
Abstract
The tumor microenvironment (TME) is made up of cells and extracellular matrix (non-cellular component), and cellular components include cancer cells and non-malignant cells such as immune cells and stromal cells. These three types of cells establish complex signals in the body and further influence tumor genesis, development, metastasis and participate in resistance to anti-tumor therapy. It has attracted scholars to study immune cells in TME due to the significant efficacy of immune checkpoint inhibitors (ICI) and chimeric antigen receptor T (CAR-T) in solid tumors and hematologic tumors. After more than 10 years of efforts, the role of immune cells in TME and the strategy of treating tumors based on immune cells have developed rapidly. Moreover, ICI have been recommended by guidelines as first- or second-line treatment strategies in a variety of tumors. At the same time, stromal cells is another major class of cellular components in TME, which also play a very important role in tumor metabolism, growth, metastasis, immune evasion and treatment resistance. Stromal cells can be recruited from neighboring non-cancerous host stromal cells and can also be formed by transdifferentiation from stromal cells to stromal cells or from tumor cells to stromal cells. Moreover, they participate in tumor genesis, development and drug resistance by secreting various factors and exosomes, participating in tumor angiogenesis and tumor metabolism, regulating the immune response in TME and extracellular matrix. However, with the deepening understanding of stromal cells, people found that stromal cells not only have the effect of promoting tumor but also can inhibit tumor in some cases. In this review, we will introduce the origin of stromal cells in TME as well as the role and specific mechanism of stromal cells in tumorigenesis and tumor development and strategies for treatment of tumors based on stromal cells. We will focus on tumor-associated fibroblasts (CAFs), mesenchymal stem cells (MSCs), tumor-associated adipocytes (CAAs), tumor endothelial cells (TECs) and pericytes (PCs) in stromal cells.
Similar content being viewed by others

Facts
-
The TME is made up of cells and extracellular matrix (non-cellular component), and cellular components include cancer cells and non-malignant cells such as immune cells and stromal cells.
-
Stromal cells is a major class of cellular components in TME, which also play a very important role in tumor metabolism, growth, metastasis, immune evasion and treatment resistance.
-
It is possible to target tumor-associated stromal cell (TASCs) to treat tumors based on the influence of TASCs on tumorigenesis and development.
Open Questions
-
So far, no specific markers have been found in stromal cells in TASC, and finding TASC-specific markers will be more helpful in identifying TASC and targeting TASC for tumor treatment.
-
TASCs such as CAFs can be divided into multiple subtypes according to different methods, however, the understanding of CAF subtypes mainly comes from the different classifications of individual experimental teams, and objective and consistent knowledge of their subtypes is required.
-
In addition to anti-angiogenic drugs targeting TECs, other strategies targeting stromal cells for tumor therapy have limited efficacy, and how to improve efficacy needs to be further explored.
Introduction
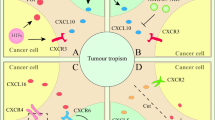
Solid tumors are composed of tumor cells and their ecosystem (namely the tumor microenvironment TME) which include various cells (immune cells, stromal cells) and extracellular matrix (ECM), blood vessels, lymphatic vessels, cytokines, mediators, and other non-cellular components [1, 2]. Complex signalings are established among tumor cells, immune cells, and stromal cells in TME thereby influencing tumor genesis, progression, and different clinical outcomes [3]. The previous strategies for treating tumors mostly targeted the cancer cells themselves such as chemotherapy, radiotherapy, and targeted therapy, but the importance of immune cells is gradually recognized in anti-tumor therapy with the discovery of immune checkpoints [4]. Strategies to target immune cells to treat tumors have seen significant clinical efficacy and are recommended by corresponding guidelines, such as T cell-based immune checkpoint inhibitors programmed cell death 1/programmed death-ligand1 (PD-1/PD-L1) antibodies, cytotoxic T lymphocyte-associated protein-4 (CTLA-4) antibodies and chimeric antigen receptor T (CAR-T) in adoptive cell therapy (ACT). Furthermore, the strategy of targeting immune cells to treat tumors also including dendritic (DC) vaccines, reduction of M2 tumor-associated macrophages (TAMs), N2 tumor-associated neutrophils (TANs), bone marrow-derived suppressor cells (MDSCs), regulatory T cells (Tregs), regulatory B cells (Bregs) and the reprogramming of TAMs and TANs into tumor killer cells, etc, and which have been introduced in another article we published [5], therefore, this part of the content will not be covered in this article. In addition, stromal cells, also present in TME (Fig. 1), have also been shown to play a crucial role in tumorigenesis, development, metastasis, and treatment resistance [6]. Each TASC in the tumor microenvironment can communicate with microenvironment components through cytokines and mediators in a paracrine manner or cell-cell interaction, thereby promoting tumor invasion, metastasis, angiogenesis, drug resistance, and recurrence [7, 8]. Therefore, cancer treatment strategies that do not consider stromal cells are not enough. Moreover, understanding the physiological role of each component is essential to understand how they affect tumor behavior and also provide ideas for finding ways to treat tumors based on stromal cells. So this article will focus on TASCs, including the origin of TASCs, the influence of stromal cells on tumorigenesis and development, the mechanism of stromal cell influence on tumors, and strategies for tumor treatment based on stromal cells. The TASCs we will highlight include tumor-associated fibroblasts (CAFs), mesenchymal stem cells (MSCs), tumor-associated adipocytes (CAAs), tumor endothelial cells (TECs), and pericytes (PCs).

Stromal cells include CAF, MSC, CAA, TEC, and PC. These three types of cells establish complex signaling pathways that affect tumor occurrence, tumor progression, and tumor resistance to treatment. Dendritic cell: DC; Natural killer cell: NK; M1/M2 tumor-associated macrophage: M1/M2 TAM; N1/N2 tumor-associated neutrophil: N1/N2 TAN; Myeloid-derived suppressor cell: MDSC; CD4+T lymphocyte/CD8+T lymphocyte/regulatory T cell: CD4+T cell/CD8+T cell/Treg; Regulatory B cell: Breg; Tumor-associated fibroblast: CAF; Mesenchymal stem cell: MSC; Tumor-associated adipocyte: CAA; Tumor endothelial cell: TECs; Pericyte: PC.
Cancer-associated fibroblasts
CAFs are the most common component of the tumor stroma, especially in the interstitium of breast cancer, prostate cancer, pancreatic cancer, and induration gastric cancer [9,10,11,12]. CAFs in TME exhibit two phenotypes: promoting tumor phenotype and inhibiting tumor phenotype, and the former phenotype can participate in tumor genesis, development, and resistance to anti-tumor therapy through multiple mechanisms [13], and this phenotype represents most CAFs groups. Based on this, targeting CAFs has become a strategy for the treatment of tumors, including direct targeting of CAFs and indirect targeting of CAFs through other therapies. In this section, we will introduce the source and classification of CAFs, the relationship with tumors, the mechanism of tumor promotion, resistance to tumor treatment, and targeted CAFs for tumor treatment.
Sources and types of cancer-associated fibroblasts
CAFs can be formed by attracting fibroblasts of adjacent tissues through transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), Fibroblast Growth Factor-2 (FGF-2), and exosomes secreted by tumor cells [14,15,16,17,18]. In addition, CAFs can also be formed by transdifferentiation of normal fibroblasts and TASCs (such as MSCs, TECs, CAAs, and PCs) in tumor tissue [15, 18,19,20,21,22,23]. For example, up to 80% of normal fibroblasts in breast tissue acquire CAFs phenotype during tumor progression [24]. CAFs are a different cell type from normal fibroblasts, such as mature fibroblasts exhibit a thin, wavy, and small spindle-shaped morphology, while CAFs are often described as immature fibroblasts and appear as large, plump spindle-shaped cells with prominent nucleoli. Moreover, CAFs exhibit dysregulation of signaling pathways and changes in protein expression compared to normal fibroblasts. Dysregulated signaling pathways include up-regulation of TGF-β, bone morphogenetic protein (BMP), Wnt, Sonic hedgehog (Shh), PDGF, Chemotokine (C-X-C motif) ligand 12 (CXCL12)/Chemotokine receptor (CXCR) 4, and integrin-mediated signaling. Changes in protein expression include up-regulation of α smooth muscle actin (α-SMA), fibroblast activating protein (FAP), fibroblast-specific protein-1 (FSP1), platelet-derived growth factor receptor-α (PDGFR-α), PDGFR-β, transcription factor Forkhead box F1 (FOXF1), wave protein, chondroitin sulfate proteoglycan glial antigen-2 (NG2), proline-4-hydroxylase, podophyllotyllin (PDPN), microfibrillary associated protein 5 (MFAP5), collagen 11-α1 (COL11A1) and interstitial matrix(IM) collagens [25, 26]. Not only that, CAFs can produce more collagen types III (PRO-C3) and VI (PRO-C6) than normal fibroblasts [27].
Normal fibroblasts are usually inhibit tumor formation while CAFs exhibit both promoting tumor phenotype and inhibiting tumor phenotype, and the former represents the majority of CAFs and involves in tumorigenesis, development, and resistance to treatment, while the inhibiting tumor phenotype can inhibit tumor proliferation and growth [13, 28]. For example, CAFs in pancreatic cancer are divided into inhibiting tumors subsets and promoting tumors subsets according to their different effects on tumors. Among them, the subsets of CAFs that inhibit tumors include Myofibroblastic (myCAFs), Meflin+CAFs, CD271+/NGFR+CAFs, Gli1+CAFs. Tumor-promoting CAFs subsets include Inflammatory CAFs (iCAFs), Zinc finger E-box binding homeobox 1 (Zeb1)+CAFs, leucine-rich replication 15+ (LRRC15+) CAFs, Serum amyloid A3+CAFs (SAA3+CAFs), FAP+/CXCL12+CAFs, CD10+/GPR77+CAFs, CD105+CAFs, Hypoxia+CAFs, Metabolic CAFs, Antigen-presenting CAFs (apCAFs) [29]. For example, myCAFs have a tumor inhibitory effect, which is located near tumor cells, and the collagen and ECM secreted by myCAFs have an important protective effect in pancreatic ductal adenocarcinoma (PDAC), while the deletion of myCAFs can reduce type I collagen content and significantly reduce tumor tissue hardness thereby leading to aggressive tumors and reducing animal survival rate [29]. Meflin+CAFs subsets also have a cancer-suppressing effect, and infiltration of Meflin+CAFs can inhibit the growth of xenograft tumors and is associated with a good prognosis for patients, while Meflin-deficient tumor tissue has poor differentiation and tumor progression is significant [6]. In the subset of tumor-promoting CAFs, iCAFs can participate in immune escape or directly act on pancreatic cancer cells by producing inflammatory cytokines such as IL-6, leukemia inhibitor factor (LIF), and CXCL1 to promote tumor progression [30]. CD105+CAFs can also promote tumor growth, while CD105-CAFs have anti-tumor immunity and tumor suppressor effects [31]. Furthermore, CAFs not only promote tumor growth in mouse models of pancreatic cancer by inhibiting CD8+T cells function [32], but also enhance the migration and invasion of cancer cells by inducing β-catenin expression and its nuclear localization [33]. In addition to the presence of CAFs subsets that have a clear effect on tumors, some new CAFs subsets are constantly being discovered. For example, complement secretory CAFs (csCAFs) express many components of the complement system [34], which may play a role in modulating immune and inflammatory responses, but further study is needed. Subtypes of unknown functions also include HoxB6+ CAFs. That is, CAFs are not a single cellular entity, but exist as distinct subpopulations and influence tumor biology at several levels.
In addition, CAFs in different tumors can also be divided into different subsets according to different methods, and different subsets have different functions. For example, CAFs can be divided into four different subgroups according to gene expression levels in breast cancer, namely CAF-S1, CAF-S2, CAF-S3, and CAF-S4, of which CAF-S1 can promote the immunosuppressive environment through multiple steps and mechanisms, and CAF-S4 can induce cancer cell invasion through the NOTCH signaling pathway [35]. Not only that the application of single-cell RNA sequencing can identify four subtypes of CAFs with different properties in gastric cancer, namely: myofibroblasts, pericytes, extracellular matrix CAFs (eCAFs), and iCAFs, in which iCAFs can attract and regulate the function of T cells by secreting Interleukin (IL)-6 and chemotokine ligand (CXCL) 12, while eCAFs can reshape ECM in TME and which can form a metastasis-friendly niche by degrading ECM and attracting M2 TAM, moreover, iCAFs and eCAFs not only exhibit enhanced pre-invasive activity but also mobilize surrounding immune cells to build a microenvironment conducive to tumor growth [36]. In addition, the single-cell RNA sequencing of stromal cells in human tumor samples showed that the CAFs subgroup had different transcriptional profiles, the first is mycofabric CAFs which are characterized by high expression of α-SMA and are located near tumor cell nests, followed by iCAFs which are located in fibroproliferative regions away from tumor cells and characterized by low expression of α-SMA and highly expression of IL-6, Leukemia Inhibitory Factor (LIF), IL-11, CXCL1, CXCL8 and platelet-derived growth factor receptor (PDGFR)-α [37]. In short, CAFs can be divided into different subgroups according to different methods, and different subsets have different phenotypes and functions, but in terms of the impact on tumors, CAFs are mainly divided into two subtypes: pro-tumor and inhibition of tumors, and in the next section we will introduce in detail the relationship between CAFs and tumors.
Cancer-associated fibroblasts and tumors
The CAFs in TME can be divided into different subtypes according to different methods, and different subtypes have differences in protein expression, paracrine signaling, tumorigenicity, and aggressiveness [26]. However, most of the CAFs population in TME have a pro-tumor phenotype and play a promoting role in the occurrence and development of tumors. For example, in breast cancer, CAFs can promote the metastasis of precancerous and malignant breast epithelial cells while normal fibroblasts promote epithelioid phenotype and inhibit metastasis [38]. Similarly, normal prostate epithelial cells cause intraepithelial neoplasia when co-injected with CAFs but not when co-injected with normal fibroblasts [39]. CAFs can also trigger nonmalignant cell malignant transformation through overexpression of estrogen, TGF-β, and Hepatocyte Growth Factor (HGF) [40, 41]. In conclusion, CAFs are involved in tumorigenesis.
CAFs are not only involved in tumorigenesis, but also play a very important role in tumor metastasis. For example, the migration potential of lung cancer cell have increased when treated with CAFs culture medium compared with normal fibroblast culture medium [42]. Co-transplantation of cervical cancer cells with CAFs into mice results in lymph node metastasis while injection without CAFs does not [43]. Moreover, CAFs can also participate in the formation of ecological niche before lung metastasis by secreting exosomes [44]. In addition, CAFs are also closely related to tumor invasion and progression [19], such as the Hyaluronan and proteoglycan link protein 1 (HAPLN1) derived from CAFs can promote tumor invasion [45]. CAFs with high expression of FOS-like antigen 2 (FOSL2) can promote angiogenesis and tumor growth [46].
In addition, IL-6 and IL-8 released by CAFs not only promote cancer cell invasion but also participate in tumor angiogenesis [47]. Conversely, blocking the IL-6/JAK2/STAT3 pathway can inhibit the progression of precancerous vocal cord (oral) leukoplakia and delay the occurrence of head and neck squamous cell carcinoma (HNSCC) tumors [48]. In conclusion, CAFs are involved in tumor invasion and metastasis.
In addition, CAFs are similar to circulating tumor cells (CTCs), such as circulating CAFs can be detected in the blood and may be related to tumor progression. For example, circulating CAFs can be detected in the blood of both breast cancer and prostate cancer with high levels of CAFs, and circulating CAFs exist in 88% of metastatic breast cancer patients and 23% of non metastatic patients (based on the expression of FAP and actin alpha 2 ACTA2) [49]. Similarly, circulating CAFs (the expression of waveform protein is positive, the expression of cytokeratin is negative) are also found in 58% of patients with metastatic prostate cancer but not in patients with nonmetastatic disease [50]. That is to say, circulating CAFs may be related to tumor progression. In short, CAFs play a very important role in the occurrence, development, and metastasis of tumors, which makes it particularly important to explore the tumor-promoting mechanism of CAFs.
Protumor mechanisms of cancer-associated fibroblasts
CAFs can promote tumorigenesis, invasion, metastasis, and resistance to treatment through a variety of mechanisms (Fig. 2) [51]. For example, compared with normal tissue fibroblasts, CAFs can increase tumor cell proliferation, ECM production, and also promote the secretion of various cytokines (such as stromal cell-derived factor-1 SDF-1; Vascular endothelial growth factor VEGF; HGF [46, 52,53,54,55]. Such as CAFs can reshape the ECM to provide a supportive microenvironment for cancer cells and become a physical barrier for drug penetration [56, 57]. In addition, CAFs can also secrete matrix metalloproteinases (MMPs) to interfere with the degradation of ECM, and initiate the migration and invasion of cancer cells in the process of promoting and degrading ECM [58].

CAFs can promote tumor growth through a variety of mechanisms: such as secreting a variety of cytokines; reshaping the extracellular matrix (ECM); promoting angiogenesis; inhibiting anti-tumor immune cells, providing metabolites (such as lactic acid, amino acids, fatty acids) to tumor cells; and participating in resistance to anti-tumor treatment, etc.
CAFs can also promote tumor occurrence, invasion, angiogenesis, and metastasis by secreting various factors [7, 59], in addition to influence tumor growth and therapeutic effect by adjusting the structure of ECM. For example, CAFs and cancer cells communicate bidirectionally through secreting of inflammatory cytokines such as IL-1A, IL-1B and Tumor Necrosis Factor (TNF) [60]. Moreover, IL-6 secreted by CAFs has a tumor-promoting effect in highly invasive tumors [61]. CXCL14 and Chemotokine ligand (CCL)2 secreted by CAFs can promote tumor cell survival, growth, and mediate Epithelial-mesenchymal transformation (EMT) [62, 63]. At the same time, α-SMA, integrin α11 and PDGF-BB secreted by CAFs can enhance the invasion potential of cancer cells [64, 65]. Moreover, the SDF-1 and PDGFC secreted by CAFs can promote cancer progression, metastasis, and angiogenesis [66,67,68]. In addition, Chitinase 3-like 1 (CHI3L1) secreted by CAFs acts on CAFs can increase the secretion of IL-8 and affect the angiogenesis of colorectal cancer tumors [69]. Simultaneously, CAFs express and secrete Collagen and calcium-binding EGF domain-1 (CCBE1) thereby promoting VEGFC maturation and lymphangiogenesis in colorectal cancer [70]. In short, CAFs can promote tumor genesis, invasion and metastasis by secreting various factors.
On the other hand, various factors secreted by CAFs can also promote tumor progression by regulating immune cells in TME [28]. For example, CAFs can inhibit natural killer (NK) cell function and the expression of NK receptors, perforin, and granzyme B by secreting cytokines, chemokines, and MMPs [71]. Moreover, CAFs can promote the recruitment of monocytes and promote their transformation into M2 TAMs by releasing IL-8, IL-10, TGF-β, and CXCL12 [28, 72, 73]. CAFs can also recruit MDSCs, and activate monocyte MDSCs (M-MDSCs) through secreting CCL2 and IL-6 by CAFs and miR-21 derived from CAFs, and further activate the production of MDSCs [74,75,76]. In addition, CAF-derived cardiotrophic factor-like cytokine 1 (CLCF1) can promote the infiltration and polarization of TANs in a paracrine manner [2]. Not only that, CAFs can also limit the intratumoral infiltration of CD8+ T cells and induce the transformation of primitive CD4+T cells into Tregs [77, 78]. To sum up, various factors secreted by CAFs can promote tumor progression by regulating immune cells in TME to form an immunosuppressive microenvironment.
