
Abstract
Differentiation of mesenchymal stem cells (MSCs) into osteoblasts is a critical process for proper skeletal development and acquisition/maintenance of bone mass. However, since this regulatory mechanism has not yet been fully elucidated, the treatment of severe osteoporosis and fractures is a challenge. Here, through a comprehensive analysis of gene expression during the differentiation of MSCs into osteoblasts, we show that the forkhead transcription factor Foxf2 is a crucial regulator of this process. Foxf2 expression transiently increased during MSC osteoblastic differentiation. Overexpression of Foxf2 in MSCs inhibited osteoblastic differentiation, and conversely, knockdown of Foxf2 expression promoted this process. Osteoprogenitor-specific Foxf2 knockout mice developed a high bone mass phenotype due to increased bone formation. RNA-seq analysis and molecular experiments revealed that Foxf2 regulation of bone formation is mediated by Wnt2b. Knockdown of Foxf2 in mouse femurs enhanced bone regeneration in vivo. FOXF2 expression was correlated with hip bone mineral density in postmenopausal women with low bone mass. Finally, inhibition of FOXF2 promoted osteoblastic differentiation of human MSCs. This study uncovers a critical role of Foxf2 in the differentiation of MSCs into osteoblasts and provides insight into the pathogenesis associated with bone-related diseases such as osteoporosis and nonunion after fracture.
Similar content being viewed by others


Introduction
Osteoblasts are cells of mesenchymal origin and play an important role in skeletal development and bone formation1,2. Thus, understanding the mechanisms that regulate osteoblast differentiation is essential for developing strategies to treat bone-related diseases such as osteoporosis and nonunion after fracture. Over the past three decades, advances in molecular and genetic research have identified various regulatory processes of bone formation. Among them, Wnt signaling plays a central role in this regulation3. Binding of Wnt to the receptor complex activates β-catenin-dependent canonical and β-catenin-independent noncanonical signaling pathways4. Canonical Wnt binds to the receptor complex of Frizzled and low-density lipoprotein receptor-related protein 5 (Lrp5) or Lrp65. This complex induces the accumulation of β-catenin in target cells4. The accumulation of β-catenin leads to its migration into the nucleus, where it interacts with the T-cell factor/lymphoid enhancer factor (Tcf/Lef) family to initiate the transcription of target genes5.
The Wnt canonical pathway is vital for bone formation because loss-of-function or gain-of-function mutations affecting LRP5 in humans or mice alter bone formation without affecting bone resorption parameters4. Regarding the clinical use of the canonical Wnt pathway, sclerostin (SOST)-neutralizing antibodies have been developed6. These antibodies are currently used to treat osteoporosis in the clinic7, since SOST, a Wnt antagonist, interacts with LRP5 and LRP6 to inhibit this pathway8. However, due to the high cost of antibody drugs, the limited continuous use period of only one year, and the occurrence of vascular adverse events9, novel therapeutic targets for this pathway are needed. In addition, with the advent of an aging society in recent years, the number of osteoporotic fractures is increasing10. Surprisingly, the incidence of nonunion after fresh osteoporotic vertebral fractures reaches 17.5% in 65- to 85-year-old women11, but there are no drugs that can be used to treat nonunion, possibly due to insufficient knowledge of how to effectively stimulate bone formation. Therefore, it is essential to clarify the mechanism of bone formation from a new perspective.
The forkhead transcription factor Foxf2 is expressed in the mesenchyme of tissues required for morphogenesis and cell differentiation12. This molecule is involved in the cascade of interactions between the epithelium and the mesenchyme12. FOXF2 promotes bone metastasis in breast cancer cells by activating the BMP4/SMAD1 signaling pathway13. Since Foxf2 is strongly expressed in limb buds during embryonic development, the importance of Foxf2 in skeletal development has been suggested12. However, the role of Foxf2 in skeletal development and bone remodeling remains unclear. Interestingly, Foxf2 is thought to be both an activator and repressor of gene expression14, but the role of Foxf2 in osteoblast differentiation is unknown.
Here, we demonstrate that Foxf2 regulates the differentiation of mesenchymal stem cells (MSCs) into osteoblasts via canonical Wnt signaling and provide experimental evidence for the role of Foxf2 in bone remodeling. These findings suggest that targeting the Foxf2-canonical Wnt axis is a promising therapeutic strategy to promote bone formation.
Materials and methods
Animals
Foxf2 flox/flox (hereafter, Foxf2f/f) mice and the paired related homeobox 1 (Prx1)-Cre mouse line (hereafter, Prx1-Cre mice) have been described previously15,16. We crossed Prx1-Cre mice with Foxf2f/f mice to obtain Prx1-Cre tg/Foxf2f/f (hereafter, Foxf2osp−/−) mice. We then crossed Foxf2osp−/− with Foxf2f/f mice, and their offspring Foxf2osp−/− mice and Foxf2f/f mice were used for the experiments. All mice were maintained under standard conditions with food and water available ad libitum under a 12 h light/dark cycle.
