a cancer-specific modality, or a way to deliver the MDM2 inhibitor selectively to cancer cells.
The MDM2-related protein MDM4 is also an important negative regulator of p53. Unlike MDM2, MDM4 does not possess intrinsic E3 ubiquitin ligase activity. However, it can facilitate E3 ligase activity of MDM2, and also can bind directly to p53 and suppress its transcriptional activity91. Like MDM2, MDM4 is amplified in many cancers, making it an appealing therapy target91. Of note, haematological malignancies, including AML and myelofibrosis, mostly retain wtp53, often together with elevated expression of MDM2 or MDM4. Unlike MDM2, MDM4 is highly expressed in leukaemic stem cells, making MDM4 inhibitors particularly promising compounds for leukaemia treatment92.
In recent years, stapled peptides have emerged as an alternative to small-molecule drugs93. Through the use of chemical hydrocarbon stapling, Bernal et al.94 developed a stapled peptide (SAH-p53-8) that is capable of blocking the interactions of both MDM2 and MDM4 with p53. However, subsequent in vitro experiments revealed that SAH-p53-8 may possess p53-independent cytotoxicity, raising concerns about its clinical utility95. Later, additional bispecific stapled peptides that target both MDM2 and MDM4 were introduced, including ALRN-6924 (ref.96). ALRN-6924 demonstrated high efficacy against multiple breast cancer cell lines with wtp53, whereas p53-mutant cells were resistant97. Similar wtp53 selectivity was also seen in AML cell lines, with greater efficacy than idasanutlin93, whereas p53 knockdown abolished these effects. The first phase I trial with ALRN-6924, initiated in 2016, revealed antitumour activity in solid tumours and lymphoma, with tolerable side effects98. At the time of writing of this Review, three active clinical trials with ALRN-6924 are ongoing (NCT04022876, NCT03725436, NCT03654716).
Recently, analysis of the crystal structure of MDM4 complexed with nutlin 3a revealed new intermolecular interactions, which were targeted to enhance the binding affinity of nutlin 3a for MDM4 (ref.99). This enabled the synthesis of improved MDM2 and MDM4 inhibitors, which could suppress the growth of colorectal cancer and lung cancer cell lines. Additional dual inhibitors, targeting MDM2 and MDM4 together, are expected to be developed.
PROTACs
‘Conventional’ small-molecule inhibitors of MDM2 and MDM4 work in a stoichiometric manner, often requiring relatively high doses. By contrast, proteolysis targeting chimeras (PROTACs) and molecular glue compounds operate catalytically, potentially rendering them effective even at low doses100. PROTACs are small heterobifunctional molecules that bind to an E3 ubiquitin ligase through one of their arms and a target protein of interest through their other arm. By positioning the target protein properly in physical proximity to the E3 ligase, PROTACs enforce the ubiquitylation and subsequent proteasomal degradation of the former. In this manner, any specific protein may potentially be eliminated selectively if a suitable PROTAC is available. As an E3 ligase, MDM2 was also employed towards degrading other oncogenic proteins via PROTAC-mediated recruitment. In particular, based on the knowledge that nutlins bind tightly to MDM2, nutlin-based PROTACs have been developed101. When applied to cancer cells carrying wtp53, such PROTACs serve as a double-edged sword, as exemplified by a nutlin-based PROTAC that targets the transcriptional and epigenetic regulator BRD4: while promoting BRD4 degradation, it also inhibits MDM2, leading to p53 activation102. Furthermore, the PROTAC methodology has also been used to target MDM2 itself. One such example is MD-224, which promoted MDM2 degradation and p53 activation when applied at a low dose to cultured leukaemia cells, and elicited tumour regression in a mouse xenograft model103. Additional nutlin-based MDM2 degraders have recently been developed104, including an MDM2 homo-PROTAC whose self-dimerization causes its auto-ubiquitylation and destruction105.
The human papillomavirus (HPV) E6 protein is another wtp53-relevant therapeutic target. E6 interacts with p53 and promotes its degradation by recruiting the ubiquitin–protein ligase E3A (E6AP); this is often the case in HPV-induced cervical cancer. RITA (reactivation of p53 and induction of tumour cell apoptosis) is a p53-binding drug first recognized as an MDM2 inhibitor, but later shown to interfere also with the interaction of p53 with E6 (ref.106). Concordantly, RITA promoted p53-dependent apoptosis in HPV-positive cervical cancer cell lines through inhibition of E6–p53 binding107. However, like several other p53-activating small molecules, RITA exerts not only p53-dependent but also p53-independent effects108,109, including increased DNA damage response110, questioning its suitability for further clinical development.
A new inhibitor of the E6–p53 interaction (‘compound 12’), identified by structural screening in silico, was shown to upregulate p53 target genes and induce cytotoxicity and replicative senescence in HPV-positive cervical cancer cells111. Interestingly, MDM4 was recently proposed to partner with E6AP to promote p53 ubiquitylation and degradation112. Accordingly, XI-011 (NSC146109), a small-molecule MDM4 inhibitor, exerted antiproliferative effects in HPV-positive cervical cancer cell lines, suggesting a potential utility of MDM4 inhibitors for treating HPV-positive cervical cancer.
Targeting truncated p53
Although most cancer-associated TP53 mutations are missense mutations, approximately 10% of TP53-mutated tumours carry nonsense mutations, yielding truncated proteins113 that are usually degraded shortly after translation by the nonsense-mediated mRNA decay (NMD) machinery. As these proteins are short lived and often devoid of much of the p53 protein sequence, attempts to reactivate them by the approaches described above are mostly probably pointless.
Accordingly, two alternative approaches have been proposed for activating the p53 signalling pathway in cancer cells that harbour p53-truncating mutations. The first approach is based on molecules that promote translational readthrough, enabling the translation machinery to bypass RNA stop codons and produce full-length p53 protein. Such compounds include the aminoglycoside antibiotic gentamicin and its derivatives, such as G418 and the new-generation synthetic derivative NB124. Treatment with these aminoglycosides rescues the synthesis of intact p53, promoting cancer cell apoptosis114,115. A complementary approach entails inhibition of the NMD process. One example is NMD14, which targets a structural pocket of SMG7, a key component of the NMD machinery116. Similar drugs, such as ataluren, have already reached phase III clinical trials for cystic fibrosis. Their efficacy as anticancer agents, particularly in the context of tumours harbouring TP53 nonsense mutations, remains to be established. Moreover, these compounds display substantial toxicity117, questioning their potential as selective p53-targeted drugs.
Targeting mutp53 GOF
Although most p53-based drug development efforts are directed towards restoring wild-type p53 activity in cancer cells, attempts have also been made to abrogate mutp53 GOF activities by targeting mutp53 for rapid degradation. This relies on the presumption that TP53-mutated cancer cells are addicted to their mutp53 protein16. Thus, given that HSP90 heat shock proteins attenuate the degradation of mutp53, Alexandrova et al.118 showed that prolonged inhibition of HSP90 increased the survival of mice carrying mutp53-expressing tumours but not those with p53-null tumours. In a complementary approach, Padmanabhan et al.119 identified a small molecule (MCB613) that selectively drives the nuclear export and lysosomal degradation of the p53(R175H) hotspot mutant, but not of other mutants. Although abrogation of mutp53 GOF via its targeted elimination remains a sensible approach, it may lose its appeal with the emergence of better drugs that restore the functionality of mutp53, thereby reconstituting tumour suppressor activities while at the same time abrogating inherent mutp53 GOF. Moreover, as the GOF effects of mutp53 are often context dependent and vary between various types of p53 mutant120,121,122,123, further elucidation of those effects may be required to stratify cancer patients for mutp53-directed treatments.
The p53-targeting small-molecule drugs era, ushered in more than 20 years ago, is showing increasing promise. However, major challenges still remain, as underscored by the unsatisfactory outcome of multiple clinical trials that have attempted to bring such molecules into the clinic. Therefore, better understanding of the mechanism of action of each drug, along with more effective ways to reduce undesirable side effects, must be attained to enable p53-targeted small molecules to successfully improve cancer management decisions and affect the lives of cancer patients.
p53-based cancer immunotherapy
Cancer immunotherapy regimens have generated great excitement in recent years, owing to their unprecedented success in several types of cancer. The renaissance of cancer immunotherapy is also kindling renewed interest in p53-based strategies, mainly those aimed at increasing the ability of the immune system to recognize and eradicate cancer cells that harbour deregulated p53. The expectation that such strategies might be effective is largely based on the fact that cancer cells that harbour TP53 missense mutations often overexpress p53, and might therefore be expected to display more p53-derived peptides on their surface through major histocompatibility complexes (MHCs).
