
Abstract
Prostate cancer is a hormone-dependent malignancy, whose onset and progression are closely related to the activity of the androgen receptor (AR) signaling pathway. Due to this critical role of AR signaling in driving prostate cancer, therapy targeting the AR pathway has been the mainstay strategy for metastatic prostate cancer treatment. The utility of these agents has expanded with the emergence of second-generation AR antagonists, which began with the approval of enzalutamide in 2012 by the United States Food and Drug Administration (FDA). Together with apalutamide and darolutamide, which were approved in 2018 and 2019, respectively, these agents have improved the survival of patients with prostate cancer, with applications for both androgen-dependent and castration-resistant disease. While patients receiving these drugs receive a benefit in the form of prolonged survival, they are not cured and ultimately progress to lethal neuroendocrine prostate cancer (NEPC). Here we summarize the current state of AR antagonist development and highlight the emerging challenges of their clinical application and the potential resistance mechanisms, which might be addressed by combination therapies or the development of novel AR-targeted therapies.
Similar content being viewed by others
Facts
-
Second-generation AR antagonists including enzalutamide, darolutamide, and apalutamide for prostate cancer treatment increase patient survival.
-
Second-generation AR antagonists only provide a temporary response and resistance eventually develops.
-
Diverse mechanisms were reported regarding the resistance to second-generation AR antagonists.
Open questions
-
What is the mechanism of treatment-induced NEPC (t-NEPC) and what is the connection between t-NEPC and second-generation AR antagonists?
-
How to address the second-generation AR antagonist-induced resistance?
-
What is the progress of alternative AR-targeted therapy?
Introduction
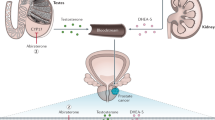
The incidence of prostate cancer ranks second among men worldwide and represents one of the leading causes of cancer death, with an estimated about 1.4 million new cases and 375,000 deaths worldwide in 2020 [1]. Prostate cancer onset and progression are closely correlated with the androgen receptor (AR) activity [2, 3] The activation of AR is mediated by androgens, whose synthesis is regulated by the hypothalamic–pituitary–testicular (HPT) axis [4]. As a result of the indispensable role of AR in prostate cancer, a number of anti-AR drugs have been developed and approved for different stages of prostate cancer in the past 30 years (Table 1). The first-generation AR antagonists included flutamide [5, 6], nilutamide [7, 8], and bicalutamide [9, 10], which were approved by the FDA in 1989, 1995, and 1996, respectively. While the patients respond to first-generation AR antagonists in the early stages of the disease, they eventually acquire resistance and progress to lethal stage castration-resistant prostate cancer (CRPC) [11]. Accumulating data indicate that restoration of AR signaling is critical for disease progression in these patients, as AR overexpression, especially due to AR genomic amplification, has been frequently observed and proven to be a principal driver of prostate cancer progression, both in clinical CRPC patients and in preclinical prostate cancer cell models [12,13,14,15,16]. The continued importance of the AR pathway in CRPC has encouraged researchers and clinicians to develop a second generation of AR antagonists with higher AR binding affinity and specificity to target aberrant AR signaling in lethal stage CRPC patients. Patient survival has indeed increased with the application of second-generation AR antagonists, which have higher AR binding affinity and inhibit AR more efficiently [17,18,19,20,21,22]. On the other hand, these agents have only provided a temporary response, due to the rapid development of resistance [23,24,25]. This review will discuss the development of AR antagonists, the limitations of current AR antagonists, and the mechanisms of resistance to these agents, and will outline emerging strategies to combat resistance and prolong patient survival.
Development of second-generation AR antagonists
FDA approved second-generation AR antagonists
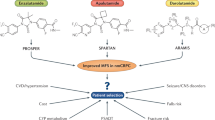
Enzalutamide (also named MDV3100) is the first FDA-approved second-generation AR antagonist for the treatment of CRPC and exhibits a much higher AR-binding affinity in comparison to the first-generation AR antagonists. It competitively binds to the ligand-binding domain (LBD) of AR and inhibits androgen binding, nuclear translocation, DNA binding, and co-activator recruitment [26, 27]. Enzalutamide significantly prolongs the overall survival and metastatic-free survival of CRPC patients [17, 28, 29], and was approved by the United States FDA for treatment of metastatic CRPC (mCRPC) and non-metastatic CRPC (nmCRPC) in 2012 and 2018, respectively. Enzalutamide was also found to markedly prolong the castration-resistant free survival time of patients with castration-sensitive prostate cancer (CSPC) [18], and was approved for the treatment of CSPC in 2019. Although enzalutamide is widely used in clinical treatment for both CSPC and CRPC, the high steady-state brain level of enzalutamide has been found in clinical practice to be associated with central nervous system (CNS)-related events such as seizure, as it can antagonize the GABAα receptor in the CNS [29,30,31,32]. Another AR antagonist with a lower steady-state brain level subsequently emerged in the form of apalutamide (ARN-509), which shares the same core structure with enzalutamide (Fig. 1), but is associated with fewer seizure side effects [33]. Apalutamide is similarly considered to be a full AR antagonist, as it has high binding affinity with the LBD of AR [33]. Apalutamide can significantly increase the metastasis-free survival of nonmetastatic CRPC as well as the overall survival of metastatic CSPC [20, 34, 35], and was approved for nmCRPC in 2018 and for mCRPC/mCSPC in 2019. Both enzalutamide and apalutamide function as AR antagonists by inhibiting multiple stages of AR-mediated transcription, including by competing with DHT for AR binding, blocking AR nuclear translocation, and blocking DNA binding and cofactor recruitment [26, 33]. In contrast to enzalutamide and apalutamide, the most recently approved second-generation AR antagonist darolutamide (ODM-201) has a different chemical structure (Fig. 1) and cannot cross the brain–blood barrier [36], suggesting a lower potential for CNS side effects. Clinical trials have indicated that darolutamide provides not only meaningful antitumor effects but also a favorable safety profile [37, 38]. A randomized, double-blind, placebo-controlled, phase 3 trial involving men with nmCRPC has demonstrated significantly longer survival with darolutamide (40.4 months) than with placebo (18.4 months) [39]. A randomized, double-blind, placebo-controlled phase 3 study of darolutamide plus ADT versus placebo plus ADT in men is ongoing to assess the efficacy and safety of darolutamide in combination with standard ADT in metastatic hormone sensitive prostate cancer (mHSPC) patients (NCT04736199) (Fig. 2). Additional in vitro data have shown that darolutamide exhibits a consistent ability to efficiently inhibit full-length AR harboring a number of different characterized gain-of-function mutations [40]. Taken together, enzalutamide, apalutamide, and darolutamide are considered “pure” AR antagonists with the ability to suppress AR activation in CSPC, mCRPC, and nmCRPC patients [41]. Several concerns still persist, such as enzalutamide-induced seizures in CNS due to its high levels in brain, and the shorter serum half-life of darolutamide that has required higher doses and more frequent administration, leading to toxic effects such as cardiovascular disease [42, 43]. More importantly, prolonged use of AR antagonists induces drug resistance that rapidly attenuates their clinical benefits, motivating scientists to further explore new AR antagonists and alternative therapeutic strategies.

The red dotted box indicates the shared structure between drugs. Drug structure resources from PubChem (https://pubchem.ncbi.nlm.nih.gov/search/search.cgi).

A All of the FDA-approved second-generation AR antagonists bind to the ligand-binding domain (LBD). Potential AR antagonists including proxalutamide (Prox), TRC253, BMS-641988, and SHR3680 bind to the LBD, while EPI-506 and EPI-7386 bind to the N-terminal domain (NTD). Among these AR antagonists, EPI-506 and EPI-7386, darolutamide, proxalutamide, and TRC253 can bind with AR harboring mutations such as F876L. B All of the AR antagonists that bind to the LBD can competitively inhibit DHT binding to AR, as well as AR nuclear translocation and binding to DNA and coactivators. Binding of EPI-506 and EPI-7386 to the NTD of AR can inhibit AR transcriptional activation. Of note, proxalutamide can also repress AR protein expression.