At the same time, exosomes derived from CAFs are also involved in tumor genesis and development. For example, CAFs can promote angiogenesis of cancer cells through the miR-135b-5p/FOXO1 axi. In addition, the downregulation of miR-214 expression in CAFs can promote cancer cell EMT in tumors [79, 80], while the expression of miR-31 upregulated and miR-1 and miR-206 downregulated can inhibite autophagy of CAFs and promote tumor migration and recruitment of TAMs [81, 82]. In addition, the long non-coding RNA (lncRNA) MIR155HG secreted by CAFs also has carcinogenic effects, which are associated with decreased apoptosis and enhanced cell growth. In addition, CAFs can also initiate the expression of lnc HOTAIR to promote EMT and tumor metastasis [83, 84]. At the same time, exosomes derived from CAFs can also provide tumor cells with mitochondrial genomes to improve tumor oxidative phosphorylation and mitochondrial metabolism and further enhance tumor drug resistance and the self-renewal ability of cancer stem cells (CSCs) [85]. In summary, exosomes derived from CAFs can participate in the occurrence and development of tumors through various mechanisms.
In addition, CAFs can also play a tumor-promoting role by adjusting the metabolism with tumor cells. For example, CAFs participate in and promote tumor metabolic reprogramming through a variety of mechanisms, such as direct/indirect export of nutrients, provision of mitochondria, and regulation of metabolic enzyme activity. For example, the aerobic glycolysis of CAFs in TME increased and futher produce a large number of metabolites pyruvate and lactic acid as a source of nutrition and energy for tumor biosynthesis [86]. CAFs can also provide amino acids needed for tumor cells growth by directly synthesizing glutamine and other amino acids [87]. At the same time, the lipid metabolism of CAFs is also reprogrammed, such as lysophosphatidylcholine (LPCs) are secreted into microenvironment and directly absorbed and utilized by tumor cells to form membrane lipids [88]. In addition, the exosomes released by CAFs also contain complete metabolites including amino acids, lipids, and tricarboxylic acid cycle (TCA) cycle intermediates, and which are widely used by cancer cells for carbon metabolism in times of nutritional deficiency or stress [89]. Moreover, exosomes secreted by CAFs can also reorganize cancer cell metabolism through the enrichment of exosomes non-coding RNA, such as upregulating the glycolytic metabolism of cancer cells [90]. In a word, CAFs can support tumor growth through multiple mechanisms, such as the production of various nutrients and metabolic reprogramming of cancer cells.
Cancer-associated fibroblasts and tumor-treatment resistance
CAFs not only participate in the occurrence and development of tumors through various mechanisms, but also participate in the resistance of tumors to various anti-tumor treatments such as chemotherapy, radiotherapy, endocrine therapy, targeted therapy, and immunotherapy. For example, the HNSCC cells are insensitive to cisplatin (CDDP) after co-culture with CAFs and HNSCC cells [91], which indicated that tumor drug resistance is closely related to CAFs. The mechanisms by which CAFs cause cancer cells to resist anti-tumor treatment involve a wide range of mechanisms, such as secreting cytokines, exosomes, and expressing different proteins (different CAFs subtypes) to participate in resistance to anti-tumor therapy. The first is the cytokines secreted by CAFs lead to anti-tumor therapy resistance, such as activated CAFs promoting cancer cell resistance to CDDP by secreting IL-6 [46]. CAF-derived IL-8 can promote gastric cancer resistance to chemotherapy by activating NF-κB and upregulating ATP-binding cassette, sub-family B (ABCB1) [92]. Not only that, CAFs can also activate the Wnt/β-catenin pathway in EOC cells (mouse microglia) through the CXCL12/ CXCR 4-axis, resulting in cancer cells resistant to CDDP [93]. In addition, CAFs-derived CCL5 can promote tumor resistance to CDDP by upregulating the expression of lncRNA HOTAIR [94]. Cytokines secreted by CAFs not only participate in tumor resistance to chemotherapy, but also participate in tumor resistance to radiotherapy. For example, CXCL1 secreted by CAFs can mediate radiation resistance by inhibiting the expression of the reactive oxygen species (ROS)-scavenging enzyme superoxide dismutase 1 or by activating the mitogen-activated proteinkinase kinase (MEK)/extracellular regulated protein kinases (ERK) pathway [95]. In addition, the insulin-like growth factor-binding proteins (IGFBP)-2, -4, and -6, insulin growth factor 2 (IFG2), and PDGF-AA produced by CAFs can also mediate tumor resistance to radiotherapy [96]. Moreover, the cytokines produced by CAFs are also involved in tumor resistance to targeted therapies, such as fibroblasts that produce HGF lead to resistance of lung cancer cells to epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) [97]. In short, CAFs can participate in the resistance of tumors to anti-tumor treatments such as chemotherapy, radiotherapy, and targeted therapy by secreting various cytokines.
At the same time, CAFs can also mediate tumor resistance to treatment by secreting exosomes. For example, chemotherapy drugs CDDP and paclitaxel inhibit arachidonate lipoxygenase by activating the Ubiquitin Specific Peptidase 7 (USP7)/heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) axis 15 (ALOX15) and reduce the accumulation of lipid-reactive oxygen species (ROS) in cancer cells and ultimately lead to a decrease in chemotherapy sensitivity [98]. CAFs derived exosomes miR-196a make advanced head and neck cancer resistant to CDDP by targeting cyclindependent kinase inhibitor 1B (CDKN1B) and inhibitor of growth family (ING5) [99]. The exosome LINC00355 also from CAFs can promote the resistance of breast cancer cells to CDDP by regulating the miR-34b-5p/ATP binding cassette, subfamily B (ABCB1) axis [100]. While CAFs-derived exosome microRNA-98-5p (miR-98-5p) can promote CDDP resistance in ovarian cancer by downregulating monoclonal antibody to cyclin-dependent kinase inhibitor 1A (CDKN1A) [101]. Furthermore, exo-miR-103a-3p derived from CAFs can promote CDDP resistance in non-small cell lung cancer (NSCLC) cells [102]. In addition, the lncRNA SNHG12 carried by CAFs-exosomes enters NSCLC cells then promotes RNA stability and X-linked inhibitor of apoptosis protein (XIAP) transcription by binding to HuR, thereby enhancing NSCLC cell resistance to CDDP [103]. Furthermore, exosomes secreted by CAFs are also involved in tumor resistance to radiotherapy in addition to mediating tumor resistance to chemotherapy drugs. For example, exosomes released by CAFs can stimulate Retinoic Acid Inducible Gene 1 Protein (RIG-1) signaling in cancer cells, and Jagged 1 (JAG1) on CAFs can activate NOTCH3 signaling on cancer cells, and these pathways work together to promote tumor resistance to radiation therapy and chemotherapy [104]. In summary, CAFs can mediate tumor resistance to antitumor therapy by secreting multiple exosomes.
In addition, CAFs can also participate in resistance to anti-tumor treatment by expressing different proteins (also known as different subtypes of CAFs). For example, CAFs expressing neuropilin 2 (NRP2) can reduce the sensitivity of gastric cancer cells to 5-fluorouracil (5-FU) [105]. α-SMA(+) CAFs can enhance hepatocellular carcinoma (HCC) resistance to chemotherapy by stimulating the HGF-MET-FRA1-HEY1 cascade reaction [106]. Furthermore, CAFs that express specific proteins are also involved in tumor resistance to targeted therapy, such as CAFs expressing high-level Neuregulin 1 (NRG1) can lead to trastuzumab resistance in HER-2+ breast cancer through the HER3/AKT pathway [107]. Furthermore, CAFs-rich tumors are also not sensitive to combined immune checkpoint inhibitors (ICB) therapy [63], indicating that CAFs are involved in tumor resistance to ICB therapy. For example, ecm-myCAF, TGF-ß- myCAF, and wound myCAF are known drivers of immunosuppressive environment and immune therapy resistance in the CAFs subtype [108], in which ecm-myCAF upregulates the levels of PD-1 and CTLA4 proteins in Tregs, and the pan-CAF subtype expresses immunosuppressive inflammatory factors CXCL12, CXCL14, and stem cell promoting factor IL-6, all of which are related to ICB resistance [109]. In summary, CAFs can not only participate in tumor resistance to treatment by secreting cytokines and exosomes, but also some subtypes of CAFs or CAFs expressing different proteins in CAFs can participate in tumor resistance to tumor treatment.
Strategies for targeting cancer-associated fibroblasts for tumor therapy
Based on the tumor-promoting effect of most CAFs in TME, the strategy of targeting CAFs as target cells for tumor treatment has become possible, including direct targeting of CAFs and indirect targeting of other treatments that affect CAFs. Direct targeting of CAFs include preventing CAFs infiltration, inhibiting CAFs activation, reducing the number of CAFs, reprogramming CAFs (restoring the phenotype of quiescent fibroblasts or converting CAFs to an inhibitory phenotype), and developing CAFs oriented vaccines and therapies. While indirect targeting of CAFs include radiotherapy and chemotherapy, targeting immune cells, targeting downstream effectors of CAFs, targeting CAFs related signaling pathways, and targeting ECM proteins derived from CAFs, which all can indirectly affect the number and activity of CAFs.
The treatment strategy of directly clearing CAFs mainly relies on the surface markers of CAFs such as FAP, α- SMA and PDGFR [110]. It has been reported that type II membrane-bound glycoprotein FAP is not expressed in normal tissues but expressed in activated CAFs in the tumor stroma, and FAPα-expressing vaccines can inhibit the growth of 4T1 tumors (breast cancer) by killing CAFs by generating FAPα-specific cytotoxic T lymphocyte (CTL) responses [111]. In addition, T cells triggered by DC/CAF fusion cells can also produce a strong CTL immune response to CAFs [112].
In addition to directly targeting CAFs to affect the number and activity of CAFs, other treatments also indirectly affect CAFs. Scriptaid (a small molecule inhibitor of histone deacetylase inhibitor (HDACs) 1/3/8) can inhibit CAFs differentiation and reduce the number of CAFs through TGF-β [113]. The drug pirfenidone (PFD) can reduce the capacity of aggressiveness and immunosuppressive mediated by CAFs in breast cancer [114]. While all-trans-retinoin and minnelide (which de-regulates the TGF-β signaling pathway) can calm the active CAFs [115, 116]. In addition, the vitamin D receptor (VDR) ligand calcipotriol can also reduce the proliferation and migration of CAFs [117], and the treatment of gastric cancer cells with calcipotriol can eliminate CAF-derived IL-8-mediated resistance to platinum oxalate by blocking the PI3K/Akt signaling pathway [118]. Moreover, tocilizumab (the inhibitor of IL-6 receptor) can inhibit the cancer-promoting effect of CAFs in breast cancer by inhibiting the STAT3/AU-rich element RNA-binding protein 1 (AUF1) pathway [114]. Not only that, eicosapentaenoic acid can inhibit angiogenesis through reducing the secretion of IL-6 and VEGF by CAFs in colon cancer [53]. In addition to drugs indirectly affecting the quantity and activity of CAFs, other treatments also indirectly affect CAFs. For example, knockout of shRNA mediated MMP2 gene can reduce the release of ECM fibers from CAFs and prevent lung metastases in breast cancer [119]. Reducing the expression of Cyclooxygenase-2 (COX-2) in cells can reduce the number of CAFs [120]. Furthermore, inhibition of CC chemokine receptor (CCR) 2 and elimination of ROS can eliminate the CAF-MDSC axis, thereby favoring the reversal of CAF-mediated immunosuppressive microenvironment [121]. Moreover, inhibition of ROS-producing enzymes nicotinamide adenine dinucleotide phosphate oxidase 4 (NOX4) expressed on CAFs can promote CD8+ T cell infiltration and enhance tumor response to ICB therapy [122]. In addition, cytokines can also affect the quantity and activity of CAFs. For example, TGF-β can downregulate CAFs by binding mothers against DPP homolog 1 (SMAD) to CCBE1 [69]. Interferon (IFN)-γ can inhibit fibroblast-leading tumor cell invasion by inhibiting fibroblast motility and their adhesion to tumor cells [58]. In short, the strategy of treating tumors with CAFs as target cells can be achieved direct action on CAFs or indirect effects such as drugs, cytokines, or reducing the expression of some enzymes.
Targeted CAFs for tumor therapy can be achieved through a variety of strategies. However, it is better to selectively reduce the tumor-promoting CAFs subsets and protect the tumor-suppressing CAFs subsets because there are both tumor-promoting and tumor-suppressing CAFs subsets in TME. For example, Meflin+CAFs can inhibit tumors, while the application of unnatural retinol Am80 can effectively induce Meflin expression of CAFs in PDAC, and Am80 administration can not only increase tumor vascular area and intratumor drug delivery, but also improve the sensitivity of PDAC to chemotherapy drugs [123]. While apCAFs are tumor-promoting CAFs subtype and derived from mesothelial cells, antibody therapy targeting the mesothelial marker mesothelin can effectively inhibit the conversion of mesothelial cells to apCAF [78]. Anti-GPR77 antibody injection can significantly reduce the infiltration of CD10+GPR77+CAFs (pro-tumor subsets of CAFs), and anti-GPR77 antibody combined with chemotherapy can significantly enhance the apoptosis of tumor cells and CAFs, and inhibit tumor growth [124]. FAP+CAFs also belong to the pro-tumor CAFs subgroup, which can secrete CXCL12 to inhibit the accumulation of cytotoxic CD8+T cells in tumors, while the use of CXCL12 receptor chemokine receptor 4 inhibitor AMD3100 not only causes rapid accumulation of CD8+T cells among cancer cells, but also enhances the efficacy of anti-PD-L1 therapy [125]. Furthermore, IL1 can induce LIF expression and activate downstream JAK/STAT to generate tumor-promoting CAFs subtype iCAFs, while TGF-β can antagonize this process by downregulating interleukin-1 receptor1 (IL1R1) expression and promoting differentiation into myCAFs [30]. In addition, the angiotensin II type 1 receptor (AGTR1) was identified as a marker of iCAF, and the inhibitor of this receptor Losartan has been shown to reduce intratumoral solid stress thereby increasing vascular perfusion and improving drug delivery [126]. In addition. diphtheria toxin (DT) can selectively deplete the pro-tumor CAFs subtype LRRC15+CAFs, and can also lead to CAFs components being recalibrated towards universal fibroblasts [127]. In short, based on the continuous discovery of CAFs subtypes in TME, reducing the tumor-promoting CAFs subsets in TME and increasing the tumor-inhibiting CAFs subsets can make the targeted CAFs treatment of tumors more accurate.
TME plays a crucial role in the occurrence, development, and metastasis of tumors, among which CAFs as the largest type of stromal cells in TME have been more well understand, including its origin, relationship with tumors, and tumor promoting mechanisms (by releasing cytokines, exosomes, and metabolites to participate in cancer cell growth and affect tumor cell resistance to treatment [128, 129]. Numerous studies have also been carried out on the treatment of CAFs, but most of these studies are in the preclinical stage, and clinical trials on CAFs and tumors mostly focus on tumor PET imaging of FAP (molecular markers of CAFs). There are only 3 clinical trials targeting CAFs for tumor treatment, including the “Phase 1 Dose escalation Trial of OMTX705, an Anti fiber last Activation Protein Antibody dry Conjugate, as Single Agent and in Combination With Pembrolizumab in Patients With Advanced Solid Tumors” (NCT05547321) and “ Phase I/II Investigator-initiated Clinical Trial of MIKE-1 With Gemcitabine and Nab-paclitaxel Combination Therapy for Unresectable Pancreatic Cancer“ (NCT05064618). In the latter clinical trial, MIKE-1 (Am80) is a synthetic unnatural retinoic acid, which can effectively convert Meflin-pCAF into Meflin+rCAF, and this clinical trial is to combine MIKE-1 with gemcitabine (GEM) and nab-paclitaxel (nab-PTX) in patients with unresectable pancreatic cancer and evaluate the safety, tolerability and efficacy of this treatment. Another clinical trial is “Basket Study to Evaluate the Therapeutic Activity of Simlukafusp Alfa as a Combination Therapy in Participants With Advanced and/or Metastatic Solid Tumors” (NCT03386721), this clinical trial is an open label, multicenter, Phase II study, in which Simlukafusp α is an immune cytokine consisting of an interleukin-2 variant (IL-2V) targeting FAP-α, and this test aims to evaluate the antitumor activity of Simlukafusp α in combination with atezolizumab (anti-PD-L1) in patients with advanced and/or metastatic solid tumors. Although a large number of preclinical studies have been carried out on CAFs for tumor treatment and some clinical trials are also underway, no CAFs-specific inhibitors have been approved so far, which may be related to the lack of specific targets for CAF and the fact that ongoing clinical trials have not yet been completed. However, since CAFs represent most cells in the tumor stroma and most CAFs have tumor-promoting properties, the treatment of tumors against CAFs will become another major and important strategy for the treatment of tumors.
Mesenchymal stem cells
MSCs are a heterogeneous population of stromal cells present in the interstitium of various tissues and organs and are also localized in various primary and metastases tumors [130,131,132]. MSCs have different roles in different stages of tumorigenesis, such as it may have the effect of inhibiting tumorigenesis and growth in the early stage of tumors, but promoting tumor development through a variety of mechanisms in the later stage of tumorigenesis [133]. Moreover, MSCs can be used as an ideal drug carrier for the treatment of tumors based on which have the property of tumor homing. We will focus on the source and homing of MSCs, the relationship between MSCs and tumors, and the strategies for treating tumors based on MSCs.
Sources and homing of mesenchymal stem cells
MSCs are plastic, adherent cells that specifically express CD73, CD90, and CD105, but do not CD34, CD45, CD14, CD11b, CD79a, CD19, and Human leukocyte antigen (HLA)-DR. It can be isolated from various tissues such as bone marrow, adipose tissue, umbilical cord [134], or induced by pluripotent stem cells [135], and can differentiate into adipocytes, osteoblasts, and chondrocytes [136, 137]. Moreover, MSCs can migrate to the tumor site through the interaction of a variety of chemokine receptors (such as CCR-1, CCR-2, CCR-3, CCR-4, CCR-5, CCR-7, CXCR-1, CXCR-2, CXCR-3, CXCR-4) on MSCs and cytokines or chemokines (such as endothelial cell selectin, MMPs, IL-8, PGDF-AB, IGF-1, VEGF) secreted by solid tumors [138,139,140]. The SDF1 receptor CXCR4 expressed by MSCs and the highly expressed SDF1 on the surface of tumor cells can migrate MSCs to the tumor through the CXCR4-SDF1 axis [141]. The chemokine CCL5 produced by MSCs combines with the homologous receptor CCR5 on human breast cancer cells (BCC) can promote lung metastasis of breast cancer [142]. Meanwhile, studies have also shown that cigarette smoke extract (CSE) and benzo[α]pyrene (B[α]P) can increase osteopontin (OPN) expression levels and promote the recruitment and adhesion of MSCs to lung cancer cells through JAK2/STAT3 signaling [132]. In addition, cytokines TNF-α, IL-6, IL-1β, and IFN-γ are involved in the adhesion of circulating MSCs to the vascular endothelial layer [143], and MSCs adhering to the vascular endothelial layer enter tumor tissue through the vascular wall [134, 140, 144]. Conversely, reducing these factors or receptors can reduce the migration and invasion of MSCs into cancer cells [145]. In conclusion, the interaction between cancer cells and MSCs promotes the homing of MSCs to tumors.
