
Abstract
The regulation of osteogenesis is important for bone formation and fracture healing. Despite advances in understanding the molecular mechanisms of osteogenesis, crucial modulators in this process are not well-characterized. Here we demonstrate that suppression of signal transducer and activator of transcription 5A (STAT5A) activates distal-less homeobox 5 (DLX5) in human bone marrow-derived stromal cells (hBMSCs) and enhances osteogenesis in vitro and in vivo. We show that STAT5A negatively regulates expression of Dlx5 in vitro and that STAT5A deletion results in increased trabecular and cortical bone mass and bone mineral density in mice. Additionally, STAT5A deletion prevents age-related bone loss. In a murine fracture model, STAT5A deletion was found to significantly enhance bone remodeling by stimulating the formation of a fracture callus. Our findings indicate that STAT5A inhibition enhances bone formation by promoting osteogenesis of BMSCs.
Similar content being viewed by others


Introduction
Human bone marrow mesenchymal stromal cells (hBMSCs) may differentiate into osteoblasts, chondrocytes, adipocytes, or tenocytes depending on culture conditions. Among these, osteoblast differentiation is important for increasing bone mass. Osteoblast differentiation of hBMSCs is a well-characterized process and proceeds as a result of the timely expression of genes, such as alkaline phosphatase (ALP), runt-related transcription factor 2 (RUNX2), distal-less homeobox 5 (DLX5), osterix (OSX), and osteocalcin (OCN), followed by extracellular matrix synthesis and mineralization1,2. RUNX2 has been characterized as the master transcription factor in osteogenic differentiation and bone formation3,4. Another transcription factor, DLX5, is also expressed in osteoblasts. DLX5 is an important regulator in the development of mineralized tissues because it induces expression of RUNX2 in the bone morphogenetic protein (BMP) signaling pathway5. DLX5 is also involved in osteoblast maturation6, and Dlx5-/- mice have been found to have severe craniofacial abnormalities with delayed ossification of the dermatocranial bones7. These findings suggest that the Dlx5 gene may play a role in bone formation and fracture healing.
Members of the signal transducers and activators of transcription (STAT) family play important roles in cell proliferation, differentiation, and survival via effects on the expression of cytokines, growth factors, and hormones8,9,10,11. Like other STAT family members, STAT5 was originally identified as a cytosolic signaling molecule involved in proliferation, differentiation, and apoptosis in cancer cell lines12,13. Following stimulation by various cytokines, STAT5 becomes phosphorylated and forms a dimer14,15. The STAT5 dimer then translocates into the nucleus where it may bind to interferon-γ-activated sequence (GAS) motifs, promoting the transcription of target genes9,16,17. There are two isoforms of STAT5: STAT5A and STAT5B18. Interestingly, STAT5A and STAT5B are encoded by separate genes, but the proteins are 90% identical at the amino acid level14. The distinct functions of these isoforms have been explored in several in vivo studies by using STAT5 global knockout mice19. The prolactin-mediated functions are blocked in the mammary gland by STAT5A-/-10,20, while STAT5B-/- disrupts the functions that are regulated by the growth hormone in the liver11,21. Mostly, the studies about STAT5 global knockout mice were conducted in hematopoietic stem cells20,22,23.
Recently, STAT5 was found to function as a negative regulator of bone resorption in osteoclasts in vitro and in vivo24. Another study reported that STAT5–RUNX2 interaction promotes osteoblast differentiation in vitro25. However, no study has yet distinguished the roles in osteoblast differentiation played by the different isoforms of STAT5. Previously, Jung et al. demonstrated that STAT5A plays a major role in adipogenesis of hBMSCs and STAT5B performs only a supportive function in these processes26. These results suggest that STAT5A plays an essential role in maintaining bone homeostasis by balancing osteogenesis and adipogenesis. In this study, we aimed to characterize the role of STAT5A in osteoblasts. By clarifying which STAT5 isoform contributes to osteoblast differentiation, our findings might identify a novel therapeutic target for bone diseases.
In this study, we found that STAT5A played an important role in bone formation and regeneration. We found that inhibition of STAT5A promoted osteoblast differentiation and bone formation through the activation of DLX5 signaling. STAT5A deletion was found to result in significantly decreased bone loss in a murine age-related osteoporosis model. Moreover, in a murine fracture model, we found that STAT5A deletion enhanced bone healing by stimulating new bone formation. These findings support an inhibitory function for STAT5A in osteogenesis and suggest therapeutic potential for STAT5A inhibition in age-related osteoporosis and fracture healing.
Results
Overexpression of STAT5A suppresses osteogenesis in hBMSCs
To investigate the possible role of STAT5 during osteogenesis, we first examined STAT5 isoform expression during osteogenesis in hBMSCs. Interestingly, STAT5A was found to increase in response to induction of osteogenesis after 6 days (Fig. 1a). However, there was no change in the expression of STAT5B during osteogenesis, while DLX5 and RUNX2, which are key osteogenic markers, were decreased in the later stage of osteogenesis. To examine the effects of STAT5A and STAT5B on osteogenesis, we overexpressed STAT5A and STAT5B in hBMSCs. On day 14 after the induction of osteogenesis, mineralization of STAT5A-overexpressed hBMSCs was significantly decreased compared to that of control hBMSCs and STAT5B-overexpressed hBMSCs (Fig. 1b, c). Following Alizarin Red S staining and quantification, we also confirmed protein levels for the osteogenic marker genes DLX5 and RUNX2. Overexpression of STAT5A in hBMSCs was found to result in significantly decreased DLX5 expression but not decreased RUNX2 expression. Overexpression of STAT5B, however, had no effect on these proteins (Fig. 1d, e). Accordingly, STAT5A decreased messenger RNA (mRNA) levels of DLX5 and its downstream genes coding for bone sialoprotein (BSP) and osteopontin (OPN) but the mRNA of RUNX2 was not changed (Fig. 1f). These findings suggest that STAT5A functions during osteogenesis of hBMSCs via regulation of DLX5 expression.