RNA-seq analysis
To characterize the transcriptome profile during osteoblast differentiation in ST2 cells, we performed RNA-seq for cells treated with BMP-2 for two days and control cells (Day 0). Total RNA extracted from these cells was sent to Hokkaido System Science Co. for library preparation and sequencing. Total RNA was sequenced at the desired depth (100×) on RNA-seq (Illumina HiSeq). To characterize the transcriptome profile in Foxf2-deleted cells, we performed RNA-seq of Foxf2osp−/− and Foxf2f/f samples. Total RNA extracted from calvarial bones was sent to Macrogen Japan Co. for library preparation and sequencing. Captured mRNA by oligo dT primers was sequenced at the desired read count (40 M reads) on RNA-seq (Illumina NovaSeq). The reads were aligned to the mouse genome with the Spliced Transcripts Alignment to a Reference alignment software. The raw read counts were generated using StringTie-count for each annotated gene. The quality and quantity of the RNA obtained was assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Cleveland, OH, USA) and analyzed with the Agilent 2100 Bioanalyzer system (Agilent Technologies, Santa Clara, CA, USA). Library preparation was performed using the TruSeq Stranded mRNA LT Sample Preparation Kit.
Pathway analysis
For identification of the pathways affected by Foxf2 deletion, significantly differentially expressed genes whose expression was increased or decreased by 2.0 times or more in the Foxf2osp−/− bones compared to the Foxf2f/f bones were functionally classified by the PANTHER database (http://pantherdb.org/) into underlying pathways.
Cell culture
Cells were purchased from the Riken Cell Bank (Tsukuba, Japan). ST2 cells were maintained in RPMI 1640 (Sigma-Aldrich, St Louis, MO, USA) containing 2 mM L-glutamine, 100 units/ml penicillin, 10 µg/ml streptomycin and 10% fetal bovine serum (FBS; Sigma-Aldrich) in 5% CO2. Primary murine bone marrow mesenchymal stem cells (BMSCs) were cultured as previously described17. Regarding osteogenic differentiation, when the culture plates became 100% confluent, the culture media of each group was changed to osteogenic media containing 10 mM β-glycerophosphate (Sigma-Aldrich) and 50 μg/ml ascorbic acid phosphate (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) with 100 nM dexamethasone (Life Technologies, Grand Island, NY, USA) or 100 ng/ml BMP-2 (Funakoshi, Tokyo, Japan). The cells were cultured for two days to examine knockdown efficiency and for seven days to examine osteoblast differentiation, except as specified.
The results are representative of more than three individual experiments.
Transfection and infection
cDNA fragments of Foxf2 and Wnt2b were chemically synthesized and then cloned into the pBapo CMV vector (TaKaRa, Tokyo, Japan) and PSF-CMV-PURO-NH2-V5 plasmid (Sigma-Aldrich). For the Foxf2 overexpression study, ST2 cells were seeded and transfected using Lipofectamine LTX reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. For the knockdown experiments, Foxf2 siRNA (ON-TARGET plus L-043398) and FOXF2 siRNA (ON-TARGET plus L-008261 SMARTpool) were purchased from Dharmacon. As a negative control, ON-TARGETplus Nontargeting Pool (Dharmacon, Chicago, IL, USA) was used. Cells were seeded one day before siRNA treatment and transfected with 20 nM siRNA using Lipofectamine RNAiMAX (Invitrogen). The siRNA sequences are shown in Supplementary Table 1.
Quantitative real-time PCR analysis
RNA from tissues and cultured cells was also extracted using TRIzol reagent (Invitrogen). Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer’s instructions. We performed a quantitative analysis of gene expression using the Mx3000p qPCR system (Agilent Technologies). Gapdh expression was used as an internal control. Primer sequences are shown in Supplementary Table 2.
ALP staining
ST2 cells were cultured in osteogenic medium (β-glycerophosphate, ascorbic acid phosphate, and dexamethasone) for 14 days. The cells were then fixed for 20 min in 4% paraformaldehyde and washed three times with PBS. Next, the cells were stained with an ALP Stain Kit (Wako) according to the manufacturer’s instructions.
Western blot analysis
For immunological detection, 20 μg of cell lysate was separated using SDS-PAGE (7.5–10% Tris gel). Then, the proteins were blotted onto a PVDF membrane, which was incubated with the PVDF blocking reagent Can Get Signal (TOYOBO, Osaka, Japan). Proteins were probed with primary antibodies against β-catenin (1/1000, Abcam, Cambridge, MA, USA), FOXF2 (1/1000, Abnova, Walnut, CA, USA), WNT2B (1/2000, Abcam), LMNB1 (1/2000, Proteintech, Rosemont, IL, USA), and GAPDH (1/2000, MBL, Aichi, Japan). A horseradish peroxidase-conjugated goat anti-rabbit antibody was then added, and secondary antibodies were detected through autoradiography using enhanced chemiluminescence (ECL Plus, General Electric Healthcare, Chicago, IL, USA).
Microcomputed tomography analysis
We obtained three-dimensional images of distal femurs via microcomputed tomography (μCT, Comscan, Kanagawa, Japan). We examined more than five mice for each group for bone morphometric analysis.
Biochemistry
Blood samples from mice were collected through cardiac puncture, kept at room temperature for 30 min, and centrifuged at 12,000 g for 15 minutes at 4 °C. Serum procollagen type 1 N-terminal propeptide (P1NP) and C-terminal telopeptide of type 1 collagen (CTX-I) levels were measured via ELISAs for mouse P1NP (Cloud-Clone, Katy, TX, USA) and CTX-I (Immunodiagnostic Systems) according to the manufacturer’s instructions, respectively.
Histomorphometric analysis
We injected mice with calcein (20 mg/kg, intraperitoneally, Sigma-Aldrich) seven and two days before euthanasia. Undecalcified sections of the distal femur were subjected to toluidine blue staining and TRAP staining, and we performed histomorphometric analyses using an OsteoMeasure Analysis System (Osteometrics Inc., Atlanta, GA, USA) as previously described18,19.