Mesenchymal stem cells and tumors
As another major class of stromal cells in TME, MSCs have different roles in different stages of tumor development. For example, in the early stage of tumorigenesis or exogenous MSCs have the effect of inhibiting tumorigenesis and growth. Such as co-culture of umbilical cord-derived MSCs with glioblastoma stem cells (CSCs) showed that the proliferation rate of CSCs was significantly reduced [146], and human cord-derived MSCs could also effectively inhibit the growth of liver cancer in mice [147]. Moreover, the systematic administration of allogeneic MSCs neither accelerates the progression of precancerous lesions nor increases the malignancy of precancerous lesions, but rather prevents the progression of oral squamous cell carcinoma (OSCC) and tumor growth [148]. At the same time, donor MSCs can also reprogram host macrophages and restore the bone marrow microenvironment, thereby inhibiting the development of leukemia and prolonging the survival of leukemia mice [149]. Moreover, exosomes secreted by MSCs are also involved in tumor inhibition. Such as exosomes derived from human MSCs can induce tumor cell apoptosis and necrosis in hepatocellular carcinoma, ovarian cancer, and kaposi’s sarcoma by activating negative cell cycle regulatory factors [150]. In addition, exosomes derived from human umbilical cord MSCs have anti-proliferation and pro-apoptosis effects in bladder cancer [151]. Exosomes derived from human adipose MSCs can also inhibit the proliferation of ovarian cancer cells and induce their apoptosis [152]. Furthermore, MSCs can also block tumor cell proliferation in G0/G1 phase of the cell cycle through cell-to-cell contact, thereby preventing tumor cells from entering S phase of the cell cycle [153]. All of the above indicated that MSCs have the effect of inhibiting tumor occurrence and growth. However, MSCs homing to the tumor site in the later stage of tumor development, which are “re-educated” by tumor cells and other cells in TME and make MSCs have the characteristics of promoting tumorigenesis, development and metastasis [19, 154].
MSCs in TME can promote the occurrence and development of tumors through mechanisms such as cell-to-cell contact, secretion of biomolecules, enhancement of angiogenesis, inhibition of immune cell activity, or conversion to CAFs (Fig. 3). For example, MSCs can be engulfed by cancer cells, and the metastasis and aggressiveness of cancer cells are enhanced after ingestion [152]. MSCs can also enhance tumor vascularization by upregulating VEGF and IL-6 [155], and promote the progression and metastasis of cancer by secreting CCL5, CCL7, and TGF-β [142, 156]. Not only that MSCs also affect anti-tumor immune function, such as inhibiting the antitumor activity of NK and DC cells [157], inducing macrophage M2 polarization [158], promoting Tregs production, and reducing B cell activation [159]. In addition, exosomes derived from MSCs can also participate in immunomodulation by regulating immune cell function and altering the secretion of inflammatory factors (such as TNF-α and IL-1β) [160]. For example, exosomes derived from MSCs can accelerate the progression of breast cancer by inducing monocytes myeloid-derived suppressor cells (M-MDSCs) to differentiate into M2 TAMs in the tumor bed [161]. Meanwhile, other exosomes derived from MSCs can also promote the proliferation, migration, and angiogenesis of cancer cells, such as miR-410, miR-130b-3p, miR-21-5p and miR-15a [162]. In addition, MSCs in TME can also differentiate into CAFs thereby indirectly promoting tumor progression [163]. In short, MSCs homing to the tumor site can promote the occurrence and development of tumors through various mechanisms such as secreting biomolecules, exosomes, and affecting anti-tumor immune function.

The mechanisms include: cell-to-cell contact, secretion of cytokines and exosomes, inhibition of immune cells activity, promotion of angiogenesis, promotion of epithelial interstitial transformation (EMT), transforming into cancer-associated fibroblasts (CAFs), etc, and ultimately promote cancer cells proliferation, tumor metastasis and anti-tumor treatments.
The tumor-promoting mechanism of MSCs can be achieved through multiple pathways. Such as MSCs promote tumor generation and progression by regulating and activating the PI3K/AKT signaling pathway and regulating the Hippo pathway [164, 165]. MSCs can also promote gastric cancer growth by upregulating c-Myc [166], and promote the progression of colorectal cancer by activating mTOR and NF-κB signaling [167]. In addition, IL-6 secreted by MSCs can promote tumor progression by activating JAK2/STAT3 signaling and upregulating NF-κB [168, 169]. At the same time, galectin3 expressed on MSCs can promote adhesion between MSCs and acute myeloid leukemia (AML) tumor cells by activating the MYC signaling pathway thereby promoting the survival of cancer cells [170]. In summary, the tumor promoting effect of MSCs can be achieved through multiple pathways.
In addition, MSCs are also involved in tumor resistance to treatment, such as co-culturing human oral squamous cell line JHU-012 cells with bone marrow MSCs can lead to resistance of JHU-012 cells to CDDP [171]. Moreover, bone marrow MSCs can also make SCC-25 cells (human tongue squamous cell carcinoma cells) resistant to paclitaxel by up-regulating BCL-2, POSTN, multiple drug resistance-related protein 1 and human adenosine triphosphate binding cassette transporter G (ABCG transporter) [172]. Moreover, exosomes derived from MSCs are also involved in the resistance of tumors to treatment. For example, MSC-derived exosomes can induce 5-FU resistance in gastric cancer cells by activating the CaM-Ks/Raf/MEK/ERK pathway [173]. In addition, the expression of S100A6 mediated by miR-21-5 in breast cancer can enhance the resistance to doxorubicin [174]. Moreover, human bone marrow microenvironment-derived MSCs can promote the proliferation of chronic myeloid leukemia (CML) cells while reducing their sensitivity to tyrosine kinase inhibitors [175]. That is to say, MSCs are involved in tumor resistance to chemotherapy and targeted therapy.
Strategies for the treatment of tumors based on mesenchymal stem cells
MSCs are considered to be an ideal carrier for cancer treatment due to their ability to homing tumors. The use of MSCs to treat tumors does not promote tumor progression despite MSCs in TME are involved in tumorigenesis and progression through multiple mechanisms such as intercellular contact, regulation of anti-tumor immunity, secretion factors, and exosomes [154]. This is because MSCs require 4 to 30 days to form a tumor-promoting phenotype (such as MSC conversion to CAF) [133], while most exogenous MSCs disappear from the body within a week due to cell death [176]. That is, endogenous mesenchymal stem cells are involved in tumorigenesis and progression, but the administration of mesenchymal stem cells may have less effect on tumor growth.
This point of view has also been proven by the facts. Such as the use of MSCs from bone marrow, adipose tissue and umbilical cord blood as carriers to deliver anti-cancer cell factors, pro-apoptotic proteins, suicide genes, oncolytic viruses (OVs), and chemotherapy drugs have all shown antitumor effects [143]. For example, the establishment of IFN-α overexpressed MSCs (IFNα-MSCs) not only eliminates tumors in situ, but also has a specific antitumor effect on distant tumors after intratumoral injection [177]. In addition, miR34a-modified MSCs can not only induce glioma cell aging, but also induce DNA damage through regulation of Sirtuin 1 (SIRT1) [178]. Similarly, human placenta-derived MSCs transduced by the HGF antagonist NK4 can inhibit tumor cell growth by inducing apoptosis [179]. At the same time, transgenic MSCs can also be used as carriers of immunomodulatory proteins to deliver them to tumor tissues and can improve the efficacy of CAR-T cells in the treatment of solid malignancies [180]. Not only that, MSCs can also be used as carriers of OVs, and which can not only protect the virus from being cleared by the immune system, but also deliver the virus to tumor lesions and release cytokines, thereby enhancing the anti-tumor immune response [181]. In addition, MSCs can also carry chemotherapy drugs such as doxorubicin, paclitaxel, and gemcitabine, and which can inhibit tumor cell growth when anticancer drug-loaded MSCs are co-cultured with tumor cells and administered locally [133]. MSCs can also be modified into suicide genes that convert non-toxic reagents into toxic antitumor drugs, such as human adipose tissue-derived MSCs expressing the suicide gene cytosine deaminase::uracil phosphoribosyltransferase (CD::UPRT) can convert relatively non-toxic 5-fluorocytosine into highly toxic antitumor 5-FU and significantly inhibit tumor growth [182]. In short, based on the tumor-homing characteristics of MSCs, MSCs can be used as a carrier of cytokines, OVs and chemotherapy drugs to target tumors and improve anti-tumor efficacy.
However, the effectiveness of using the tumor homing properties of MSCs to treat tumors will be affected by insufficient homing ability thereby resulting in insufficient targeting and affecting therapeutic efficacy [183]. In order to improve the tumor homing characteristics of MSCs, the P-selectin ligand PSGL-1 mRNA, E-/L-selectin ligand SLeX mRNA, or CXCR4 mRNA were transfected into MSCs, resulting in the rolling and adhesion of the modified MSCs in vascular endothelial cells was enhanced, thereby increasing the homing of MSCs [144]. In addition, overexpression of chemokine receptors (CCR2, CCR3, and CCR4) can enhance the homing ability of MSCs to targets by increasing the migration of MSCs to chemokines [184]. Not only that, loading iron oxide nanoparticles into MSCs can improve the homing ability of MSCs to targets through magnetic guidance [185]. In conclusion, improving the homing ability of MSCs can be achieved through a variety of strategies based on the homing of MSCs involves multiple mechanisms and steps [144].
In addition to using the homing ability of MSCs as a carrier for the treatment of tumors, MSCs also have important value in stem cell transplantation therapy, such as helpful hematopoietic reconstitution after hematopoietic stem cell transplantation, especially for the treatment of leukemia, multiple myeloma and lymphoma [186]. In addition, the immunosuppressive function of bone marrow MSCs can also be utilized to alleviate the possibility of graft versus host disease (GVHD) caused by allogeneic transplantation [187]. Furthermore, allogeneic bone marrow MSCs transplantation rarely causes rejection due to the bone marrow MSCs express low levels of major histocompatibility complex (MHC) class I, MHCII class I, and co-stimulatory molecules (CD40, CD80, and CD86) [188, 189]. Therefore, therapies based on MSCs are a promising way to support hematopoietic stem cell or bone marrow transplantation. In summary, MSCs can not only be used as vectors to target tumors thereby improving anti-tumor efficacy, but also contribute to hematopoietic reconstruction and alleviate GVHD after hematopoietic stem cell transplantation.
A large number of clinical trials have also been carried out on the application of MSCs in tumors. So far, 39 trials have been registered in the ClinicalTrials database, of which 18 trials are aimed at using MSCs to treat cancer (including glioma, myelodysplastic syndrome (MDS), ovarian cancer, head and neck cancer, lung cancer, etc.), 11 trials aimed to use MSCs to treat side effects caused by anti-tumor treatment (including bone marrow suppression, acute kidney injury, cardiomyopathy, erectile dysfunction after rectal cancer surgery, and xerostomia caused by radiotherapy), another 10 clinical trials related to MSCs and hematopoietic stem cell transplantation (5 aimed to investigate the prevention of GVHD after hematopoietic stem cell transplantation by MSCs, and 4 aimed to observe the feasibility or effectiveness of co-infusion of hematopoietic stem cells and MSCs) (Table 1). 12 out of 39 clinical trials on MSCs and tumors have been completed (MSCs for cancer treatment: 4 items, MSCs for reducing side effects of anti-tumor treatment: 2 items, MSCs and hematopoietic stem cell transplantation: 6 items). We have seen the potential of using MSCs therapy in tumors in the published experimental results, such as in the clinical trial “Phase 1 Trial of Celyvir in Children and Adults With Metastatic and Refractory Solid Tumors.” (NCT01844661), where MSCs are used to transport oncolytic adenoviruses (OAds) to the tumor site (referred to as Celyvir therapy). The results showed that OAd MSCs treatment could significantly reduce the tumor growth of osteosarcoma in vivo, and the infiltration of immune cells (especially tumor-infiltrating lymphocytes) in the tumor was higher after treatment. Another clinical trial with published results is “A Phase I/II Trial in Treating Patients With Graft Versus Host Disease by the Infusion of Expanded in Vitro Allogenic Mesenchymal Stem Cell” (NCT00447460), which evaluated the feasibility and efficacy of MSCs infusion with human serum augmentation in the treatment of refractory acute and chronic graft-versus-host disease. The results showed that among the 10 patients treated, 3 patients achieved complete remission, 6 patients achieved partial remission, while only 3 patients did not respond to MSC infusion [190]. The above clinical trial results indicate that the use of MSCs by tumor patients can bring benefits to patients and has broad application prospects.
MSCs as another large group of cells in TASCs which have tumor homing ability and play different roles at different stages of tumors, such as exogenous MSCs have the effect of inhibiting tumorigenesis and growth, but MSCs homing to tumor sites can promote tumor occurrence and development through a variety of mechanisms. Moreover, due to the ability of MSCs to spontaneously invade tumors guided by chemokines in TME, MSCs can be used as a carrier to target tumors to exert anti-tumor effects through delivering anti-cancer cell factors, pro-apoptotic proteins, suicide genes, OVs and chemotherapy drugs. In addition, MSCs can also help hematopoietic reconstruction, alleviate GVHD, and alleviate the side effects of anti-tumor therapy after hematopoietic stem cell transplantation, so using some of the properties of mesenchymal stem cells to treat tumors is a promising strategy.
Cancer-associated adipocytes
Adipocytes are considered to be an inert cell population, however, adipocytes located in TME are “activated” by tumor cells with a pro-tumor phenotype, and these adipocytes are called “tumor-associated adipocytes (CAAs)”. CAAs are mainly derived from MSCs or undifferentiated adipocyte precursors in adipose tissue matrix, and a small number of CAAs can also come from CSCs [191]. CAAs can promote tumor occurrence and development through the secretion of adipokines, inflammatory factors, and the production of fatty acids [192]. It is possible to target CAAs for tumor treatment based on the tumor-promoting effect of CAAs. We will further introduce the relationship between CAAs and tumors, the mechanism of CAAs promoting tumors, and the strategies of targeting CAAs for the treatment of tumors.
Cancer-associated adipocytes and tumors
CAAs are an important component of cellular composition in TME, especially in tumors such as breast cancer, ovarian cancer, prostate cancer, kidney cancer, gastric cancer, and colon cancer [193]. The crosstalk between adipocytes and cancer cells can lead to changes in the phenotype and function of adipocytes, such as tumor-derived soluble factors TNF-α, IL-6, plasminogen activator inhibitors 1, Wnt3a, and exosomes microRNAs (such as miR-144, miR-126, and miR-155) can act on adipocytes at the forefront of tumor invasion and further induce the formation of CAAs [2]. Moreover, activated CAAs differ from normal adipocytes in morphology and function, they exhibit decreased adipocytes/macrophage fatty acid-binding protein 2 (Ap2) and fatty acid-binding protein 4 (FABP 4), while the expression of MMP11 and the release of IL-6, IL-1β, IGFBP-2 increased [194, 195].
Cancer cells are more likely to recruit adipocytes compared with normal tissues, and the adipocytes recruited into TME participate in tumorigenesis, promoting tumor growth and invasion [196, 197]. For example, the accumulation of adipose tissue adjacent to tumor tissue is related to the increase of incidence rate, progress, and metastasis of breast cancer [198]. Mature adipocytes in vitro can significantly promote the proliferation of breast cancer cells (MCF-7) and normal breast cells (184B5) [199]. At the same time, co-culture of mature adipocytes and breast cancer cells can promote cancer cell growth [200] and mediate EMT by enhancing the expression of MCF7 cell Forkhead Box C2 (FOXC2), twist family bHLH transcription factor 1 (TWIST1), and N- and E-cadherin [201]. In addition, adipocytes co-cultured with breast cancer cells can promote the invasion of tumor cells by overexpressing the inflammatory cytokines such as IL-6, TNFα, and MMPs [202]. Moreover, the production of IL-8 and fatty acid-binding protein 4 by adipocytes increased after co-culture of human adipocytes and ovarian cancer cells thereby promoting the homing, migration, and invasion of cancer cells [203]. In conclusion, adipocytes play an important role in the occurrence, development and metastasis of cancer, especially in breast cancer with rich adipose tissue [204]. At the same time, adipocytes also promote tumor cell resistance to anti-tumor treatment, such as adipose stromal cells inhibit the tolerance of prostate cancer cells to docetaxel, cabazitaxel and CDDP [205]. The exosome microsomal triglyceride transfer protein (MTTP) derived from adipocyte can inhibit iron ptosis in colorectal cancer and promote tumor resistance to chemotherapy [206].
Protumor mechanisms of cancer-associated adipocytes
CAAs can promote tumorigenesis and development by secreting a large number of cytokines (e.g., IL-6, IL-8, and chemokines), adipokines (leptin, adiponectin, autotaxin, and resistin), lipid metabolites (free fatty acids and β-hydroxybutyric acid), and exosomes [192, 207] (Fig. 4). Among them, cytokines derived from adipocyte, such as leptin, globular adiponectin, resistin, IGFBP-2, and CCL5, can be used as paracrine signals of cancer cells to upregulate invasion related proteins such as calcium binding protein S100A7, MMP-9, and urokinase type plasmin activator (UPA), thereby promoting the invasion and migration of cancer cells [208,209,210]. In addition, CAAs-induced TGF-β, secreted IL-8, and produced leptin can mediate EMT in cancer cells, promote tumor spread and tumor angiogenesis, respectively [211,212,213,214].

Activated CAAs showed a decrease in adipocyte markers such as adipocytes/macrophage fatty acid-binding protein 2 (Ap2) and fatty acid-binding protein 4 (FABP4), while the expression of MMP11 and the release of IL-6, IL-1β, and IGFBP-2 increased. CAAs secrete cytokines, adipokines, lipid metabolites, and exosomes to promote the proliferation of cancer cells, regulate extracellular matrix (ECM) structure, form an immunosuppressive microenvironment, promote tumor angiogenesis and epithelial interstitial transformation (EMT), and ultimately promote tumor growth, metastasis and resistance to anti-tumor treatment.
CAAs can also promote tumor cell invasion by upregulating the levels of versican and leptin in renal cancer cell lines [215]. In addition, the high expression of epidermal FABP5 in adipocytes can also lead to the development and metastasis of cancer [216]. CAAs can also support tumor progression by secreting lipid metabolites in addition to secreting cytokines and expressing invasion proteins to promote tumor development and metastasis. For example, cancer cells stimulate the breakdown of fat into fatty acids (FA) in CAAs, and FA enters cancer cells through specific fatty acid receptors and binding proteins (such as CD36 and Fatty Acid Transport Protein 1) for membrane synthesis and energy metabolism (β-oxidation), or lipid-derived cell signaling (derivatives of arachidonic acid and linolenic acid) [197]. Among them, CAAs release essential fatty acids (FFA) that are absorbed by cancer cells to promote growth and proliferation [217, 218]. The released free FA can also activate and regulate other cells such as macrophages, vascular endothelial cells and muscle cells, which is conducive to the formation of the original cancer microenvironment, and FABP4 released by CAAs is an energy source carrier for cell invasion [219]. In summary, CAAs can participate in the occurrence and development of tumors through mechanisms such as the secretion of cytokines, lipid metabolites, and expression of invasion proteins.