a Protein levels of STAT5A, STAT5B, DLX5, and RUNX2 during osteogenesis of hBMSCs. b Alizarin Red S staining demonstrating the effect of STAT5 overexpression on the osteogenesis at day 14. Scale bar, 60 μm. c Quantification of Alizarin Red S staining. d Western blot analysis of protein expression of osteogenic master genes, DLX5 and Runx2 after STAT5 overexpression. GAPDH was used as a control. e Quantification of DLX5 and RUNX2 protein levels. f Gene expression of DLX5 and expression of downstream genes of DLX5 after 4 days with STAT5 overexpression in hBMSCs using real-time PCR. All mRNA and protein levels were normalized with GAPDH. Each experiment was performed in triplicate (n = 3). All error bars indicate ± SEM. *P < 0.01; **P < 0.01; ***P < 0.001
Suppression of STAT5A promotes osteogenesis in hBMSCs
To clarify the distinct roles of STAT5A and STAT5B in osteogenesis of hBMSCs, we knocked down STAT5A and STAT5B using small interfering RNAs (siRNAs), and we also treated hBMSCs with a STAT5 inhibitor (sc-355979). Suppression of STAT5A by siRNA had no effect on mRNA and protein expression of STAT5B (Supplementary Figures 1a and b). The siRNA-mediated knockdown of STAT5A promoted osteoblast differentiation, while siRNA-mediated knockdown of STAT5B had no effect (Fig. 2a, b). We then tested whether STAT5A regulates the expression of DLX5. The siRNA-mediated knockdown of STAT5A increased DLX5 protein expression (Fig. 2c, d). Consistently, a STAT5 inhibitor-induced mineral accumulation in a dose-dependent manner at day 14 after the induction of osteogenesis (Fig. 2e). At the dose for which the STAT5 inhibitor exhibited its maximal effect (10 μM), mineral accumulation was increased about 2.5-fold over that in vehicle control-treated cells (Fig. 2f). Accordingly, the STAT5 inhibitor increased DLX5 mRNA and protein expression in a dose-dependent manner in hBMSCs (Fig. 2g, h).

a Alizarin Red S staining to detect suppression of STAT5A and STAT5B on osteogenesis using targeted siRNA. Staining was performed on day 14 of osteogenesis. Scale bar, 30 μm. b Quantification of Alizarin Red S staining. c Western blot analysis of DLX5 protein expression after silencing of STAT5A in hBMSCs. d Quantification of DLX5 protein level compared with GAPDH expression level. e Alizarin Red S staining for the effect of STAT5 inhibitor during osteogenesis of hBMSCs. STAT5 inhibitor (sc-355797) was used at concentrations of 0, 2, 6, and 10 μM. Staining was performed at day 14 of osteogenesis. Scale bar, 30 μm. f Quantification of Alizarin Red S staining. g Analysis of DLX5 mRNA level after 10 μM STAT5 inhibitor treatment. mRNA level was checked on day 5 of osteogenesis. h Western blot analysis of DLX5 protein expression after STAT5 inhibitor treatment. Protein level was detected on day 5 of osteogenesis. Each experiment was performed in triplicate (n = 3). All error bars indicate ±SEM. *P < 0.01; **P < 0.01; ***P < 0.001
STAT5A directly regulates DLX5 in hBMSCs
To examine the relationship between STAT5A and the osteogenic transcriptional regulator DLX5 luciferase assays for each regulator were performed at day 5 after induction of osteogenesis in hBMSCs. STAT5A reduced DLX5 promoter activity in a dose-dependent manner (Supplementary Figure 2a). Furthermore, DLX5 promoter assays showed that the STAT5 inhibitor partially restored DLX5 promoter activity that had been decreased by STAT5A (Supplementary Figure 3b).
To characterize the mechanism by which STAT5A suppresses DLX5 promoter activity, we first predicted a STAT5A-binding site within the DLX5 promoter region (Supplementary Figure 3c, asterisk) using the transcription element search system (TESS). We then designed truncated constructs of the DLX5 promoter (Supplementary Figure 3c). We observed significantly decreased DLX5 promoter activity in constructs containing the STAT5A-binding site relative to vector control, vs. no change was seen in the activities of constructs without a STAT5A-binding site (Supplementary Figure 3d). Furthermore, chromatin immuno-precipitation (ChIP) assays demonstrated that osteogenic stimulation for 5 days induced recruitment of STAT5A to the DLX5 promoter, supporting the occurrence of direct regulation of DLX5 expression by STAT5A (Supplementary Figure 3e).
Deletion of STAT5A enhances bone formation in mice
To determine the in vivo role of STAT5A in skeletal development, we bred mice up to the seventh generation. Using whole-mount Alizarin Red/Alcian Blue-stained skeletal preparations, general aspects of skeletal development were not found to differ between E19.5 embryos of wild-type and Stat5a-/- mice (Supplementary Figure S2). However, postnatal histologic analysis using Masson’s Trichrome staining revealed that femur trabecular bone mass was markedly increased in Stat5a-/- mice compared to that in wild-type mice (Fig. 3a). Microcomputed tomography (μCT) analysis of femurs also showed that Stat5a deletion resulted in increased trabecular and cortical bone mass relative to wild-type mice (Fig. 3b, c). We found that multiple μCT parameters for trabecular bone were increased in Stat5a-/- mice compared to wild-type mice (Fig. 3d–h). Also, we analyzed the difference between 10 and 40 weeks in the two genotypes. In Stat5a-/- mice, the bone mineral density (BMD) reduction rate between 10 and 40 weeks was lower than that of wild-type mice. We found that Stat5a deletion reversed age-related reductions in bone mass and BMD in 40-week-old mice, suggesting that Stat5a deletion had a protective effect against age-related osteoporosis. Additionally, cortical thickness, one of the most important factors in determining mechanical bone strength, was significantly higher in Stat5a-/- mice compared to wild-type mice (Fig. 3i). Cortical bone volume and cortical cross-sectional area were also higher in Stat5a-/- mice compared to wild-type mice (Fig. 3j, k). Femoral shaft biomechanical properties were assessed by three-point bending, and Stat5a deletion resulted in significantly increased bone strength during bending compared to wild-type mice (Fig. 3l). Taken together, our results suggest a role for STAT5A in the maintenance of bone mass.