Emerging AR antagonists in clinical trials
Several clinical trials are currently underway investigating novel AR antagonists with the potential to conquer the shortcomings of the present AR antagonists (Table 1). Proxalutamide (GT-0918) is a full AR antagonist which has 3-fold higher binding affinity in comparison with enzalutamide [44, 45]. It can also down-regulate the protein level of AR in CRPC. More importantly, luciferase reporter assays have shown that proxalutamide has the same effect on wild-type AR and on AR with clinically observed mutations (including F877L, W747C, and H875Y) that confer resistance to 1st or 2nd generation AR antagonists [44]. A phase I dose-escalation study of proxalutamide to evaluate its safety, pharmacokinetics, and antitumor efficacy in 16 patients with CRPC has shown a high degree of tolerance and promising antitumor activity in CRPC [44]. An open-label, randomized, expanded/phase II study is currently recruiting subjects with mCRPC who have progressed after either abiraterone or enzalutamide treatment in order to evaluate the safety and tolerability of proxalutamide and determine the dose level for phase III and/or other confirming studies (NCT03899467). BMS-641988 is another promising AR antagonist which was first reported by Salcati’s laboratory [46, 47]. BMS-641988 has comparable AR-binding affinity with proxalutamide and has shown an antitumor effect superior to bicalutamide in CWR22-BMSLD1 and LNCaP tumor xenograft models [46, 47]. In a phase I clinical trial to define the safety and tolerability of oral BMS-641988 in patients with CRPC, the therapeutic dose of BMS-641988 exhibited promising anti-tumor activity but was associated with an episode of seizure activity that led to study closure [48]. Although BMS-641988 did not progress further in clinical trials, it may be possible to design novel AR antagonists based on its core structure that have a reduced ability to concentrate in the brain, similar to the relationship between enzalutamide and apalutamide. While enzalutamide and apalutamide share the same core structure, the seizure side effect associated with enzalutamide is dramatically decreased with apalutamide because of its shorter half-life and lower steady-state level in the brain. Another novel AR antagonist, SHR3680, preclinically has shown anti-tumor potency comparable to enzalutamide but with a reduced distribution in the brain and significantly decreased risk to induce a seizure. A phase 1/2 study of patients with progressive mCRPC has shown that SHR3680 is well tolerated and safe, with promising anti-tumor activity across all doses tested in patients [49].
Agents that target regions of AR other than the LBD are in development as well, with the potential to counteract constitutively activating AR splice variants and AR point mutations. EPI-7386, for instance, is a second-generation NTD inhibitor (aniten) that is more active and more metabolically stable than EPI-506 (EPI-002 pro-drug) and has demonstrated a 20-fold improvement in AR-driven cellular potency compared to EPI-002 [50]. EPI-7386 inhibits cell proliferation across a panel of prostate cancer cell lines, including those driven by the AR variant AR-V7, can control tumor growth and induce tumor regression in several CRPC xenograft models, and is well tolerated in animal models [50, 51]. A phase 1 dose-escalation clinical trial of EPI-7386 in mCRPC patients is underway to assess its safety and to find a dose that can be given without unacceptable side effects (NCT04421222), as well as a phase 1/2 clinical trial of EPI-7386 in combination with enzalutamide in patients with mCRPC progressing on the standard of care therapies including second-generation anti-androgens (NCT05075577). TRC-253 is another novel AR antagonist that functions as a high-affinity competitive inhibitor of both wild-type AR and AR harboring mutations within the LBD [52]. A multi-center, first-in-human, open-label, Phase 1/2A dose-escalation study conducted in eligible mCRPC patients has indicated that high doses of TRC-253 are associated with some adverse events such as anemia (NCT02987829). It is not yet clear whether combination therapy of low dose of TRC-253 with other androgen inhibitors such as abiraterone can lower its toxicity and improve the anti-tumor effect. A more recent study has developed several AR antagonists based on the structure of darolutamide, from which “compound 28t” has been found to show superior efficacy against two resistant mutants (AR-F876L and AR-T877A) relative to darolutamide [53]. Further clinical trials are needed to assess its safety, pharmacokinetics, and efficiency. Additional candidates are anticipated as the next generation of AR antagonists emergent and enter clinical trials.
Challenges of second-generation AR antagonists
Off-target effects of AR antagonists
Although second-generation AR antagonists have become mainstays for the treatment of both CSPC and CRPC patients, their clinical benefits have been limited by potential side effects and especially by induced drug resistance. As mentioned above, the long half-lives and high levels of enzalutamide and BMS-641988 in the CNS may induce seizures in a small proportion of prostate cancer patients, as these AR antagonists can competitively bind and inhibit GABA-α activity [18, 32, 48, 54, 55]. A retrospective observational study has reported falls as a CNS-related event in patients with metastatic prostate cancer receiving enzalutamide (4.6%) [31]. Additionally, the clinical application of second-generation AR antagonists may increase the risk of cardiovascular events [56, 57]. A recent meta-analysis involving 4110 nmCRPC patients treated with enzalutamide, darolutamide, or apalutamide has indicated that the application of second-generation AR antagonists is associated with significantly increased risk of cardiovascular events including stroke, heart failure, and peripheral vascular disease [42]. This is consistent with previous enzalutamide studies, especially among prostate cancer patients with pre-existing cardiovascular disease, for whom enzalutamide may increase the risk of hypertension, likely driven by mineralocorticoid excess [58,59,60,61]. At the same time, the incidence of cardiovascular disease associated with second-generation AR antagonists is significantly decreased in comparison with other AR inhibitors such as abiraterone (CYP17 inhibitor) [62].
AR antagonists-induced drug resistance and cancer evolution
Overall, administration of second-generation AR antagonists to patients in different stages of disease has resulted in a moderate survival benefit. However, several studies have indicated that about 30–60% of patients who receive second-generation AR antagonists eventually progress to death [17, 18, 20, 28, 29, 34, 39]. A proportion of these patients are primarily resistant to the treatment, which may be caused by AR heterogeneity in prostate cancer [63, 64] and other alterations in enzymes crucial for the conversion of extragonadal precursors to potent androgens, such as the 3βHSD1 germline variant [4]. In addition to de novo resistance, patients who receive second-generation AR antagonists inevitably develop acquired resistance within a variable period of time [65], which represents the greatest challenge of AR antagonists in prostate cancer treatment. Treatment-induced lethal NEPC (t-NEPC) progression is increased by the application of AR antagonists, especially in patients who have undergone enzalutamide treatment. De novo NEPC accounts for <2% of all prostate cancer at the time of diagnosis [66, 67], but the incidence of NEPC has significantly increased with the clinical application of AR inhibitors [68,69,70,71]. The current incidence of NEPC accounts for 18–20% of patients with CRPC, coinciding with the widespread clinical use of AR antagonists [68, 71].
Emerging mechanisms of resistance to AR antagonists
AR alterations and dysregulation in CRPC
Genomic analyses have indicated that 15–20% of CRPC patients harbor AR mutations [72, 73]. Collectively, over 150 mutations have been reported by the Androgen Receptor Gene Mutations Database within the LBD domain of AR in the context of prostate cancer, including single point mutations, pre-termination, deletions, and insertions. L702H, T878A, H875Y, W742C, and W743L are the most prevalent mutations reported in clinical prostate cancer patients (Fig. 3) [74, 75]. These point mutations in the LBD may result in lower AR antagonist binding affinity or even conversion of the AR antagonist into an agonist. Studies have demonstrated that multiple point mutations confer resistance to enzalutamide and apalutamide, including A587V, F876L, F877L, G684A, K631T, L595M, Q920R, R630Q, T576A, and T878A [76, 77]. F876L in particular triggers an antagonist-to-agonist switch that drives phenotypic resistance to enzalutamide [76]. Additionally, enzalutamide can act as a weak partial agonist in CRPC patients who harbor F877L, and becomes a strong agonist in patients harboring both F877L and T878A mutations [78]. Such mutations do not appear to be prevalent among clinical prostate cancer patients, and although anti-androgen withdrawal syndrome (AAWS) frequently occurs after discontinuation of first-generation anti-androgen therapy, but it is rarely observed in enzalutamide treated patients [79,80,81].

Mutations in red are the most prevalent mutations in patients [74, 75], while those in black are enzalutamide- and apalutamide-resistant mutations [76, 77]. AR-v7 lacks exons 4/5/6/7/8 and differs by 16aa at the C-terminus compared with AR-FL. Exons 5/6/7 are excluded in ARv567es compared with AR-FL.