Luciferase reporter assay
ST2 cells were plated into 24-well plates and incubated overnight at 37 °C. The next day, the cells were transfected with siRNA-Foxf2 or siRNA-negative control (15 pmol each), pTOP-FLASH or pFOP-FLASH reporter plasmids (0.5 μg each), and pGL4.74 hRluc/TK Renilla plasmid using Lipofectamine LTX. Twenty-four hours later, luciferase activity was examined.
Chromatin immunoprecipitation (ChIP)
ST2 cells were crosslinked with 1% formaldehyde for 10 min and quenched with 0.125 M glycine for 5 min. The cells were washed and lysed with lysis buffer (10 mM Tris-HCl, pH 7.4; 10 mM NaCl; 5 mM MgCl2; 0.2% NP-40) and resuspended with 100 μl of glycerol buffer (10 mM Tris-HCl, pH 7.4; 0.1 mM EDTA; 5 mM MgAc2; 25% glycerol) and 100 μl of reaction buffer (100 mM Tris-HCl, pH 7.4; 50 mM KCl; 8 mM MgCl2; 2 mM CaCl2). Chromatin was sheared by micrococcal nuclease, the reaction was stopped by EGTA, and the samples were diluted with 1,000 μl of IP buffer (25 mM Tris-HCl, pH 8.0; 150 mM NaCl; 2 mM EDTA; 1% Triton X-100; 0.1% SDS). The chromatin solution was incubated with anti-V5 antibody (Invitrogen)-conjugated Dynabeads Protein G (Invitrogen) at 4 °C for 3 h. Beads were washed with low salt wash buffer (20 mM Tris-HCl, pH 8.0; 150 mM NaCl; 2 mM EDTA; 1% Triton X-100; 0.1% SDS) four times and high salt wash buffer (20 mM Tris-HCl, pH 8.0; 500 mM NaCl; 2 mM EDTA; 1% Triton X-100; 0.1% SDS), and immune complexes were eluted from beads with ChIP elution buffer (50 mM Tris-HCl, pH 8.0; 10 mM EDTA; 1% SDS) at 65 °C. Eluates were additionally incubated at 65 °C to reverse crosslinking and then incubated with proteinase K at 55 °C. DNA was purified by a QIAquick PCR purification kit (Qiagen, Valencia, CA, USA). Aliquots and whole-cell extracts (serving as input samples) were analyzed by PCR amplification. The proximal promoter regions of candidate Foxf2 target genes in the resulting DNA fragments were PCR amplified using the primers listed in Supplementary Table 3.
Preparation of patient-derived bone samples and human BMSCs
We enrolled 11 patients who underwent spinal surgeries at the Orthopedic Department of the Tokyo Medical and Dental University Hospital. We obtained written informed consent from all the patients. The inclusion criteria were as follows: (a) postmenopausal women with low bone mineral density (T score ≤ −1.0) who had undergone spinal surgery for a lumbar degenerative disease (e.g., lumbar spondylolisthesis and lumbar spinal stenosis), (b) subjects who had not been treated with antiosteoporosis drugs affecting bone metabolism (e.g., bisphosphonates, selective estrogen receptor modulators, teriparatide, or denosumab), (c) subjects with no systemic disease affecting bone metabolism (e.g., secondary osteoporosis, osteogenesis imperfecta), (d) subjects with no fresh osteoporotic fracture, and (e) subjects with no severe liver or kidney dysfunction. We collected vertebral bone samples.
Human BMSCs were cultured as previously described with modifications20. After informed consent was obtained, human BMSCs were cultured from bone marrow aspirates of female patients who had received spine surgery at Tokyo Medical and Dental University under a protocol that was approved by the institutional review board. First, 2 ml of bone marrow aspirate was obtained from the iliac crest of each patient using a bone marrow biopsy needle (Cardinal Health, Dublin, OH, USA). Next, the aspirate was added to 20 ml of standard medium (Dulbecco’s modified Eagle’s medium (DMEM), Sigma-Aldrich) containing 10% fetal bovine serum (Life Technologies) and 1% antibiotic-antimycotic (10,000 U/ml penicillin G sodium, 10,000 μg/ml streptomycin sulfate and 25 μg/ml amphotericin B; Life Technologies) that contained 200 IU sodium heparin (Mochida Pharmaceutical Co., Ltd., Tokyo, Japan) and then centrifuged to remove the fat layer. Bone marrow cells were then resuspended in a standard medium. Subsequently, the bone marrow cells were plated into a 100-cm2 dish. The cells were then cultured in each medium at 37 °C in a humidified atmosphere containing 95% air and 5% CO2, and the medium was replaced every three days. When primary cultures became nearly confluent, the cells were detached with 0.25% trypsin containing 1 mM EDTA (Life Technologies) and subsequently replated for each assay. The collected hBMSCs were either cultured or preserved separately; the cells from individual donors were assayed independently to prevent cross-contamination of hBMSCs from different donors. Regarding osteogenic differentiation, when the culture plates became 100% confluent, the culture medium of each group was changed to osteogenic medium containing 10 mM β-glycerophosphate (Sigma-Aldrich) and 50 μg/ml ascorbic acid phosphate (Wako) with 100 nM dexamethasone (Life Technologies).
Bone marrow ablation
Mice were anesthetized intraperitoneally with ketamine and xylazine. After removal of the hair from both hind limbs, the bone marrow of both femora on each animal was ablated as described previously21. Briefly, bilateral longitudinal incisions were made on the knees of each mouse to expose the femoral condyle by dislocation of the patella. Next, a hole was made at the intercondylar notch of the femur using a dental drill. A 0.6 mm diameter Kirschner wire was inserted in the proximal end of the femur to confirm the completion of marrow ablation by radiography. Then, 20 µmol of siRNA-Foxf2 or the negative control was injected into the bone marrow. After injection, the dislocated patella was repositioned, and the skin was sutured. Mice were euthanized at day seven after ablation, and microcomputed tomography analyses were performed.