a Masson’s Trichrome staining of longitudinal sections of 10-week-old and 40-week-old male wild-type (WT) and Stat5a-/- (KO) femurs. Scale bar: 500 μm (top) and 200 μm (bottom) (10-weeks: n = 8 and 40-weeks: n = 6 per each group). b Representative µCT images of trabecular bone from 10-week-old and 40-week-old male wild-type and Stat5a-/- femurs. Scale bars, 0.5 mm. c Representative µCT images of cortical bone at 10 and 40 weeks of age in male wild-type and Stat5a-/- femurs. Scale bars, 0.5 mm. d–h Quantitative µCT analysis of trabecular bone parameters for 10-week-old and 40-week-old male wild-type (WT) and Stat5a-/- (KO) femurs, including (d) bone volume fraction ratio (BV/TV), (e) volumetric BMD of trabecular bone, (f) trabecular thickness (Tb.Th), (g) trabecular number (Tb.N), and (h) trabecular separation (Tb.Sp) are shown. (i–k) Quantitative µCT analysis of cortical bone parameters for 10-week-old and 40-week-old male wild-type (WT) and Stat5a-/- (KO) femurs, with (i) cross-sectional thickness (Cs.Th), (j) bone volume, and (k) cross-sectional area (Cs.Area) are shown. l Maximum load on femurs from postnatal 10-week-old mice (n = 8 for each group). All error bars indicate ±SEM. *P <0.01; **P < 0.01; ***P < 0.001
STAT5A deletion promotes osteogenesis in mBMSCs
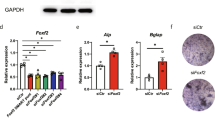
To investigate the osteoblast activity of mouse bone marrow-derived stromal cells (mBMSCs), we isolated mBMSCs from wild-type and Stat5a-/- mice. Stat5a-/- mBMSCs showed significantly higher baseline ALP activity compared to wild-type mice (Fig. 4a, b). Regardless of age, mineral accumulations in Stat5a-/- mBMSCs were markedly higher compared to wild-type mice at day 8 after the induction of osteogenesis (Fig. 4c). Interestingly, mineral accumulations in 30-week-old Stat5a-/- mBMSCs were decreased compared to 10-week-old Stat5a-/- mBMSCs, but comparable to those of 10-week-old wild-type mice (Fig. 4d). Accordingly, DLX5 protein and mRNA expression levels were higher in Stat5-/- mBMSCs compared to those in wild-type mice (Fig. 4e, f). Stat5a deletion led to significant increases in mRNA expression of the osteogenic genes Alp, Bsp, Opn, and Ocn (Fig. 4g). Next, we examined whether STAT5A overexpression reversed DLX5 expression in Stat5a-/- mBMSCs. STAT5A overexpression in Stat5a-/- mBMSCs significantly decreased Dlx5 mRNA and protein expression, which had been increased by STAT5A deletion (Fig. 4h–j). In accordance with these findings, Stat5a overexpression decreased mineral accumulation and calcification in Stat5a-/- mBMSCs (Fig. 4k, l). Conversely, DLX5 suppression in Stat5a-/- mBMSCs resulted in decreased osteogenesis (Supplementary Figure 4). These results demonstrate that STAT5A control osteogenic differentiation by regulating of DLX5.

a ALP staining of 10-week-old wild-type and Stat5a-/- mBMSCs at day 0, as indicated. Scale bar, 60 μm. b Relative ALP activity assay results at days 0 in wild-type and Stat5a-/- mBMSCs. c and d Alizarin Red S staining (c) and quantitative analysis (d) of at day 8 after induction of osteogenesis in 10-week-old, 20-week-old, and 30-week-old wild-type and Stat5a-/- mBMSCs. e Protein levels of DLX5 in 10-week-old, 20-week-old, and 30-week-old male wild-type and Stat5a-/- mBMSCs using western blotting at 5 days after osteogenesis. f mRNA levels of Dlx5 in wild-type and Stat5a-/- mBMSCs at day 3 after the induction of osteogenesis. g mRNA levels of osteoblast-related genes Alp, Bsp, and Opn at day 3 and mRNA level of Ocn at day 7 after the induction of osteogenesis. h–j Relative mRNA (h) and protein levels (I and j) of DLX5 depending on exogenously increased Stat5a expression in wild-type and Stat5a-/- mBMSCs. pcDNA-mStat5a was transfected in Stat5a-/- mBMSCs. mRNA and protein levels of Dlx5 were checked at day 3 and 5 after osteogenesis, respectively. Osteogenesis was induced after overexpression of Stat5a by transfection. k The osteoblast differentiation of Stat5a-/- mBMSCs upon overexpression of STAT5A using Alizarin Red S and Von Kossa staining at day 8 after induction of osteogenesis. l Quantification of Alizarin Red S staining in Stat5a-/- mBMSCs. Each experiment was performed in triplicate (n = 3). All error bars indicate ±SEM. *P < 0.01; **P < 0.01; ***P < 0.001
STAT5A deletion promotes bone fracture healing
We next investigated the possible roles of STAT5A in fracture healing using a murine fracture model. In this experiment, we generated femur fractures in 6-week-old wild-type and Stat5a-/- mice as previously described27. At 2 and 4 weeks post-procedure, we then examined callus formation using μCT (Fig. 5a). Analysis of μCT images found that callus area in Stat5a-/- mice was increased at 2 weeks post-fracture and decreased at 4 weeks post-fracture, as compared to wild-type mice (Fig. 5b). Interestingly, the reduction rate of callus bone and cartilaginous callus in Stat5a-/- mice were significantly higher than wild-type mice (Fig. 5b, c). Accordingly, cartilaginous callus stained with Safranin O and total callus stained with Masson’s Trichrome showed similar patterns to those found by μCT, suggesting that Stat5a deletion accelerated the fracture healing process in our model (Fig. 5c–f). Furthermore, immunohistochemical staining demonstrated that DLX5 expression was increased at the fracture site of Stat5a-/- mice. Notably, DLX5 was highly expressed in periosteal bone at the fracture site in Stat5a-/- mice (Fig. 5g). Overall, these results indicate that Stat5a deletion promotes bone regeneration and healing under fracture conditions.