AR alternative splicing (AS) events resulting in the absence of the LBD from AR isoforms is another major resistance mechanism relevant to prostate cancer [82]. The human AR gene is comprised of eight canonical exons (Fig. 3) encoding the NTD, DNA-binding domain (DBD), hinge region, and LBD [83]. At least 18 splicing variants have been reported, as summarized by Lu et al. [84] and Snow et al. [85], most of which do not contain the LBD targeted by second-generation AR antagonists. Importantly, AR variants such as AR-v7 and ARv567es arise from intricate AR genomic arrangements and dysregulation of AS factors [63, 64], and have been shown to be constitutively active in CRPC patients and prostate cancer cell models [86,87,88,89]. AR-v7 and ARv567es can induce the expression of genes that regulate cell growth and survival independently of their interaction with full-length AR (AR-FL) [90,91,92]. On the other hand, constitutively active AR-v7 and ARv567es also promote the function of AR-FL by facilitating its nuclear localization and DNA binding in the absence of androgen or even in the presence of enzalutamide [90, 91, 93, 94].
AR overexpression is also considered a mechanism of resistance to AR antagonists, and can result from AR gene amplification, enhanced transcription activation, or increased AR stability at the mRNA/protein level. Studies have shown that about 80% of tumors overexpressing AR exhibit AR amplification [95, 96]. Systematic analysis across different CRPC patients has shown that ~30% have amplification of the AR locus, and that AR overexpression is sufficient to confer resistance to AR antagonists in clinical practice [65, 74, 97]. Notably, AR amplification is more common in patients who have progressed on enzalutamide compared to abiraterone or other agents (53% vs. 17% or 21%) [98]. Enhanced AR transcription or increased stability of AR protein/mRNA is sufficient to upregulate AR levels without AR gene amplification [99,100,101], and can facilitate tumor growth despite minimal androgen [102]. Interestingly, a recent study has found that MYB interaction with AR can sustain its ligand-independent activation and promote castration resistance in prostate cancer [101].
Reprogramming of AR transcriptional activity by AR antagonists
Although second-generation AR antagonists are typically considered “pure” antagonists, our recent studies have found that these agents function as partial agonists as well [103, 104]. We have demonstrated that in the presence of enzalutamide or darolutamide, AR is enriched in the distal elements of cancer-related genes such as NR3C1 (encoding GR) and SLC7A11, and upregulates their expression in both ADPC and CPRC cell models. Transcriptome analysis further demonstrates that enzalutamide induces global upregulation of a number of cancer-related genes. Mechanistic studies have revealed that this process is assisted by the pioneer factor GATA2 and the mediator complex [104,105,106,107]. Interestingly, comparative analysis of the cistrome and transcriptome profiles of AR and GR has shown a high degree overlap [108, 109]. These results are consistent with earlier findings from Sawyer’s lab that GR upregulation contributes to enzalutamide and apalutamide resistance in LNCaP and VCaP cell models [12, 108]. ChIP-seq studies have revealed that agonist-liganded AR and antagonist-liganded AR bind to two different motifs, leading to distinct transcriptional outcomes in prostate cancer cells [103]. In conclusion, second-generation agents previously thought to function as pure AR antagonists might also perform a partial agonist function that reprograms AR transcriptional activity to transcribe oncogenes that counteract their AR antagonist role. Targeting GATA2, which mediates the agonist role of enzalutamide, with a small molecular inhibitor can re-sensitize both ADPC and CRPC cell models to enzalutamide treatment [104]. This indicates that prostate cancer treatment may benefit from combining AR antagonist therapy with inhibitors targeting the AR co-factors that facilitate antagonist-induced reprogramming of AR transcriptional activity. Further experiments are necessary to identify the most critical AR antagonist-specific co-factors.
Heterogenetic evaluation independent of AR
Tumor heterogeneity is one of the major drivers of cancer progression and represents one of the primary challenges in cancer treatment. Tumor heterogeneity exists both in nascent prostate cancers and following antagonist-driven evolution, and contributes to CRPC progression and drug resistance [110,111,112,113,114]. As described above, heterogeneity within the AR locus alone can range from AR LBD point mutations to alternative splicing events, to overexpression that increases the sensitivity of AR to hormone stimulation, to loss of the antagonist binding region or other changes that mediate antagonist-agonist switching of the second-generation AR-targeted therapies [63, 64]. Outside of its impacts on the AR gene, tumor heterogeneity can also contribute to AR antagonist resistance through other pathways independent of AR.
Lineage plasticity is driven by alterations in PTEN, RB1, TP53, or SOX2 enables tumors to become AR independent and activates neuroendocrine differentiation, which is emerging as an increasingly recognized mechanism of resistance to AR-targeted therapies. It has not been established whether these alternations pre-exist within a subset of prostate epithelium cells that are intrinsically resistant to AR-targeted therapies or whether they are induced during the course of treatment with AR-targeted therapies. A recent whole-exome sequencing and immunohistochemistry (IHC) study in 37 prostate cancer patients before ADT has shown that loss of chromosome 10q (containing PTEN) and alterations to TP53 are predictive of poor response to enzalutamide. A subset of prostate cancer exhibits greater histologic and genomic diversity, accompanied by a higher fitness to resist therapy [112]. In addition to the pre-existing heterogeneity of the tumor, the application of second-generation AR antagonists is associated with treatment-induced heterogenetic evolution [115]. A related fact is that the increasing use of AR antagonists such as enzalutamide in CRPC settings has favored the increase in incidence of t-NEPC [70, 115]. Recurrent amplification or loss of function of genes such as PTEN, RB1, and TP53 are characteristic of treatment-induced NEPC patients [116,117,118]. Experiments in cellular models also demonstrate that overexpression of MYC or knockdown of PTEN/RB1/TP53 drives lineage plasticity models to shift from CRPC to NEPC [119,120,121,122].
Activation or deactivation of other pathways including Wnt-β-catenin, PI3K–AKT-mTOR, and DNA repair are also reported to be associated with resistance to AR antagonists, as summarized by Schmidt et al. [24, 123]. Stromal reactivity (SR) surrounding tumors can also shape the dynamics of prostate cancer evolution and tumor aggressiveness [124]. Further studies are needed to determine the extent to which AR antagonists can drive the cross-talk of intricate intercellular signaling networks between the tumor and stromal cells. Although the source of tumor heterogeneity in CRPC remains unclear, it is increasingly recognized that this phenomenon contributes to second-generation AR antagonist resistance and NEPC progression. In summary, the evolution of CRPC resistance and progression results from the combined contributions of both AR-dependent and AR-independent pathways (Fig. 4).

The partial agonist role of second-generation AR antagonists induces the expression of cancer-related genes including GR. GR in turn regulates the expression of a set of genes that overlaps with AR downstream pathways. AR alterations can include alternative splicing, point mutation and overexpression. Other AR signaling-independent mechanisms such as PTEN/TP53/RB1 loss of function and MYCN/SOC2 activation can mediate CRPC progression and contribute to AR antagonist resistance in CRPC.
Strategies to overcome resistance to AR Antagonists
Development of novel AR-targeted therapies
In reviewing the mechanisms of AR antagonist resistance, changes in the AR signaling pathway stand out as one of the primary reasons, with accumulating studies demonstrating that events including AR point mutation, rearrangement, amplification, and transcriptional upregulation of AR variants can result in the failure of AR antagonist treatment and in some cases can mediate antagonist–agonist switching. Additionally, side effects such as seizure and cardiovascular disease have limited the clinical benefits of second-generation AR antagonists. Although darolutamide and proxalutamide show clinical efficacy in CRPC patients harboring point mutations within the AR LBD [40, 44], these antagonists cannot target AR variants such as AR-v7 or ARv567es that lack the LBD and contribute to prostate cancer progression and resistance to AR antagonists [93, 125,126,127,128]. As a result, drug development strategies are increasingly recognizing the need to screen for novel agents that can target both AR-FL harboring clinical mutations and AR variants that lack the LBD [129, 130]. As a result, AR-targeted therapies focusing on the N-terminal domain or DBD of AR have become a subject of intense interest as a potentially promising strategy to overcome AR heterogeneity in prostate cancer. Several N-terminal inhibitors and DBD inhibitors, such as EPI-7386 and SBF-1 have been reported in preclinical studies with promising ability to overcome many known mechanisms of resistance to existing hormonal therapies [131,132,133,134]. The highly selective N-terminal domain inhibitor EPI-7386 is currently in phase 1 and phase 1/2 clinical trials to evaluate the safety, tolerability, and preliminary efficacy of EPI-7386 alone and in combination with enzalutamide in mCRPC (NCT05075577 and NCT04421222). Another recent study has found that the well-characterized antitumor agent SBF-1 can selectively bind to the AR-DBD and block the transcription of AR target genes, and has been proven to repress prostate cancer growth both in vitro and in vivo [134]. Beyond targeting alternative AR domains, another strategy to overcome resistance conferred by point mutations in the LBD is the structure-based design of novel AR antagonists to specifically disrupt LBD dimerization, as AR transactivation potential requires LBD-mediated homodimer formation, regardless of the presence or absence of LBD point mutations [135].