Statistics
All data are presented as the mean ± SEM (n ≥ 3). We performed statistical analysis using Student’s t test or one-way ANOVA and Tukey’s HSD test. P < 0.05 was considered statistically significant.
Study approval
All animal experiments were performed with the approval of the Animal Study Committee of Tokyo Medical and Dental University and conformed to relevant guidelines and laws. Regarding patient-derived bone samples and human BMSCs, all protocols were approved by the Ethics Committee of Tokyo Medical and Dental University (G2020-031). Written informed consent was received prior to participation.
Results
Foxf2 regulates osteoblastic differentiation of MSCs
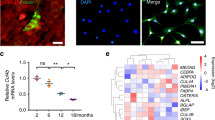
To investigate the genes involved in MSC differentiation into osteoblasts, we first attempted to identify the genes expressed in this process, particularly those genes whose expression changes during osteoblast differentiation. To this end, we administered BMP-2 to pluripotent ST2 mesenchymal progenitors for 2 days, an established model for studying osteoblast differentiation22, and analyzed gene expression by RNA-seq. Alp and Sp7, markers of osteoblast differentiation, showed upregulated expression with BMP-2 treatment, confirming that the experiment was conducted correctly (Fig. 1a). To identify genes that are expressed in undifferentiated MSCs and whose expression is increased by osteogenic differentiation stimulation, we investigated genes with FPKM >10 on Day 0 and whose expression was increased by BMP-2 treatment. Among the genes whose expression increased more than 2-fold by the stimulation of osteoblast differentiation, Kazald1, Hpgd, and Smad6 have been previously reported to be associated with osteoblast differentiation and/or activity (Fig. 1a)23,24,25. Accordingly, among the genes whose function in osteoblast differentiation is not well understood, we selected Foxf2 as the gene with the highest fold change (Fig. 1a). Then, to investigate the changes in Foxf2 expression during MSC differentiation into osteoblasts, we administered BMP-2 to ST2 cells for seven days and analyzed gene expression by qPCR analysis. While the expression of Alp, an osteoblastic differentiation marker, was increased by BMP-2 administration over time, Foxf2 expression was transiently increased on Day 3 and then decreased on Day 7 (Fig. 1b). The protein levels of FOXF2 were also transiently increased on Day 3 during BMP2-induced osteoblast differentiation (Fig. 1c). The change in the expression of Foxf2 during osteoblastic differentiation of ST2 cells prompted us to investigate whether Foxf2 regulates osteoblastic differentiation of MSCs. First, we tested which siRNA would most effectively reduce Foxf2 mRNA expression. SiRNA#2 resulted in the greatest decrease in the expression of Foxf2, so we selected siRNA#2 for use in the following experiments (Fig. 1d). Regarding osteoblast differentiation, knockdown of Foxf2 significantly induced osteoblastic differentiation of ST2 cells, as demonstrated by the increase in Alp and Bglap expression (Fig. 1e). We then investigated the effect of Foxf2 knockdown on the osteogenic potential of ST2 cells by performing ALP staining. Foxf2 knockdown increased ALP staining compared to the control treatment (Fig. 1f). Since inhibition of Foxf2 accelerated osteoblastic differentiation, we next investigated whether increasing Foxf2 expression would inhibit their differentiation. In the transient DNA transfection assay, Foxf2 repressed the osteoblastic differentiation of ST2 cells (Fig. 1g). Collectively, these results demonstrate that Foxf2 physiologically regulates the osteoblastic differentiation of MSCs.

a Changes in gene expression were examined between Days 0 and 2 after BMP-2 stimulation by RNA-seq analysis. Only representative genes with FPKM > 10 on Day 0 and whose expression showed a 2-fold increase by BMP-2 treatment are shown. Although ALP and Sp7 did not meet the requirements, they are included in the table as markers of osteoblast differentiation. b Changes in Foxf2 mRNA expression induced by BMP-2 treatment during ST2 cell differentiation, as determined by qPCR. ANOVA and Tukey HSD test. *, P < 0.05. c Changes in the expression of FOXF2 during osteoblast differentiation as determined at the protein level with Western blot analysis. d Foxf2 knockdown efficiency. ST2 cells were cultured and transiently transfected with siRNA-Foxf2. Among the siRNAs tested, #2 resulted in the greatest reduction in Foxf2 mRNA expression. *, P < 0.05. e Effect of Foxf2 knockdown on osteoblastic differentiation. ST2 cells were cultured and transiently transfected with siRNA-Foxf2. Note the significant increase in osteoblast differentiation markers in the Foxf2 knockdown cells. *, P < 0.05. f Foxf2 knockdown enhanced the osteogenesis of ST2 cells. Representative images of ALP staining of the ST2 cells transfected with siRNA-Foxf2. N = 3. g Effect of Foxf2 overexpression on osteoblast differentiation. ST2 cells were cultured and transiently transfected with Foxf2. Note the significant decrease in osteoblast differentiation markers in the Foxf2-expressing cells. *, P < 0.05.