a Representative longitudinal sections of fractured femurs from wild-type and Stat5a-/- mice at 2 and 4 weeks post-fracture. 6-week-old male mice were used for the fracture model. Scale bar, 4 mm (n = 16 per each group). b Quantitative analysis of newly formed callus volume at 2 and 4 weeks post-fracture. c–f Representative histological analysis of paraffin sections of calluses from wild-type and Stat5a-/- mice at 2 and 4 weeks post-fracture stained with (c) Safranin O/Fast Green staining for cartilaginous bone callus. d Quantitative analysis of remaining cartilaginous callus area at 2 and 4 weeks post-fracture (n = 8 per each group). Scale bar, 0.5 mm. e Masson’s Trichrome staining for total callus. f Quantitative analysis of total callus area at 2 and 4 weeks post-fracture (n = 8 per each group). Scale bar, 0.5 mm. g Immunohistochemistry against DLX5 at 2 weeks post-fracture of wild-type and Stat5a-/- mice in the fractured femoral section. Scale bars, 100 μm (n = 5 for each group). All error bars indicate ±SEM. *P <0.01; **P <0.01; ***P <0.001
STAT5A deletion increased osteoclastogenesis in vivo
Next, we examined the effect of STAT5A on osteoclast differentiation in mouse bone marrow monocytes (mBMMs). mBMMs from wild-type and Stat5a-/- mice were cultured for 3–4 days in α-minimum essential medium (α-MEM) with receptor activator of nuclear factor-kappa B ligand (RANKL) and macrophage colony stimulating factor (M-CSF). On day 4 after the induction of osteoclastogenesis, osteoclast differentiation ability was measured by counting tartrate-resistant acid phosphatase (TRAP) positive multinucleated osteoclasts (defined as those with ≥3 nuclei. Stat5a deletion, however, had no effect on osteoclast differentiation. The numbers of TRAP-positive multinucleated osteoclasts showed no difference between Stat5a-/- and wild-type specimens (Supplementary Figures S5a and b). Next, multinuclear osteoclasts were identified in fractured femoral H&E section28,29. At 2 weeks post-fracture, the number of multinuclear osteoclasts were higher in Stat5a-/- mice than wild-type. Intriguingly, at 4 weeks post-fracture, the multinuclear osteoclasts were lower in Stat5a-/- mice than wild-type (Supplementary Figures S6a and b). To confirm these observations, we performed immunohistochemistry against dendritic cell-specific transmembrane protein (DC-STAMP). DC-STAMP has been reported as an inducer of osteoclastogenesis and core factor for cell–cell fusion of osteoclast30,31. As a result, at 4 weeks post-fracture, the DC-STAMP-positive cells were decreased in Stat5a-/- mice group (Supplementary Figures S6c). These finding supported that the smaller callus at 4 weeks in Stat5a-/- mice suggests a rapid callus remodeling.
Discussion
Emerging evidence suggests that Janus-activated kinase (JAK)-STAT signaling plays an important role in osteoblast differentiation and bone formation27,32,33. Tajima et al. have reported that inhibition of STAT1 accelerates osteoblast differentiation and bone fracture healing by regulation of OSX27, and Levy et al. reported that inhibition of STAT3 results in the augmentation of BMP-induced osteogenic differentiation32. Furthermore, chemical inhibitors of STAT1 and STAT3 have been proposed for the treatment of fractures. Also, as Zhou suggested, the inactivation of stat3 by mutation in osteoblast/osteocyte-specific reduced bone formation34. Dieudonne et al. showed that STAT5 inhibitor abrogated in vitro osteogenesis induced by E3 ubiquitin-protein ligase CBL (c-CBL) silencing in hBMSCs25. They, however, did not explore the isoform-specific functions of STAT5A and STAT5B during osteoblast differentiation. In the present study, we sought to clarify isoform-specific functions of STAT5A and STAT5B and investigate in vivo role of STAT5 in the bone. Surprisingly, we found that inhibition of STAT5A significantly increased osteoblast differentiation in hBMSCs, whereas inhibition of STAT5B showed no effect on osteoblast differentiation (Fig. 2a, b). Thus, we investigated the relationship between STAT5A and osteogenic transcriptional factors and in vivo role of STAT5A for bone formation and fracture healing in murine models.
One study reported that DLX5 is a central regulator of bone homeostasis through direct regulation of osteogenesis35. Previous studies have reported that DLX5 is required for RUNX2 expression under BMP signaling36,37,38. We found that ALP activity and DLX5 expression in Stat5a-/- mBMSCs were higher than those in wild-type mice during osteogenic differentiation (Fig. 4b–f). STAT5A overexpression in Stat5a-/- mBMSCs was found to decrease DLX5 expression and impair osteogenic differentiation (Fig. 4). Our results further indicated that STAT5A inhibits DLX5 expression and thereby acts as a negative regulator in osteogenesis.
Interestingly, we found seemingly contradictory protein levels of STAT5A and DLX5 during osteogenesis. Specifically, expression of STAT5A was found to increase throughout the process of osteoblast differentiation (Fig. 1a). This increase in STAT5A seems paradoxical because of its negative effects on osteogenesis. The reason for this pattern and its biological significance have not been previously described. An earlier study found that expression of DLX5 is normally decreased in the late stages of osteogenesis and disappears in fully matured bone39. Expression of DLX5 is known to impair the ability of hBMSCs to proceed to the last stages of osteogenesis through suppression of osteocalcin, a marker of terminal osteogenic differentiation6,40,41. Consistent with this previous report39, our results showed that the expression of DLX5 decreased as osteogenesis progressed (Fig. 1a). Taken together, these findings suggest that the increased expression of STAT5A during osteogenesis may provide a homeostatic mechanism that counterbalances the suppressive effects of excessive DLX5 activity during the late stages of osteogenesis.
In our murine model, the skeletal phenotype of Stat5a-/- embryos was not found to differ from wild-type controls (Supplementary Figure S2), but postnatal Stat5a-/- mice were found to have greater bone mass than postnatal wild-type mice. The finding of increased bone mass in Stat5a-/- mice was maintained with aging. Age-related bone loss was partially protected against in Stat5a-/- mice, and the bone mineral densities of 40-week-old Stat5a-/- mice were comparable to those of young wild-type mice (Fig. 3d, e). These findings are consistent with several previous studies that have reported that DLX5 expression is related to postnatal bone developments and regulation of bone homeostasis in adulthood5,42. Our results suggest that Stat5 deletion affects postnatal bone formation via regulation of DLX5.
In addition, Stat5 deletion increased not only trabecular bone density but also cortical bone formation (Fig. 3i–k). The strength of Stat5a-/- femurs was found to be greater than that of wild-type mice during biomechanical testing (Fig. 3l). We hypothesize that increased DLX5 expression resulting from Stat5 deletion may especially target periosteal bone modeling and lead to the observed increase in mechanical strength in Stat5a-/- mouse bones compared to wild-type specimens. This speculation is supported by our results showing that DLX5 expression predominates in periosteal bone (Fig. 5g), possibly due to the presence of greater numbers of osteoblasts at a specific stage of differentiation7,43,44. The balance between bone formation and bone resorption is essential for bone homeostasis. In osteoclast differentiation, a DC-STAMP is a crucial factor in osteoclast fusion. Deletion of STAT5A had no effect on osteoclast differentiation in vitro, but the increase in number of osteoclasts and DC-STAMP positive cells in Stat5a-/- mice had resulted in rapid callus remodeling (Supplementary Figure 6). Overall, our results demonstrated that STAT5A deletion promotes osteoblast activity and that STAT5A plays an important role in regulating bone formation and bone remodeling.