One major caveat, though, is that although partly driven by increased expression and translation of the p53 mRNA, the abundance of mutp53 proteins in cancer cells is mainly due to their inefficient degradation by the ubiquitin–proteasome system. As MHC-displayed peptides are produced by proteolytic degradation in the immunoproteasome, it stands to reason that the inefficient degradation of mutp53 may actually restrict, rather than augment, the presentation of p53-derived peptides. Nevertheless, studies performed over the past two decades raise hope that p53-based immunotherapy may eventually gain clinical relevance.
Broadly speaking, the overexpression of missense mutp53 proteins in cancer cells is expected to increase the presentation of various peptides, derived from all over the p53 protein. Although at least some of these peptides might be shared with wtp53, the selectivity of the immune system for the cancer cells will rely on the fact that normal cells express only very low amounts of p53 (Fig. 4). The feasibility of this approach is supported by the observation that the T cell response to p53 is not restricted by natural self-tolerance124. However, the assumption that healthy cells will not be affected is risky: indeed, differentiated cells may possess extremely low amounts of p53 mRNA and hence hardly synthesize any p53 protein, but this may not hold for rapidly proliferating normal progenitor cells, in which TP53 mRNA expression is more substantial77.

a | In the non-proliferative compartment of normal tissues, TP53 is usually silent, and therefore cells do not produce p53 protein and do not present p53-derived peptides on their surface major histocompatibility complex (MHC) class I. b | Proliferating normal cells produce low amounts of p53 protein and present small amounts of p53-derived peptides on their MHC class I (MHC-I). c | In cancer cells expressing wild-type p53 (wtp53), oncogenic stress upregulates p53 mRNA synthesis and translation, causing a more pronounced presentation of p53-derived peptides. These differences in quantity and quality of presented peptides provide the rationale for developing antibody-based strategies to target selectively the cancer cells. T cell receptor (TCR)-like antibodies recognize p53-derived epitopes displayed on MHC-I on the cell surface, triggering an immune response. d | In cancer cells that harbour TP53 missense mutations, p53 protein levels are even more elevated; consequently, such cells may present increased amounts of peptides derived from non-mutated regions of the protein (wtp53 peptides in blue), along with neopeptides comprising the mutated sequence (mutp53 peptide, in red). Bispecific antibodies can be engineered to recognize a neoantigen derived from mutp53 and the TCR–CD3 complex on CD8+ T cells, which results in selective cytotoxicity against mutp53-expressing cancer cells.
p53-based vaccines
Vaccination attempts, aimed to raise cellular immunity against cancer cells that contain highly excessive amounts of p53, were initiated in the 1990s125,126,127. The sequences of the peptides used in those attempts were derived from regions of the wtp53 protein that are rarely mutated in cancer, and thus are shared with cancer-associated mutp53. However, selectivity for cancer cells was enabled by the fact that normal cells possess very low amounts of p53, and thus are not expected to be recognized and attacked by the immune system of the vaccinated host. Subsequently, a synthetic long peptide (SLP) vaccine comprising ten overlapping peptides from the wtp53 sequence (together representing amino acids 70–248), injected twice at a 3 week interval, was shown to elicit a T cell response predominated by CD4+ T cells in metastatic colorectal cancer128. Adverse events were relatively mild: toxicity was limited to grade 1/2, mostly at the vaccination site128. In patients with ovarian cancer, p53 immunogenicity was potentiated by low-dose cyclophosphamide treatment before SLP vaccination129. However, a clinical trial failed to show benefit of SLP vaccination over historical controls130. A modified vaccinia virus ankara (MVA) vaccine encoding wtp53 was also tested in early-phase clinical trials in patients with refractory gastrointestinal cancer and ovarian cancer, and could induce CD8+ and CD4+ T cell responses in six and five patients, respectively, out of a total of 11 patients131,132. Importantly, patients who had an immune response after p53 vaccination had significantly longer progression-free survival compared with patients with no CD8+ T cell expansion131. Further clinical trials using the MVAp53 vaccine together with anti-PD1 antibody pembrolizumab are currently ongoing (NCT03113487, NCT02432963). In a complementary approach, autologous dendritic cells (DCs) were modified to express p53 peptides on MHC class I and II133. Such a DC–p53 vaccine triggered a p53-specific immune response in 16 patients with SCLC, out of the 28 patients treated134. Importantly, of 21 patients who received secondary chemotherapy after p53 vaccination, 13 patients showed an objective clinical response135. Disappointingly, a phase II randomized trial of paclitaxel following DC–p53 vaccine or control revealed no differences in overall response rate136.
The success of mRNA vaccination during the coronavirus disease 2019 (COVID-19) pandemic raises new hopes for a p53 mRNA vaccine. Of note, this avenue was already explored in the past. Remarkably, after introduction of autologous TP53 mRNA-transfected DCs into patients with breast cancer, 13 of 18 patients with tumours expressing high levels of p53 displayed a p53-specific interferon gamma (IFNγ) T cell response in vitro; this was in striking contrast to the p53-specific IFNγ T cell response in healthy donors and in patients with breast cancer with low p53 expression (1 of 10 and 2 of 18, respectively)137. This approach is likely to see a revival with recent advanced methodologies.
p53-specific antibodies
Other p53-based immunotherapy approaches are also emerging. T cell receptor mimic (TCRm) antibodies, also called TCR-like antibodies, are a potential strategy to target intracellular proteins. These antibodies, usually generated by phage display library screening or by hybridoma screening, recognize epitopes displayed by MHC class I on the cell surface, similarly to recognition of such epitopes by T cells via their TCR, enabling the recognition of peptides derived from intracellular proteins (Fig. 4). Accordingly, Li et al.138 developed a novel TCRm antibody that recognized a p53-derived epitope that is displayed selectively on MHC class I by cancer cells but not by normal peripheral blood mononuclear cells. Importantly, this p53 TCRm antibody elicited tumour regression in mice carrying breast cancer xenografts. Likewise, Low et al.139 generated a p53-specific TCR-like antibody designated P1C1TM. Although designed on the basis of a wtp53 peptide, P1C1TM elicited selective antibody-dependent cellular cytotoxicity towards cancer cells that harboured several different p53 mutations, presumably owing to their high p53 abundance. As an additional therapeutic advantage, P1C1TM also facilitated drug delivery into p53-mutated cancer cells via antibody–drug conjugates139.
An alternative scenario relies on the notion that peptides that comprise the mutated amino acid of a p53 missense mutant, when displayed by MHC molecules, will constitute neoantigens. The extensive diversity of TP53 missense mutations in human cancer presents a wealth of potential neoantigens. An immune response elicited against such neoantigens will be specific to cancer cells harbouring particular mutations, and will not endanger rapidly proliferating normal cells. The attractiveness of mutp53-derived peptides as targets for cancer-specific immunotherapy was noted long ago140. Recent work has confirmed that mutp53 may give rise to neoantigens that are presented by MHC molecules and activate a mutp53-specific immune response141,142,143. Notably, cancer patients who mounted a tumour-infiltrating lymphocyte response to mutp53-derived neoantigens also had mutp53-specific reactive T cells in their peripheral blood, raising hope that such peripheral blood T cells might potentially be used for adoptive cell therapy of patients whose tumours carry the same TP53 mutation144.