In addition, adipocytes can also support tumor progression by regulating immune cells in TME, such as the levels of exosome microRNA (miRNA)-155 increased during co-culture of tumor cells and adipocytes, thereby recruiting macrophages and promoting their differentiation into TAMs that support tumor development [220,221,222] In addition, visfatin secreted by CAAs can also induce M2 macrophage polarization and accelerate the glycolysis process in malignant tumor cells [223]. Not only that adipocytes can also acquire myofibroblast and macrophage-like features through metabolic reprogramming and dedifferentiation (that is “adipocytes-stromal switch”) thereby promoting tumor progression [224]. Adipocytes co-cultured with gastric cancer cells can also be converted into CAFs to promote tumor progression and peritoneal metastasis [225]. In addition, CAAs can also differentiate into PCs and participate in the formation of blood vessel walls [226]. Moreover, the interaction between various lipids, cytokines, and adipokines secreted by bone marrow adipocytes and cancer cells can promote bone metastasis in solid tumors, especially prostate cancer, breast cancer, lung cancer, and multiple myeloma [227]. In summary, CAAs can participate in the progression of tumors through various mechanisms.
The tumor-promoting mechanism of CAAs can be realized by multiple signaling pathways, such as IL-6 and leptin derived from CAAs can promote tumor metastasis by activating the JAK/STAT3 and PI3K/AKT signaling pathways to promote the expression of lysine hydroxylase (PLOD2) [228] And the cytokines LIF secreted by CAAs can promote the migration and invasion of breast cancer cells through the STAT3 signaling pathway [229]. In addition, the elevated exosome miRNA-155 during the co-culture of tumor cells and adipocytes can promote the production and release of adipocytes CCL2 and CCL5 by targeting the SOCS6/STAT3 pathway, thereby regulating the function and polarity of macrophages and promoting tumor progression [221]. In summary, CAAs can participate in tumor progression and metastasis by secreting cytokines, adipokines, providing metabolites, and transdifferentiation into other cells, and the tumor-promoting mechanism of CAAs can be realized through multiple signaling pathways.
Strategies for targeting cancer-associated adipocytes to treat tumors
Transforming CAAs into normal adipocytes, inhibiting related bioactive molecules, and exosomes are effective methods for targeting CAAs in the treatment of tumors. For example, metformin can exert anti-tumor effects by regulating adipocyte leptin and reversing dysfunctional adipocytes and normalizing them [230]. In recent years, it has also been found that metformin has a significant inhibitory effect on the growth and adipocyte differentiation of human adipose stromal cells (ADSCs) [231]. In addition, treatment targeting bioactive molecules secreted by CAAs can also inhibit tumors, such as Peroxisome proliferator-activated receptor-γ (PPAR-γ) agonists can inhibit tumors by reversing disorders of bioactive molecules such as upregulation of adiponectin, and PPAR-γ agonists such as rosiglitazone and pioglitazone can also promote adipocyte differentiation by downregulating factors such as leptin, IL-6, and TNF-α [232, 233]. Moreover, peptide analogues located at the binding site of leptin and leptin receptor (ObR) can selectively inhibit the interaction between leptin and leptin receptor, thereby inhibiting tumor occurrence and metastasis [234]. In addition, inhibiting the exosomes miRNA-155 of tumor cells can reduce the levels of CCL2 and CCL5 in CAAs co-culture thereby inhibiting tumor growth [221]. At the same time, blocking fatty acid-derived lipid uptake or lipid-related metabolic pathways in cancer cells is also an effective therapeutic strategy for lipid-rich cancers [197]. In summary, targeted CAAs for tumor therapy can be achieved through a variety of strategies.
As an important component of the tumor matrix, CAAs can promote the survival, proliferation, and migration of tumor cells through mechanisms such as secretion of tumor-related adipocytokines, inflammatory factors, and production of fatty acids. They can also work with other stromal cells to promote tumor progression. In addition, the interaction between adipocytes and tumor cells can also increase resistance to anti-tumor therapy. In summary, the activated CAAs in TME promote tumor progression, so strategies such as converting CAAs into normal adipocytes, inhibiting related bioactive molecules, and exosomes are an option for treating tumors based on CAAs.
Tumor-associated endothelial cells
The formation of blood vessels in tumors is essential for tumor growth and metastasis, and tumor blood vessels not only provide oxygen and nutrients for tumors to support tumor growth but also provide a channel for tumor metastasis [235]. Among them, vascular endothelial cells are important tissues for maintaining hemoperfusion, and TECs interact with tumor cells to form neovascularization thereby supporting tumor development, promoting tumor metastasis and participating in the resistance of anti-tumor therapy [236]. Therefore, it is possible to target vascular endothelial cells to treat tumors based on these effects of TECs on tumors, such as macromolecular anti-angiogenic drugs (AADs) represented by bevacizumab have been widely used in clinical practice. In addition, a variety of small-molecule kinase inhibitors have also seen significant efficacy. Next, we will introduce endothelial cells in tumors in detail.
Tumor blood vessels and tumor-associated endothelial cells
Angiogenesis is strictly regulated by the balance of angiogenic factors and antiangiogenic factors [237], but there is an imbalance between these two factors in tumors, with levels of angiogenic activators higher than those of angiogenic inhibitors [238]. For example, VEGF upregulateion due to hypoxia, activation of oncogenes and mutation of tumor suppressor genes in tumors, which activates ECs through paracrine signals to stimulate ECs proliferation, induce angiogenesis and enhance vascular permeability [239]. In addition to VEGF, cancer cells also secrete other angiogenic factors such as basic FGF, angiopoietin (Ang), HGF, epidermal growth factor, PDGF, and placental-derived growth factor [239], and all of which can significantly increase the proliferation, migration, and vascular formation of ECs [236, 238]. These overexpressed proangiogenic factors can induce the transition of ECs from a quiescent state to an active state, thereby enabling them to acquire a more migratory and invasive phenotype [240]. At the same time, the expression of angiogenesis inhibitory genes are downregulated in TME such as thrombospondin-1 (TSP-1) [241]. In conclusion, angiogenic factors are elevated in tumors and angiogenesis inhibitory genes are downregulated, and elevated angiogenic factors can induce tumor vascularization by promoting the proliferation and migration of ECs.
Tumor blood vessels are different from normal blood vessels in phenotype and morphology, and tumor blood vessels are chaotic, unhierarchical, abnormally dilated, and have the characteristics of high penetration and low perfusion. In addition, these vessels do not serve as normal barriers due to the lack of proper perivascular coverage and tight EC connections [242,243,244]. Not only that, the TECs that make up tumor blood vessels are also significantly different from normal endothelial cells (NECs), which are fragile, leaky, and highly proliferative and angiogenic [245]. Moreover, TECs are irregular in shape and size [237], and cells proliferate and migrate faster [229]. At the same time, TECs also manifest as cytogenetic abnormalities, such as both TECs and circulating TECs show chromosome instability characteristics such as aneuploidy, chromosome translocation and chromosome deletion [243]. Furthermore, TECs are also different from NECs at the molecular level, for example, TECs express embryonic markers such as renal transcription factor Paired Box2 (PAX2) [246] and CSCs markers such as stem cell antigen 1 (SCA-1) and CD90, and upregulate multidrug resistance (MDR) 1, Aldehyde dehydrogenase (ALDH), VEGF and VEGFR2 [247]. In addition, TECs also have metabolic plasticity and reprogramming. For example, TECs have increased glucose uptake and higher glycolysis rate than NECs [248, 249]. Not only that, fatty acid (FA) and serine biosynthesis pathways have also undergone changes [243], such as upregulation of FASN, PHGDH, and phosphoserine aminotransferase 1 (PSAT1) [238, 250]. Moreover, the phenotype of TECs also changes with tumor progression, such as TECs isolated from metastatic tumors have more chromosomal abnormalities, higher proliferation index and invasion potential, expression of more vascular secretory factors, more ability to attract and adhere to tumor cells, and resistance to anticancer drugs (5-FU, paclitaxel) than TECs isolated from non-metastatic tumors [238, 243]. In summary, tumor blood vessels differ from normal blood vessels in phenotype and morphology, and the TECs that make up tumor blood vessels exhibit significant differences from NECs in morphology, chromosome, molecular level, and metabolism.
The mechanisms that cause TECs to be abnormal may be related to the source of TECs (Fig. 5). For example, TECs can be transdifferentiated and formed by inducing tumor cells, CSCs, or vascular progenitor cells (VPCs) through mechanisms such as autophagy, and ROS activation of Akt/inhibitor of kappa B kinase (IKK) signaling pathways, such as CSCs of glioblastoma, breast, and ovaries all can differentiate into ECs in morphology and function [242, 243, 251, 252]. In addition, TECs can also be formed by the fusion of NECs with malignant tumor cells. ECs can also take up apoptotic bodies or exosomes released by tumor cells thereby taking up tumor oncogenes. In addition, the mechanism that causes TECs to be abnormal is also related to the genetic instability of TECs due to the influence of growth factors or cytokines in TME [243]. In summary, a variety of mechanisms lead to TECs being different from NECs thus resulting in TECs having different properties from NECs.

TECs can be formed by transdifferentiation of tumor cells, cancer stem cells (CSCs), or vascular progenitor cells (VPCs), and TECs can also be formed by fusion of normal endothelial cells (NECs) with malignant tumor cells or VPCs. TECs can also absorb apoptotic bodies or exosomes released by tumor cells, which can absorb tumor oncogenes. In addition, growth factors or cytokines in the tumor microenvironment can lead to genetic instability of TECs, thereby leading to TECs abnormalities.
Tumor-associated endothelial cells and tumors
TECs can participate in the occurrence, development, and metastasis of tumors by secreting cytokines such as interleukin, VEGF-A, and Heat Shock 70 kDa Protein 12B HSPA12B, as well as exosomes to activate receptors on tumor cells, support tumor metabolism, or suppress anti-tumor immune responses [236, 253,254,255,256,257]. In addition, TECs can promote cancer cell progression and metastasis by expressing endothelial protein C receptor (EPCR) and NOTCH1 [7, 258]. At the same time, TECs can also recruit macrophages and induce them to differentiate into M2 TAM, thereby inhibiting anti-tumor immune responses [257]. Moreover, TECs can also use MMPs to destroy the basement membrane and cause pericyte separation, endothelial cell migration, and degradation of extracellular matrix, thus contributing to tumor growth and metastasis [243]. In summary, TECs can participate in the occurrence, development, and metastasis of tumors through various mechanisms.
In addition, TECs are also involved in tumor resistance to antitumor treatments, such as kidney cancer-derived TECs resistant to vincristine [259], and liver cancer-derived TECs resistant to 5-FU and doxorubicin [260]. TECs can also resist paclitaxel therapy by upregulating P-glycoprotein (P-gp, ABCB1) [261]. The mechanism of TECs leading to tumor drug resistance may be related to the cytogenetic abnormalities of TECs leading to genetic instability [243]. In addition, exosomes secreted by cancer cells can also induce TECs to resist antitumor drugs, such as miRNA-1246 secreted by cancer cells can induce TECs to be resistant to 5-FU [262]. In summary, TECs are not only involved in the progression of tumors but also in the resistance of tumors to anti-tumor therapy.
Therapeutic strategies targeting tumor-associated endothelial cells
The strategy of anti-angiogenesis therapy for tumors is already relatively mature in clinical practice. Up to now, hundreds of AADs have been developed, among which TECs are the main targets of anti-angiogenic therapy. AADs can be divided into three categories according to the mechanism of action of AADs: anti-angiogenesis monoclonal antibody (AA-MA), anti-angiogenesis tyrosine kinase inhibitor (AA-TKI) and endogenous angiogenesis inhibitor. Among them, AA-MA mainly binds to VEGFA or vascular endothelial growth factor receptor (VEGFR) 2, while AA-TKI mainly inhibits targets such as VEGFR, PDGFR, and c-Kit. Endogenous angiogenesis inhibitors mainly exert anti-angiogenic effects by downregulating the expression of VEGF and its receptors. Currently, more than 10 anti-angiogenic drugs are approved by the FDA or NMPA, including AA-MA: Bevacizumab, Aflibercept, and Ramucirumab. AA-TKI: Sorafenib, Sunitinib, Pazopanib, etc, and endogenous angiogenesis inhibitors: Endostar. These drugs all can exert antitumor effects by acting on TECs [236]. For example, Bevacizumab inhibits endothelial mitosis, Aflibercept reduces vascular endothelial permeability [263], Endostar induces endothelial apoptosis [264], Sorafenib inhibits endothelial cell proliferation [265], etc. Table 2 lists the current AADs approved by the Food and Drug Administration (FDA) and the National Medical Products Administration (NMPA) for the treatment of cancer.
In addition to acting on VEGF/VEGFR affecting ECs in the treatment of tumors, peroxisome proliferator-activated receptors (PPARs) and vitamin D receptors (VDR) are also targets for the treatment of tumors targeting ECs, such as PPARα ligands have a powerful effect in inhibiting endothelial cell proliferation and angiogenesis, and the PPARα agonist fenofibrate can inhibit endothelial cell proliferation and VEGF production thereby inhibiting tumor growth [266]. In addition, calcitriol, which acts on VDR, also has an inhibitory proliferative effect on TECs but has no inhibitory effect on normal ECs [267]. Not only that, treatment targeting 17β-estradiol can also treat tumors by affecting ECs since 17β-estradiol can increase tumor vascular density and stabilize the vascular endothelial system [268].
In addition to acting on targets such as VEGF, PPARs, VDR, and 17β-estradiol to affect ECs to treat tumors, silencing-related genes can also treat tumors through ECs. For example, ablation of the A Disintegrin And Metalloprotease (ADAM) 17 gene in ECs and drugs that inhibit ADAM17 can prevent long-term metastases formation in the lung [269]. At the same time, knockout of Homologous to the E6-associated protein carboxyl terminus domain containing 3 (HECTD3) in ECs can also significantly inhibit lung colonization of tumor cells [270]. In addition, based on the fact that Notch signaling is essential for vascular development and tumor angiogenesis, inhibiting Notch ligand Delta like 4 or inhibiting Notch signaling in endothelial cells can lead to endothelial dysfunction, disrupt new angiogenesis, and limit tumor growth [271, 272].
Moreover, ECs L-type amino acid transporter 1 (LAT1)-mediated amino acid transport is the basis for supporting ECs proliferation and in vitro translation initiation, so therapeutic inhibition against LAT1 can also inhibit angiogenesis [273]. In addition, tumor progression can also be inhibited by regulating the metabolism of ECs. For example, inhibition of COX-2 in ECs can induce normalization of glucose metabolism thereby inhibiting tumor progression [274]. At the same time, the fatty acid synthase (FASN) blocker orlistat can also inhibit ECs proliferation, and knocking out FASN in endothelial cells can prevent vascular sprouting by reducing ECs proliferation [275]. In short, ECs can be inhibited by targeting receptors, silencing genes, inhibiting signaling pathways, and regulating ECs metabolism, thereby inhibiting their tumor-promoting effects.
TECs are another type of stromal cells in TME, in which tumor cells interact with TECs to form tumor blood vessels that are different from normal blood vessels in phenotype and morphology, and ultimately promote tumor development and metastasis. Not only that, TECs are also involved in drug resistance to anti-tumor treatment. Based on the influence of TECs on tumors and anti-tumor therapy, the treatment targeting vascular endothelial cells has become an effective anti-cancer treatment. For example, most of the current anti-angiogenic therapies target endothelial cells, such as large molecule antibodies and small molecule kinase inhibitors that are widely used in clinical practice. In addition, it can also inhibit ECs thereby inhibiting tumors by targeting other targets such as PPARs, VDR, 17β-estradiol, etc, as well as silencing genes and inhibiting signaling pathways. In summary, weakening the complex relationship between ECs and cancer cells, and finding therapeutic strategies that can target TECs with little interference with the normal vascular system is the direction of targeting ECs to treat tumors.
Tumor-associated pericytes
Pericytes (PCs) also known as parietal cells which are located on the inner surface of blood vessels and interact with newly proliferated ECs to play an important role in regulating vascular stability, permeability and maintaining the integrity of the blood-brain barrier. In addition, PCs are directly involved in the supply of oxygen, nutrient, and removal of waste [276]. In terms of tumor promoting mechanisms, PCs not only regulate angiogenesis and remodeling but also participate in tumor progression through various mechanisms, such as regulating immune responses in TME, secreting soluble factors, and transforming into other stromal cells [276]. We will introduce PCs in TME below.
Pericytes and tumors
PCs do not have specific surface markers and mainly express PDGFRβ, CD146 (also known as the melanoma cell adhesion molecule MCAM), Neuron-glial antigen 2 (NG2), CD13 (aminopeptidase N), and α-SMA, but these markers are also expressed on other types of cells such as endothelial cells and smooth muscle cells [277]. During tumorigenesis, PCs can be formed by TGF-β-induced epithelial cell transformation or by transdifferentiation of tumor cells or activated fibroblasts in TME [278, 279]. PCs themselves are also highly plastic, they can differentiate into different cell populations such as fibroblasts, fat cells, myoblasts, smooth muscle cells, and bone and chondrocytes [280]. For example, PCs expressing chondroitin sulfate proteoglycans (NG2 proteoglycans) may be the origin cells of stromal tumors such as bone and soft tissue sarcomas [281].
During angiogenesis, ECs can recruit PCs into new blood vessels through activating PDGFR signal by secreting PDGF β [282]. PCs in tumors are important mediators for maintaining the integrity of tumor blood vessels, and they interact with ECs to form a dysfunctional, leaky, and dysfunctional tumor vascular system [276]. Moreover, the coverage of PCs in tumor blood vessels is relatively low, which leads to tumor cell extravasation, increased plasma volume in the tumor interior/interstitium, and increased local pressure, thereby promoting tumor progression and metastasis [283]. In addition, PCs also play an important role in the formation of niche before cancer metastasis, such as PCs leaving blood vessels under the action of primary tumor factors in lung metastasis, and PCs detach from pulmonary blood vessels after primary tumor implantation into the lung and expand into stromal producing cells in the lung parenchyma before metastasis [284] thereby forming a microenvironment that is easy to be colonized by cancer cells [285]. Furthermore, the PCs in tumors are different from those in normal tissues, the PCs in tumors exhibit abnormalities and functional disorders [286, 287]. In addition, the glycolysis driven by hexokinase 2 (HK2) is increased in tumor PCs, and which up-regulates their ROCK2-MLC2 mediated contractility leading to impaired blood vessel supporting function [288]. In summary, TECs are different from PCs in normal tissues, and TECs can interact with ECs to form dysfunctional tumor blood vessels and participate in tumor progression and metastasis.
Pro-tumor mechanism and resistance to antitumor therapy of Pericytes
PCs as essential components of TME, which not only work with ECs to promote tumor growth and metastasis, but also promote tumor growth and progression by regulating the immune response in TME, secreting soluble factors, and converting to other stromal cells [276]. For example, PCs can affect the number of immune cells, and studies have shown that the coverage of PCs is related to the migration and infiltration of immune cells [289], and PCs can attract innate white blood cells through microvenous outflow by upregulating the expression of the adhesion molecule Intercellular Cell Adhesion Molecule (ICAM)-1 and releasing the chemokine macrophage migrationinhibitory factor (MIF) [290]. More and more evidence show that PCs in TME have regulatory effects on both innate and adaptive immunity (Fig. 6). For example, PCs can recruit MDSCs, and PCs-derived IL-33, CXCL12, and CXCL14 can promote macrophage recruitment and polarize towards M2 like phenotype [276, 291, 292]. In addition, human malignant glioma-derived pericytes (HMGPs) can inhibit T cell proliferation by releasing Prostaglandin E2 (PGE2), nitrous oxide (NO), serum human leukocyte antigen G (sHLA-G), and HGF, and can enhance the expression of anti-inflammatory cytokines TGF-β and IL-10 by upregulating chaperone-mediated autophagy (CMA), thereby recruiting Tregs and inhibiting the activity of T cells and antigen-presenting cells [293]. CD90-positive PCs in malignant gliomas are inhibitors of leukocyte and CD8+ T cells infiltration [293]. Moreover, PCs are also negative regulators of CD4+ T cells, which can inhibit the proliferation and activation of CD4+ T cells [276]. Furthermore, PCs activated by cancer cells can also inhibit anti-tumor immune responses by downregulating the expression of CD80, CD86 co-stimulatory molecules and MHC-II, as well as upregulating the expression of PD-L1 [294, 295]. In summary, PCs can affect immune cells in TME and inhibit anti-tumor immune responses through various mechanisms.