In conclusion, we have described a novel mechanism of STAT5A in bone homeostasis through regulation of the DLX5. In addition, we demonstrated, for the first time, the STAT5A function in hBMSCs during osteogenesis and confirmed its function in in vivo. Taken all together, our findings contribute to understand the mechanism of STAT5A in controlling bone homeostasis and promoting fracture healing. Furthermore, this work may offer a new therapeutic strategy for preventing age-related osteoporosis.
Materials and methods
Cell culture and differentiation conditions
Selection of hBMSCs was performed by selecting them according to the cells’ natural tendency to adhere to the culture dish. Primary hBMSCs were cultured in growth medium, Dulbecco’s modified Eagle’s medium-low glucose (DMEM-LG; Gibco, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS; Gibco), 1% antibiotic–antimycotic solution (Invitrogen, Grand Island, NY, USA), and confluence was achieved within 7 days in 5% CO2 at 37 °C. For osteogenesis, cells were cultured in DMEM-LG with 1 μM dexamethasone (Sigma, St. Louis, MO, USA), 10 mM β-glycerophosphate (Sigma), and 50 μM ascorbic acid (Sigma) for 14 days. Mouse bone marrow-derived MSCs (mBMSCs) were isolated from the femurs and tibias of 3–5 mice between 10 and 30 weeks of age. mBMSCs were cultured in growth medium with α-MEM (Gibco) with 10% FBS, 1% antibiotic–antimycotic solution, and 2 mM l-glutamine (Invitrogen, Carlsbad, CA, USA) in 5% CO2 at 37 °C45. To induce osteogenic differentiation, cells were cultured in growth medium with 10 mM β-glycerophosphate and 50 μM ascorbic acid for 8 days. For osteoclastogenesis, mouse bone marrow monocyte cells (mBMMs) were cultured for 3–4 days in α-MEM containing 15 ng/ml mRANKL (R&D systems, MN, USA) and 40 ng/ml mMCSF (R&D systems)46.
Plasmid constructs
Recombinant pcDNA-mStat5a and pcDNA-mStat5b plasmids were kindly gifted by Dr. Hiroko Yamashita (Nagoya City University, Nagoya, Japan). pcDNA-hSTAT5A, pcDNA-hSTAT5B, and pcDNA-hDLX5 were constructed by amplifying each coding region from cDNA of HeLa cells. All promoter vectors were obtained with polymerase chain reaction (PCR) of human genomic DNA. Human DLX5 and STAT5A promoter regions spanning −2208/+142 base pairs (bp) and −2121/+88 bp were inserted into a pGL3 basic vector (Addgene, Cambridge, MA, USA) and named pGL-DLX5 Full and pGL-STAT5A, respectively. pGL-DLX5 Del1–pGL-DLX5 Del4, containing 5′ serial deletions fragments of the DLX5 promoter were cloned by amplifying the regions from −2003 to +142, −1751 to +142, −1254 to +142, and −645 to +142 bp, respectively.
Gene transfection and luciferase reporter assays
Recombinant plasmids were transfected into hBMSCs using Lipofectamine LTX and Plus Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The medium was then replaced with 2 ml DMEM-LG with 10% FBS and 1% antibiotic–antimycotic solution or osteogenic medium. After 24 h, the cells were lysed with 200 µl of 1 × passive lysis buffer (Promega, Madison, WI, USA) per tube, and cell debris was removed. Luciferase activity was measured using the Dual-Luciferase Reporter Assay Kit (Promega, Madison, WI, USA). Relative luciferase activity was normalized by renilla activities to adjust for transfection efficiency.
Inhibition of STAT5A and STAT5B
Synthetic siRNAs for STAT5A, STAT5B, and DLX5 mRNA were purchased from Bioneer (Bioneer, Daejeon, South Korea). 80% confluence hBMSCs were transfected with each siRNA using Lipofectamine LTX (Invitrogen). The cells were then collected and prepared for analysis. The silencing effects of siSTAT5A and siSTAT5B were checked by measuring reduced protein expression of STAT5A and STAT5B. Negative control duplexes (Bioneer, Korea) were used as negative controls. A STAT5 inhibitor which inhibits phosphorylation of STAT5 via binding to the SH2 domain of STAT5 was purchased from Santa Cruz Biotechnology (sc-355979, Dallas, TX, USA).
Isolation of mRNA and real-time (RT)-PCR analysis
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Venlo, Netherlands) according to the manufacturer’s protocol. For quantitative RT-PCR, complementary DNA (cDNA) was synthesized by oligo dT18 primer (Invitrogen) using the isolated RNA as a template. The Omniscript Reverse-Transcription Kit (Qiagen) was used to synthesize the cDNA, according to the manufacturer’s instructions. RT-PCR was then performed using 2× qPCRBIO SyGreen Mix Hi-Rox (PCR Biosystems, London, UK) and analyzed using the Applied Biosystems (ABI) StepOne Plus System (ABI, Carlsbad, CA, USA). All primers were purchased from Bioneer. The relative expression of each gene was normalized by GAPDH expression. The specific primer pairs are shown in Supplementary Table S1. All reactions were performed in triplicate.