Foxf2 deficiency in osteoblast progenitors results in high bone volume in vivo
To investigate the role of Foxf2 in bone metabolism, we generated conditional mesenchymal osteoblast progenitor-specific Foxf2 knockout mice because germline Foxf2-null mice die shortly after birth26. To achieve this, we crossed Foxf2f/f mice with Prx1-Cre mice to generate Foxf2osp−/− mice. Prx1 is expressed in mesenchymal osteoblast progenitors in limb buds and the craniofacial mesenchyme27. Prx1+ cells have also been reported as mesenchymal osteoblast progenitors residing in both the periosteum of cortical bone and the bone marrow in adult mice28,29,30. Foxf2osp−/− mice were born at the expected Mendelian ratio, indicating that embryonic development can proceed without Foxf2 expression in mesenchymal osteoblast progenitors in the limb buds and craniofacial mesenchyme. Before analyzing the Foxf2osp−/− mice, we confirmed that the Prx1-cre transgene did not result in a notable phenotype at 3 months of age (not shown). Genetic compensation, a change in the Foxf1 expression level, was not observed upon deletion of Foxf2 in the mouse calvaria (Fig. 2a). The deletion of FOXF2 was confirmed by Western blotting (Fig. 2b). Micro-CT analysis of femoral trabecular bone of the 3-month-old Foxf2osp−/− mice demonstrated a 43% increase in bone mass compared to that of the Foxf2f/f mice as measured by BV/TV (Fig. 2c). Furthermore, micro-CT analysis indicated that bone mineral density and trabecular number were increased, and trabecular separation was decreased in the long bones of the Foxf2osp−/− mice compared to the long bones of the Foxf2f/f mice (Fig. 2d). Accordingly, the serum level of P1NP, a biomarker correlating with bone formation31, was increased in the Foxf2osp−/− mice, although CTX-I, a serum bone resorption marker31, showed no significant difference between the two groups (Fig. 2e). In addition, toluidine blue staining of femoral sections showed an increase in the number of osteoblasts in the Foxf2osp−/− mice compared to the Foxf2f/f mice (Fig. 2f), whereas the number of osteoclasts was not affected (Fig. 2g). Last, calcein labeling revealed that the bone formation rate was significantly increased in the Foxf2osp−/− mice compared to the Foxf2f/f mice (Fig. 2h). These results indicate that Foxf2 expression in osteoprogenitors regulates bone formation in vivo.

a Gene expression of Foxf2f/f and Foxf2osp−/− mouse calvaria (P2). The expression of Foxf2 was significantly decreased in the Foxf2osp−/− mice, while no significant difference was observed in Foxf1, as demonstrated by qPCR analysis. *, P < 0.05 versus the Foxf2f/f mice. ns, not significant. b FOXF2 protein levels in the Foxf2osp−/− mice based on Western blot analysis. Calvarial bones were isolated from the Foxf2f/f (control) or Foxf2osp−/− mice. c Micro-CT analysis of 3-month-old Foxf2osp−/− and Foxf2f/f mice. Representative 3D images of femoral trabecular bone from both groups are shown. *, P < 0.05 versus the Foxf2f/f mice. d Micro-CT analysis of 3-month-old Foxf2osp−/− and Foxf2f/f mice. *, P < 0.05 versus the Foxf2f/f mice. e Serum P1NP and CTX-I levels measured in 3-month-old female mice. P1NP was increased in the Foxf2osp−/− mice. *, P < 0.05 versus the Foxf2f/f mice. ns, not significant. (f and g) Bone histomorphometric analysis of 3-month-old Foxf2f/f and Foxf2osp−/− mice. The number of osteoblasts was increased in the Foxf2osp−/− mice, whereas the number of osteoclasts was not affected. Representative images of toluidine blue staining (f) and TRAP staining (g). White arrowheads show osteoblasts, and black arrows show osteoclasts. Scale bars, 100 μm. *, P < 0.05 versus the Foxf2f/f mice. ns not significant. h Representative images of calcein labeling. *, P < 0.05 versus the Foxf2f/f mice. All data represent the mean ± SEM.
Inhibition of Foxf2 enhances the differentiation of MSCs into osteoblasts via activation of the canonical Wnt signaling pathway
To determine whether the increase in bone formation in the Foxf2osp−/− mice was cell-autonomous, we isolated BMSCs from the Foxf2f/f and Foxf2osp−/− mice. In accordance with the in vivo observations, the Foxf2osp−/− BMSCs showed an increase in osteoblastic differentiation, as demonstrated by an increase in the expression of osteoblast markers (Fig. 3a). To understand the mechanistic role of Foxf2 in osteoprogenitors, we performed an in-depth analysis of the effect of Foxf2 deficiency on the genomic transcriptional network of osteoprogenitors. Whole-transcriptome RNA-seq was performed on calvarial bones derived from either the Foxf2f/f or Foxf2osp−/− mice. RNA-seq analysis revealed that loss of Foxf2 in osteoprogenitors significantly upregulated the expression of 57 genes while downregulating the expression of 633 genes based on fold change and p value of the comparison pairs (Fig. 3b and Supplementary Tables 4, 5 and 6). To identify target genes of Foxf2, we relied on a computational approach using an established database (PANTHER database). The PANTHER pathway analysis revealed altered expression of many genes associated with inflammation mediated by the chemokine and cytokine signaling pathway, nicotinic acetylcholine receptor signaling pathway, and Wnt signaling pathway in the comparison between the Foxf2osp−/− and Foxf2f/f mice (Fig. 3c). Among the many pathways predicted to be potential pathways by the PANTHER database, we focused on the Wnt signaling pathway because that pathway has an important role in osteoblast differentiation and proliferation4. Indeed, several of the genes with upregulated or downregulated expression are associated with canonical Wnt signaling, e.g., Wnt2b and Lgr5 (Fig. 3d). We then validated the results of RNA-seq analysis by qPCR using RNAs from the Foxf2osp−/− and Foxf2f/f mouse calvaria. The expression of Foxf2 was downregulated and that of Wnt2b was upregulated in the Foxf2osp−/− mouse calvaria, reproducing the results of RNA-seq analysis (Figs. 2a and 3e). Furthermore, the increase in WNT2B protein levels in the Foxf2f/f mouse calvaria was confirmed by Western blotting (Fig. 3f).