PCs can induce the expression of IL-6 thereby increasing the migration of bone marrow-derived suppressor cells (MDSCs). PC can also increase the recruitment of regulatory T cells (Treg) and B cells. Not only that, PCs may be an inhibitor of CD8+ T cells infiltration. It is also a negative regulator of CD4+ T cells, inducing the incompetence of CD4+ T cells. PCs can also inhibit T cells proliferation by releasing PGE2, NO, HGF, etc.
In addition to their immunomodulatory effects, PCs can also influence tumor progression through mechanisms such as transforming into other stromal cells and secreting cytokines. For example, PCs are considered to be one of the main sources of CAFs, and research has shown that cancer cells can induce the transformation of PCs into CAFs by activating the PI3K/AKT and MEK/ERK pathways, thereby promoting tumor invasion and metastasis [296]. In addition, PCs also have chemical attraction, adhesion, and proliferation-promoting effects on cancer cells by secreting soluble factors such as CCL2, CCL3, CXCL1, IFN- γ, and IL-8 [276, 285]. Among which PC-derived CCL2 can also improve tumor cell survival and tumor growth by stimulating MEK1-ERK1/2-Rho-associated coiled spiral protein kinase 2 (ROCK2)-dependent signaling in tumor cells [286]. Moreover, PCs can also promote local adhesion formation of tumor cells by secreting ECM, and exhibit migratory and invasive phenotypes by inhibiting intercellular adhesion and E-cadherin expression [297]. PCs can also promote EMT by secreting TGF-β1, which in turn contributes to cancer cell invasion [298]. Not only that PCs also play a crucial role in the occurrence and development of brain metastases, such as peribrain cells have a chemotaxis effect on cancer cells by secreting a large number of extracellular matrix proteins, and can also enhance the adhesion of cancer cells, and promote the proliferation of cancer cells by secreting IGF2 [4]. In conclusion, cytokines secreted by PCs can promote tumor progression and invasion through a variety of mechanisms.
In addition, PCs also mediate the resistance of tumors to anti-tumor therapy. For example, during the process of tumor growth, the proliferation of PCs leads to vascular abnormalities that affect the delivery of anti-tumor drugs. Moreover, PCs can also participate in tumor resistance to treatment by secreting cytokines. For example, PCs in gliomas can promote DNA repair and induce resistance to temozolomide (TMZ) drugs through secreting CCL5 and binding to CCR5 expressed on glioblastoma multiforme (GBM) cells, conversely, silencing the CCL5-CCR5 signal can improve the efficacy of TMZ [299]. In addition, PCs can also protect ECs from the toxicity of chemotherapy and mediate the efficacy of anti-angiogenic drugs and other molecular targeted therapies [300, 301]. For example, PCs can adaptively increase their coverage around tumor vessels (BVs) and induce tumors to resist antiangiogenic therapy by secreting proangiogenic factors or other soluble factors [276]. In summary, PCs can participate in tumor resistance to anti-tumor treatment through multiple mechanisms.
Therapy with targeted pericytes
PCs can promote tumor growth, metastasis, and resistance to anti-tumor treatment through a variety of mechanisms, so treatments of PCs can also treat tumors. For example, targeting glioma stem cell-derived PCs in gliomas can disrupt the blood tumor barrier (BTB) in GBM thereby leading to increased leakage of chemotherapy drugs and improving drug delivery and anti-tumor efficacy [289]. In addition, inhibiting the glycolytic activator fructose-2,6-diphosphatase 3 (PFKFB3) in ECs not only induces normalization of tumor blood vessels, but also increases the coverage of PCs and the adhesion of tumor blood vessels to ECs, thereby increasing the delivery of chemotherapy drugs [248]. Furthermore, due to the high proportion of hexokinase 2 (HK2)-positive PCs in tumors, reducing HK2/ROCK2 through shRNA or inhibitors can reduce the contractility of tumor PCs to rebuild the vascular system and thus improve drug delivery. For example, the combination of HK2 inhibitor 3-bromopyruvate and doxorubicin therapy can enhance the inhibitory effect of chemotherapy drugs on tumor growth in animal models of lung and liver cancer [302, 303].
In addition to influencing PCs by acting on metabolism-related enzymes, the inactivation of some genes and the inhibition of corresponding signals can also inhibit tumor growth through PCs. For example, inactivation of the Kruppel like factor 4 (KLF4) gene in PCs can reduce colonization (metastasis) of lung tumor cells by reducing the formation of extracellular fibrobinding proteins [284]. In addition, silencing IGF2 signaling or inhibiting IGF2 signaling with matrine (PPP) can block the promoting effect of PCs on breast cancer cell proliferation [304]. At the same time, based on PCs protecting ECs against angiogenic drugs, tyrosine kinase inhibitors which eliminate PDGFRβ-positive PCs combined with VEGF inhibitors are more effective at blocking tumor angiogenesis than anti-VEGF alone [305]. Such as a tyrosine kinase inhibitor imatinib targeted the PDGFRβ receptor upregulated by PCs, combined with antiangiopoietin-2 can control lung metastases [306]. In summary, many strategies can affect the growth and metastasis of tumors by targeting PCs, such as targeting metabolic enzymes, inhibiting genes, or inhibiting signaling and pathways.
PCs are important cellular components in TME and are associated with tumor angiogenesis, tumor metastasis, resistance to anti-tumor therapy, and patient mortality [283]. In terms of tumor-promoting mechanisms, it not only cooperates with ECs to regulate angiogenesis, but also promotes tumor growth and progression through various mechanisms such as regulating immune responses in TME, secreting soluble factors, and transforming into other stromal cells. Based on its tumor-promoting effect, some studies have also been carried out on the treatment of tumors by PCs, and some therapeutic strategies have seen efficacy in cell and preclinical animal experiments. However, there are some reports to the contrary, such as studies showing that the higher PC coverage in some patients, the better prognosis [307], and in some cases targeted PCs can even promote tumor metastasis [308], suggesting that the regulatory role of PCs in TME is far more complex than we think. In addition, due to the lack of effective methods to isolate human PCs, most in vitro PCs studies are carried out with bovine/mouse periretinal cells or mouse peribrain cells [288], and there is a lack of cell models to study the biology of human PCs, this difference will affect our understanding of PCs in human tumors, so the research on PCs still needs to go a lot.
Conclusions and perspectives
TASCs are mainly composed of CAFs, MSCs, CAAs, TECs, and PCs, and the source of which can be formed by the recruitment of adjacent endogenous stromal cells or transdifferentiation from cancer cells or other stromal cells in TME. TASCs together with immune cells and tumor cells constitute the cellular components of TME, of which TASCs account for about 50% of the total number of tumor tissue cells [309]. Complex signals have been established among tumor cells, immune cells, and stromal cells, and each TASCs can communicate with components of the microenvironment in a paracrine manner through cytokines and mediators, or in a cell-cell interaction, thereby participating in the entire process of tumorigenesis and development. For example, stromal cells play a role in inhibiting tumors in the early stage of tumor development or in normal tissues, but as the tumor progresses stromal cells in TME are edited by cancer cells and other components to form a phenotype that promotes tumor development. TASCs that obtain a tumor promoting phenotype can promote the occurrence, development, and resistance to tumor therapy by secreting various factors and exosomes, participating in tumor angiogenesis and tumor metabolism, participating in tumor immune escape, and regulating extracellular matrix. Based on this, targeting TASCs is also a strategy for cancer treatment, including but not limited to preventing stromal cells from entering TME, reducing tumor-promoting stromal cells and the physical barriers they formed, reprogramming tumor promoting stromal cells into tumor inhibiting stromal cells or normal cells, and blocking communication between stromal cells and cancer cells.
CAFs are the most abundant stromal cells in the TMEs of a variety of tumors and also the most studied and the most in-depth stromal cells [37]. It can be formed by transdifferentiation of normal fibroblasts in tumor tissue, or by transformation of other TASCs and immune cells. CAFs can be divided into multiple subtypes and different subtypes have different functions and spatial distribution, but most phenotypes have pro-tumor effects, they can release growth factors, chemokines, exosomes, and metabolites, thereby reshaping the extracellular matrix, promoting cancer cell growth, promoting angiogenesis, and influencing tumor cell resistance to antitumor therapy. The strategy of using CAFs as target cells to treat tumors include direct targeting of CAFs and indirect targeting of other therapies that affect CAFs. In addition, due to the presence of both tumor-promoting and tumor-inhibiting CAF subpopulations in TME, reducing the tumor-promoting CAFs subpopulations in TME and increasing the tumor-inhibiting CAFs subpopulations can make targeted CAFs more accurate in treating tumors. Some of these therapeutic strategies have seen anti-tumor effects in preclinical experiments. However, few clinical trials targeting CAFs to treat tumors have been carried out, and no specific inhibitor of CAFs has been approved for tumor treatment so far. However, since CAFs represent the majority of cells in the tumor stroma and most CAFs have pro-tumor properties, targeting CAFs to treat tumors will become a promising and important strategy for treating tumors.
In addition, MSCs are another large class of stromal cells in TME, which can be isolated from various tissues such as bone marrow, adipose tissue, umbilical cord, or induced by pluripotent stem cells. MSCs can migrate to the tumor site under the action of a variety of cytokines and chemokines in TME, and assume different roles at different stages of tumor development. For example, in vitro cell studies on MSCs have shown that MSCs have the effect of inhibiting tumorigenesis and growth, but MSCs nesting to the tumor site are “re-educated” by tumor cells and other cells in TME, which makes MSCs have the effect of promoting tumorigenesis, development, and metastasis. For example, MSCs can promote the occurrence and development of tumors through mechanisms such as cell-to-cell contact, secretion of biomolecules, enhancement of angiogenesis, inhibition of immune cell activity, or conversion to CAFs. That is to say, endogenous MSCs are involved in the occurrence and development of tumors, but exogenous MSCs, such as administration of MSCs have less effect on promoting tumor growth. Based on this and the characteristic of tumor homing ability of MSCs, MSCs can be used as carriers for the treatment of tumors with a variety of anti-tumor substances. In addition, MSCs also contribute to hematopoietic reconstruction after hematopoietic stem cell transplantation and reduce the side effects caused by anti-tumor therapy. A large number of clinical trials have been conducted on the application of MSCs in tumors, and some trial results have also shown that the use of MSCs can bring benefits to cancer patients. Therefore, using some characteristics of mesenchymal stem cells to treat tumors is a promising tumor treatment strategy.
CAAs are also an important component of cellular composition in TME, which can be derived from MSCs or undifferentiated adipocyte precursors in adipose tissue matrix, and a small portion of CAAs can also be derived from CSCs. CAAs can participate in tumor progression, metastasis, and resistance to antitumor therapy by secreting cytokines, adipokines, providing metabolites, and transdifferentiation into other cells. Therefore, targeting CAA is also a means of treating tumors, and specific strategies include converting CAAs into normal fat cells, inhibiting related bioactive molecules and exosomes, etc. But so far, there have been no approved drugs or registered clinical trials targeting CAA for tumor treatment. However, based on the tumor-promoting effect of CAAs, targeting CAA will surely become an option for tumor treatment.
In addition, the tumor vascular system is essential in multiple stages of tumor growth and metastasis. The tumor vascular system formed by the interaction between TECs and PCs in TME participates in tumor oxygen, nutrient supply, waste removal and provides a channel for tumor metastasis, and the formed vascular system is chaotic, unhierarchical, abnormally dilated, high penetration and low perfusion, which affects the delivery of drugs to the interior of the tumor and becomes a problem for the treatment of tumors. Furthermore, TECs and PCs not only support tumor growth and metastasis through the formation of blood vessels, but also participate in tumor occurrence, development, metastasis, and resistance to anti-tumor therapy through various pathways such as secretion of soluble factors and extracellular vesicles, inhibition of anti-tumor immune responses, and transformation into other stromal cells. which makes TECs become an important target cell of anti-tumor therapy. Among the many strategies based on TECs for the treatment of tumors, anti-angiogenic therapy is the most studied, and it is also the only drug approved for clinical application in the strategy of targeting stromal cells for tumor treatment, and there are currently more than 10 anti-angiogenic drugs approved by FDA or NMPA, which has become a mature and effective anti-cancer treatment. In addition, TECs can also be inhibited by targeting certain targets and pathways of EC, but most of these strategies are in the preclinical trial stage.
At the same time, as an essential component of TME, PCs not only coordinate with ECs to regulate angiogenesis and promote tumor growth and metastasis, but also promote tumor growth and progression through mechanisms such as regulating immune responses in TME, secreting soluble factors, and transforming into other stromal cells. Based on this, treatments targeting PCs can also affect tumor growth and metastasis, and specific strategies including acting on metabolism-related enzymes, inhibiting certain genes or inhibiting signals, pathways, etc, and some of these treatment strategies have already shown efficacy in cellular and preclinical animal experiments. However, few clinical trials have been carried out. A clinical trial registered in the clinicaltrials.gov database on targeted PC for the treatment of tumors is “Nintedanib (BIBF1120) in Thyroid Cancer” (NCT01788982), in which nintedanib is a triple angiogenesis inhibitor that may act not only on endothelial cells but also on pericyte and smooth muscle cells. The primary endpoint of this clinical trial is progression free survival (PFS). With the in-depth research on PCs, we also look forward to the development of various clinical trials targeting PC for tumor treatment.
It is possible to target TASCs to treat tumors based on the influence of TASCs on tumorigenesis and development. However, since there are no specific markers for each stromal cell in TASCs, until now the other strategies targeting stromal cells for tumor treatment have not been approved in clinical in addition to anti-angiogenic drugs targeting TECs. But it is not enough to not take into account the strategy of anti-TASCs for tumor treatment based on the influence of stromal cells on tumorigenesis, development and treatment in TME. At the same time, the use of single-cell RNA sequencing (scRNA-seq), which not only improves the resolution of stromal cells in healthy and disease tissues, but also finds that each stromal cell can be divided into a variety of subtypes. Further research on different subtypes and strategies based on subtype therapy for tumors will inevitably improve the accuracy and effectiveness of tumor treatment. Therefore, targeting TASCs to treat tumors will become an important direction for tumor treatment.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
Dai J, Su Y, Zhong S, Cong L, Liu B, Yang J, et al. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct Target Ther. 2020;5:145.
Frisbie L, Buckanovich RJ, Coffman L. Carcinoma-associated mesenchymal stem/stromal cells: architects of the pro-tumorigenic tumor microenvironment. Stem Cells. 2022;40:705–15.
Garcia-Gomez A, Rodriguez-Ubreva J, Ballestar E. Epigenetic interplay between immune, stromal and cancer cells in the tumor microenvironment. Clin Immunol. 2018;196:64–71.
Xue R, Zhang Q, Cao Q, Kong R, Xiang X, Liu H, et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature. 2022;612:141–7.
Zhao Y, Bai Y, Shen M, Li Y. Therapeutic strategies for gastric cancer targeting immune cells: future directions. Front Immunol. 2022;13:992762.
Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol. 2018;15:366–81.
Nilendu P, Sarode SC, Jahagirdar D, Tandon I, Patil S, Sarode GS, et al. Mutual concessions and compromises between stromal cells and cancer cells: driving tumor development and drug resistance. Cell Oncol (Dordr). 2018;41:353–67.
Mao YKE, Garfield DH, Shen K, Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013;32:303–15.
Zheng S, Zou Y, Tang Y, Yang A, Liang JY, Wu L, et al. Landscape of cancer-associated fibroblasts identifies the secreted biglycan as a protumor and immunosuppressive factor in triple-negative breast cancer. Oncoimmunology. 2022;11:2020984.
Wang S, Du P, Cao Y, Ma J, Yang X, Yu Z, et al. Cancer associated fibroblasts secreted exosomal miR-1290 contributes to prostate cancer cell growth and metastasis via targeting GSK3beta. Cell Death Discov. 2022;8:371.
Yuan M, Tu B, Li H, Pang H, Zhang N, Fan M, et al. Cancer-associated fibroblasts employ NUFIP1-dependent autophagy to secrete nucleosides and support pancreatic tumor growth. Nat Cancer. 2022;3:945–60.
Ucaryilmaz Metin C, Ozcan G. Comprehensive bioinformatic analysis reveals a cancer-associated fibroblast gene signature as a poor prognostic factor and potential therapeutic target in gastric cancer. BMC Cancer. 2022;22:692.
Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–54.
Song M, He J, Pan QZ, Yang J, Zhao J, Zhang YJ, et al. Cancer-associated fibroblast-mediated cellular crosstalk supports hepatocellular carcinoma progression. Hepatology. 2021;73:1717–35.
Aoto K, Ito K, Aoki S. Complex formation between platelet-derived growth factor receptor beta and transforming growth factor beta receptor regulates the differentiation of mesenchymal stem cells into cancer-associated fibroblasts. Oncotarget. 2018;9:34090–102.
Santolla MF, Vivacqua A, Lappano R, Rigiracciolo DC, Cirillo F, Galli GR, et al. GPER mediates a feedforward FGF2/FGFR1 paracrine activation coupling CAFs to cancer cells toward breast tumor progression. Cells. 2019;8:223.
Ji Q, Zhou L, Sui H, Yang L, Wu X, Song Q, et al. Primary tumors release ITGBL1-rich extracellular vesicles to promote distal metastatic tumor growth through fibroblast-niche formation. Nat Commun. 2020;11:1211.
Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. 2014;211:1503–23.
Hill BS, Sarnella A, D’Avino G, Zannetti A. Recruitment of stromal cells into tumour microenvironment promote the metastatic spread of breast cancer. Semin Cancer Biol. 2020;60:202–13.
Sobierajska K, Ciszewski WM, Sacewicz-Hofman I, Niewiarowska J. Endothelial cells in the tumor microenvironment. Adv Exp Med Biol. 2020;1234:71–86.
Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem. 2007;101:830–9.
Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67:10123–8.
Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res. 2011;1:482–97.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401.
Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 2016;30:1002–19.
Nissen NI, Karsdal M, Willumsen N. Collagens and cancer associated fibroblasts in the reactive stroma and its relation to cancer biology. J Exp Clin Cancer Res. 2019;6:115.
Nissen NI, Johansen AZ, Chen I, Johansen JS, Pedersen RS, Hansen CP, et al. Collagen biomarkers quantify fibroblast activity in vitro and predict survival in patients with pancreatic ductal adenocarcinoma. Cancers (Basel). 2022;6:819.
Saw PE, Chen J, Song E. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer. 2022;8:527–55.
Brichkina APP, Sharma SD, Visestamkul N, Lauth M. A quick guide to CAF subtypes in pancreatic cancer. Cancers (Basel). 2023;4:2614.
Biffi GOT, Spielman B, Hao Y, Elyada E, Park Y, Preall J, et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9:282–301.
Hutton C, Heider F, Blanco-Gomez A, Banyard A, Kononov A, Zhang X, et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell. 2021;39:1227–44.e20.
Krishnamurty AT, Shyer JA, Thai M, Gandham V, Buechler MB, Yang YA, et al. LRRC15(+) myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature. 2022;611:148–54.