Western blot analysis and antibodies
Collected cells were washed twice with 1× phosphate buffered saline (PBS) and lysed by the addition of 100 µl of whole cell lysis buffer (60 mM Tris–HCl pH 6.8, 1% SDS) for 10 min at 100 °C. Collected supernatants, following centrifugation at 13,000 rotations/min (rpm) for 10 min, were quantified using a BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA). For western blot analysis, 25 µg of protein were separated on 10% SDS–PAGE under reducing conditions. Subsequently, proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Amersham Pharmacia, Piscataway, NJ, USA) in transfer buffer (1.4% glycine, 20% methanol and 25 mM Tris–HCl, pH 8.3 for 90 min at 70 V). After that, membranes were incubated in blocking solution (1× TBST (50 mM Tris–HCl, 150 mM NaCl, and 0.1% Tween-20) containing 5% skimmed milk powder) for 1 h at room temperature. Then blocked membranes were incubated in 1% skimmed milk powder solution including primary antibodies overnight at 4 °C. Membranes were then washed three times with 1× TBST, followed by incubation with secondary antibodies for 1 h at room temperature. Primary antibodies used for blotting were anti-STAT5 (1:1000, Abcam; ab68465), anti-STAT5A (1:1000, Abcam; ab32043), anti-STAT5B (1:1000, Abcam; ab178941), anti-DLX5 (1:500, Abcam; ab109737), and anti-RUNX2 (1:1000, Millipore; 05–1478). All secondary antibody complexes were visualized by autoradiography using ECL Plus and ECL Western Blotting Detection Systems (Amersham BioSciences, Buckinghamshire, UK). Signal quantification was performed by scanning the autoradiographs. Signal intensities were determined by densitometry analysis using Image J software.
Chromatin immunoprecipitation (ChIP) analysis
ChIP analysis was performed using a ChIP Assay Kit (Upstate Biotechnology, Charlottesville, VA) according to the manufacturer’s instructions. Briefly, hBMSCs were induced to form osteoblasts for 3 days and the differentiated cells were then cross-linked by the addition of 1% formaldehyde solution for 10 min at room temperature. The cross-linked cells were lysed with SDS lysis buffer and sonicated. DNA–protein complexes were bound using antibodies against STAT5A (1:1000, Abcam; ab32043) or an unspecific rabbit IgG (1:1000, Santa Cruze; sc2027), and these immune complexes were collected by binding with protein A-agarose after an overnight incubation at 4 °C. After the complexes were washed with 1× PBS, DNA was extracted using phenol/chloroform alcohol precipitation and used as a template for amplification of the DNA fragments. The specific primer pairs for the DLX5 promoter are shown in Supplemental Table 2.
ALP staining and activity
Differentiated cells were fixed in a mixture of citrate working solution (Sigma Aldrich, 854c) and acetone (2:3) for 30 s and washed in distilled water for 1 min. ALP staining was performed using a Fast Violet B Salt Kit (Sigma Aldrich, 815). Cells were placed in ALP staining solution for 30 min and then washed two times with distilled water. ALP activity was measured using a colorimetric assay at day 0. For this assay, cells were washed two times with ice-cold PBS and harvested in 0.5 ml 50 mM Tris–HCl, pH 7.6. After sonication and centrifugation for 15 min at 12,000 rpm, ALP activity of the supernatant was measured by the quantity of p-nitrophenylphosphate released/min/µg total protein using a microplate reader (VersaMax™ Microplate Leader, CA, USA) at 405 nm. Total cellular protein amount was determined using the BCA Assay Kit.
Von kossa staining and Alizarin Red S staining
Differentiated cells were washed twice with PBS and fixed in a mixture of acetone and methanol (1:1) or 70% ethanol for Von Kossa staining or Alizarin Red S staining, respectively. After fixation, cells were washed two times with distilled water and then stained with either 3% silver nitrate solution or 2% Alizarin Red S solution for 30 min, respectively. For quantification of Alizarin Red S staining, the stained samples were distained by 10% (weight/volume) cetylpyridinium chloride for 30 min at room temperature, and then the supernatants were measured using a microplate reader (VersaMax™ Microplate Leader) at 595 nm.
Mice
Stat5a general knockout mice10 were gifted from Dr. Lothar Hennighausen (National Institutes of Health, MD, USA). Deletion of the Stat5a gene was confirmed by PCR around the substituted neomycin gene site using the specific primer. The study population of Stat5a-/- mice was generated by intercrossing Stat5a heterozygous knockout (Stat5a+/-) mice. 10, 20, 30, and 40 weeks old male mice were used in this study.
μCT analysis
For μCT imaging, soft tissue was dissected from mouse femurs and the bones were fixed in 70% ethanol for 24 h at room temperature. The fixed femurs were then analyzed using high-resolution μCT (Skyscan-1076, Skyscan, Kontich, Belgium). Image reconstruction and analysis was then performed using reconstruction software NRecon (version 1.6.6.0, Skyscan) and CT-analyzer software CTAn (version 1.13.2.1, Skyscan), respectively. Measured parameters for epiphyseal trabecular bone were analyzed using three-dimensional (3D) model visualization software CT-Vol (version 2.0, Skyscan). The acquisition setting conditions were followed by an X-ray source voltage of 70 kVp and a current 140 μA. Beam hardening reduction depended on a 0.5 mm-thick aluminum filter. The pixel size was 18 μm: exposure time was 14.7 s; the rotation step was 0.5°; and full rotation occurred over 360°. The quantitative analysis for the trabecular parameters and cortical parameter were measured at the femoral metaphysis (a total of 300 slices) and femoral diaphysis (a total of 100 slices), respectively. The measured 3D bone parameters of 10-week-old mice included the following: total tissue volume (TV), which included volume measurement of both trabecular and cortical bone; trabecular bone volume per tissue volume (BV/TV); trabecular number (Tb. N); trabecular thickness (Tb. Th); trabecular separation (Tb. Sp); BMD; cortical bone volume (bone volume); cortical cross-sectional thickness (Cs. Th); and cortical cross-sectional area (Cs. area).
Murine fracture model
A standardized mid-diaphyseal fracture was induced in 24 wild-type mice and 24 Stat5a-/- mice at 6 weeks of age. Mice were anesthetized with Zoletil® (30 mg/kg body weight, Virbac, Carros, France) and Rompun® (10 mg/kg body weight, Bayer Ontario, Canada), administered by intraperitoneal injection. The right hind leg was shaved and disinfected. The femur was then exposed through lateral incision, and severed in the mid-diaphysis using a scalpel. The fractured femur was then fixed using 22-gauge needle. After fixation the surgical wound was irrigated with normal saline and closed by nylon suture. Immediately after fracture creation, post-surgical condition of the femur was measured using µCT. Mice were then sacrificed at 2 and 4 weeks post-procedure (n = 16 and n = 32 per time point, respectively). There were no complications or animal deaths during surgery or postoperative management. At 2 and 4 weeks post-procedure (n = 8 from the Stat5a-/- group and n = 8 from the wild-type group per time point) were used for BMD measurements and µCT analysis of callus formation and mineralization. The other subjects (n = 8 in each group) were used for mechanical testing at 4 weeks post-procedure. Calcified callus volume was determined by subtracting cortical bone volume from the total volume. Before mechanical analysis and imaging, femurs were dissected out from the soft tissue and intramedullary pins were removed. The fracture model protocol was approved by the Committee on the Ethics of Animal Experiments.