The emergence of gene-targeted therapies for different diseases may be a more straightforward approach to confronting AR splicing variants and point mutations. PROteolysis-TArgeting Chimeras (PROTACs) have been recognized as a promising technology to chemically knock down targeted genes at the protein level, particularly in the context of cancer [136]. Preliminary clinical data on PROTAC ARV-110, which flags AR for degradation, have shown safety and efficacy in men with mCRPC (NCT03888612) [137, 138]. Gene knockdown technologies such as CRISPR-Cas9 directed gene deletion have shown promise but have not yet found clinical applications, as CRISPR-Cas9 directed DNA editing may cause unreversible and unpredictable mutations in chromatin in vivo and in vitro [139,140,141]. Although the approval of the first siRNA drug by the US FDA in 2018 marks the beginning of a new era of RNAi therapeutics, numerous studies demonstrated widespread off-target effects of siRNA-mediated gene silencing have similarly limited its clinical implementation [142,143,144]. The more recently-reported CRISPR-Cas13 system might circumvent these limitations, as it has been proven to target RNA with high specificity and efficiency [145, 146], and several studies have demonstrated that targeting driver oncogenes using CRISPR-Cas13 can repress the growth of different types of cancer both in vitro and in vivo [147,148,149]. However, further investigation is needed to evaluate the specificity and efficiency of CRISPR-Cas13 for AR targeted therapy specifically in the context of prostate cancer. in summary, accumulating studies indicate that the development of novel AR antagonists recognizing the NTD or LBD of AR and the gene-targeted therapies hold great promise to overcome the shortcomings of current AR antagonists (Table 2).
Combined therapies with AR antagonists
Although second-generation AR antagonists have prolonged prostate cancer survival time, side effects and the rapid evolution of drug resistance remain stumbling blocks associated with the use of AR antagonists in clinical practice. One possible contributor to both the side effects and the induction of drug resistance is the high dose of AR antagonists currently administered to patients. Indeed, our study has found that 25 μM of enzalutamide that imitates the real dose of enzalutamide in patients induced higher expression of cancer-related genes such as GR and SLC7A11, in comparison with 10 μM of enzalutamide [104]. Therefore, multipoint targeting of the AR signaling pathway may accomplish the same or even greater antitumor effect while reducing side-effects, which may slow down the induction of resistance and cancer progression. A meta-analysis of two phase 3 trials has shown that abiraterone and prednisolone, which target androgen synthesis, can combine with enzalutamide to significantly improve metastasis-free survival in high-risk non-metastatic prostate cancer [150]. A phase IB/IIA study of the pan-BET inhibitor ZEN-3694 in combination with enzalutamide showed acceptable tolerability and potential efficacy in patients with androgen-signaling inhibitor-resistant mCRPC [151]. Other promising combined therapies involving immunotherapy, CDK inhibitors and radiotherapy are summarized in Table 3. Notably, we and other groups have demonstrated that enzalutamide and darolutamide can induce the expression of ferroptosis-related genes in both ADPC and CRPC [104, 152], which have proven to be correlated with prostate cancer recurrence [153, 154]. Targeting ferroptosis might be a novel therapeutic strategy for advanced prostate cancer, as ferroptosis inducers significantly decrease prostate cancer cell growth and migration in vitro and delay tumor growth of treatment-resistant prostate cancer in vivo, with no measurable side effects [155, 156]. Further clinical trials are needed to test the potential of this therapeutic strategy.
Sequencing treatment strategies with different inhibitors have achieved initial success in CRPC. A phase II clinic trial has shown that using a sequencing strategy of abiraterone acetate followed by enzalutamide in CRPC patients provides a clinical benefit [157], although further exploration is needed to determine whether these findings apply to patients who have previously received one of the androgen-directed agents in a hormone-sensitive setting [158, 159]. Adaptive therapy to cycle drug selection using real-time data to limit the length of exposure to one selective pressure should also be considered [160]. Although no trials of adaptive therapy to reduce resistance to second-generation AR antagonists are currently underway, a pilot study to assess adaptive abiraterone monotherapy has supported the potential of the adaptive therapy approach with AR antagonists (Table 3) [161].
Notably, although GR pathway activation has been considered one of the principal mechanisms of resistance to AR antagonists [23, 24, 41], a phase I/II clinical trial for enzalutamide and the GR antagonist mifepristone in mCRPC (NCT02012296) has shown that the combined treatment is safe and well tolerated, but does not delay time to PSA, radiographic or clinical PSA progression-free survival [162]. These preliminary results indicate that the development of more specific GR antagonists should be explored in combination with AR antagonists.
Conclusions and perspectives
In conclusion, three second-generation AR antagonists have been developed through interdisciplinary efforts during the past decade, and their approval for prostate cancer treatment has significantly improved survival and decreased prostate cancer-related death worldwide, particularly in patients with mCSPC and CRPC. At the same time, drawbacks of these AR antagonists have gradually emerged, especially the ability of second-generation AR antagonists to induce resistance and progression of patients from CRPC to t-NEPC. Mechanistic studies indicate that AR alteration, reprogramming of AR transcriptional activity by induced AR antagonists, and both pre-existing and therapy-driven tumor heterogeneity contribute to prostate cancer resistance and tumor progression. These obstacles might be addressable through the joint efforts of both clinical doctors and basic researchers to develop novel inhibitors or other technologies targeting AR and to explore combination/sequencing therapeutic strategies.
Data availability
All relevant data are included in this manuscript.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Schatzl G, Madersbacher S, Gsur A, Preyer M, Haidinger G, Haitel A, et al. Association of polymorphisms within androgen receptor, 5alpha-reductase, and PSA genes with prostate volume, clinical parameters, and endocrine status in elderly men. Prostate 2002;52:130–8.
Lee D. High androgen receptor levels are predictive of decreased survival in prostate cancer. Clin Prostate Cancer. 2003;2:13–4.
Desai K, McManus JM, Sharifi N. Hormonal therapy for prostate cancer. Endocr Rev. 2021;42:354–73.
Airhart RA, Barnett TF, Sullivan JW, Levine RL, Schlegel JU. Flutamide therapy for carcinoma of the prostate. South Med J. 1978;71:798–801.
Jacobo E, Schmidt JD, Weinstein SH, Flocks RH. Comparison of flutamide (SCH-13521) and diethylstilbestrol in untreated advanced prostatic cancer. Urology 1976;8:231–3.
Navratil H. Double-blind study of Anandron versus placebo in stage D2 prostate cancer patients receiving buserelin. Results on 49 cases from a multicentre study. Prog Clin Biol Res. 1987;243A:401–10.
Namer M, Amiel J, Toubol J. Anandron (RU 23908) associated with orchiectomy in stage D prostate cancer. Preliminary results of a randomized, double-blind study. Am J Clin Oncol. 1988;11:S191–6.
Eri LM, Tveter KJ. A prospective, placebo-controlled study of the antiandrogen Casodex as treatment for patients with benign prostatic hyperplasia. J Urol. 1993;150:90–4.
Cockshott ID, Cooper KJ, Sweetmore DS, Blacklock NJ, Denis L. The pharmacokinetics of Casodex in prostate cancer patients after single and during multiple dosing. Eur Urol. 1990;18:10–7.
Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–61.
Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9.
Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–6.
Tan J, Sharief Y, Hamil KG, Gregory CW, Zang DY, Sar M, et al. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol. 1997;11:450–9.
Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, Claassen E, et al. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. 1990;173:534–40.
Klein KA, Reiter RE, Redula J, Moradi H, Zhu XL, Brothman AR, et al. Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice. Nat Med. 1997;3:402–8.
Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97.
Davis ID, Martin AJ, Stockler MR, Begbie S, Chi KN, Chowdhury S, et al. Enzalutamide with Standard First-Line Therapy in metastatic prostate cancer. N Engl J Med. 2019;381:121–31.
Penson DF, Armstrong AJ, Concepcion R, Agarwal N, Olsson C, Karsh L, et al. Enzalutamide versus bicalutamide in castration-resistant prostate cancer: the STRIVE Trial. J Clin Oncol. 2016;34:2098–106.
Smith MR, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. 2018;378:1408–18.
Rajaram P, Rivera A, Muthima K, Olveda N, Muchalski H, Chen QH. Second-generation androgen receptor antagonists as hormonal therapeutics for three forms of prostate cancer. Molecules. 2020;25:2448. https://doi.org/10.3390/molecules25102448.
Fizazi K, Shore N, Tammela TL, Ulys A, Vjaters E, Polyakov S, et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. N Engl J Med. 2019;380:1235–46.
Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–11.
Schmidt KT, Huitema ADR, Chau CH, Figg WD. Resistance to second-generation androgen receptor antagonists in prostate cancer. Nat Rev Urol. 2021;18:209–26.
Crawford ED, Schellhammer PF, McLeod DG, Moul JW, Higano CS, Shore N, et al. Androgen receptor targeted treatments of prostate cancer: 35 years of progress with antiandrogens. J Urol. 2018;200:956–66.
Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–90.
Gim HJ, Park J, Jung ME, Houk KN. Conformational dynamics of androgen receptors bound to agonists and antagonists. Sci Rep. 2021;11:15887.
Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:424–33.
Hussain M, Fizazi K, Saad F, Rathenborg P, Shore N, Ferreira U, et al. Enzalutamide in men with nonmetastatic, castration-resistant prostate cancer. N Engl J Med. 2018;378:2465–74.
Higano CS, Beer TM, Taplin ME, Efstathiou E, Hirmand M, Forer D, et al. Long-term safety and antitumor activity in the Phase 1–2 study of enzalutamide in pre- and post-docetaxel castration-resistant prostate cancer. Eur Urol. 2015;68:795–801.
Pilon D, Behl AS, Ellis LA, Robitaille MN, Lefebvre P, Dawson NA. Assessment of real-world central nervous system events in patients with advanced prostate cancer using abiraterone acetate, bicalutamide, enzalutamide, or chemotherapy. Am Health Drug Benefits. 2017;10:143–53.
Foster WR, Car BD, Shi H, Levesque PC, Obermeier MT, Gan J, et al. Drug safety is a barrier to the discovery and development of new androgen receptor antagonists. Prostate 2011;71:480–8.
Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72:1494–503.
Chi KN, Agarwal N, Bjartell A, Chung BH, Pereira de Santana Gomes AJ, Given R, et al. Apalutamide for metastatic, castration-sensitive prostate cancer. N Engl J Med. 2019;381:13–24.
Smith MR, Antonarakis ES, Ryan CJ, Berry WR, Shore ND, Liu G, et al. Phase 2 study of the safety and antitumor activity of apalutamide (ARN-509), a potent androgen receptor antagonist, in the high-risk nonmetastatic castration-resistant Prostate Cancer Cohort. Eur Urol. 2016;70:963–70.
Moilanen AM, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep. 2015;5:12007.
Fizazi K, Massard C, Bono P, Jones R, Kataja V, James N, et al. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. Lancet Oncol. 2014;15:975–85.
Shore ND, Tammela TL, Massard C, Bono P, Aspegren J, Mustonen M, et al. Safety and antitumour activity of ODM-201 (BAY-1841788) in chemotherapy-naive and CYP17 inhibitor-naive patients: follow-up from the ARADES and ARAFOR trials. Eur Urol Focus. 2018;4:547–53.
Fizazi K, Shore N, Tammela TL, Ulys A, Vjaters E, Polyakov S, et al. Nonmetastatic, castration-resistant prostate cancer and survival with darolutamide. N Engl J Med. 2020;383:1040–9.
Lallous N, Snow O, Sanchez C, Parra Nunez AK, Sun B, Hussain A, et al. Evaluation of darolutamide (ODM201) efficiency on androgen receptor mutants reported to date in prostate cancer patients. Cancers (Basel). 2021;13:2939. https://doi.org/10.3390/cancers13122939.
Lokeshwar SD, Klaassen Z, Saad F. Treatment and trials in non-metastatic castration-resistant prostate cancer. Nat Rev Urol. 2021;18:433–42.
Rizzo A, Merler S, Sorgentoni G, Oderda M, Mollica V, Gadaleta-Caldarola G, et al. Risk of cardiovascular toxicities and hypertension in nonmetastatic castration-resistant prostate cancer patients treated with novel hormonal agents: a systematic review and meta-analysis. Expert Opin Drug Metab Toxicol. 2021;17:1237–43.
Morgans AK, Shore N, Cope D, McNatty A, Moslehi J, Gomella L, et al. Androgen receptor inhibitor treatments: cardiovascular adverse events and comorbidity considerations in patients with non-metastatic prostate cancer. Urol Oncol. 2021;39:52–62.
Zhou T, Xu W, Zhang W, Sun Y, Yan H, Gao X, et al. Preclinical profile and phase I clinical trial of a novel androgen receptor antagonist GT0918 in castration-resistant prostate cancer. Eur J Cancer. 2020;134:29–40.
Youzhi Tong CC, Wu J, Yang J, Zhang H, Wu X, Duan Y, et al. Abstract 614: proxalutamide (GT0918), a potent androgen receptor pathway inhibitor. Cancer Res. 2014;74:614.
Balog A, Rampulla R, Martin GS, Krystek SR, Attar R, Dell-John J, et al. Discovery of BMS-641988, a novel androgen receptor antagonist for the treatment of prostate cancer. ACS Med Chem Lett. 2015;6:908–12.
Attar RM, Jure-Kunkel M, Balog A, Cvijic ME, Dell-John J, Rizzo CA, et al. Discovery of BMS-641988, a novel and potent inhibitor of androgen receptor signaling for the treatment of prostate cancer. Cancer Res. 2009;69:6522–30.
Rathkopf D, Liu G, Carducci MA, Eisenberger MA, Anand A, Morris MJ, et al. Phase I dose-escalation study of the novel antiandrogen BMS-641988 in patients with castration-resistant prostate cancer. Clin Cancer Res. 2011;17:880–7.
Qin X, Ji D, Gu W, Han W, Luo H, Du C, et al. Activity and safety of SHR3680, a novel antiandrogen, in patients with metastatic castration-resistant prostate cancer: a phase I/II trial. BMC Med. 2022;20:84.
Ronan Le Moigne CAB, Mawji NR, Tam T, Wang J, Jian K, Andersen RJ, et al. Abstract B117: treatment of castrated resistant prostate cancer with EPI-7386, a second generation N-terminal domain androgen receptor inhibitor. Mol Cancer Ther. 2019;18:B117. https://doi.org/10.1158/1535-7163.TARG-19-B117.
Hong NH, Sun S, Virsik P, Cesano A, Mostaghel EA, Plymate SR, et al. Comprehensive in vitro characterization of the mechanism of action of EPI-7386, an androgen receptor Nterminal domain inhibitor. Cancer Research 2021;81:1209.
Dellis AE, Papatsoris AG. Perspectives on the current and emerging chemical androgen receptor antagonists for the treatment of prostate cancer. Expert Opin Pharmacother. 2019;20:163–72.
Xu Q, Zhang Z, Huang C, Bao Q, Zhang R, Wu M, et al. Development of novel androgen receptor antagonists based on the structure of darolutamide. Bioorg Chem. 2022;124:105829.