Yang YWH, Fan S, Bi Y, Hao M, Shang J. Cancer‑associated fibroblast‑derived LRRC15 promotes the migration and invasion of triple‑negative breast cancer cells via Wnt/β‑catenin signalling pathway regulation. Mol Med Rep. 2022;25:2.
Chen KWQ, Li M, Guo H, Liu W, Wang F, Tian X, et al. Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. EBioMedicine. 2021;66:103315.
Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33:463–79.e10.
Li X, Sun Z, Peng G, Xiao Y, Guo J, Wu B, et al. Single-cell RNA sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics. 2022;12:620–38.
Tanaka M. Crosstalk of tumor stromal cells orchestrates invasion and spreading of gastric cancer. Pathol Int. 2022;72:219–33.
Dumont N, Liu B, Defilippis RA, Chang H, Rabban JT, Karnezis AN, et al. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia. 2013;15:249–62.
Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–11.
Shekhar MP, Werdell J, Santner SJ, Pauley RJ, Tait L. Breast stroma plays a dominant regulatory role in breast epithelial growth and differentiation: implications for tumor development and progression. Cancer Res. 2001;61:1320–6.
Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci USA. 2004;101:4966–71.
Wang L, Cao L, Wang H, Liu B, Zhang Q, Meng Z, et al. Cancer-associated fibroblasts enhance metastatic potential of lung cancer cells through IL-6/STAT3 signaling pathway. Oncotarget. 2017;8:76116–28.
Murata T, Mekada E, Hoffman RM. Reconstitution of a metastatic-resistant tumor microenvironment with cancer-associated fibroblasts enables metastasis. Cell Cycle. 2017;16:533–5.
Kong J, Tian H, Zhang F, Zhang Z, Li J, Liu X, et al. Extracellular vesicles of carcinoma-associated fibroblasts creates a pre-metastatic niche in the lung through activating fibroblasts. Mol Cancer. 2019;18:175.
Zhang T, Li X, He Y, Wang Y, Shen J, Wang S, et al. Cancer-associated fibroblasts-derived HAPLN1 promotes tumour invasion through extracellular matrix remodeling in gastric cancer. Gastric Cancer. 2022;25:346–59.
Tong Y, Yang L, Yu C, Zhu W, Zhou X, Xiong Y, et al. Tumor-Secreted Exosomal lncRNA POU3F3 Promotes Cisplatin Resistance in ESCC by Inducing Fibroblast Differentiation into CAFs. Mol Ther Oncolytics. 2020;18:1–13.
Eiro N, Gonzalez L, Martinez-Ordonez A, Fernandez-Garcia B, Gonzalez LO, Cid S, et al. Cancer-associated fibroblasts affect breast cancer cell gene expression, invasion and angiogenesis. Cell Oncol (Dordr). 2018;41:369–78.
Fang Y, Chen M, Li G, Yang Y, He P, Chen J, et al. Cancer-associated fibroblast-like fibroblasts in vocal fold leukoplakia suppress CD8(+)T cell functions by inducing IL-6 autocrine loop and interacting with Th17 cells. Cancer Lett. 2022;546:215839.
Ao Z, Shah SH, Machlin LM, Parajuli R, Miller PC, Rawal S, et al. Identification of cancer-associated fibroblasts in circulating blood from patients with metastatic breast cancer. Cancer Res. 2015;75:4681–7.
Jones ML, Siddiqui J, Pienta KJ, Getzenberg RH. Circulating fibroblast-like cells in men with metastatic prostate cancer. Prostate. 2013;73:176–81.
Biffi G, Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev. 2021;101:147–76.
Kay EJ, Paterson K, Riera-Domingo C, Sumpton D, Dabritz JHM, Tardito S, et al. Cancer-associated fibroblasts require proline synthesis by PYCR1 for the deposition of pro-tumorigenic extracellular matrix. Nat Metab. 2022;4:693–710.
Wang Y, Lan W, Xu M, Song J, Mao J, Li C, et al. Cancer-associated fibroblast-derived SDF-1 induces epithelial-mesenchymal transition of lung adenocarcinoma via CXCR4/beta-catenin/PPARdelta signalling. Cell Death Dis. 2021;12:214.
Ando N, Hara M, Shiga K, Yanagita T, Takasu K, Nakai N, et al. Eicosapentaenoic acid suppresses angiogenesis via reducing secretion of IL‑6 and VEGF from colon cancer‑associated fibroblasts. Oncol Rep. 2019;42:339–49.
Stadler M, Pudelko K, Biermeier A, Walterskirchen N, Gaigneaux A, Weindorfer C, et al. Stromal fibroblasts shape the myeloid phenotype in normal colon and colorectal cancer and induce CD163 and CCL2 expression in macrophages. Cancer Lett. 2021;520:184–200.
Bughda R, Dimou P, D’Souza RR, Klampatsa A. Fibroblast activation protein (FAP)-targeted CAR-T cells: launching an attack on tumor stroma. Immunotargets Ther. 2021;10:313–23.
Pothula SP, Xu Z, Goldstein D, Pirola RC, Wilson JS, Apte MV. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. 2016;381:194–200.
Kaushik S, Pickup MW, Weaver VM. From transformation to metastasis: deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016;35:655–67.
Liu X, Zhu L, Wang R, Lou X, Yao X, Ni C, et al. IFNgamma inhibits fibroblast-leading tumor cell invasion through downregulating N-cadherin. Biochem Biophys Res Commun. 2019;512:544–51.
Ishimoto T, Miyake K, Nandi T, Yashiro M, Onishi N, Huang KK, et al. Activation of transforming growth factor beta 1 signaling in gastric cancer-associated fibroblasts increases their motility, via expression of rhomboid 5 homolog 2, and ability to induce invasiveness of gastric cancer cells. Gastroenterology. 2017;153:191–204.e16.
Heichler C, Scheibe K, Schmied A, Geppert CI, Schmid B, Wirtz S, et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut. 2020;69:1269–82.
Kuziel G, Thompson V, D’Amato JV, Arendt LM. Stromal CCL2 signaling promotes mammary tumor fibrosis through recruitment of myeloid-lineage cells. Cancers (Basel). 2020;12:2083.
Zhang Q, Zhou N, Wang W, Zhou S. A novel autocrine CXCL14/ACKR2 axis: the achilles’ heel of cancer metastasis? Clin Cancer Res. 2019;25:3476–8.
Sun X, He X, Zhang Y, Hosaka K, Andersson P, Wu J, et al. Inflammatory cell-derived CXCL3 promotes pancreatic cancer metastasis through a novel myofibroblast-hijacked cancer escape mechanism. Gut. 2022;71:129–47.
Primac I, Maquoi E, Blacher S, Heljasvaara R, Van Deun J, Smeland HY, et al. Stromal integrin alpha11 regulates PDGFR-beta signaling and promotes breast cancer progression. J Clin Invest. 2019;129:4609–28.
Yoon H, Tang CM, Banerjee S, Yebra M, Noh S, Burgoyne AM, et al. Cancer-associated fibroblast secretion of PDGFC promotes gastrointestinal stromal tumor growth and metastasis. Oncogene. 2021;40:1957–73.
Wei L, Ye H, Li G, Lu Y, Zhou Q, Zheng S, et al. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 2018;9:1065.
Pelon F, Bourachot B, Kieffer Y, Magagna I, Mermet-Meillon F, Bonnet I, et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat Commun. 2020;11:404.
Watanabe K, Shiga K, Maeda A, Harata S, Yanagita T, Suzuki T, et al. Chitinase 3-like 1 secreted from cancer-associated fibroblasts promotes tumor angiogenesis via interleukin-8 secretion in colorectal cancer. Int J Oncol. 2022;60:3.
Song J, Chen W, Cui X, Huang Z, Wen D, Yang Y, et al. CCBE1 promotes tumor lymphangiogenesis and is negatively regulated by TGFbeta signaling in colorectal cancer. Theranostics. 2020;10:2327–41.
An Y, Liu F, Chen Y, Yang Q. Crosstalk between cancer-associated fibroblasts and immune cells in cancer. J Cell Mol Med. 2020;24:13–24.
Zhang R, Qi F, Zhao F, Li G, Shao S, Zhang X, et al. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019;10:273.
Chen S, Morine Y, Tokuda K, Yamada S, Saito Y, Nishi M, et al. Cancer‑associated fibroblast‑induced M2‑polarized macrophages promote hepatocellular carcinoma progression via the plasminogen activator inhibitor‑1 pathway. Int J Oncol. 2021;59:59.
Lin Y, Cai Q, Chen Y, Shi T, Liu W, Mao L, et al. CAFs shape myeloid-derived suppressor cells to promote stemness of intrahepatic cholangiocarcinoma through 5-lipoxygenase. Hepatology. 2022;75:28–42.
Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 2016;76:4124–35.
Zhao Q, Huang L, Qin G, Qiao Y, Ren F, Shen C, et al. Cancer-associated fibroblasts induce monocytic myeloid-derived suppressor cell generation via IL-6/exosomal miR-21-activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett. 2021;518:35–48.
Jenkins L, Jungwirth U, Avgustinova A, Iravani M, Mills A, Haider S, et al. Cancer-associated fibroblasts suppress CD8+ T-cell infiltration and confer resistance to immune-checkpoint blockade. Cancer Res. 2022;82:2904–17.
Huang H, Wang Z, Zhang Y, Pradhan RN, Ganguly D, Chandra R, et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell. 2022;40:656–73.e7.
Dai X, Xie Y, Dong M. Cancer-associated fibroblasts derived extracellular vesicles promote angiogenesis of colorectal adenocarcinoma cells through miR-135b-5p/FOXO1 axis. Cancer Biol Ther. 2022;23:76–88.
Wang R, Sun Y, Yu W, Yan Y, Qiao M, Jiang R, et al. Downregulation of miRNA-214 in cancer-associated fibroblasts contributes to migration and invasion of gastric cancer cells through targeting FGF9 and inducing EMT. J Exp Clin Cancer Res. 2019;38:20.
Yang X, Xu X, Zhu J, Zhang S, Wu Y, Wu Y, et al. miR-31 affects colorectal cancer cells by inhibiting autophagy in cancer-associated fibroblasts. Oncotarget. 2016;7:79617–28.
Shen H, Yu X, Yang F, Zhang Z, Shen J, Sun J, et al. Reprogramming of Normal Fibroblasts into Cancer-Associated Fibroblasts by miRNAs-Mediated CCL2/VEGFA Signaling. PLoS Genet. 2016;12:e1006244.
Qin Y, Liu X, Pan L, Zhou R, Zhang X. Long noncoding RNA MIR155HG facilitates pancreatic cancer progression through negative regulation of miR-802. J Cell Biochem. 2019;120:17926–34.
Ren Y, Jia HH, Xu YQ, Zhou X, Zhao XH, Wang YF, et al. Paracrine and epigenetic control of CAF-induced metastasis: the role of HOTAIR stimulated by TGF-ss1 secretion. Mol Cancer. 2018;17:5.
Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc Natl Acad Sci USA. 2017;114:E9066–E75.
Li Z, Sun C, Qin Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics. 2021;11:8322–36.
Bertero T, Oldham WM, Grasset EM, Bourget I, Boulter E, Pisano S, et al. Tumor-stroma mechanics coordinate amino acid availability to sustain tumor growth and malignancy. Cell Metab. 2019;29:124–40.e10.
Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, et al. A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov. 2019;9:617–27.
Achreja A, Zhao H, Yang L, Yun TH, Marini J, Nagrath D. Exo-MFA - A 13C metabolic flux analysis framework to dissect tumor microenvironment-secreted exosome contributions towards cancer cell metabolism. Metab Eng. 2017;43:156–72.
Li Y, Zhao Z, Liu W, Li X. SNHG3 functions as miRNA sponge to promote breast cancer cells growth through the metabolic reprogramming. Appl Biochem Biotechnol. 2020;191:1084–99.
Steinbichler TB, Metzler V, Pritz C, Riechelmann H, Dudas J. Tumor-associated fibroblast-conditioned medium induces CDDP resistance in HNSCC cells. Oncotarget. 2016;7:2508–18.
Zhai J, Shen J, Xie G, Wu J, He M, Gao L. et al. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019;454:37–43.
Zhang F, Cui JY, Gao HF, Yu H, Gao FF, Chen JL, et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition and cisplatin resistance in ovarian cancer via CXCL12/CXCR4 axis. Future Oncol. 2020;16:2619–33.
Sun X, Chen Z. Cancer-associated fibroblast-derived CCL5 contributes to cisplatin resistance in A549 NSCLC cells partially through upregulation of lncRNA HOTAIR expression. Oncol Lett. 2021;22:696.
Zhang H, Yue J, Jiang Z, Zhou R, Xie R, Xu Y, et al. CAF-secreted CXCL1 conferred radioresistance by regulating DNA damage response in a ROS-dependent manner in esophageal squamous cell carcinoma. Cell Death Dis. 2017;8:e2790.
Chu TY, Yang JT, Huang TH, Liu HW. Crosstalk with cancer-associated fibroblasts increases the growth and radiation survival of cervical cancer cells. Radiat Res. 2014;181:540–7.
Wang W, Li Q, Yamada T, Matsumoto K, Matsumoto I, Oda M, et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2009;15:6630–8.
Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer. 2020;19:43.
Qin X, Guo H, Wang X, Zhu X, Yan M, Wang X, et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol. 2019;20:12.
Luo G, Zhang Y, Wu Z, Zhang L, Liang C, Chen X. Exosomal LINC00355 derived from cancer-associated fibroblasts promotes bladder cancer cell resistance to cisplatin by regulating miR-34b-5p/ABCB1 axis. Acta Biochim Biophys Sin (Shanghai). 2021;53:558–66.
Guo H, Ha C, Dong H, Yang Z, Ma Y, Ding Y. Cancer-associated fibroblast-derived exosomal microRNA-98-5p promotes cisplatin resistance in ovarian cancer by targeting CDKN1A. Cancer Cell Int. 2019;19:347.
Wang H, Huang H, Wang L, Liu Y, Wang M, Zhao S, et al. Cancer-associated fibroblasts secreted miR-103a-3p suppresses apoptosis and promotes cisplatin resistance in non-small cell lung cancer. Aging (Albany NY). 2021;13:14456–68.
Tan D, Li G, Zhang P, Peng C, He B. LncRNA SNHG12 in extracellular vesicles derived from carcinoma-associated fibroblasts promotes cisplatin resistance in non-small cell lung cancer cells. Bioengineered. 2022;13:1838–57.
Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. 2014;159:499–513.
Yang Y, Ma Y, Yan S, Wang P, Hu J, Chen S, et al. CAF promotes chemoresistance through NRP2 in gastric cancer. Gastric Cancer. 2022;25:503–14.
Lau EY, Lo J, Cheng BY, Ma MK, Lee JM, Ng JK, et al. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 signaling. Cell Rep. 2016;15:1175–89.
Guardia C, Bianchini G, Arpi LO, Menendez S, Casadevall D, Galbardi B, et al. Preclinical and clinical characterization of fibroblast-derived neuregulin-1 on trastuzumab and pertuzumab activity in HER2-positive breast cancer. Clin Cancer Res. 2021;27:5096–108.
Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov. 2020;10:1330–51.
Galbo PM Jr, Zang X, Zheng D. Molecular features of cancer-associated fibroblast subtypes and their implication on cancer pathogenesis, prognosis, and immunotherapy resistance. Clin Cancer Res. 2021;27:2636–47.
Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20:131.
Xia Q, Zhang FF, Geng F, Liu CL, Xu P, Lu ZZ, et al. Anti-tumor effects of DNA vaccine targeting human fibroblast activation protein alpha by producing specific immune responses and altering tumor microenvironment in the 4T1 murine breast cancer model. Cancer Immunol Immunother. 2016;65:613–24.
Qian L, Tang Z, Yin S, Mo F, Yang X, Hou X, et al. Fusion of dendritic cells and cancer-associated fibroblasts for activation of anti-tumor cytotoxic T lymphocytes. J Biomed Nanotechnol. 2018;14:1826–35.
Kim DJ, Dunleavey JM, Xiao L, Ollila DW, Troester MA, Otey CA, et al. Suppression of TGFbeta-mediated conversion of endothelial cells and fibroblasts into cancer associated (myo)fibroblasts via HDAC inhibition. Br J Cancer. 2018;118:1359–68.
Aboulkheyr EsH, Zhand S, Thiery JP, Warkiani ME. Pirfenidone reduces immune-suppressive capacity of cancer-associated fibroblasts through targeting CCL17 and TNF-beta. Integr Biol (Camb). 2020;12:188–97.
Han X, Li Y, Xu Y, Zhao X, Zhang Y, Yang X, et al. Reversal of pancreatic desmoplasia by re-educating stellate cells with a tumour microenvironment-activated nanosystem. Nat Commun. 2018;9:3390.
Dauer P, Zhao X, Gupta VK, Sharma N, Kesh K, Gnamlin P, et al. Inactivation of cancer-associated-fibroblasts disrupts oncogenic signaling in pancreatic cancer cells and promotes its regression. Cancer Res. 2018;78:1321–33.
Gorchs L, Ahmed S, Mayer C, Knauf A, Fernandez Moro C, Svensson M, et al. The vitamin D analogue calcipotriol promotes an anti-tumorigenic phenotype of human pancreatic CAFs but reduces T cell mediated immunity. Sci Rep. 2020;10:17444.
Zhao ZX, Zhang YQ, Sun H, Chen ZQ, Chang JJ, Wang X, et al. Calcipotriol abrogates cancer-associated fibroblast-derived IL-8-mediated oxaliplatin resistance in gastric cancer cells via blocking PI3K/Akt signaling. Acta Pharm Sin. 2023;44:178–88.
Bates AL, Pickup MW, Hallett MA, Dozier EA, Thomas S, Fingleton B. Stromal matrix metalloproteinase 2 regulates collagen expression and promotes the outgrowth of experimental metastases. J Pathol. 2015;235:773–83.
Krishnamachary B, Stasinopoulos I, Kakkad S, Penet MF, Jacob D, Wildes F, et al. Breast cancer cell cyclooxygenase-2 expression alters extracellular matrix structure and function and numbers of cancer associated fibroblasts. Oncotarget. 2017;8:17981–94.
Xiang H, Ramil CP, Hai J, Zhang C, Wang H, Watkins AA, et al. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol Res. 2020;8:436–50.
Ford K, Hanley CJ, Mellone M, Szyndralewiez C, Heitz F, Wiesel P, et al. NOX4 inhibition potentiates immunotherapy by overcoming cancer-associated fibroblast-mediated CD8 T-cell exclusion from tumors. Cancer Res. 2020;80:1846–60.
Iida TMY, Esaki N, Ponik SM, Burkel BM, Weng L, Kuwata K, et al. Pharmacologic conversion of cancer-associated fibroblasts from a protumor phenotype to an antitumor phenotype improves the sensitivity of pancreatic cancer to chemotherapeutics. Oncogene. 2022;41:2764–77.
Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10+GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. 2018;172:841–56.
Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20212–7.
Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019;9:1102–23.
Krishnamurty AT, Shyer JA, Thai M, Gandham V, Buechler MB, Yang YA, et al. LRRC15+ myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature. 2022;611:148–54.
Monteran L, Erez N. The dark side of fibroblasts: cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front Immunol. 2019;10:1835.
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20:174–86.
Salem HK, Thiemermann C. Mesenchymal stromal cells: current understanding and clinical status. Stem Cells. 2010;28:585–96.
Tu Z, Karnoub AE. Mesenchymal stem/stromal cells in breast cancer development and management. Semin Cancer Biol. 2022;86:81–92.