Histology and immunohistochemistry
The fractured femurs were removed from eight mice in each group and fixed in 10% formalin solution for 5–7 days at room temperature. Femurs were decalcified in Calci-Clear RapidTM (National Diagnostics Inc., GA, USA) solution for 3 days at room temperature. Decalcified femurs were then embedded in paraffin blocks. The paraffin sections were dehydrated by passage through an ethanol series, cleared twice in xylene, and embedded in paraffin, after which 5 mm sections were cut using a rotary microtome. Decalcified femoral sections were stained with Masson’s Trichrome, Safranin O, and Fast Green. For immunohistochemistry, antigen retrieval was performed using pH 6.0 citrate buffer for the deparaffinized sections. Sections were blocked with 5% normal goat serum, without PBS and without 0.1% Tween 20 for 1 h at room temperature. Sections were then incubated with DLX5 antibody (1:100, EPR4488, Abcam) and anti-DC-STAMP (1:100, Millipore; MABF39) for 1 h at room temperature and with anti-rabbit and anti-mouse secondary antibodies for 1 h at room temperature. Then the DAB Substrate Kit (ab64238, Abcam) was used for detection of DLX5 and DC-STAMP positive cells on tissue sections.
Statistical analysis
The numbers of mice used for in vivo experiments were based on our pilot experiments and µCT analysis was performed in a blinded fashion. For checking the normal distributions of the groups, we first performed normality testing using the Shapiro–Wilk method. If normality tests were passed, two-tailed, unpaired Student’s t-tests were used for the comparisons between two groups. If normality tests were failed, Mann–Whitney tests were used for the comparisons between two groups. For comparisons between three or four groups, we used one-way ANOVA if normality tests were passed, followed by Tukey’s post hoc test for all pairs of groups. If normality tests were failed, Kruskal–Wallis testing was performed and followed with Dunn’s post hoc test. GraphPad PRISM Software (version 5.0) was used for statistical analysis. P < 0.05 was considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001. All experiments were performed in biological triplicate.
Study approval
hBMSCs were obtained from the posterior iliac crest of 10 adult donors (age range: 21–51 years) after receiving approval from the Institutional Review Board. All experiments with mice were performed in an Animal Care Center and in accordance with Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International Guidelines. All animal care and use protocols were approved by the Committee on the Ethics of Animal Experiments.
References
Aubin, J. E. Regulation of osteoblast formation and function. Rev. Endocr. Metab. Disord. 2, 81–94 (2001).
Lian, J. B. et al. Regulatory controls for osteoblast growth and differentiation: role of Runx/Cbfa/AML factors. Crit. Rev. Eukaryot. Gene Expr. 14, 1–41 (2004).
Franceschi, R. T. & Xiao, G. Regulation of the osteoblast-specific transcription factor, Runx2: responsiveness to multiple signal transduction pathways. J. Cell. Biochem. 88, 446–454 (2003).
Karsenty, G. Transcriptional control of skeletogenesis. Annu. Rev. Genom. Hum. Genet. 9, 183–196 (2008).
Holleville, N., Mateos, S., Bontoux, M., Bollerot, K. & Monsoro-Burq, A. H. Dlx5 drives Runx2 expression and osteogenic differentiation in developing cranial suture mesenchyme. Dev. Biol. 304, 860–874 (2007).
Ryoo, H. M. et al. Stage-specific expression of Dlx-5 during osteoblast differentiation: involvement in regulation of osteocalcin gene expression. Mol. Endocrinol. 11, 1681–1694 (1997).
Acampora, D. et al. Craniofacial, vestibular and bone defects in mice lacking the Distal-less-related gene Dlx5. Development 126, 3795–3809 (1999).
Agaisse, H., Petersen, U. M., Boutros, M., Mathey-Prevot, B. & Perrimon, N. Signaling role of hemocytes in Drosophila JAK/STAT-dependent response to septic injury. Dev. Cell. 5, 441–450 (2003).
Ivashkiv, L. B. & Hu, X. Signaling by STATs. Arthritis Res. Ther. 6, 159–168 (2004).
Liu, X. et al. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 11, 179–186 (1997).
Udy, G. B. et al. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc. Natl Acad. Sci. USA 94, 7239–7244 (1997).
Cumaraswamy, A. A. & Gunning, P. T. Progress towards direct inhibitors of Stat5 protein. Horm. Mol. Biol. Clin. Investig. 10, 281–286 (2012).
Liao, Z. et al. Structure-based screen identifies a potent small molecule inhibitor of stat5a/b with therapeutic potential for prostate cancer and chronic myeloid leukemia. Mol. Cancer Ther. 14, 1777–1793 (2015).
Grimley, P. M., Dong, F. & Rui, H. Stat5a and Stat5b: fraternal twins of signal transduction and transcriptional activation. Cytokine Growth Factor Rev. 10, 131–157 (1999).
Gouilleux, F., Wakao, H., Mundt, M. & Groner, B. Prolactin induces phosphorylation of Tyr694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO J. 13, 4361–4369 (1994).
Meyer, T. & Vinkemeier, U. Nucleocytoplasmic shuttling of STAT transcription factors. Eur. J. Biochem. 271, 4606–4612 (2004).
Zeng, R., Aoki, Y., Yoshida, M., Arai, K. & Watanabe, S. Stat5B shuttles between cytoplasm and nucleus in a cytokine-dependent and -independent manner. J. Immunol. 168, 4567–4575 (2002).
Wang, D., Stravopodis, D., Teglund, S., Kitazawa, J. & Ihle, J. N. Naturally occurring dominant negative variants of Stat5. Mol. Cell. Biol. 16, 6141–6148 (1996).
Cui, Y. et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol. Cell. Biol. 24, 8037–8047 (2004).
Bradley, H. L., Hawley, T. S. & Bunting, K. D. Cell intrinsic defects in cytokine responsiveness of STAT5-deficient hematopoietic stem cells. Blood 100, 3983–3989 (2002).
Teglund, S. et al. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell 93, 841–850 (1998).
Bunting, K. D. et al. Reduced lymphomyeloid repopulating activity from adult bone marrow and fetal liver of mice lacking expression of STAT5. Blood 99, 479–487 (2002).