Slovin S, Clark W, Carles J, Krivoshik A, Park JW, Wang F, et al. Seizure rates in enzalutamide-treated men with metastatic castration-resistant prostate cancer and risk of seizure: the UPWARD Study. JAMA Oncol. 2018;4:702–6.
Ryan C, Wefel JS, Morgans AK. A review of prostate cancer treatment impact on the CNS and cognitive function. Prostate Cancer Prostatic Dis. 2020;23:207–19.
Lai LY, Oerline MK, Caram MEV, Tsao PA, Kaufman SR, Hollenbeck BK, et al. Risk of metabolic and cardiovascular adverse events with abiraterone or enzalutamide among men with advanced prostate cancer. J Natl Cancer Inst. 2022. djac081. https://doi.org/10.1093/jnci/djac081. Online ahead of print.
Wilk M, Wasko-Grabowska A, Szmit S. Cardiovascular complications of prostate cancer treatment. Front Pharm. 2020;11:555475.
Zhu J, Liao R, Su C, Liang D, Wu J, Qiu K, et al. Toxicity profile characteristics of novel androgen-deprivation therapy agents in patients with prostate cancer: a meta-analysis. Expert Rev Anticancer Ther. 2018;18:193–8.
Moreira RB, Debiasi M, Francini E, Nuzzo PV, Velasco G, Maluf FC, et al. Differential side effects profile in patients with mCRPC treated with abiraterone or enzalutamide: a meta-analysis of randomized controlled trials. Oncotarget 2017;8:84572–8.
Iacovelli R, Ciccarese C, Bria E, Romano M, Fantinel E, Bimbatti D, et al. The cardiovascular toxicity of abiraterone and enzalutamide in prostate cancer. Clin Genitourin Cancer. 2018;16:e645–e53.
Armstrong AJ, Lin P, Tombal B, Saad F, Higano CS, Joshua AM, et al. Five-year survival prediction and safety outcomes with enzalutamide in men with chemotherapy-naive metastatic castration-resistant prostate cancer from the PREVAIL Trial. Eur Urol. 2020;78:347–57.
Kulkarni AA, Rubin N, Tholkes A, Shah S, Ryan CJ, Lutsey PL, et al. Risk for stroke and myocardial infarction with abiraterone versus enzalutamide in metastatic prostate cancer patients. ESMO Open. 2021;6:100261.
Stockley J, Markert E, Zhou Y, Robson CN, Elliott DJ, Lindberg J, et al. The RNA-binding protein Sam68 regulates expression and transcription function of the androgen receptor splice variant AR-V7. Sci Rep. 2015;5:13426.
Kawamura N, Nimura K, Saga K, Ishibashi A, Kitamura K, Nagano H, et al. SF3B2-mediated RNA splicing drives human prostate cancer progression. Cancer Res. 2019;79:5204–17.
Buttigliero C, Tucci M, Bertaglia V, Vignani F, Bironzo P, Di Maio M, et al. Understanding and overcoming the mechanisms of primary and acquired resistance to abiraterone and enzalutamide in castration resistant prostate cancer. Cancer Treat Rev. 2015;41:884–92.
Zaffuto E, Pompe R, Zanaty M, Bondarenko HD, Leyh-Bannurah SR, Moschini M, et al. Contemporary incidence and cancer control outcomes of primary neuroendocrine prostate cancer: a SEER Database Analysis. Clin Genitourin Cancer. 2017;15:e793–e800.
Vlachostergios PJ, Puca L, Beltran H. Emerging variants of castration-resistant prostate cancer. Curr Oncol Rep. 2017;19:32.
Alanee S, Moore A, Nutt M, Holland B, Dynda D, El-Zawahry A, et al. Contemporary incidence and mortality rates of neuroendocrine prostate cancer. Anticancer Res. 2015;35:4145–50.
Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell. 2017;32:474–89 e6.
Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a Multi-institutional Prospective Study. J Clin Oncol. 2018;36:2492–503.
Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci USA. 2019;116:11428–36.
Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28.
Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63:920–6.
Ledet EM, Lilly MB, Sonpavde G, Lin E, Nussenzveig RH, Barata PC, et al. Comprehensive analysis of AR alterations in circulating tumor DNA from patients with advanced prostate cancer. Oncologist. 2020;25:327–33.
Jacob A, Raj R, Allison DB, Myint ZW. Androgen receptor signaling in prostate cancer and therapeutic strategies. Cancers (Basel). 2021;13:5417. https://doi.org/10.3390/cancers13215417.
Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013;3:1030–43.
Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–9.
Prekovic S, van Royen ME, Voet AR, Geverts B, Houtman R, Melchers D, et al. The effect of F877L and T878A mutations on androgen receptor response to enzalutamide. Mol Cancer Ther. 2016;15:1702–12.
Rodriguez-Vida A, Bianchini D, Van Hemelrijck M, Hughes S, Malik Z, Powles T, et al. Is there an antiandrogen withdrawal syndrome with enzalutamide? BJU Int. 2015;115:373–80.
Annala M, Vandekerkhove G, Khalaf D, Taavitsainen S, Beja K, Warner EW, et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov. 2018;8:444–57.
von Klot CA, Kramer MW, Boker A, Herrmann TR, Peters I, Kuczyk MA, et al. Is there an anti-androgen withdrawal syndrome for enzalutamide? World J Urol. 2014;32:1171–6.
Kanayama M, Lu C, Luo J, Antonarakis ES. AR splicing variants and resistance to AR targeting agents. Cancers (Basel). 2021;13:2563. https://doi.org/10.3390/cancers13112563.
Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–15.
Lu C, Luo J. Decoding the androgen receptor splice variants. Transl Androl Urol. 2013;2:178–86.
Snow O, Lallous N, Singh K, Lack N, Rennie P, Cherkasov A. Androgen receptor plasticity and its implications for prostate cancer therapy. Cancer Treat Rev. 2019;81:101871.
Nyquist MD, Li Y, Hwang TH, Manlove LS, Vessella RL, Silverstein KA, et al. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc Natl Acad Sci USA. 2013;110:17492–7.
Li Y, Alsagabi M, Fan D, Bova GS, Tewfik AH, Dehm SM. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res. 2011;71:2108–17.
Li Y, Hwang TH, Oseth LA, Hauge A, Vessella RL, Schmechel SC, et al. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene 2012;31:4759–67.
Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat Commun. 2016;7:13668.
Zhan Y, Zhang G, Wang X, Qi Y, Bai S, Li D, et al. Interplay between cytoplasmic and nuclear androgen receptor splice variants mediates castration resistance. Mol Cancer Res. 2017;15:59–68.
Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Investig. 2010;120:2715–30.
Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–62.
Cao B, Qi Y, Zhang G, Xu D, Zhan Y, Alvarez X, et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget 2014;5:1646–56.
Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W, et al. Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res. 2015;75:3663–71.
Leversha MA, Han J, Asgari Z, Danila DC, Lin O, Gonzalez-Espinoza R, et al. Fluorescence in situ hybridization analysis of circulating tumor cells in metastatic prostate cancer. Clin Cancer Res. 2009;15:2091–7.
Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, Folkerd E, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27:3742–8.
Jernberg E, Bergh A, Wikstrom P. Clinical relevance of androgen receptor alterations in prostate cancer. Endocr Connect. 2017;6:R146–R61.
Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, Bell RH, et al. Androgen receptor gene aberrations in circulating cell-free DNA: biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin Cancer Res. 2015;21:2315–24.
Sharma A, Yeow WS, Ertel A, Coleman I, Clegg N, Thangavel C, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Investig. 2010;120:4478–92.
Lin PC, Chiu YL, Banerjee S, Park K, Mosquera JM, Giannopoulou E, et al. Epigenetic repression of miR-31 disrupts androgen receptor homeostasis and contributes to prostate cancer progression. Cancer Res. 2013;73:1232–44.
Srivastava SK, Khan MA, Anand S, Zubair H, Deshmukh SK, Patel GK, et al. MYB interacts with androgen receptor, sustains its ligand-independent activation and promotes castration resistance in prostate cancer. Br J Cancer. 2022;126:1205–14.
Waltering KK, Helenius MA, Sahu B, Manni V, Linja MJ, Janne OA, et al. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res. 2009;69:8141–9.
Chen Z, Lan X, Thomas-Ahner JM, Wu D, Liu X, Ye Z, et al. Agonist and antagonist switch DNA motifs recognized by human androgen receptor in prostate cancer. EMBO J. 2015;34:502–16.