Jiang YJ, Chao CC, Chang AC, Chen PC, Cheng FJ, Liu JF, et al. Cigarette smoke-promoted increases in osteopontin expression attract mesenchymal stem cell recruitment and facilitate lung cancer metastasis. J Adv Res. 2022;41:77–87.
Takayama Y, Kusamori K, Nishikawa M. Mesenchymal stem/stromal cells as next-generation drug delivery vehicles for cancer therapeutics. Expert Opin Drug Deliv. 2021;18:1627–42.
Wu HH, Zhou Y, Tabata Y, Gao JQ. Mesenchymal stem cell-based drug delivery strategy: from cells to biomimetic. J Control Release. 2019;294:102–13.
Zhang L, Ma XJ, Fei YY, Han HT, Xu J, Cheng L, et al. Stem cell therapy in liver regeneration: focus on mesenchymal stem cells and induced pluripotent stem cells. Pharm Ther. 2022;232:108004.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006;8:315–7.
Cheng S, Nethi SK, Rathi S, Layek B, Prabha S. Engineered mesenchymal stem cells for targeting solid tumors: therapeutic potential beyond regenerative therapy. J Pharm Exp Ther. 2019;370:231–41.
de Miranda MC, Melo MIA, Cunha PDS, Gentilini JJ, Faria J, Rodrigues MA, et al. Roles of mesenchymal stromal cells in the head and neck cancer microenvironment. Biomed Pharmacother. 2021;144:112269.
Stone MJ, Hayward JA, Huang C, E Huma Z, Sanchez J. Mechanisms of regulation of the chemokine-receptor network. Int J Mol Sci. 2017;18:342.
Nitzsche F, Muller C, Lukomska B, Jolkkonen J, Deten A, Boltze J. Concise review: MSC adhesion cascade-insights into homing and transendothelial migration. Stem Cells. 2017;35:1446–60.
Zheng XB, He XW, Zhang LJ, Qin HB, Lin XT, Liu XH, et al. Bone marrow-derived CXCR4-overexpressing MSCs display increased homing to intestine and ameliorate colitis-associated tumorigenesis in mice. Gastroenterol Rep (Oxf). 2019;7:127–38.
Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–63.
Lan T, Luo M, Wei X. Mesenchymal stem/stromal cells in cancer therapy. J Hematol Oncol. 2021;14:195.
Ullah M, Liu DD, Thakor AS. Mesenchymal stromal cell homing: mechanisms and strategies for improvement. iScience. 2019;15:421–38.
Watts TL, Cui R, Szaniszlo P, Resto VA, Powell DW, Pinchuk IV. PDGF-AA mediates mesenchymal stromal cell chemotaxis to the head and neck squamous cell carcinoma tumor microenvironment. J Transl Med. 2016;14:337.
Bajetto A, Pattarozzi A, Corsaro A, Barbieri F, Daga A, Bosio A, et al. Different effects of human umbilical cord mesenchymal stem cells on glioblastoma stem cells by direct cell interaction or via released soluble factors. Front Cell Neurosci. 2017;11:312.
Liu Y, Ren H, Zhou Y, Shang L, Zhang Y, Yang F, et al. The hypoxia conditioned mesenchymal stem cells promote hepatocellular carcinoma progression through YAP mediated lipogenesis reprogramming. J Exp Clin Cancer Res. 2019;38:228.
Bruna F, Plaza A, Arango M, Espinoza I, Conget P. Systemically administered allogeneic mesenchymal stem cells do not aggravate the progression of precancerous lesions: a new biosafety insight. Stem Cell Res Ther. 2018;9:137.
Xia C, Wang T, Cheng H, Dong Y, Weng Q, Sun G, et al. Mesenchymal stem cells suppress leukemia via macrophage-mediated functional restoration of bone marrow microenvironment. Leukemia. 2020;34:2375–83.
Bruno S, Collino F, Deregibus MC, Grange C, Tetta C, Camussi G. Microvesicles derived from human bone marrow mesenchymal stem cells inhibit tumor growth. Stem Cells Dev. 2013;22:758–71.
Wu S, Ju GQ, Du T, Zhu YJ, Liu GH. Microvesicles derived from human umbilical cord Wharton’s jelly mesenchymal stem cells attenuate bladder tumor cell growth in vitro and in vivo. PLoS ONE. 2013;8:e61366.
Chen YC, Gonzalez ME, Burman B, Zhao X, Anwar T, Tran M, et al. Mesenchymal stem/stromal cell engulfment reveals metastatic advantage in breast cancer. Cell Rep. 2019;27:3916–26.e5.
Sarmadi VH, Tong CK, Vidyadaran S, Abdullah M, Seow HF, Ramasamy R. Mesenchymal stem cells inhibit proliferation of lymphoid origin haematopoietic tumour cells by inducing cell cycle arrest. Med J Malays. 2010;65:209–14.
Zheng Z, Li P, Shen F, Shi Y, Shao C. Mesenchymal stem/stromal cells in cancer: from initiation to metastasis. Arch Med Res. 2022;53:785–93.
Zhang T, Lee YW, Rui YF, Cheng TY, Jiang XH, Li G. Bone marrow-derived mesenchymal stem cells promote growth and angiogenesis of breast and prostate tumors. Stem Cell Res Ther. 2013;4:70.
Xu Z, Gao H, Zhang Y, Feng W, Miao Y, Xu Z, et al. CCL7 and TGF-beta secreted by MSCs play opposite roles in regulating CRC metastasis in a KLF5/CXCL5-dependent manner. Mol Ther. 2022;30:2327–41.
Yigitbilek F, Ozdogan E, Abrol N, Park WD, Hansen MJ, Dasari S, et al. Liver mesenchymal stem cells are superior inhibitors of NK cell functions through differences in their secretome compared to other mesenchymal stem cells. Front Immunol. 2022;13:952262.
Ren W, Hou J, Yang C, Wang H, Wu S, Wu Y, et al. Extracellular vesicles secreted by hypoxia pre-challenged mesenchymal stem cells promote non-small cell lung cancer cell growth and mobility as well as macrophage M2 polarization via miR-21-5p delivery. J Exp Clin Cancer Res. 2019;38:62.
Radmanesh F, Mahmoudi M, Yazdanpanah E, Keyvani V, Kia N, Nikpoor AR, et al. The immunomodulatory effects of mesenchymal stromal cell-based therapy in human and animal models of systemic lupus erythematosus. IUBMB Life. 2020;72:2366–81.
Liu H, Liang Z, Wang F, Zhou C, Zheng X, Hu T, et al. Exosomes from mesenchymal stromal cells reduce murine colonic inflammation via a macrophage-dependent mechanism. JCI Insight. 2019;4:e131273.
Biswas S, Mandal G, Roy Chowdhury S, Purohit S, Payne KK, Anadon C, et al. Exosomes produced by mesenchymal stem cells drive differentiation of myeloid cells into immunosuppressive M2-polarized macrophages in breast cancer. J Immunol. 2019;203:3447–60.
Weng Z, Zhang B, Wu C, Yu F, Han B, Li B, et al. Therapeutic roles of mesenchymal stem cell-derived extracellular vesicles in cancer. J Hematol Oncol. 2021;14:136.
Ryan D, Koziol J, ElShamy WM. Targeting AXL and RAGE to prevent geminin overexpression-induced triple-negative breast cancer metastasis. Sci Rep. 2019;9:19150.
Liu C, Feng X, Wang B, Wang X, Wang C, Yu M, et al. Bone marrow mesenchymal stem cells promote head and neck cancer progression through Periostin-mediated phosphoinositide 3-kinase/Akt/mammalian target of rapamycin. Cancer Sci. 2018;109:688–98.
Ai J, Ketabchi N, Verdi J, Gheibi N, Khadem Haghighian H, Kavianpour M. Mesenchymal stromal cells induce inhibitory effects on hepatocellular carcinoma through various signaling pathways. Cancer Cell Int. 2019;19:329.
Chen B, Yu J, Wang Q, Zhao Y, Sun L, Xu C, et al. Human bone marrow mesenchymal stem cells promote gastric cancer growth via regulating c-Myc. Stem Cells Int. 2018;2018:9501747.
Wu XB, Liu Y, Wang GH, Xu X, Cai Y, Wang HY, et al. Mesenchymal stem cells promote colorectal cancer progression through AMPK/mTOR-mediated NF-kappaB activation. Sci Rep. 2016;6:21420.
Avnet S, Di Pompo G, Chano T, Errani C, Ibrahim-Hashim A, Gillies RJ, et al. Cancer-associated mesenchymal stroma fosters the stemness of osteosarcoma cells in response to intratumoral acidosis via NF-kappaB activation. Int J Cancer. 2017;140:1331–45.
Zhang X, Hu F, Li G, Li G, Yang X, Liu L, et al. Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2018;9:25.
Ruvolo PP, Ruvolo VR, Burks JK, Qiu Y, Wang RY, Shpall EJ, et al. Role of MSC-derived galectin 3 in the AML microenvironment. Biochim Biophys Acta Mol Cell Res. 2018;1865:959–69.
Wang J, Cui R, Clement CG, Nawgiri R, Powell DW, Pinchuk IV, et al. Activation PDGFR-alpha/AKT mediated signaling pathways in oral squamous cell carcinoma by mesenchymal stem/stromal cells promotes anti-apoptosis and decreased sensitivity to cisplatin. Front Oncol. 2020;10:552.
Liu C, Billet S, Choudhury D, Cheng R, Haldar S, Fernandez A, et al. Bone marrow mesenchymal stem cells interact with head and neck squamous cell carcinoma cells to promote cancer progression and drug resistance. Neoplasia 2021;23:118–28.
Ji R, Zhang B, Zhang X, Xue J, Yuan X, Yan Y, et al. Exosomes derived from human mesenchymal stem cells confer drug resistance in gastric cancer. Cell Cycle. 2015;14:2473–83.
Luo T, Liu Q, Tan A, Duan L, Jia Y, Nong L, et al. Mesenchymal stem cell-secreted exosome promotes chemoresistance in breast cancer via enhancing miR-21-5p-mediated S100A6 expression. Mol Ther Oncolytics. 2020;19:283–93.
Zhang X, Yang Y, Yang Y, Chen H, Tu H, Li J. Exosomes from bone marrow microenvironment-derived mesenchymal stem cells affect cml cells growth and promote drug resistance to tyrosine kinase inhibitors. Stem Cells Int. 2020;2020:8890201.
Dhada KS, Hernandez DS, Suggs LJ. In vivo photoacoustic tracking of mesenchymal stem cell viability. ACS Nano. 2019;13:7791–9.
Zhang T, Wang Y, Li Q, Lin L, Xu C, Xue Y, et al. Mesenchymal stromal cells equipped by IFNalpha empower T cells with potent anti-tumor immunity. Oncogene. 2022;41:1866–81.
Li Q, Wang C, Cai L, Lu J, Zhu Z, Wang C, et al. miR‑34a derived from mesenchymal stem cells stimulates senescence in glioma cells by inducing DNA damage. Mol Med Rep. 2019;19:1849–57.
Jabbarpour Z, Kiani J, Keshtkar S, Saidijam M, Ghahremani MH, Ahmadbeigi N. Effects of human placenta-derived mesenchymal stem cells with NK4 gene expression on glioblastoma multiforme cell lines. J Cell Biochem. 2020;121:1362–73.
Hombach AA, Geumann U, Gunther C, Hermann FG, Abken H. IL7-IL12 engineered mesenchymal stem cells (MSCs) improve a CAR T cell attack against colorectal cancer cells. Cells. 2020;9:873.
Wang X, Zhao X, He Z. Mesenchymal stem cell carriers enhance anti-tumor efficacy of oncolytic virotherapy. Oncol Lett. 2021;21:238.
Cavarretta IT, Altanerova V, Matuskova M, Kucerova L, Culig Z, Altaner C. Adipose tissue-derived mesenchymal stem cells expressing prodrug-converting enzyme inhibit human prostate tumor growth. Mol Ther. 2010;18:223–31.
De Becker A, Riet IV. Homing and migration of mesenchymal stromal cells: How to improve the efficacy of cell therapy? World J Stem Cells. 2016;8:73–87.
Ponte AL, Marais E, Gallay N, Langonne A, Delorme B, Herault O, et al. The in vitro migration capacity of human bone marrow mesenchymal stem cells: comparison of chemokine and growth factor chemotactic activities. Stem Cells. 2007;25:1737–45.
Yun WS, Aryal S, Ahn YJ, Seo YJ, Key J. Engineered iron oxide nanoparticles to improve regenerative effects of mesenchymal stem cells. Biomed Eng Lett. 2020;10:259–73.
Fernandez-Garcia M, Yanez RM, Sanchez-Dominguez R, Hernando-Rodriguez M, Peces-Barba M, Herrera G, et al. Mesenchymal stromal cells enhance the engraftment of hematopoietic stem cells in an autologous mouse transplantation model. Stem Cell Res Ther. 2015;6:165.
Vianello F, Dazzi F. Mesenchymal stem cells for graft-versus-host disease: a double edged sword? Leukemia. 2008;22:463–5.
Christodoulou I, Goulielmaki M, Devetzi M, Panagiotidis M, Koliakos G, Zoumpourlis V. Mesenchymal stem cells in preclinical cancer cytotherapy: a systematic review. Stem Cell Res Ther. 2018;9:336.
Zhang J, Huang X, Wang H, Liu X, Zhang T, Wang Y, et al. The challenges and promises of allogeneic mesenchymal stem cells for use as a cell-based therapy. Stem Cell Res Ther. 2015;6:234.
Pérez-Simon JA, López-Villar O, Andreu EJ, Rifón J, Muntion S, Diez Campelo M, et al. Mesenchymal stem cells expanded in vitro with human serum for the treatment of acute and chronic graft-versus-host disease: results of a phase I/II clinical trial. Haematologica. 2011;96:1072–6.
Osman A, Afify SM, Hassan G, Fu X, Seno A, Seno M. Revisiting cancer stem cells as the origin of cancer-associated cells in the tumor microenvironment: a hypothetical view from the potential of iPSCs. Cancers (Basel). 2020;2:879.
Zhu Z, Zhu X, Yang S, Guo Z, Li K, Ren C, et al. Yin-yang effect of tumour cells in breast cancer: from mechanism of crosstalk between tumour-associated macrophages and cancer-associated adipocytes. Am J Cancer Res. 2020;10:383–92.
Duong MN, Geneste A, Fallone F, Li X, Dumontet C, Muller C. The fat and the bad: Mature adipocytes, key actors in tumor progression and resistance. Oncotarget. 2017;8:57622–41.
Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18:84.
Poltavets V, Kochetkova M, Pitson SM, Samuel MS. The role of the extracellular matrix and its molecular and cellular regulators in cancer cell plasticity. Front Oncol. 2018;8:431.
Xie H, Li L, Zhu G, Dang Q, Ma Z, He D, et al. Infiltrated pre-adipocytes increase prostate cancer metastasis via modulation of the miR-301a/androgen receptor (AR)/TGF-beta1/Smad/MMP9 signals. Oncotarget. 2015;6:12326–39.
Mukherjee A, Bilecz AJ, Lengyel E. The adipocyte microenvironment and cancer. Cancer Metastasis Rev. 2022;41:575–87.
Lyu X, Zhang Q, Fares HM, Wang Y, Han Y, Sun L. Contribution of adipocytes in the tumor microenvironment to breast cancer metabolism. Cancer Lett. 2022;534:215616.
Bougaret L, Delort L, Billard H, Le Huede C, Boby C, De la Foye A, et al. Adipocyte/breast cancer cell crosstalk in obesity interferes with the anti-proliferative efficacy of tamoxifen. PLoS ONE. 2018;13:e0191571.
Manabe Y, Toda S, Miyazaki K, Sugihara H. Mature adipocytes, but not preadipocytes, promote the growth of breast carcinoma cells in collagen gel matrix culture through cancer-stromal cell interactions. J Pathol. 2003;201:221–8.
Yao-Borengasser A, Monzavi-Karbassi B, Hedges RA, Rogers LJ, Kadlubar SA, Kieber-Emmons T. Adipocyte hypoxia promotes epithelial-mesenchymal transition-related gene expression and estrogen receptor-negative phenotype in breast cancer cells. Oncol Rep. 2015;33:2689–94.
Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011;71:2455–65.
Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498–503.
Mentoor I, Engelbrecht AM, van Jaarsveld PJ, Nell T. Chemoresistance: intricate interplay between breast tumor cells and adipocytes in the tumor microenvironment. Front Endocrinol (Lausanne). 2018;9:758.
Su F, Ahn S, Saha A, DiGiovanni J, Kolonin MG. Adipose stromal cell targeting suppresses prostate cancer epithelial-mesenchymal transition and chemoresistance. Oncogene. 2019;38:1979–88.
Zhang Q, Deng T, Zhang H, Zuo D, Zhu Q, Bai M, et al. Adipocyte-derived exosomal MTTP suppresses ferroptosis and promotes chemoresistance in colorectal cancer. Adv Sci (Weinh). 2022;9:e2203357.
Zhou X, Zhang J, Lv W, Zhao C, Xia Y, Wu Y, et al. The pleiotropic roles of adipocyte secretome in remodeling breast cancer. J Exp Clin Cancer Res. 2022;41:203.
D’Esposito V, Liguoro D, Ambrosio MR, Collina F, Cantile M, Spinelli R, et al. Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget. 2016;7:24495–509.
Li K, Wei L, Huang Y, Wu Y, Su M, Pang X, et al. Leptin promotes breast cancer cell migration and invasion via IL-18 expression and secretion. Int J Oncol. 2016;48:2479–87.
Sakurai M, Miki Y, Takagi K, Suzuki T, Ishida T, Ohuchi N, et al. Interaction with adipocyte stromal cells induces breast cancer malignancy via S100A7 upregulation in breast cancer microenvironment. Breast Cancer Res. 2017;19:70.
Park J, Scherer PE. Adipocyte-derived endotrophin promotes malignant tumor progression. J Clin Invest. 2012;122:4243–56.
Al-Khalaf HH, Al-Harbi B, Al-Sayed A, Arafah M, Tulbah A, Jarman A, et al. Interleukin-8 activates breast cancer-associated adipocytes and promotes their angiogenesis- and tumorigenesis-promoting effects. Mol Cell Biol. 2019;39:e00332–18.
Vazquez Rodriguez G, Abrahamsson A, Jensen LDE, Dabrosin C. Adipocytes promote early steps of breast cancer cell dissemination via interleukin-8. Front Immunol. 2018;9:1767.
Dykes SS, Hughes VS, Wiggins JM, Fasanya HO, Tanaka M, Siemann D. Stromal cells in breast cancer as a potential therapeutic target. Oncotarget. 2018;9:23761–79.
Campo-Verde-Arbocco F, Lopez-Laur JD, Romeo LR, Giorlando N, Bruna FA, Contador DE, et al. Human renal adipose tissue induces the invasion and progression of renal cell carcinoma. Oncotarget. 2017;8:94223–34.
Senga S, Kobayashi N, Kawaguchi K, Ando A, Fujii H. Fatty acid-binding protein 5 (FABP5) promotes lipolysis of lipid droplets, de novo fatty acid (FA) synthesis and activation of nuclear factor-kappa B (NF-kappaB) signaling in cancer cells. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1863:1057–67.
Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC, et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017;5:1.
Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis. 2016;5:e189.
Correa LH, Correa R, Farinasso CM, de Sant’Ana Dourado LP, Magalhaes KG. Adipocytes and macrophages interplay in the orchestration of tumor microenvironment: new implications in cancer progression. Front Immunol. 2017;8:1129.