Bispecific antibodies are a very promising approach to cancer immunotherapy145. Indeed, an engineered single-chain mutp53-based bispecific antibody, recognizing a neoantigen derived from the p53(R175H) hotspot mutant and the TCR–CD3 complex, was recently generated146. Usually, the low density of such neoantigens on the surface of the cancer cells hinders their immune elimination. However, by binding with high affinity to both the p53(R175H) peptide–HLA complex on the cancer cells and the TCR–CD3 complex on T cells, this bispecific antibody could overcome the paucity of neoantigen presentation and selectively redirect T cells to recognize cancer cells presenting the mutant peptide (Fig. 4). This resulted in remarkable selective cytotoxicity against p53(R175H)-expressing cancer cells, both in vitro and in vivo146. This and additional mutp53-selective immunotherapy approaches are likely to gain increasing popularity in coming years.
p53 and the tumour microenvironment
Beyond the targeted attempts to develop p53-specific immunotherapy modalities, recent studies have underscored a broader connection between p53 and cancer immunotherapy. Thus, p53 status in cancer cells can affect the immune landscape in the TME23,147 (Fig. 5); functional wtp53 in cancer cells tends to favour a cancer-restrictive TIME, whereas loss of wtp53 tilts the balance towards a more cancer-supportive TIME. Furthermore, some missense mutp53 proteins, as part of their GOF activities, may further restrict the ability of the immune system to attack cancer cells. For example, wtp53 can reduce the levels of PDL1 indirectly via upregulation of miR-34a148, and induce the expression of the natural killer (NK) cell-activating ligands UL16-binding protein 1 (ULBP1) and ULBP2. These transcriptional effects of wtp53, mediated by direct binding of p53 to the corresponding target genes149, render cancer cells more amenable to attack by cytotoxic T cells and NK cells, respectively. Moreover, through regulation of cytokine expression, wtp53 can confer antitumour effects by changing the composition of the TIME24. Interestingly, using several MDM2 inhibitors, Zhou et al.150 reported that p53 activation orchestrates a tumour-suppressive TIME through activation of endogenous retroviruses, leading to increased IFNγ signalling and sensitizing the tumour to immune checkpoint inhibitors150. Conversely, by altering the secretion of cytokines, as well as the physical properties of the TME, hotspot p53 mutants may exert GOF effects on the TIME that go beyond the mere impact of wtp53 loss25,151. Hence, the TP53 status of a tumour could potentially be important for patient management decisions related to immunotherapy. Furthermore, drugs that restore or boost p53 functionality in cancer cells, as discussed in this Review, might also be considered for administration in combination with immunotherapy regimens to increase their likelihood of success. Indeed, in July 2022, PMV Pharmaceuticals announced a clinical trial collaboration with Merck of PC14586 (see above ‘Targeting TP53 missense mutant tumours’) in combination with the immune checkpoint inhibitor pembrolizumab (KEYTRUDA), in patients with advanced solid tumours that carry the p53(Y220C) mutation152.

In p53-wild-type (wtp53) tumours, the tumour immune microenvironment has a predominantly anti-tumoural effect. This can be attributed to several mechanisms. For example, wtp53 can upregulate the expression of UL16-binding protein 1 (ULBP1) and ULBP2 and microRNA miR-34a. ULBP1 and ULBP2 are activating ligands for natural killer (NK) cells, which enhances their cytotoxic activity. miR-34 blocks the translation of PDL1 mRNA and drives its degradation; consequently, PDL1 protein levels are downregulated in cancer cells that retain wtp53, sensitizing these tumour cells to CD8+ T cell-mediated killing. Moreover, wtp53 can promote the secretion of anti-tumoural cytokines such as tumour necrosis factor (TNF), which further enhances the protective effects of the tumour immune microenvironment. In TP53-mutated cells, ULBP1 and ULBP2 are downregulated and PDL1 is upregulated, causing resistance to killing by NK and CD8+ T cells, respectively. Furthermore, loss of p53 function can drive secretion of WNT ligands, which bind to their cognate receptors on macrophages, causing them to secrete IL-1β, which attracts pro-tumoural cells such as neutrophils. Mutp53 proteins promote the secretion of exosomes that can deliver microRNAs such as miR-1246, which convert anti-tumoural M1 macrophages to pro-tumoural M2 macrophages. Such pro-tumoural macrophages secrete IL-1β, which attracts pro-tumoural regulatory T (Treg) cells that dampen the anti-tumoural response and protect the tumour from immune elimination. MHC-I, major histocompatibility complex class I; RE, response element; TCR, T cell receptor.
Of note, non-cancer cells in the TME retain wtp53. Hence, drugs that boost p53 activity may also augment the non-autonomous cancer-suppressive functions of p53 in the TME, as exemplified by nutlin treatment of stromal fibroblasts153. Likewise, enhancement of p53 activity in immune cells may contribute directly to a tumour-suppressive TIME. This is illustrated by a study in which the MDM2 inhibitor APG-115 exerted antitumour effects in wtp53- and mutp53-expressing hepatoma and colon carcinoma syngeneic models, respectively154. In the latter case, the effects were attributed to the ability of APG-115 to promote M1 macrophage polarization, presumably through modulation of their endogenous wtp53. Of note, M1 macrophages promote a cancer-inhibitory TIME, as opposed to M2 macrophages, which are associated with a more tumour-protective TIME. By contrast, CD4+ T cell activation and CD8+ T cell infiltration relied on activation of p53 in the cancer cells. Reassuringly, combination therapy with APG-115 and a PDL1 inhibitor conferred enhanced antitumour immunity relative to each treatment alone, but this effect was abolished in Trp53-knockout mice, further underscoring the importance of p53 activation in the TME154. A combination of APG-115 and pembrolizumab was tested in a clinical trial in metastatic melanoma and advanced-stage solid tumours, demonstrating good tolerability and preliminary indications of antitumour activity155. Moreover, beneficial effects of combining p53 activation with immunotherapy were observed also with gene therapy modules, including nanoparticles156 and adenovirus–p53 (ref.157). Given the great interest in cancer immunotherapy, further studies on such combination treatments will most certainly continue and intensify.
p53-based genetic therapies
Gene therapy has seen renewed enthusiasm in recent years158,159. Remarkably, the first gene therapy ever to be approved for clinical use is p53 based. Gendicine, a recombinant human p53 adenovirus developed by Shenzhen SiBiono GeneTech, was approved in 2003 by the China Food and Drug Administration (CFDA) for HNSCC. Since then, gendicine has been administered to thousands of patients in China, and was reported to achieve significantly higher response rates than standard of care when combined with chemotherapy or radiotherapy160,161,162,163. Additional adenovirus-based p53 gene therapies, including advexin and SCH-58500 (refs.164,165), showed promising results in clinical trials166, but unlike gendicine, they have not been approved for clinical use. However, it should be noted that serious concerns have been raised regarding the likelihood that such approaches will be effective, particularly if p53 reconstitution is not combined with other therapeutic targets167. Moreover, there is still debate about the rate of success of gendicine and its correct use in cancer treatment168. With the introduction of newer, more sophisticated viral vectors, p53 gene therapy may hopefully become more effective and more broadly feasible, conceivably as part of combination therapy regimens169.
Nanoparticles have also been explored as p53 gene therapy vehicles (Fig. 6). Unlike viruses, nanoparticles have low immunogenicity and are thus refractory to inhibitory antibodies, thereby extending their circulation time while diminishing immune-related adverse side effects. Moreover, by intravenous administration, nanoparticles are more suitable for treating distant metastases than intratumoural injection, the main route for gendicine administration. Importantly, the improvement of methods for delivering gene products specifically to cancer cells has greatly increased the efficiency and specificity of nanoparticles, improving their ability to selectively restore p53 expression in cancer cells and enforce more robust anticancer effects170,171,172 both in vitro and in several xenograft models, such as those of hepatocellular carcinoma (HCC) and breast cancer. For example, SGT-53, a cationic liposome developed by SynerGene Therapeutics, carrying wtp53-encoding DNA that homes selectively to tumour cells via an anti-transferrin single-chain antibody fragment, sensitized glioblastoma cells to temozolomide both in vitro and in vivo and improved survival in a glioblastoma mouse model173. In a phase I clinical trial with 11 patients harbouring different types of advanced solid tumour, SGT-53 delivered the TP53 transgene to metastatic lesions and exerted anticancer effects174. Moreover, seven of those 11 patients demonstrated stable disease at a 6-week assessment175. Ongoing clinical trials should determine the benefit of SGT-53 relative to alternative approaches.

Wild-type p53 (wtp53)-encoding DNA and RNA can be introduced into cancer cells by several approaches, including recombinant viruses and nanoparticles. This drives p53 expression and transcription of wtp53 target genes, resulting in anticancer effects. In TP53-mutated tumours, delivery of CRISPR–Cas9 together with suitable guide RNA (gRNA) might potentially enable base editing, restoring wild-type TP53 sequence.