Yuan F, Hankey W, Wu D, Wang H, Somarelli J, Armstrong AJ, et al. Molecular determinants for enzalutamide-induced transcription in prostate cancer. Nucleic Acids Res. 2019;47:10104–14.
He B, Lanz RB, Fiskus W, Geng C, Yi P, Hartig SM, et al. GATA2 facilitates steroid receptor coactivator recruitment to the androgen receptor complex. Proc Natl Acad Sci USA. 2014;111:18261–6.
Wu D, Sunkel B, Chen Z, Liu X, Ye Z, Li Q, et al. Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res. 2014;42:3607–22.
Jeronimo C, Robert F. The mediator complex: at the nexus of RNA polymerase II transcription. Trends Cell Biol. 2017;27:765–83.
Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013;155:1309–22.
Sahu B, Laakso M, Pihlajamaa P, Ovaska K, Sinielnikov I, Hautaniemi S, et al. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013;73:1570–80.
Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015;520:353–7.
Taavitsainen S, Engedal N, Cao S, Handle F, Erickson A, Prekovic S, et al. Single-cell ATAC and RNA sequencing reveal pre-existing and persistent cells associated with prostate cancer relapse. Nat Commun. 2021;12:5307.
Wilkinson S, Ye H, Karzai F, Harmon SA, Terrigino NT, VanderWeele DJ, et al. Nascentrostate cancer heterogeneity drives evolution and resistance to intense hormonal therapy. Eur Urol. 2021;80:746–57.
Ketola K, Kaljunen H, Taavitsainen S, Kaarijarvi R, Jarvela E, Rodriguez-Martin B, et al. Subclone eradication analysis identifies targets for enhanced cancer therapy and reveals L1 retrotransposition as a dynamic source of cancer heterogeneity. Cancer Res. 2021;81:4901–9.
Martens-Uzunova ES, Kusuma GD, Crucitta S, Lim HK, Cooper C, Riches JE, et al. Androgens alter the heterogeneity of small extracellular vesicles and the small RNA cargo in prostate cancer. J Extracell Vesicles. 2021;10:e12136.
Small EJ, Huang J, Youngren J, Sokolov A, Aggarwal RR, Thomas G, et al. Characterization of neuroendocrine prostate cancer (NEPC) in patients with metastatic castration resistant prostate cancer (mCRPC) resistant to abiraterone (Abi) or enzalutamide (Enz): Preliminary results from the SU2C/PCF/AACR West Coast Prostate Cancer Dream Team (WCDT). ASCO Annual Meeting. J Clin Oncol. 2015;33.5003. https://doi.org/10.1200/jco.2015.33.15_suppl.5003.
Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res. 2014;20:890–903.
Chedgy EC, Vandekerkhove G, Herberts C, Annala M, Donoghue AJ, Sigouros M, et al. Biallelic tumour suppressor loss and DNA repair defects in de novo small-cell prostate carcinoma. J Pathol. 2018;246:244–53.
Mei W, Lin X, Kapoor A, Gu Y, Zhao K, Tang D. The contributions of prostate cancer stem cells in prostate cancer initiation and metastasis. Cancers (Basel). 2019;11:434. https://doi.org/10.3390/cancers11040434.
Yin Y, Xu L, Chang Y, Zeng T, Chen X, Wang A, et al. N-Myc promotes therapeutic resistance development of neuroendocrine prostate cancer by differentially regulating miR-421/ATM pathway. Mol Cancer. 2019;18:11.
Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017;355:78–83.
Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355:84–8.
Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, et al. N-Myc drives neuroendocrine prostate cancer initiated from human prostate epithelial cells. Cancer Cell. 2016;29:536–47.
Zhu YP, Wan FN, Shen YJ, Wang HK, Zhang GM, Ye DW. Reactive stroma component COL6A1 is upregulated in castration-resistant prostate cancer and promotes tumor growth. Oncotarget 2015;6:14488–96.
Frankenstein Z, Basanta D, Franco OE, Gao Y, Javier RA, Strand DW, et al. Stromal reactivity differentially drives tumour cell evolution and prostate cancer progression. Nat Ecol Evol. 2020;4:870–84.
Liu Y, Yu C, Shao Z, Xia X, Hu T, Kong W, et al. Selective degradation of AR-V7 to overcome castration resistance of prostate cancer. Cell Death Dis. 2021;12:857.
Di Lorenzo G, Zappavigna S, Crocetto F, Giuliano M, Ribera D, Morra R, et al. Assessment of total, PTEN(−), and AR-V7(+) circulating tumor cell count by flow cytometry in patients with metastatic castration-resistant prostate cancer receiving enzalutamide. Clin Genitourin Cancer. 2021;19:e286–e98.
Sekine Y, Nakayama H, Miyazawa Y, Arai S, Koike H, Matsui H, et al. Ratio of the expression levels of androgen receptor splice variant 7 to androgen receptor in castration refractory prostate cancer. Oncol Lett. 2021;22:831.
Lu S, Dong Z. Proliferating cell nuclear antigen directly interacts with androgen receptor and enhances androgen receptormediated signaling. Int J Oncol. 2021;59:41. https://doi.org/10.3892/ijo.2021.5221.
Dalal K, Roshan-Moniri M, Sharma A, Li H, Ban F, Hessein M, et al. Selectively targeting the DNA-binding domain of the androgen receptor as a prospective therapy for prostate cancer. J Biol Chem. 2014;289:26417–29.
Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–46.
Yang YC, Banuelos CA, Mawji NR, Wang J, Kato M, Haile S, et al. Targeting androgen receptor activation function-1 with EPI to overcome resistance mechanisms in castration-resistant prostate cancer. Clin Cancer Res. 2016;22:4466–77.
Sadar MD. Discovery of drugs that directly target the intrinsically disordered region of the androgen receptor. Expert Opin Drug Discov. 2020;15:551–60.
Maurice-Dror C, Le Moigne R, Vaishampayan U, Montgomery RB, Gordon MS, Hong NH, et al. A phase 1 study to assess the safety, pharmacokinetics, and anti-tumor activity of the androgen receptor n-terminal domain inhibitor epi-506 in patients with metastatic castration-resistant prostate cancer. Invest N Drugs. 2022;40:322–9.
Elgehama A, Sun L, Yu B, Guo W, Xu Q. Selective targeting of the androgen receptor-DNA binding domain by the novel antiandrogen SBF-1 and inhibition of the growth of prostate cancer cells. Invest N Drugs. 2021;39:442–57.
Nadal M, Prekovic S, Gallastegui N, Helsen C, Abella M, Zielinska K, et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat Commun. 2017;8:14388.
Li X, Pu W, Zheng Q, Ai M, Chen S, Peng Y. Proteolysis-targeting chimeras (PROTACs) in cancer therapy. Mol Cancer. 2022;21:99.
Proof-of-Concept with PROTACs in Prostate Cancer. Cancer Discov. 2020;10:1084.
Nguyen TT, Kim JW, Choi HI, Maeng HJ, Koo TS. Development of an LC-MS/MS method for ARV-110, a PROTAC molecule, and applications to pharmacokinetic studies. Molecules. 2022;27:1977. https://doi.org/10.3390/molecules27061977.
Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. 2018;24:927–30.
Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. 2018;24:939–46.
Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018;36:765–71.
Fedorov Y, Anderson EM, Birmingham A, Reynolds A, Karpilow J, Robinson K, et al. Off-target effects by siRNA can induce toxic phenotype. RNA 2006;12:1188–96.
Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–7.
Lin X, Ruan X, Anderson MG, McDowell JA, Kroeger PE, Fesik SW, et al. siRNA-mediated off-target gene silencing triggered by a 7 nt complementation. Nucleic Acids Res. 2005;33:4527–35.
Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, Hsu PD. Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell 2018;173:665–76 e14.
Zhou H, Su J, Hu X, Zhou C, Li H, Chen Z, et al. Glia-to-neuron conversion by CRISPR-CasRx alleviates symptoms of neurological disease in mice. Cell 2020;181:590–603 e16.
Yue H, Huang R, Shan Y, Xing D. Delivery of Cas13a/crRNA by self-degradable black phosphorus nanosheets to specifically inhibit Mcl-1 for breast cancer therapy. J Mater Chem B 2020;8:11096–106.