Habanjar O, Diab-Assaf M, Caldefie-Chezet F, Delort L. The impact of obesity, adipose tissue, and tumor microenvironment on macrophage polarization and metastasis. Biology (Basel). 2022;11:339.
Li B, Liu S, Yang Q, Li Z, Li J, Wu J, et al. Macrophages in tumor-associated adipose microenvironment accelerate tumor progression. Adv Biol (Weinh). 2023;7:e2200161.
Wen D, Liang T, Chen G, Li H, Wang Z, Wang J, et al. Adipocytes encapsulating telratolimod recruit and polarize tumor-associated macrophages for cancer immunotherapy. Adv Sci (Weinh). 2022;10:e2206001.
Merino M, Briones L, Palma V, Herlitz K, Escudero C. Role of adenosine receptors in the adipocyte-macrophage interaction during obesity. Endocrinol Diabetes Nutr. 2017;64:317–27.
Zhu Q, Zhu Y, Hepler C, Zhang Q, Park J, Gliniak C, et al. Adipocyte mesenchymal transition contributes to mammary tumor progression. Cell Rep. 2022;40:111362.
Hamabe-Horiike T, Harada SI, Yoshida K, Kinoshita J, Yamaguchi T, Fushida S. Adipocytes contribute to tumor progression and invasion of peritoneal metastasis by interacting with gastric cancer cells as cancer associated fibroblasts. Cancer Rep (Hoboken). 2023;6:e1647.
Zhang Y, Daquinag AC, Amaya-Manzanares F, Sirin O, Tseng C, Kolonin MG. Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Res. 2012;72:5198–208.
Otley MOC, Sinal CJ. Adipocyte-cancer cell interactions in the bone microenvironment. Front Endocrinol (Lausanne). 2022;13:903925.
He JY, Wei XH, Li SJ, Liu Y, Hu HL, Li ZZ, et al. Adipocyte-derived IL-6 and leptin promote breast Cancer metastasis via upregulation of Lysyl Hydroxylase-2 expression. Cell Commun Signal. 2018;16:100.
Zhou C, He X, Tong C, Li H, Xie C, Wu Y, et al. Cancer-associated adipocytes promote the invasion and metastasis in breast cancer through LIF/CXCLs positive feedback loop. Int J Biol Sci. 2022;18:1363–80.
Tebbe C, Chhina J, Dar SA, Sarigiannis K, Giri S, Munkarah AR, et al. Metformin limits the adipocyte tumor-promoting effect on ovarian cancer. Oncotarget. 2014;5:4746–64.
Teufelsbauer M, Rath B, Plangger A, Staud C, Nanobashvili J, Huk I, et al. Effects of metformin on adipose-derived stromal cell (ADSC)—breast cancer cell lines interaction. Life Sci. 2020;261:118371.
Park J, Euhus DM, Scherer PE. Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocr Rev. 2011;32:550–70.
Lee J, Imm JY, Lee SH. beta-catenin mediates anti-adipogenic and anticancer effects of arctigenin in preadipocytes and breast cancer cells. J Agric Food Chem. 2017;65:2513–20.
Otvos L Jr., Kovalszky I, Scolaro L, Sztodola A, Olah J, Cassone M, et al. Peptide-based leptin receptor antagonists for cancer treatment and appetite regulation. Biopolymers. 2011;96:117–25.
Zhao L, Guo Y, Guo Y, Ji X, Fan D, Chen C, et al. Effect and mechanism of circRNAs in tumor angiogenesis and clinical application. Int J Cancer. 2022;150:1223–32.
Choi H, Moon A. Crosstalk between cancer cells and endothelial cells: implications for tumor progression and intervention. Arch Pharm Res. 2018;41:711–24.
Ciesielski O, Biesiekierska M, Panthu B, Vialichka V, Pirola L, Balcerczyk A. The epigenetic profile of tumor endothelial cells. effects of combined therapy with antiangiogenic and epigenetic drugs on cancer progression. Int J Mol Sci. 2020;21:2606.
Hida K, Maishi N, Annan DA, Hida Y. Contribution of tumor endothelial cells in cancer progression. Int J Mol Sci. 2018;19:1272.
Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–5.
Elebiyo TC, Rotimi D, Evbuomwan IO, Maimako RF, Iyobhebhe M, Ojo OA, et al. Reassessing vascular endothelial growth factor (VEGF) in anti-angiogenic cancer therapy. Cancer Treat Res Commun. 2022;32:100620.
Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994;265:1582–4.
Movahed ZG, Yarani R, Mohammadi P, Mansouri K. Sustained oxidative stress instigates differentiation of cancer stem cells into tumor endothelial cells: Pentose phosphate pathway, reactive oxygen species and autophagy crosstalk. Biomed Pharmacother. 2021;139:111643.
Garcia-Caballero M, Sokol L, Cuypers A, Carmeliet P. Metabolic reprogramming in tumor endothelial cells. Int J Mol Sci. 2022;23:11052.
Giordo R, Wehbe Z, Paliogiannis P, Eid AH, Mangoni AA, Pintus G. Nano-targeting vascular remodeling in cancer: Recent developments and future directions. Semin Cancer Biol. 2022;86:784–804.
Hegde M, Bhat SM, Guruprasad KP, Moka R, Ramachandra L, Satyamoorthy K, et al. Human breast tumor derived endothelial cells exhibit distinct biological properties. Biol Cell. 2022;114:73–85.
Fonsato V, Buttiglieri S, Deregibus MC, Puntorieri V, Bussolati B, Camussi G. Expression of Pax2 in human renal tumor-derived endothelial cells sustains apoptosis resistance and angiogenesis. Am J Pathol. 2006;168:706–13.
Ohmura-Kakutani H, Akiyama K, Maishi N, Ohga N, Hida Y, Kawamoto T, et al. Identification of tumor endothelial cells with high aldehyde dehydrogenase activity and a highly angiogenic phenotype. PLoS ONE. 2014;9:e113910.
Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, et al. Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell. 2016;30:968–85.
Rohlenova K, Veys K, Miranda-Santos I, De Bock K, Carmeliet P. Endothelial cell metabolism in health and disease. Trends Cell Biol. 2018;28:224–36.
Li X, Sun X, Carmeliet P. Hallmarks of endothelial cell metabolism in health and disease. Cell Metab. 2019;30:414–33.
Yasui H, Hideshima T, Richardson PG, Anderson KC. Novel therapeutic strategies targeting growth factor signalling cascades in multiple myeloma. Br J Haematol. 2006;132:385–97.
Zhao Z, Gao J, Li C, Xu X, Hu Y, Huang S. Reactive oxygen species induce endothelial differentiation of liver cancer stem-like sphere cells through the activation of Akt/IKK signaling pathway. Oxid Med Cell Longev. 2020;2020:1621687.
Cui G, Liu H, Laugsand JB. Endothelial cells-directed angiogenesis in colorectal cancer: Interleukin as the mediator and pharmacological target. Int Immunopharmacol. 2023;114:109525.
Lacal PM, Graziani G. Therapeutic implication of vascular endothelial growth factor receptor-1 (VEGFR-1) targeting in cancer cells and tumor microenvironment by competitive and non-competitive inhibitors. Pharm Res. 2018;136:97–107.
Zhou J, Zhang A, Fan L. HSPA12B secreted by tumor-associated endothelial cells might induce M2 polarization of macrophages via activating PI3K/Akt/mTOR signaling. Onco Targets Ther. 2020;13:9103–11.
Njock MS, O’Grady T, Nivelles O, Lion M, Jacques S, Cambier M, et al. Endothelial extracellular vesicles promote tumour growth by tumour-associated macrophage reprogramming. J Extracell Vesicles. 2022;11:e12228.
Yang D, Guo P, He T, Powell CA. Role of endothelial cells in tumor microenvironment. Clin Transl Med. 2021;11:e450.
Perurena N, Zandueta C, Martinez-Canarias S, Moreno H, Vicent S, Almeida AS, et al. EPCR promotes breast cancer progression by altering SPOCK1/testican 1-mediated 3D growth. J Hematol Oncol. 2017;10:23.
Bussolati B, Deambrosis I, Russo S, Deregibus MC, Camussi G. Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J. 2003;17:1159–61.
Xiong YQ, Sun HC, Zhang W, Zhu XD, Zhuang PY, Zhang JB, et al. Human hepatocellular carcinoma tumor-derived endothelial cells manifest increased angiogenesis capability and drug resistance compared with normal endothelial cells. Clin Cancer Res. 2009;15:4838–46.
Kikuchi H, Maishi N, Annan DA, Alam MT, Dawood RIH, Sato M, et al. Chemotherapy-induced IL8 upregulates MDR1/ABCB1 in tumor blood vessels and results in unfavorable outcome. Cancer Res. 2020;80:2996–3008.
Torii C, Maishi N, Kawamoto T, Morimoto M, Akiyama K, Yoshioka Y, et al. miRNA-1246 in extracellular vesicles secreted from metastatic tumor induces drug resistance in tumor endothelial cells. Sci Rep. 2021;11:13502.
Kim ID, Cave JW, Cho S. Aflibercept, a VEGF (Vascular Endothelial Growth Factor)-trap, reduces vascular permeability and stroke-induced brain swelling in obese mice. Stroke 2021;52:2637–48.
Yang FHY. [Recombinant human endostatin induces apoptosis of human umbilical venous endothelial cells]. Nan Fang Yi Ke Da Xue Xue Bao. 2006;26:978–80.
Ishihara S, Onoda N, Noda S, Asano Y, Tauchi Y, Morisaki T, et al. Sorafenib inhibits vascular endothelial cell proliferation stimulated by anaplastic thyroid cancer cells regardless of BRAF mutation status. Int J Oncol. 2019;55:1069–76.
Panigrahy D, Kaipainen A, Huang S, Butterfield CE, Barnes CM, Fannon M, et al. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc Natl Acad Sci USA. 2008;105:985–90.
Cheng HS, Lee JXT, Wahli W, Tan NS. Exploiting vulnerabilities of cancer by targeting nuclear receptors of stromal cells in tumor microenvironment. Mol Cancer. 2019;18:51.
Pequeux C, Raymond-Letron I, Blacher S, Boudou F, Adlanmerini M, Fouque MJ, et al. Stromal estrogen receptor-alpha promotes tumor growth by normalizing an increased angiogenesis. Cancer Res. 2012;72:3010–9.
Bolik J, Krause F, Stevanovic M, Gandrass M, Thomsen I, Schacht SS, et al. Inhibition of ADAM17 impairs endothelial cell necroptosis and blocks metastasis. J Exp Med. 2022;219:e20201039.
Li F, Liang H, You H, Xiao J, Xia H, Chen X, et al. Targeting HECTD3-IKKalpha axis inhibits inflammation-related metastasis. Signal Transduct Target Ther. 2022;7:264.
Funahashi Y, Hernandez SL, Das I, Ahn A, Huang J, Vorontchikhina M, et al. A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res. 2008;68:4727–35.
Patenaude A, Fuller M, Chang L, Wong F, Paliouras G, Shaw R, et al. Endothelial-specific Notch blockade inhibits vascular function and tumor growth through an eNOS-dependent mechanism. Cancer Res. 2014;74:2402–11.
Quan L, Ohgaki R, Hara S, Okuda S, Wei L, Okanishi H, et al. Amino acid transporter LAT1 in tumor-associated vascular endothelium promotes angiogenesis by regulating cell proliferation and VEGF-A-dependent mTORC1 activation. J Exp Clin Cancer Res. 2020;39:266.
Zhang L, Li S, Li L, Chen Z, Yang Y. COX‑2 inhibition in the endothelium induces glucose metabolism normalization and impairs tumor progression. Mol Med Rep. 2018;17:2937–44.
Bruning U, Morales-Rodriguez F, Kalucka J, Goveia J, Taverna F, Queiroz KCS, et al. Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab. 2018;28:866–80.e15.
Sun R, Kong X, Qiu X, Huang C, Wong PP. The emerging roles of pericytes in modulating tumor microenvironment. Front Cell Dev Biol. 2021;9:676342.
Cathery W, Faulkner A, Maselli D, Madeddu P. Concise review: the regenerative journey of pericytes toward clinical translation. Stem Cells. 2018;36:1295–310.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582–98.
Guerra DAP, Paiva AE, Sena IFG, Azevedo PO, Silva WN, Mintz A, et al. Targeting glioblastoma-derived pericytes improves chemotherapeutic outcome. Angiogenesis 2018;21:667–75.
Sato S, Tang YJ, Wei Q, Hirata M, Weng A, Han I, et al. Mesenchymal tumors can Derive from Ng2/Cspg4-expressing pericytes with beta-catenin modulating the neoplastic phenotype. Cell Rep. 2016;16:917–27.
Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215.
Garza Trevino EN, Gonzalez PD, Valencia Salgado CI, Martinez Garza A. Effects of pericytes and colon cancer stem cells in the tumor microenvironment. Cancer Cell Int. 2019;19:173.
Murgai M, Ju W, Eason M, Kline J, Beury DW, Kaczanowska S, et al. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat Med. 2017;23:1176–90.
Paiva AE, Lousado L, Guerra DAP, Azevedo PO, Sena IFG, Andreotti JP, et al. Pericytes in the premetastatic niche. Cancer Res. 2018;78:2779–86.
Wong PP, Munoz-Felix JM, Hijazi M, Kim H, Robinson SD, De Luxan-Delgado B, et al. Cancer burden is controlled by mural cell-beta3-integrin regulated crosstalk with tumor cells. Cell. 2020;181:1346–63.e21.
Figueiredo AM, Villacampa P, Dieguez-Hurtado R, Jose Lozano J, Kobialka P, Cortazar AR, et al. Phosphoinositide 3-kinase-regulated pericyte maturation governs vascular remodeling. Circulation. 2020;142:688–704.
Meng YM, Jiang X, Zhao X, Meng Q, Wu S, Chen Y, et al. Hexokinase 2-driven glycolysis in pericytes activates their contractility leading to tumor blood vessel abnormalities. Nat Commun. 2021;12:6011.
Zhang XN, Yang KD, Chen C, He ZC, Wang QH, Feng H, et al. Pericytes augment glioblastoma cell resistance to temozolomide through CCL5-CCR5 paracrine signaling. Cell Res. 2021;31:1072–87.
Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Bruhl ML, et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and ‘instruct’ them with pattern-recognition and motility programs. Nat Immunol. 2013;14:41–51.
Wang Y, Sun Q, Ye Y, Sun X, Xie S, Zhan Y, et al. FGF-2 signaling in nasopharyngeal carcinoma modulates pericyte-macrophage crosstalk and metastasis. JCI Insight. 2022;7:e157874.
Hong J, Tobin NP, Rundqvist H, Li T, Lavergne M, Garcia-Ibanez Y, et al. Role of tumor pericytes in the recruitment of myeloid-derived suppressor cells. J Natl Cancer Inst. 2015;107:djv209.
Valdor R, Garcia-Bernal D, Riquelme D, Martinez CM, Moraleda JM, Cuervo AM, et al. Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc Natl Acad Sci USA. 2019;116:20655–65.
Valdor R, Garcia-Bernal D, Bueno C, Rodenas M, Moraleda JM, Macian F, et al. Glioblastoma progression is assisted by induction of immunosuppressive function of pericytes through interaction with tumor cells. Oncotarget. 2017;8:68614–26.
Navarro R, Compte M, Alvarez-Vallina L, Sanz L. Immune regulation by pericytes: modulating innate and adaptive immunity. Front Immunol. 2016;7:480.
Ning X, Zhang H, Wang C, Song X. Exosomes released by gastric cancer cells induce transition of pericytes into cancer-associated fibroblasts. Med Sci Monit. 2018;24:2350–9.
Fedele M, Cerchia L, Chiappetta G. The epithelial-to-mesenchymal transition in breast cancer: focus on basal-like carcinomas. Cancers (Basel). 2017;9:134.
Navarro R, Tapia-Galisteo A, Martin-Garcia L, Tarin C, Corbacho C, Gomez-Lopez G, et al. TGF-beta-induced IGFBP-3 is a key paracrine factor from activated pericytes that promotes colorectal cancer cell migration and invasion. Mol Oncol. 2020;14:2609–28.
Zhou W, Chen C, Shi Y, Wu Q, Gimple RC, Fang X, et al. Targeting glioma stem cell-derived pericytes disrupts the blood-tumor barrier and improves chemotherapeutic efficacy. Cell Stem Cell. 2017;21:591–603.
Franco M, Roswall P, Cortez E, Hanahan D, Pietras K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood. 2011;118:2906–17.
Prete A, Lo AS, Sadow PM, Bhasin SS, Antonello ZA, Vodopivec DM, et al. Pericytes elicit resistance to vemurafenib and sorafenib therapy in thyroid carcinoma via the TSP-1/TGFbeta1 axis. Clin Cancer Res. 2018;24:6078–97.
DeWaal D, Nogueira V, Terry AR, Patra KC, Jeon SM, Guzman G, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. 2018;9:446.
Fan T, Sun G, Sun X, Zhao L, Zhong R, Peng Y. Tumor energy metabolism and potential of 3-bromopyruvate as an inhibitor of aerobic glycolysis: implications in tumor treatment. Cancers (Basel). 2019;11:317.
Molnar K, Meszaros A, Fazakas C, Kozma M, Gyori F, Reisz Z, et al. Pericyte-secreted IGF2 promotes breast cancer brain metastasis formation. Mol Oncol. 2020;14:2040–57.
Chen Z, Xu XH, Hu J. Role of pericytes in angiogenesis: focus on cancer angiogenesis and anti-angiogenic therapy. Neoplasma. 2016;63:173–82.
Keskin D, Kim J, Cooke VG, Wu CC, Sugimoto H, Gu C, et al. Targeting vascular pericytes in hypoxic tumors increases lung metastasis via angiopoietin-2. Cell Rep. 2015;10:1066–81.
Mezheyeuski A, Bradic Lindh M, Guren TK, Dragomir A, Pfeiffer P, Kure EH, et al. Survival-associated heterogeneity of marker-defined perivascular cells in colorectal cancer. Oncotarget 2016;7:41948–58.
Cooke VG, LeBleu VS, Keskin D, Khan Z, O’Connell JT, Teng Y, et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell. 2012;21:66–81.
Xing Y, Zhao S, Zhou BP, Mi J. Metabolic reprogramming of the tumour microenvironment. FEBS J. 2015;282:3892–8.
Funding
This work was supported by Jilin University Bethune Program (No. 2023B16) and the “Medicine + X” cross-innovation team of Bethune Medical Department of Jilin University “Leading the Charge with Open Competition” construction project (No. 2022JBGS04).
Author information
Authors and Affiliations
Contributions
YB and YL offered the main direction and significant guidance of this manuscript. YZ and MS drafted the manuscript. LW, HY, YY revised the manuscript., QY, JD, LL illustrated the figures. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Hans-Uwe Simon
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, Y., Shen, M., Wu, L. et al. Stromal cells in the tumor microenvironment: accomplices of tumor progression?. Cell Death Dis 14, 587 (2023). https://doi.org/10.1038/s41419-023-06110-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-023-06110-6
This article is cited by
-
Cancer-associated fibroblasts drive colorectal cancer cell progression through exosomal miR-20a-5p-mediated targeting of PTEN and stimulating interleukin-6 production
BMC Cancer (2024)
-
Single-cell aggrephagy-related patterns facilitate tumor microenvironment intercellular communication, influencing osteosarcoma progression and prognosis
Apoptosis (2024)
-
RNA modification regulator DDC in endometrial cancer affects the tumor microenvironment and patient prognosis
Scientific Reports (2023)