a Gene expression in BMSCs isolated from the Foxf2f/f and Foxf2osp−/− mice by qPCR. Foxf2osp−/− BMSCs showed a significant increase in osteogenic markers. *, P < 0.05 versus the Foxf2f/f mice. b, c, and d RNA-seq data from calvaria of the Foxf2f/f and Foxf2osp−/− mice. b Loss of Foxf2 in osteoprogenitors significantly upregulated the expression of 57 genes while downregulating the expression of 633 genes. c Bar chart of pathways with potentially differentially expressed genes extracted from calvarial bone expression data. The PANTHER server with default parameters for pathway analysis was used for pathway analysis. The length of each bar shows how many genes have been assigned to a given pathway. d There were significant changes in the expression of Wnt2b and Lgr5, which are associated with canonical Wnt signaling. e Gene expression in calvaria bones isolated from the Foxf2f/f and Foxf2osp−/− mice by qPCR. Foxf2osp−/− calvaria showed a significant increase in Wnt2b expression. f WNT2B protein levels in the Foxf2osp−/− mice based on Western blot analysis. Calvaria bones were isolated from the Foxf2f/f (control) or Foxf2osp−/− mice. *, P < 0.05 versus the Foxf2f/f mice. All data represent the mean ± SEM.
Wnt2b is a molecular target of Foxf2 in MSCs
Pathway analysis and the significant increase in Wnt2b expression suggest that the Wnt pathway is enhanced in the osteoprogenitors of the Foxf2osp−/− mice. As expected, the deletion of Foxf2 increased the cytoplasmic expression of WNT2B and nuclear expression of β-catenin (Fig. 4a). Furthermore, TOPFlash activity was increased by 210% in the ST2 cells with Foxf2 knockdown (Fig. 4b). Collectively, these results indicate that Foxf2 acts upstream of the canonical Wnt signaling pathway to repress the osteoblastic differentiation of MSCs. Since Wnt2b is a candidate target of Foxf2, we investigated whether Wnt2b is a direct target gene of Foxf2. ChIP-PCR analysis showed that the FOXF2 protein bound to the Wnt2b promoter regions containing the putative Foxf2 binding sequence in ST2 cells (Fig. 4c). In addition, knockdown of Foxf2 promoted Wnt2b expression, whereas overexpression of Foxf2 suppressed Wnt2b expression in ST2 cells (Fig. 4d, e). To further test the concept of Foxf2 as a critical regulator of Wnt signaling in MSCs, we overexpressed Wnt2b in the ST2 cells overexpressing Foxf2 to rescue the effects of Foxf2 overexpression. Indeed, the decreased Alp and Bglap expression upon Foxf2 overexpression was reversed by overexpression of the Wnt2b plasmid in 7-day-differentiated ST2 cells, which further suggests that Wnt2b is a downstream mediator of Foxf2 signaling during osteoblastic differentiation of MSCs (Fig. 4f). Finally, to investigate the importance of Wnt2b in osteoblast differentiation, we tested whether Wnt2b overexpression induced osteoblast differentiation. ALP staining and qPCR analysis showed that overexpression of Wnt2b promoted osteoblastic differentiation of ST2 cells (Fig. 4g). Collectively, our results suggest that Foxf2 represses the osteoblastic differentiation of MSCs via the Wnt2b/β-catenin signaling pathway, at least in part.

a Protein levels in the Foxf2f/f and Foxf2osp−/− mouse BMSCs. Note the increase in β-catenin and WNT2B protein levels in the Foxf2osp−/− mice. b ST2 cells were transfected with TOPFlash or FOPFlash and siRNA as indicated. TOPFlash activity was increased in the Foxf2 knockdown cells. *, P < 0.05 versus the control. c The binding of FOXF2 protein to the Wnt2b promoter regions containing the putative Foxf2 binding sequence was assessed by ChIP PCR in the ST2 cells transfected with PSF-CMV-PURO-NH2-V5-Foxf2 using an anti-V5 antibody or IgG control. d Knockdown of Foxf2 increased the expression of Wnt2b in ST2 cells. *, P < 0.05 versus the control. e Transient overexpression of Foxf2 decreased Wnt2b expression in ST2 cells. *, P < 0.05 versus the control. f Wnt2b rescued the inhibitory effect of Foxf2 on osteoblast differentiation. ST2 cells overexpressing Foxf2 were cotransfected with either the Wnt2b-expressing plasmid or control plasmid. Subsequently, the expression of osteoblast differentiation markers was analyzed by qPCR. Note that the inhibitory effect of Foxf2 overexpression on osteoblast differentiation was reversed by coexpression of Wnt2b. *, P < 0.05. ns, not significant. g Transient overexpression of Wnt2b promoted osteoblast differentiation in ST2 cells. Representative images of ALP staining of the ST2 cells transfected with Wnt2b or negative control (left). Note the significant increase in osteoblast differentiation markers in the Wnt2b-expressing cells by qPCR (right). All data represent the mean ± SEM.