Zhu, Y. et al. Cutting edge: IL-5 primes Th2 cytokine-producing capacity in eosinophils through a STAT5-dependent mechanism. J. Immunol. 173, 2918–2922 (2004).
Hirose, J. et al. Bone resorption is regulated by cell-autonomous negative feedback loop of Stat5-Dusp axis in the osteoclast. J. Exp. Med. 211, 153–163 (2014).
Dieudonne, F. X. et al. Promotion of osteoblast differentiation in mesenchymal cells through Cbl-mediated control of STAT5 activity. Stem Cells 31, 1340–1349 (2013).
Jung, H. S. et al. Peroxisome proliferator-activated receptor gamma/signal transducers and activators of transcription 5A pathway plays a key factor in adipogenesis of human bone marrow-derived stromal cells and 3T3-L1 preadipocytes. Stem. Cells Dev. 21, 465–475 (2012).
Tajima, K. et al. Inhibition of STAT1 accelerates bone fracture healing. J. Orthop. Res. 28, 937–941 (2010).
Gross, C. et al. Osteoclast profile of medication-related osteonecrosis of the jaw secondary to bisphosphonate therapy: a comparison with osteoradionecrosis and osteomyelitis. J. Transl. Med. 15, 128 (2017).
Tomankova, T. et al. Comparison of periprosthetic tissues in knee and hip joints: differential expression of CCL3 and DC-STAMP in total knee and hip arthroplasty and similar cytokine profiles in primary knee and hip osteoarthritis. Osteoarthr. Cartil. 22, 1851–1860 (2014).
Yagi, M. et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 202, 345–351 (2005).
Chiu, Y. H. & Ritchlin, C. T. DC-STAMP: a key regulator in osteoclast differentiation. J. Cell. Physiol. 231, 2402–2407 (2016).
Levy, O., Ruvinov, E., Reem, T., Granot, Y. & Cohen, S. Highly efficient osteogenic differentiation of human mesenchymal stem cells by eradication of STAT3 signaling. Int. J. Biochem. Cell. Biol. 42, 1823–1830 (2010).
Levy, O., Dvir, T., Tsur-Gang, O., Granot, Y. & Cohen, S. Signal transducer and activator of transcription 3-A key molecular switch for human mesenchymal stem cell proliferation. Int. J. Biochem. Cell. Biol. 40, 2606–2618 (2008).
Zhou, H. et al. Osteoblast/osteocyte-specific inactivation of Stat3 decreases load-driven bone formation and accumulates reactive oxygen species. Bone 49, 404–411 (2011).
Samee, N. et al. Dlx5, a positive regulator of osteoblastogenesis, is essential for osteoblast-osteoclast coupling. Am. J. Pathol. 173, 773–780 (2008).
Merlo, G. R. et al. Multiple functions of Dlx genes. Int. J. Dev. Biol. 44, 619–626 (2000).
Hassan, M. Q. et al. Dlx3 transcriptional regulation of osteoblast differentiation: temporal recruitment of Msx2, Dlx3, and Dlx5 homeodomain proteins to chromatin of the osteocalcin gene. Mol. Cell. Biol. 24, 9248–9261 (2004).
Ryoo, H. M., Lee, M. H. & Kim, Y. J. Critical molecular switches involved in BMP-2-induced osteogenic differentiation of mesenchymal cells. Gene 366, 51–57 (2006).
Muraglia, A. et al. DLX5 overexpression impairs osteogenic differentiation of human bone marrow stromal cells. Eur. J. Cell Biol. 87, 751–761 (2008).
Mark, M. P., Butler, W. T., Prince, C. W., Finkelman, R. D. & Ruch, J. V. Developmental expression of 44-kDa bone phosphoprotein (osteopontin) and bone gamma-carboxyglutamic acid (Gla)-containing protein (osteocalcin) in calcifying tissues of rat. Differentiation 37, 123–136 (1988).
Stein, G. S., Lian, J. B. & Owen, T. A. Relationship of cell growth to the regulation of tissue-specific gene expression during osteoblast differentiation. FASEB J. 4, 3111–3123 (1990).
Miyama, K. et al. A BMP-inducible gene, dlx5, regulates osteoblast differentiation and mesoderm induction. Dev. Biol. 208, 123–133 (1999).
Zhang, J. et al. Phenotypic analysis of Dlx5 overexpression in post-natal bone. J. Dent. Res. 87, 45–50 (2008).
Samee, N. et al. Increased bone resorption and osteopenia in Dlx5 heterozygous mice. J. Cell. Biochem. 107, 865–872 (2009).
Naot, D. et al. Differential gene expression in cultured osteoblasts and bone marrow stromal cells from patients with Paget’s disease of bone. J. Bone Miner. Res. 22, 298–309 (2007).
Park, K. H. et al. Zinc inhibits osteoclast differentiation by suppression of Ca2 + -Calcineurin-NFATc1 signaling pathway. Cell Commun. Signal. 11, 74 (2013).
Acknowledgements
This work was supported by the Mid-career Researcher Program (NRF-2018R1A2B6007376) and by the Basic Science Research Program (NRF-2016R1D1A1B03933182) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education.
Author information
Authors and Affiliations
Contributions
K.M.L. and K.H.P. conceived and designed the study and wrote the manuscript. K.M.L., K.H.P., J.S.H., H.S.J., D.S.Y., and H.A.R. performed the experiments and analyzed the data. K.W.P., M.L., and C.Y.M. contributed to the in vivo experiments including fracture surgery. J. K. performed the revised experiments and analyzed the data. S.H.K., W.J.C., and S.W.P. provided input into clinical significance and interpretation of experimental work. J.W.L. conceived and oversaw the project and wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, KM., Park, K.H., Hwang, J.S. et al. Inhibition of STAT5A promotes osteogenesis by DLX5 regulation. Cell Death Dis 9, 1136 (2018). https://doi.org/10.1038/s41419-018-1184-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-1184-7
This article is cited by
-
RUNX transcription factors: biological functions and implications in cancer
Clinical and Experimental Medicine (2024)
-
Downregulation of the RNA-binding protein PUM2 facilitates MSC-driven bone regeneration and prevents OVX-induced bone loss
Journal of Biomedical Science (2023)
-
Osteomodulin positively regulates osteogenesis through interaction with BMP2
Cell Death & Disease (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}