In tune with the increased interest in mRNA delivery, p53 mRNA nanoparticles are also being developed. In a recent study, p53 mRNA administration via redox-responsive nanoparticles caused a significant decrease in the viability of p53-null lung cancer cells and a robust reduction in tumour size in HCC and non-small-cell lung cancer (NSCLC) mouse models, which was further enhanced when combined with mTOR inhibitors176. Similarly, in an HCC mouse model, administration of p53 mRNA nanoparticles combined with immunotherapy resulted in an improved anticancer effect compared with each treatment alone, supporting further attempts to explore the efficacy of combining p53 mRNA therapy with immune checkpoint blockade177. It will not be surprising to see a growing number of p53 mRNA–nanoparticle formulations in the near future.
One obvious question is to what extent the efficacy of p53 gene therapy may depend on TP53 status in the cancer cells. True p53-null tumours are theoretically the perfect targets, but they represent only a small minority of cases. Tumours carrying truncating or frameshift TP53 mutations, which are more common, also represent attractive targets. Yet, many tumours harbour TP53 missense mutations. On the one hand, such tumours might have been selected for loss of wtp53 activity, and as such should respond more vigorously to wtp53 restoration. On the other hand, however, many of those tumours accumulate excessive mutp53 protein, which may act in a dominant-negative manner to neutralize the reintroduced wtp53. Interestingly, a meta-analysis of patients with recurrent HNSCC treated with adenovirus–p53 gene therapy showed that all responders had only weak p53 staining by immunohistochemistry (IHC) and no TP53 missense mutations178. Hence, abundant mutp53 might indeed restrict the efficacy of wtp53 gene therapy, suggesting strong p53 IHC staining as an exclusion criterion for such treatments. Obviously, more studies and more compelling clinical trial data are still required to draw firm conclusions about the long-term utility of wtp53 gene therapy.
In the context of p53-targeted genetic therapy, one may also consider delivery of synthetic small interfering RNA (siRNA) oligonucleotides that target specific mutations within p53 mRNA, to abrogate the GOF effects of mutp53. Indeed, Martinez et al.179 showed that a single base difference in the siRNA can discriminate between wtp53 and mutp53. Delivery of an mRNA targeting the p53(R248W) mutant resulted in decreased cancer cell survival and increased apoptosis. More recently, Ubby et al.180 devised siRNAs specific for four different hotspot p53 mutants and showed that they decreased the viability of patient-derived xenografts in a mutant-specific manner with no organ toxicity and no effect on wtp53 mRNA. Although siRNA-based treatments are still in their infancy, the above findings and the rapid advances in RNA delivery may encourage further exploration of mutant-specific p53 siRNA for cancer therapy.
The power of CRISPR–Cas9 has kindled great excitement, raising the question of whether it can also be applied to p53-targeted cancer therapy181. As one example, in a study introducing CRISPR–Cas9 base editing — which provides increased specificity and fewer indels — HCC1954 breast cancer cells were treated with a base editor to convert a TP53 missense mutation into a wild-type sequence. Upon base editing, correction of the TP53 mutation was achieved in 7.6% of cells, while only 0.7% of the cells displayed evidence of indel formation182. Although that study did not investigate the biological effects on the conversion, it is tempting to think that correction of TP53 mutations by base editing might become clinically feasible in the future. Notably, CRISPR–Cas9 elicits DNA damage-induced p53 activation in p53-wild-type cells, causing cell cycle arrest or cell death, thereby selecting for survival of cells with TP53 mutations183,184,185. By the same logic, TP53-mutant cancer cells in which conversion into wild-type TP53 is successfully achieved might be even more effectively eliminated owing to concurrent DNA damage signalling. Yet, a major caveat is that, to have clinical benefit, successful conversion must be achieved in the vast majority of cancer cells. Time will tell whether this is feasible in humans and whether CRISPR–Cas9 TP53 base editing may become a valid cancer therapy option.
Challenges and concerns
Although constant progress is being made towards better p53-based cancer therapy, many challenges remain and the search for efficient and selective drugs that will eventually be able to enter the clinic is still ongoing.
As with all other anticancer treatments, a major concern regarding p53-based therapy is the emergence of resistance. Experience-based knowledge is still scarce; yet, some potential scenarios are predictable. Thus, an obvious mechanism of resistance to MDM2 inhibitors is mutation of the TP53 gene, which has already been demonstrated experimentally upon prolonged nutlin treatment186. Others include activation of anti-apoptotic genes, intrinsic resistance to apoptosis and upregulation of MDM4 (refs.2,187). It remains to be seen to what extent this may reduce the efficacy of p53-based drugs.
Furthermore, p53-based drugs are unlikely to enter the clinic as monotherapies. Numerous studies have attempted to identify promising relevant drug combinations. For example, combining a BET inhibitor with nutlin 3a had an additive effect in a wtp53 AML model, which involved enhanced activation of p53 by the BET inhibitor188. Likewise, a combination of the BCL-2 inhibitor venetoclax with idasanutlin yielded promising results in primary AML cells, in which it overcame inherent resistance to either monotherapy, presumably by lowering the apoptotic threshold189. This combination has subsequently gone into clinical trials190, including an ongoing trial in young adults and children with relapsed/refractory leukaemia and solid tumours (NCT04029688). Additionally, as discussed earlier, combining p53 activation with immunotherapy is also appealing. Such combined treatments might reduce the required doses and may even overcome resistance in some instances.
Another concern arises from the fact that the in vivo testing of p53-based drugs is performed primarily in mice. Although mouse models remain a standard tool for drug discovery, many differences exist between mice and humans, including interspecies differences in the sequences of the p53, MDM2 and MDM4 proteins, as well as differences in the p53 signalling pathway191. Advanced experimental methodologies, such as organ-on-a-chip192 and other ex vivo models193, will hopefully become increasingly more useful in bypassing these interspecies differences, towards accelerating the transition of p53-targeted drugs to the clinic.
Finally, perhaps the biggest challenge is our incomplete understanding of human biology and of the complex processes that take place within a cancer cell upon administration of a drug. Thus, although our screening methods are constantly improving and are yielding a growing list of p53-targeted compounds, we still lack sufficient knowledge to predict reliably off-target effects and even undesirable on-target effects, resulting in the unwelcome situation that unacceptable toxicity is discovered only when a drug is administered to patients. There is therefore an urgent need for deeper understanding and more definitive testing models.
Conclusion
The fact that efforts to develop p53-based therapies have been ongoing for almost three decades raises concerns about their cost effectiveness. However, the centrality of p53 alterations in human cancer, and thus the promise that success may have an exceptionally high impact on cancer management, provides an assurance that such efforts will not cease. Hence, one may view targeting p53 as a ‘high risk/high gain’ endeavour, with potential to revolutionize cancer treatment if truly successful. The progress achieved in recent years is gradually dismissing the idea that p53 is undruggable, but much remains to be explored before p53-targeted therapies can become part of standard-of-care protocols. Yet, encouragement can perhaps be drawn from the recent exciting development in KRAS inhibition. Like p53, KRAS has also come to be considered by many as insufficiently druggable, in view of repeated failures. However, a specific KRAS(G12C) inhibitor194 has recently received FDA approval, raising hopes that other ‘difficult’ targets such as p53 may eventually follow the path to success.
As our knowledge grows, it is becoming increasingly clear that a one-p53-drug-fits-all approach is bound to fail, just as the KRAS(G12C) inhibitor does not benefit patients whose cancers carry other KRAS mutations. Therefore, careful patient–drug matching will be essential. Fortunately, owing to the prevalence of p53 dysfunction in human cancer, even regimens that show efficacy in only a small percentage of p53-dysfunctional cases could benefit significant numbers of patients.
References
Barbosa, A. et al. Gene panel tumor testing in ovarian cancer patients significantly increases the yield of clinically actionable germline variants beyond BRCA1/BRCA2. Cancers 12, 2834 (2020).
Hoe, K. K., Verma, C. S. & Lane, D. P. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 13, 217–236 (2014).
Sabapathy, K. & Lane, D. P. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 15, 13–30 (2018).
Bykov, V. J. N., Eriksson, S. E., Bianchi, J. & Wiman, K. G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 182, 89–102 (2017).
Cheok, C. F. & Lane, D. P. Exploiting the p53 pathway for therapy. Cold Spring Harb. Perspect. Med. 7, a026310 (2017).