Knockdown of Foxf2 promotes bone formation in vivo, and inhibition of FOXF2 promotes osteoblastic differentiation of human MSCs
To address the functional role of Foxf2 in vivo, we ablated the bone marrow of mouse femurs and then treated them with siRNA-Foxf2 or control scrambled siRNA. Treatment with siRNA-Foxf2 enhanced bone regeneration compared with that of the mice treated with control scrambled siRNA at Day 7 after bone marrow ablation (Fig. 5a, b and Supplementary Fig. 1). These results indicate that Foxf2 also represses the bone regeneration process in adult mice.

a Representative micro-CT images of bone regeneration after femoral bone marrow ablation in the negative control and siFoxf2-treated mice at three months. b Bone volume/tissue volume (BV/TV; %). Measurement areas are indicated in (a). *, P < 0.05. c Correlation analysis demonstrated that FOXF2 mRNA expression was decreased along with the decreasing hip bone mineral density levels in postmenopausal female patients with low bone mass. n = 11. d Knockdown of FOXF2 increased the expression of WNT2B in human BMSCs. *, P < 0.05. e Effect of FOXF2 knockdown on the osteoblastic differentiation of human BMSCs. Note the significant increase in osteoblast differentiation markers in the FOXF2 knockdown cells. *, P < 0.05. f A proposed mechanism through which Foxf2 in MSCs inhibits Wnt signaling in osteoprogenitors. All data are expressed as the mean ± SEM.
To investigate the importance of FOXF2 expression in humans, we then investigated the correlation between FOXF2 expression in bone samples of patients with low bone mass (T score ≤1.0) undergoing spinal surgery and bone mass. As a result, COL1A1 mRNA expression in bone tissue was shown to have a significant correlation with ALP mRNA expression and serum P1NP concentration (p = 0.01 and 0.01, respectively). Interestingly, FOXF2 expression was correlated with femoral bone mineral density (r = 0.63, p = 0.04) (Fig. 5c and Tables 1 and 2). Finally, to investigate the clinical importance of modulating FOXF2 expression in bone formation, we knocked down FOXF2 in human MSCs. The expression of WNT2B was increased by 30% by suppressing FOXF2 (Fig. 5d). Furthermore, FOXF2 knockdown significantly promoted the differentiation of MSCs into osteoblasts, as the expression of ALP, COL1A1, and SP7 was increased after seven days of culture (Fig. 5e). These results suggest that FOXF2 represses the differentiation of MSCs into osteoblasts via the Wnt pathway not only in mice but also in humans.
Discussion
The differentiation of MSCs into osteoblasts is essential in bone remodeling, especially bone formation32. From a clinical point of view, reduced bone formation has a profound impact on the development of osteoporosis and nonunion after fracture33,34. In this study, we found that Foxf2 was one of the top genes with upregulated expression during MSC osteoblast differentiation, and therefore, we investigated the function of Foxf2 during this process. Overexpression of Foxf2 in MSCs inhibited their osteoblastic differentiation, and conversely, knockdown of Foxf2 expression promoted osteoblast differentiation. Osteoprogenitor-specific Foxf2 knockout mice developed a high bone mass phenotype due to increased bone formation. RNA-seq analysis and molecular experiments identified Wnt2b as a target of Foxf2. Knockdown of Foxf2 in the mouse femur enhanced bone regeneration in vivo. FOXF2 expression was correlated with femoral bone mineral density in postmenopausal women with low bone mass. Finally, inhibition of FOXF2 promoted the osteoblastic differentiation of human MSCs. Collectively, Foxf2 plays a crucial role in the differentiation of MSCs into osteoblasts (Fig. 5f).
Deletion of Foxf2 in osteoprogenitors in mice resulted in a high bone volume phenotype, which is characterized by increased bone formation. The depletion of Foxf2 in the Foxf2osp−/− mouse model is restricted to multipotent mesenchymal progenitors in the appendicular skeleton29, suggesting that Foxf2 affects bone mass by regulating the osteoblastic differentiation of multipotent mesenchymal progenitors. Furthermore, our in vitro assessment showed that the ST2 cells transfected with siRNA-Foxf2 and Foxf2osp−/− MSCs were prone to differentiate into osteoblasts, suggesting that regulation of Foxf2 expression in MSCs is vital in regulating their differentiation into osteoblasts. Of note, a previous study showed that forced expression of Foxf2 mildly stimulates adipocyte differentiation35, suggesting that the expression of Foxf2 may be important in the determination of MSC differentiation into osteoblasts or adipocytes.
Foxf2 suppression was shown to activate Wnt signaling36, but the detailed mechanism of this effect has not been elucidated. In addition, WNT2B was shown to promote osteoblast differentiation of human MSCs37. Our study indicated that deletion of Foxf2 increases Wnt2b expression and that Foxf2 binds directly to the Wnt2b promoter in ST2 mesenchymal progenitors. Biochemical assays suggested that the FOXF2 protein contains an activation domain and may function as a transcriptional activator38. However, more recent studies, such as the recent findings that Foxf2 mediated transcriptional repression of target genes during Tgfβ-induced epithelial-mesenchymal transition39 and FOXF2 recruited nuclear receptor corepressor1 (NCoR1) and histone deacetylase 3 to the FOXQ1 promoter to inhibit its translation in BLBC cells40, suggest that Foxf2 may act as both activator and repressor14,41. RNA-seq analysis showed that loss of Foxf2 in osteoprogenitors significantly upregulated the expression of 57 genes while downregulating the expression of 633 genes. Since Foxf2 deficiency reduced the expression of many genes, Foxf2 may basically act to activate gene expression. However, it may be important for the osteoblastic differentiation of MSCs that Foxf2 binds directly to the Wnt2b promoter and acts to directly or indirectly repress Wnt2b expression. Gene regulation by members of the FOX family is not only achieved through cell-specific expression but is also fine-tuned through myriad post-translational modifications and interactions with specific cofactors, such as NCoR1, NCoR2, and Groucho41. Our study showed that FOXF2 binds directly to the promoter of Wnt2b, but it is still unclear what corepressor cooperates with FOXF2 to reduce Wnt2b expression. Accordingly, the detailed molecular mechanisms that mediate the activator and repressor functions of Foxf2 in vivo need to be clarified in the future. Interestingly, although genetic defects in Foxf2 are involved in the development of cleft palate42, overexpression of Wnt2b has also been reported to cause cleft palate in vivo43. Therefore, modulating Wnt2b expression may lead to cleft palate treatment caused by Foxf2 gene defects.