Gao J, Luo T, Lin N, Zhang S, Wang J. A new tool for CRISPR-Cas13a-based cancer gene therapy. Mol Ther Oncolytics. 2020;19:79–92.
Palaz F, Kalkan AK, Can O, Demir AN, Tozluyurt A, Ozcan A, et al. CRISPR-Cas13 system as a promising and versatile tool for cancer diagnosis, therapy, and research. ACS Synth Biol. 2021;10:1245–67.
Attard G, Murphy L, Clarke NW, Cross W, Jones RJ, Parker CC, et al. Abiraterone acetate and prednisolone with or without enzalutamide for high-risk non-metastatic prostate cancer: a meta-analysis of primary results from two randomised controlled phase 3 trials of the STAMPEDE platform protocol. Lancet 2022;399:447–60.
Aggarwal RR, Schweizer MT, Nanus DM, Pantuck AJ, Heath EI, Campeau E, et al. A Phase Ib/IIa study of the Pan-BET inhibitor ZEN-3694 in combination with enzalutamide in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. 2020;26:5338–47.
Tousignant KD, Rockstroh A, Poad BLJ, Talebi A, Young RSE, Taherian Fard A, et al. Therapy-induced lipid uptake and remodeling underpin ferroptosis hypersensitivity in prostate cancer. Cancer Metab. 2020;8:11.
Lv Z, Wang J, Wang X, Mo M, Tang G, Xu H, et al. Identifying a ferroptosis-related gene signature for predicting biochemical recurrence of prostate cancer. Front Cell Dev Biol. 2021;9:666025.
Liu H, Gao L, Xie T, Li J, Zhai TS, Xu Y. Identification and validation of a prognostic signature for prostate cancer based on ferroptosis-related genes. Front Oncol. 2021;11:623313.
Ghoochani A, Hsu EC, Aslan M, Rice MA, Nguyen HM, Brooks JD, et al. Ferroptosis inducers are a novel therapeutic approach for advanced prostate cancer. Cancer Res. 2021;81:1583–94.
Zhou X, Zou L, Chen W, Yang T, Luo J, Wu K, et al. Flubendazole, FDA-approved anthelmintic, elicits valid antitumor effects by targeting P53 and promoting ferroptosis in castration-resistant prostate cancer. Pharm Res. 2021;164:105305.
Khalaf DJ, Annala M, Taavitsainen S, Finch DL, Oja C, Vergidis J, et al. Optimal sequencing of enzalutamide and abiraterone acetate plus prednisone in metastatic castration-resistant prostate cancer: a multicentre, randomised, open-label, phase 2, crossover trial. Lancet Oncol. 2019;20:1730–9.
Sidaway P. Sequence of AR inhibitors affects outcome. Nat Rev Clin Oncol. 2020;17:69.
Maughan BL, Antonarakis ES. Does sequencing order of antiandrogens in prostate cancer matter? Nat Rev Urol. 2020;17:197–8.
West J, You L, Zhang J, Gatenby RA, Brown JS, Newton PK, et al. Towards Multidrug Adaptive Therapy. Cancer Res. 2020;80:1578–89.
Zhang J, Cunningham JJ, Brown JS, Gatenby RA. Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nat Commun. 2017;8:1816.
Serritella AV, Shevrin D, Heath EI, Wade JL, Martinez E, Anderson A, et al. Phase I/II trial of enzalutamide and mifepristone, a glucocorticoid receptor antagonist, for metastatic castration-resistant prostate cancer. Clin Cancer Res. 2022;28:1549–59.
van Royen ME, van Cappellen WA, de Vos C, Houtsmuller AB, Trapman J. Stepwise androgen receptor dimerization. J Cell Sci. 2012;125:1970–9.
Radaeva M, Ban F, Zhang F, LeBlanc E, Lallous N, Rennie PS, et al. Development of novel inhibitors targeting the D-box of the DNA binding domain of androgen receptor. Int J Mol Sci. 2021;22.
Antonarakis ES, Chandhasin C, Osbourne E, Luo J, Sadar MD, Perabo F. Targeting the N-terminal domain of the androgen receptor: a new approach for the treatment of advanced prostate cancer. Oncologist. 2016;21:1427–35.
Salem AF, Gambini L, Billet S, Sun Y, Oshiro H, Zhao M, et al. Prostate cancer metastases are strongly inhibited by agonistic Epha2 ligands in an orthotopic mouse model. Cancers (Basel). 2020;12.
Centenera MM, Carter SL, Gillis JL, Marrocco-Tallarigo DL, Grose RH, Tilley WD, et al. Co-targeting AR and HSP90 suppresses prostate cancer cell growth and prevents resistance mechanisms. Endocr Relat Cancer. 2015;22:805–18.
Moon SJ, Jeong BC, Kim HJ, Lim JE, Kim HJ, Kwon GY, et al. Bruceantin targets HSP90 to overcome resistance to hormone therapy in castration-resistant prostate cancer. Theranostics 2021;11:958–73.
Xiao L, Parolia A, Qiao Y, Bawa P, Eyunni S, Mannan R, et al. Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature 2022;601:434–9.
Rao A, Moka N, Hamstra DA, Ryan CJ. Co-inhibition of androgen receptor and parp as a novel treatment paradigm in prostate cancer-where are we now? Cancers (Basel). 2022;157:103199. https://doi.org/10.1016/j.critrevonc.2020.103199.
Brighi N, Conteduca V, Lolli C, Gurioli G, Schepisi G, Palleschi M, et al. The cyclin-dependent kinases pathway as a target for prostate cancer treatment: rationale and future perspectives. Crit Rev Oncol Hematol. 2021;157:103199.
Maughan BL, Kessel A, McFarland TR, Sayegh N, Nussenzveig R, Hahn AW, et al. Radium-223 plus enzalutamide versus enzalutamide in metastatic castration-refractory prostate cancer: final safety and efficacy results. Oncologist. 2021;26:1006–e2129.
McDermott RS, Greene J, McCaffrey J, Parker I, Helanova S, Baird AM, et al. Radium-223 in combination with enzalutamide in metastatic castration-resistant prostate cancer: a multi-centre, phase II open-label study. Ther Adv Med Oncol. 2021;13:17588359211042691.
Zhao H, Howard LE, De Hoedt AM, Terris MK, Amling CL, Kane CJ, et al. Safety of concomitant therapy with radium-223 and abiraterone or enzalutamide in a real-world population. Prostate 2021;81:390–7.
Kim SI, Szeto AH, Morgan KP, Brower B, Dunn MW, Khandani AH, et al. A real-world evaluation of radium-223 in combination with abiraterone or enzalutamide for the treatment of metastatic castration-resistant prostate cancer. PLoS ONE. 2021;16:e0253021.
Onal C, Kose F, Ozyigit G, Aksoy S, Oymak E, Muallaoglu S, et al. Stereotactic body radiotherapy for oligoprogressive lesions in metastatic castration-resistant prostate cancer patients during abiraterone/enzalutamide treatment. Prostate 2021;81:543–52.
Acknowledgements
We extend a sincere apology to those whose work was not discussed or cited in this review because of space limitations. FY was supported by Topnotch Personnel from Shanghai University of Traditional Chinese Medicine.
Author information
Authors and Affiliations
Contributions
FY, YC and QZ wrote the article. FY and XF provided the idea. FY, YC, QZ and WH revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This research was based on the review of published/publicly reported literature and did not require ethical approval.
COMPETING INTERESTS
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Francesca Pentimalli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Y., Zhou, Q., Hankey, W. et al. Second generation androgen receptor antagonists and challenges in prostate cancer treatment. Cell Death Dis 13, 632 (2022). https://doi.org/10.1038/s41419-022-05084-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-05084-1
This article is cited by
-
DHODH inhibition represents a therapeutic strategy and improves abiraterone treatment in castration-resistant prostate cancer
Oncogene (2024)
-
Rezvilutamide: First Approval
Drugs (2023)
-
The androgen receptor-targeted proteolysis targeting chimera and other alternative therapeutic choices in overcoming the resistance to androgen deprivation treatment in prostate cancer
Clinical and Translational Oncology (2022)