Levine, A. J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 20, 471–480 (2020).
Kastan, M. B., Canman, C. E. & Leonard, C. J. p53, cell cycle control and apoptosis: implications for cancer. Cancer Metastasis Rev. 14, 3–15 (1995).
Williams, A. B. & Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 6, a026070 (2016).
Fridman, J. S. & Lowe, S. W. Control of apoptosis by p53. Oncogene 22, 9030–9040 (2003).
Kastenhuber, E. R. & Lowe, S. W. Putting p53 in context. Cell 170, 1062–1078 (2017).
Speidel, D. Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol. 20, 14–24 (2010).
Vaseva, A. V. & Moll, U. M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 1787, 414 (2009).
Giorgi, C. et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl Acad. Sci. USA 112, 1779–1784 (2015).
Maiuri, M. C. et al. Autophagy regulation by p53. Curr. Opin. Cell Biol. 22, 181–185 (2010).
Tan, X. et al. p53 loss activates prometastatic secretory vesicle biogenesis in the Golgi. Sci. Adv. 7, eabf4885 (2021).
Mantovani, F., Collavin, L. & Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 26, 199–212 (2018).
Sicari, D. et al. Mutant p53 improves cancer cells’ resistance to endoplasmic reticulum stress by sustaining activation of the UPR regulator ATF6. Oncogene 38, 6184–6195 (2019).
Walerych, D. et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 18, 897–909 (2016).
Capaci, V. et al. Mutant p53 induces Golgi tubulo-vesiculation driving a prometastatic secretome. Nat. Commun. 11, 3945 (2020).
Weisz, L., Oren, M. & Rotter, V. Transcription regulation by mutant p53. Oncogene 26, 2202–2211 (2007).
Kim, M. P. & Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 25, 161–168 (2018).
Amit, M. et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature 578, 449–454 (2020).
Blagih, J., Buck, M. D. & Vousden, K. H. p53, cancer and the immune response. J. Cell Sci. 133, jcs237453 (2020).
Wellenstein, M. D. et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 572, 538–542 (2019). This paper shows that p53 loss in cancer cells promotes a tumour-supportive immune microenvironment in multiple mouse models of breast cancer.
Cooks, T. et al. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 9, 771 (2018).
Pilley, S., Rodriguez, T. A. & Vousden, K. H. Mutant p53 in cell-cell interactions. Genes Dev. 35, 433–448 (2021).
Olivier, M., Hollstein, M. & Hainaut, P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2, a001008 (2010).
Shchors, K. et al. Using a preclinical mouse model of high-grade astrocytoma to optimize p53 restoration therapy. Proc. Natl Acad. Sci. USA 110, E1480–E1489 (2013).
Martins, C. P., Brown-Swigart, L. & Evan, G. I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 127, 1323–1334 (2006).
Xue, W. et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 (2007).
Ventura, A. et al. Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665 (2007).
Sánchez-Rivera, F. J. et al. Mitochondrial apoptotic priming is a key determinant of cell fate upon p53 restoration. Proc. Natl Acad. Sci. USA 118, e2019740118 (2021).
Foster, B. A., Coffey, H. A., Morin, M. J. & Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 286, 2507–2510 (1999).
Wang, W., Takimoto, R., Rastinejad, F. & El-Deiry, W. S. Stabilization of p53 by CP-31398 inhibits ubiquitination without altering phosphorylation at serine 15 or 20 or MDM2 binding. Mol. Cell. Biol. 23, 2171–2181 (2003).
Rippin, T. M. et al. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 21, 2119–2129 (2002).
Bykov, V. J. N. et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 8, 282–288 (2002). This is the first report of PRIMA-1, a small molecule that restores wtp53-like functionality to multiple p53 mutants and induces apoptosis in cancer cells.
Lambert, J. M. R. et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 15, 376–388 (2009).
Bykov, V. J. N. et al. Targeting of mutant p53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front. Oncol. 6, 21 (2016).
Chipuk, J. E., Maurer, U., Green, D. R. & Schuler, M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell 4, 371–381 (2003).
Saha, M. N., Chen, Y., Chen, M. H., Chen, G. & Chang, H. Small molecule MIRA-1 induces in vitro and in vivo anti-myeloma activity and synergizes with current anti-myeloma agents. Br. J. Cancer 110, 2224–2231 (2014).
Zache, N. et al. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2, 70–80 (2008).
Bou-Hanna, C. et al. Acute cytotoxicity of MIRA-1/NSC19630, a mutant p53-reactivating small molecule, against human normal and cancer cells via a caspase-9-dependent apoptosis. Cancer Lett. 359, 211–217 (2015).
Tal, P. et al. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget 7, 11817–11837 (2016).
Ali, D. et al. APR-246 exhibits anti-leukemic activity and synergism with conventional chemotherapeutic drugs in acute myeloid leukemia cells. Eur. J. Haematol. 86, 206–215 (2011).
Zandi, R. et al. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 17, 2830–2841 (2011).
Liu, X. et al. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 41, 6034–6044 (2013).
Bykov, V. J. N. et al. PRIMA-1MET synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 24, 3484–3491 (2005).
Mohell, N. et al. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 6, e1794 (2015).
Fransson, Å. et al. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant high-grade serous ovarian cancer. J. Ovarian Res. 9, 27 (2016).
Sallman, D. A. et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant myelodysplastic syndromes. J. Clin. Oncol. 39, 1584–1594 (2021).
Cluzeau, T. et al. Eprenetapopt plus azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia: a phase II study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 39, 1575–1583 (2021).
Aprea https://ir.aprea.com/news-releases/news-release-details/aprea-therapeutics-announces-positive-results-phase-2-trial (2021).
Kravchenko, J. E. et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl Acad. Sci. USA 105, 6302–6307 (2008).
Zhang, S. et al. Small-molecule NSC59984 restores p53 pathway signaling and antitumor effects against colorectal cancer via p73 activation and degradation of mutant p53. Cancer Res. 75, 3842–3852 (2015).
Xu, J. et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 7, 285–295 (2011).
Zhang, S., Zhou, L. & El-Deiry, W. S. Small-molecule NSC59984 induces mutant p53 degradation through a ROS–ERK2–MDM2 axis in cancer cells. Mol. Cancer Res. 20, 622–636 (2022).
Wang, G. & Fersht, A. R. Propagation of aggregated p53: cross-reaction and coaggregation vs. seeding. Proc. Natl Acad. Sci. USA 112, 2443 (2015).
Soragni, A. et al. A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell 29, 90–103 (2016).
Palanikumar, L. et al. Protein mimetic amyloid inhibitor potently abrogates cancer-associated mutant p53 aggregation and restores tumor suppressor function. Nat. Commun. 12, 3962 (2021).
Joerger, A. C., Ang, H. C. & Fersht, A. R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl Acad. Sci. USA 103, 15056–15061 (2006).
Boeckler, F. M. et al. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl Acad. Sci. USA 105, 10360–10365 (2008).
ASCO. https://meetings.asco.org/abstracts-presentations/207594 (2022).
Yu, X., Vazquez, A., Levine, A. J. & Carpizo, D. R. Allele-specific p53 mutant reactivation. Cancer Cell 21, 614–625 (2012).
Yu, X. et al. Thiosemicarbazones functioning as zinc metallochaperones to reactivate mutant p53. Mol. Pharmacol. 91, 567–575 (2017).
Salim, K. Y., Maleki Vareki, S., Danter, W. R. & Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 7, 41363–41379 (2016).
Lindemann, A. et al. COTI-2, a novel thiosemicarbazone derivative, exhibits antitumor activity in HNSCC through p53-dependent and -independent mechanisms. Clin. Cancer Res. 25, 5650–5662 (2019).
Westin, S. N. et al. Abstract CT033: Safety and early efficacy signals for COTI-2, an orally available small molecule targeting p53, in a phase I trial of recurrent gynecologic cancer. Cancer Res. 78, CT033 (2018).
Chen, S. et al. Arsenic trioxide rescues structural p53 mutations through a cryptic allosteric site. Cancer Cell 39, 225–239.e8 (2021). This paper reports that ATO, used for treating acute promyelocytic leukaemia, can reactivate structural p53 mutants and induce p53-dependent tumour suppression.
Oliner, J. D., Kinzler, K. W., Meltzer, P. S., George, D. L. & Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 358, 80–83 (1992).
Oliner, J. D., Saiki, A. Y. & Caenepeel, S. The role of MDM2 amplification and overexpression in tumorigenesis. Cold Spring Harb. Perspect. Med. 6, a026336 (2016).
Vassilev, L. T. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 (2004).
Vu, B. et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 4, 466–469 (2013).
Ray-Coquard, I. et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 13, 1133–1140 (2012).
Andreeff, M. et al. Results of the phase 1 trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin. Cancer Res. 22, 868 (2016).
Kim, E. S. & Shohet, J. M. Reactivation of p53 via MDM2 inhibition. Cell Death Dis. 6, e1936 (2015).
Iancu-Rubin, C. et al. Activation of p53 by the MDM2 inhibitor RG7112 impairs thrombopoiesis. Exp. Hematol. 42, 137–145.e5 (2014).
Xue, Y. et al. Bortezomib stabilizes and activates p53 in proliferative compartments of both normal and tumor tissues in vivo. Cancer Res. 79, 3595–3607 (2019).
Yee, K. et al. Murine double minute 2 inhibition alone or with cytarabine in acute myeloid leukemia: results from an idasanutlin phase 1/1b study. Leuk. Res. 100, 106489 (2021).
Montesinos, P. et al. MIRROS: a randomized, placebo-controlled, phase III trial of cytarabine ± idasanutlin in relapsed or refractory acute myeloid leukemia. Future Oncol. 16, 807–815 (2020).
Konopleva, M. Y. et al. Idasanutlin plus cytarabine in relapsed or refractory acute myeloid leukemia: results of the MIRROS trial. Blood Adv. 6, 4147–4156 (2022).
Mascarenhas, J. et al. Oral idasanutlin in patients with polycythemia vera. Blood 134, 525–533 (2019).
Mascarenhas, J. et al. Safety and efficacy of idasanutlin in patients (pts) with hydroxyurea (HU)-resistant/intolerant polycythemia vera (PV): results of an international phase II study. Blood 136, 29–31 (2020).
Fang, D. D. et al. MDM2 inhibitor APG-115 exerts potent antitumor activity and synergizes with standard-of-care agents in preclinical acute myeloid leukemia models. Cell Death Disco. 7, 90 (2021).
Yi, H. et al. A novel small molecule inhibitor of MDM2-p53 (APG-115) enhances radiosensitivity of gastric adenocarcinoma. J. Exp. Clin. Cancer Res. 37, 97 (2018).
Sun, D. et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J. Med. Chem. 57, 1454–1472 (2014).
Canon, J. et al. The MDM2 inhibitor AMG 232 demonstrates robust antitumor efficacy and potentiates the activity of p53-inducing cytotoxic agents. Mol. Cancer Ther. 14, 649–658 (2015).
Verstovsek, S. et al. BOREAS: a global phase 3 study of KRT-232, a first-in-class murine double minute 2 (MDM2) inhibitor in TP53WT relapsed/refractory (R/R) myelofibrosis (MF). J. Clin. Oncol. 39 (suppl.15), TPS7057 (2021).
Erba, H. P. et al. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 3, 1939–1949 (2019).
Jeay, S. et al. Dose and schedule determine distinct molecular mechanisms underlying the efficacy of the p53-MDM2 inhibitor HDM201. Cancer Res. 78, 6257–6267 (2018).
Gounder, M. M. et al. A phase 1 study of the MDM2 inhibitor DS-3032b in patients (pts) with advanced solid tumors and lymphomas. J. Clin. Oncol. 34 (suppl.15), 2581 (2016).
Wade, M., Li, Y. C. & Wahl, G. M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 13, 83–96 (2013).
Carvajal, L. A. et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 10, 3003 (2018).
Tan, Y. S., Lane, D. P. & Verma, C. S. Stapled peptide design: principles and roles of computation. Drug. Discov. Today 21, 1642–1653 (2016).
Bernal, F. et al. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell 18, 411–422 (2010).
Li, Y. C. et al. A versatile platform to analyze low-affinity and transient protein-protein interactions in living cells in real time. Cell Rep. 9, 1946–1958 (2014).
Chang, Y. S. et al. Stapled α-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl Acad. Sci. USA 110, E3445–E3454 (2013).
Pairawan, S. et al. First in class dual MDM2/MDMX inhibitor ALRN-6924 enhances antitumor efficacy of chemotherapy in TP53 wild-type hormone receptor-positive breast cancer models. Breast Cancer Res 23, 29 (2021).
Meric-Bernstam, F. et al. Phase I trial of a novel stapled peptide ALRN-6924 disrupting MDMX- and MDM2-mediated inhibition of WT p53 in patients with solid tumors and lymphomas. J. Clin. Oncol. 35, 2505 (2017).
Cheng, X. et al. Leveraging the multivalent p53 peptide-MdmX interaction to guide the improvement of small molecule inhibitors. Nat. Commun. 13, 1087 (2022).
Mares, A. et al. Extended pharmacodynamic responses observed upon PROTAC-mediated degradation of RIPK2. Commun. Biol. 3, 140 (2020).
Schneekloth, A. R., Pucheault, M., Tae, H. S. & Crews, C. M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 18, 5904–5908 (2008).
Hines, J., Lartigue, S., Dong, H., Qian, Y. & Crews, C. M. MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res. 79, 251–262 (2019).
Li, Y. et al. Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J. Med. Chem. 62, 448–466 (2019).
Wang, B. et al. Development of MDM2 degraders based on ligands derived from Ugi reactions: lessons and discoveries. Eur. J. Med. Chem. 219, 113425 (2021).
He, S. et al. Homo-PROTAC mediated suicide of MDM2 to treat non-small cell lung cancer. Acta Pharm. Sin. B 11, 1617–1628 (2021).
Issaeva, N. et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 10, 1321–1328 (2004).
Zhao, C. Y., Szekely, L., Bao, W. & Selivanova, G. Rescue of p53 function by small-molecule RITA in cervical carcinoma by blocking E6-mediated degradation. Cancer Res. 70, 3372–3381 (2010).
Weilbacher, A., Gutekunst, M., Oren, M., Aulitzky, W. E. & Van Der Kuip, H. RITA can induce cell death in p53-defective cells independently of p53 function via activation of JNK/SAPK and p38. Cell Death Dis. 5, e1318 (2014).
Wiegering, A. et al. Reactivating p53 and inducing tumor apoptosis (RITA) enhances the response of RITA-sensitive colorectal cancer cells to chemotherapeutic agents 5-fluorouracil and oxaliplatin. Neoplasia 19, 301–309 (2017).
Yang, J. et al. Small-molecule activation of p53 blocks hypoxia-inducible factor 1alpha and vascular endothelial growth factor expression in vivo and leads to tumor cell apoptosis in normoxia and hypoxia. Mol. Cell. Biol. 29, 2243–2253 (2009).
Celegato, M. et al. A novel small-molecule inhibitor of the human papillomavirus E6–p53 interaction that reactivates p53 function and blocks cancer cells growth. Cancer Lett. 470, 115–125 (2020).
Zhang, J. et al. A small-molecule inhibitor of MDMX suppresses cervical cancer cells via the inhibition of E6–E6AP–p53 axis. Pharmacol. Res. 177, 106128 (2022).
Donehower, L. A. et al. Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep. 28, 1370–1384.e5 (2019).
Bidou, L., Bugaud, O., Belakhov, V., Baasov, T. & Namy, O. Characterization of new-generation aminoglycoside promoting premature termination codon readthrough in cancer cells. RNA Biol. 14, 378–388 (2017).
Floquet, C., Deforges, J., Rousset, J. P. & Bidou, L. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res. 39, 3350–3362 (2011).
Martin, L. et al. Identification and characterization of small molecules that inhibit nonsense-mediated rna decay and suppress nonsense p53 mutations. Cancer Res. 74, 3104–3113 (2014).
Dabrowski, M., Bukowy-Bieryllo, Z. & Zietkiewicz, E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 24, 25 (2018).
Alexandrova, E. M. et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 523, 352–356 (2015).
Padmanabhan, A. et al. USP15-dependent lysosomal pathway controls p53-R175H turnover in ovarian cancer cells. Nat. Commun. 9, 1270 (2018).
Hassin, O. et al. Different hotspot p53 mutants exert distinct phenotypes and predict outcome of colorectal cancer patients. Nat. Commun. 13, 2800 (2022).
Mukherjee, S. et al. Cross-talk between mutant p53 and p62/SQSTM1 augments cancer cell migration by promoting the degradation of cell adhesion proteins. Proc. Natl Acad. Sci. USA 119, e2119644119 (2022).
Kennedy, M. C. & Lowe, S. W. Mutant p53: it’s not all one and the same. Cell Death Differ. 29, 983–987 (2022).
Alvarado-Ortiz, E. et al. Mutant p53 gain-of-function: role in cancer development, progression, and therapeutic approaches. Front. Cell Dev. Biol. 8, 607670 (2021).
Lauwen, M. M. et al. Self-tolerance does not restrict the CD4+ T-helper response against the p53 tumor antigen. Cancer Res. 68, 893–900 (2008).
Chikamatsu, K. et al. Generation of anti-p53 cytotoxic T lymphocytes from human peripheral blood using autologous dendritic cells. Clin. Cancer Res. 5, 1281–1288 (1999).
Röpke, M. et al. Spontaneous human squamous cell carcinomas are killed by a human cytotoxic T lymphocyte clone recognizing a wild-type p53-derived peptide. Proc. Natl Acad. Sci. USA 93, 14704–14707 (1996).
Vierboom, M. P. M. et al. Tumor eradication by wild-type p53-specific cytotoxic T lymphocytes. J. Exp. Med. 186, 695–704 (1997).
Speetjens, F. M. et al. Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin. Cancer Res. 15, 1086–1095 (2009).
Vermeij, R. et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: a single-arm phase II study. Int. J. Cancer 131, E670–E680 (2012).
Leffers, N. et al. Long-term clinical and immunological effects of p53-SLP® vaccine in patients with ovarian cancer. Int. J. Cancer 130, 105–112 (2012).
Hardwick, N. R. et al. p53MVA therapy in patients with refractory gastrointestinal malignancies elevates p53-specific CD8+ T-cell responses. Clin. Cancer Res. 20, 4459–4470 (2014).
Hardwick, N. R. et al. p53-Reactive T cells are associated with clinical benefit in patients with platinum-resistant epithelial ovarian cancer after treatment with a p53 vaccine and gemcitabine chemotherapy. Clin. Cancer Res. 24, 1315–1325 (2018).
Barfoed, A. M. et al. Cytotoxic T-lymphocyte clones, established by stimulation with the HLA-A2 binding p5365-73 wild type peptide loaded on dendritic cells In vitro, specifically recognize and lyse HLA-A2 tumour cells overexpressing the p53 protein. Scand. J. Immunol. 51, 128–133 (2000).
Antonia, S. J. et al. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin. Cancer Res. 12, 878–887 (2006).
Chiappori, A. A., Soliman, H., Janssen, W. E., Antonia, S. J. & Gabrilovich, D. I. INGN-225: a dendritic cell-based p53 vaccine (Ad.p53-DC) in small cell lung cancer: observed association between immune response and enhanced chemotherapy effect. Expert. Opin. Biol. Ther. 10, 983–991 (2010).
Chiappori, A. A. et al. Randomized-controlled phase II trial of salvage chemotherapy after immunization with a TP53-transfected dendritic cell-based vaccine (Ad.p53-DC) in patients with recurrent small cell lung cancer. Cancer Immunol. Immunother. 68, 517–527 (2019).
Met, Ö., Balslev, E., Flyger, H. & Svane, I. M. High immunogenic potential of p53 mRNA-transfected dendritic cells in patients with primary breast cancer. Breast Cancer Res. Treat. 125, 395–406 (2010).
Li, D. et al. Development of a T-cell receptor mimic antibody against wild-type p53 for cancer immunotherapy. Cancer Res. 77, 2699–2711 (2017).
Low, L., Goh, A., Koh, J., Lim, S. & Wang, C. I. Targeting mutant p53-expressing tumours with a T cell receptor-like antibody specific for a wild-type antigen. Nat. Commun. 10, 5382 (2019).
Cheever, M. A. et al. The prioritization of cancer antigens: a National Cancer Institute pilot project for the acceleration of translational research. Clin. Cancer Res. 15, 5323–5337 (2009).
Balachandran, V. et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, 512–516 (2017).
Deniger, D. C. et al. T-cell responses to TP53 ‘Hotspot’ mutations and unique neoantigens expressed by human ovarian cancers. Clin. Cancer Res. 24, 5562–5573 (2018).
Malekzadeh, P. et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Invest. 129, 1109–1114 (2019).
Malekzadeh, P. et al. Antigen experienced T cells from peripheral blood recognize p53 neoantigens. Clin. Cancer Res. 26, 1267–1276 (2020).
You, G. et al. Bispecific antibodies: a smart arsenal for cancer immunotherapies. Vaccines 9, 724 (2021).
Hsiue, E. H. C. et al. Targeting a neoantigen derived from a common TP53 mutation. Science 371, eabc8697 (2021). This paper reports a bispecific antibody, highly specific to the p53(R175H) mutation, which activates T cells to kill cancer cells presenting a p53(R175H)-derived neoantigen in vitro and in vivo.
Levine, A. J. P53 and the immune response: 40 years of exploration-a plan for the future. Int. J. Mol. Sci. 21, 541 (2020).
Cortez, M. A. et al. PDL1 regulation by p53 via miR-34. J. Natl Cancer Inst. 108, djv303 (2015). This paper reports that wtp53 can downregulate PDL1 in cancer cells, suggesting a mechanism whereby loss of wtp53 can render such cells more refractory to immune attack.
Textor, S. et al. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res. 71, 5998–6009 (2011).
Zhou, X. et al. Pharmacological activation of p53 triggers viral mimicry response thereby abolishing tumor immune evasion and promoting anti-tumor immunity. Cancer Discov. 11, 3090–3105 (2021).
Maddalena, M. et al. TP53 missense mutations in PDAC are associated with enhanced fibrosis and an immunosuppressive microenvironment. Proc. Natl Acad. Sci. USA 118, e2025631118 (2021).
PMVPharma. PMV Pharmaceuticals Announces a Clinical Trial Collaboration with Merck to Evaluate PC14586 in Combination with KEYTRUDA® (pembrolizumab) in Patients with Advanced Solid Tumors. GlobeNewswire (18 July 2022); go.nature.com/3fyQKDo
Moskovits, N., Kalinkovich, A., Bar, J., Lapidot, T. & Oren, M. p53 Attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer Res. 66, 10671–10676 (2006).
Fang, D. D. et al. MDM2 inhibitor APG-115 synergizes with PD-1 blockade through enhancing antitumor immunity in the tumor microenvironment. J. Immunother. Cancer 7, 327 (2019). This paper describes the enhanced antitumour effect of combining a p53 activating molecule (APG-115) with an anti-PD1 antibody, attributed to the effect of APG-115 on the tumour immune microenvironment.
Tolcher, A. W. et al. Preliminary results of a phase II study of alrizomadlin (APG-115), a novel, small-molecule MDM2 inhibitor, in combination with pembrolizumab in patients (pts) with unresectable or metastatic melanoma or advanced solid tumors that have failed immuno-oncologic (I-O) drugs. J. Clin. Oncol. 39, 2506 (2021).
Kim, S.-S., Harford, J. B., Moghe, M., Rait, A. & Chang, E. H. Combination with SGT-53 overcomes tumor resistance to a checkpoint inhibitor. Oncoimmunology 7, e1484982 (2018).
Chada, S. et al. Tumor suppressor immune gene therapy to reverse immunotherapy resistance. Cancer Gene Ther. 29, 825–834 (2022).
Wang, D. & Wang, K. An overview of development in gene therapeutics in China. Gene Ther. 27, 338–348 (2020).
Goswami, R. et al. Gene therapy leaves a vicious cycle. Front. Oncol. 9, 297 (2019).
Zhang, W. W. et al. The first approved gene therapy product for cancer ad-p53 (gendicine): 12 years in the clinic. Hum. Gene Ther. 29, 160–179 (2018).
Xia, Y., Du, Z., Wang, X. & Li, X. Treatment of uterine sarcoma with rAd-p53 (gendicine) followed by chemotherapy: clinical study of TP53 gene therapy. Hum. Gene Ther. 29, 242–250 (2018).
Li, Y. et al. Selective intra-arterial infusion of rAd-p53 with chemotherapy for advanced oral cancer: a randomized clinical trial. BMC Med. 12, 16 (2014).
Liu, S. et al. Randomized, controlled phase II study of post-surgery radiotherapy combined with recombinant adenoviral human p53 gene therapy in treatment of oral cancer. Cancer Gene Ther. 20, 375–378 (2013).
Gabrilovich, D. I. INGN 201 (Advexin): adenoviral p53 gene therapy for cancer. Expert Opin. Biol. Ther. 6, 823–832 (2006).
Atencio, I. A. et al. Biological activities of a recombinant adenovirus p53 (SCH 58500) administered by hepatic arterial infusion in a phase 1 colorectal cancer trial. Cancer Gene Ther. 13, 169–181 (2006).
Buller, R. E. et al. A phase I/II trial of rAd/p53 (SCH 58500) gene replacement in recurrent ovarian cancer. Cancer Gene Ther. 9, 553–566 (2002).
Zeimet, A. G. & Marth, C. Why did p53 gene therapy fail in ovarian cancer? Lancet Oncol. 4, 415–422 (2003).
Li, Y. et al. Expert consensus on the clinical application of recombinant adenovirus human p53 for head and neck cancers. Int. J. Oral. Sci. 13, 38 (2021).
Tamura, R. E. et al. Induction of oxidants distinguishes susceptibility of prostate carcinoma cell lines to p53 gene transfer mediated by an improved adenoviral vector. Hum. Gene Ther. 28, 639–653 (2017).
Chen, S. et al. A surface charge-switchable and folate modified system for co-delivery of proapoptosis peptide and p53 plasmid in cancer therapy. Biomaterials 77, 149–163 (2016).
Misra, S. K., Naz, S., Kondaiah, P. & Bhattacharya, S. A cationic cholesterol based nanocarrier for the delivery of p53-EGFP-C3 plasmid to cancer cells. Biomaterials 35, 1334–1346 (2014).
Rejeeth, C. & Kannan, S. p53 Gene therapy of human breast carcinoma: using a transferrin-modified silica nanoparticles. Breast Cancer 23, 101–110 (2016).
Kim, S. S., Rait, A., Kim, E., Pirollo, K. F. & Chang, E. H. A tumor-targeting p53 nanodelivery system limits chemoresistance to temozolomide prolonging survival in a mouse model of glioblastoma multiforme. Nanomedicine 11, 301–311 (2015).
Senzer, N. et al. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol. Ther. 21, 1096–1103 (2013).
Pirollo, K. F. et al. Safety and efficacy in advanced solid tumors of a targeted nanocomplex carrying the p53 gene used in combination with docetaxel: a phase 1b study. Mol. Ther. 24, 1697–1706 (2016).
Kong, N. et al. Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci. Transl. Med. 11, 1565 (2019). This paper describes the use of synthetic p53 mRNA as a potential anticancer treatment.
Xiao, Y. et al. Combining p53 mRNA nanotherapy with immune checkpoint blockade reprograms the immune microenvironment for effective cancer therapy. Nat. Commun. 13, 758 (2022).
Sobol, R. E. et al. Meta-analysis of adenoviral p53 gene therapy clinical trials in recurrent head and neck squamous cell carcinoma. medRxiv https://doi.org/10.1101/2021.01.06.20248743 (2021).
Martinez, L. A. et al. Synthetic small inhibiting RNAs: efficient tools to inactivate oncogenic mutations and restore p53 pathways. Proc. Natl Acad. Sci. USA 99, 14849–14854 (2002).
Ubby, I. et al. Cancer therapeutic targeting using mutant–p53-specific siRNAs. Oncogene 38, 3415–3427 (2019).
Mirgayazova, R. et al. Therapeutic editing of the TP53 gene: is CRISPR/Cas9 an option? Genes 11, 704 (2020).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Haapaniemi, E., Botla, S., Persson, J., Schmierer, B. & Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 24, 927–930 (2018).
Ihry, R. J. et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 24, 939–946 (2018).
Enache, O. M. et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 52, 662–668 (2020).
Michaelis, M. et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2, e243 (2011).
Chapeau, E. A. et al. Resistance mechanisms to TP53-MDM2 inhibition identified by in vivo piggyBac transposon mutagenesis screen in an Arf−/− mouse model. Proc. Natl Acad. Sci. USA 114, 3151–3156 (2017).
Latif, A. L. et al. BRD4-mediated repression of p53 is a target for combination therapy in AML. Nat. Commun. 12, 241 (2021).
Pan, R. et al. Synthetic lethality of combined Bcl-2 inhibition and p53 activation in AML: mechanisms and superior antileukemic efficacy. Cancer Cell 32, 748–760.e6 (2017).
Daver, N. G. et al. Safety, efficacy, pharmacokinetic (PK) and biomarker analyses of BCL2 inhibitor venetoclax (Ven) plus MDM2 inhibitor idasanutlin (idasa) in patients (pts) with relapsed or refractory (R/R) AML: a phase Ib, non-randomized, open-label study. Blood 132, 767–767 (2018).
Fischer, M. Mice are not humans: the case of p53. Trends Cancer 7, 12–14 (2021).
Horejs, C. Organ chips, organoids and the animal testing conundrum. Nat. Rev. Mater. 6, 372–373 (2021).
Gavert, N. et al. Ex vivo organotypic cultures for synergistic therapy prioritization identify patient-specific responses to combined MEK and Src inhibition in colorectal cancer. Nat. Cancer 3, 219–231 (2022).
Hong, D. S. et al. KRAS G12C inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 383, 1207–1217 (2020).
Degtjarik, O. et al. Structural basis of reactivation of oncogenic p53 mutants by a small molecule: methylene quinuclidinone (MQ). Nat. Commun. 12, 7057 (2021).
Acknowledgements
Work in the authors’ lab is supported in part by grants from the Dr Miriam and Sheldon G. Adelson Medical Research Foundation, a Center of Excellence from the Israel Science Foundation, the Israel Science Foundation within the Israel Precision Medicine Partnership programme, the Rising Tide Foundation, Quintrigen, The Robert Bosch Stiftung GmbH and the Berthold Leibinger Stiftung GmbH, the United States–Israel Binational Science Foundation (BSF), Jerusalem, Israel, and the Moross Integrated Cancer Center.
Author information
Authors and Affiliations
Contributions
Both authors contributed equally to all aspects of this Review.
Corresponding author
Ethics declarations
Competing interests
M.O. consults for Quintrigen.
Peer review
Peer review information
Nature Reviews Drug Discovery thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Michael acceptor
-
Michael addition is a process of addition of a carbanion or another nucleophile to an α,β-unsaturated carbonyl compound. Michael acceptors are the substituent groups on the activated unsaturated compound. The ability of Michael acceptors to form conjugates with peptides bearing nucleophilic groups provides them with a broad spectrum of potential biological effects.
- Stapled peptides
-
Short peptides locked in a specific conformation by an external brace formed by covalent binding between two amino acid side chains. This allows the molecule to be kept in an α-helical structure with high hydrophobicity, facilitating the crossing of biological membranes.
- Bispecific antibodies
-
Artificial antibodies that possess two binding sites, designed to recognize two different epitopes or antigens. One of the major applications of bispecific antibodies is to bridge two different cell types.
- Indels
-
Insertions or deletions of one or more nucleotides in a DNA sequence.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hassin, O., Oren, M. Drugging p53 in cancer: one protein, many targets. Nat Rev Drug Discov 22, 127–144 (2023). https://doi.org/10.1038/s41573-022-00571-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-022-00571-8
This article is cited by
-
Genome-wide CRISPR screen identifies ESPL1 limits the response of gastric cancer cells to apatinib
Cancer Cell International (2024)
-
p53 biology and reactivation for improved therapy in MDS and AML
Biomarker Research (2024)
-
Harnessing adenovirus in cancer immunotherapy: evoking cellular immunity and targeting delivery in cell-specific manner
Biomarker Research (2024)
-
Harnessing ferroptosis for enhanced sarcoma treatment: mechanisms, progress and prospects
Experimental Hematology & Oncology (2024)
-
DCAF13 inhibits the p53 signaling pathway by promoting p53 ubiquitination modification in lung adenocarcinoma
Journal of Experimental & Clinical Cancer Research (2024)