We found that knockdown of Foxf2 enhanced bone regeneration compared with that of the mice treated with negative control siRNA at Day 7 after bone marrow ablation. Bone marrow ablation induces a sequence of events, including forming a blood clot with inflammation and capillary invasion, mesenchymal cell migration, osteoblast proliferation, and new trabecular medullary bone formation, followed by osteoclastic bone resorption and bone marrow reconstitution44. Of these, the bone formation phase occurs between 6 and 10 days after surgery44. Therefore, the increase in bone mass seven days after surgery indicates that the local administration of siFoxf2 promoted bone formation. Thus, expanding on our research, local administration of a FOXF2 inhibitor to a fractured site may promote bone formation, leading to accelerated fracture healing.
The correlation analysis revealed that FOXF2 expression in bone tissue showed a significant correlation with femoral bone mineral density in postmenopausal women with a low bone mass. Given that Foxf2 is a transcription factor that represses the differentiation of MSCs into osteoblasts, it is interesting that FOXF2 expression in bone tissue was lower in patients with lower bone mass. Furthermore, based on the results that mice lacking Foxf2 in osteoprogenitors exhibit high bone mass, there is a discrepancy between human and mouse results. We speculate that this discrepancy may be due to differences in genetic background and the fact that the mice analyzed were young (3 months old), while the humans analyzed were elderly females. In a study investigating changes in gene expression in postmenopausal and premenopausal females, 29 of the 118 genes investigated were significantly altered45. Accordingly, the regulation of FOXF2 expression by estrogen will need to be investigated in the future. Of note, serum P1NP concentration, a marker of bone formation, was shown to be negatively correlated with the bone mineral density of postmenopausal females with low bone mass46,47,48. These results suggest that bone formation was enhanced in inverse proportion to bone mass in postmenopausal women with low bone mass. Consistent with the results of previous studies, ALP and COL1A1 expression in bone tissue, markers of osteoblastic differentiation, tended to show negative correlations with femoral bone mineral density in our study, although these relationships were not significant, possibly due to the small sample size (p = 0.08 and 0.19). Collectively, these results suggest that in postmenopausal women with low bone mass, a mechanism may be already in operation that suppresses the expression of FOXF2, a repressor of bone formation, to promote bone formation in response to low bone mineral density. How FOXF2 expression is regulated in postmenopausal women needs to be clarified in the future. Furthermore, it will be necessary to compare the expression level of FOXF2 in premenopausal female and male bone tissues with bone mineral density in the future.
The fact that FOXF2 expression in bone tissue showed a stronger association with femoral bone mineral density than the serum concentration of TRACP-5b and P1NP, the representative bone turnover markers currently in use, suggests that examining the expression of FOXF2 in bone tissue may be a sensitive indicator of osteoporosis. Furthermore, if the expression of FOXF2 can be systematically reduced by medication to promote bone formation, this strategy may be helpful in the treatment of bone-related diseases such as osteoporosis. Note that since the sample size of patient specimens in this study was small, the possibility of a type II error cannot be excluded. Considering future clinical applications, validation with larger sample sizes is essential, and further research is needed.
In conclusion, we demonstrated that Foxf2 represses osteoblastic differentiation of MSCs. Thus, from a clinical perspective, systemic or local suppression of FOXF2 expression may be a promising strategy for treating bone-related diseases such as osteoporosis and fractures.
References
Florencio-Silva, R., Sasso, G. R., Sasso-Cerri, E., Simões, M. J. & Cerri, P. S. Biology of bone tissue: structure, function, and factors that influence bone cells. Biomed. Res. Int. 2015, 421746 (2015).
Long, F. & Ornitz, D. M. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 5, a008334 (2013).
Manolagas, S. C. Wnt signaling and osteoporosis. Maturitas 78, 233–237 (2014).
Baron, R. & Rawadi, G. Targeting the Wnt/β-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology 148, 2635–2643 (2007).
MacDonald, B. T. & He, X. Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harb. Perspect. Biol. 4, a007880 (2012).
van Dinther, M. et al. Anti-sclerostin antibody inhibits internalization of sclerostin and sclerostin-mediated antagonism of Wnt/LRP6 signaling. PLoS One 8, e62295 (2013).
Cosman, F. et al. Romosozumab treatment in postmenopausal women with osteoporosis. N. Engl. J. Med. 375, 1532–1543 (2016).
Semënov, M., Tamai, K. & He, X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 280, 26770–26775 (2005).
Asadipooya, K. & Weinstock, A. Cardiovascular outcomes of romosozumab and protective role of Alendronate. Arterioscler. Thromb. Vasc. Biol. 39, 1343–1350 (2019).
Ensrud, K. E. Epidemiology of fracture risk with advancing age. J. Gerontol. A Biol. Sci. Med. Sci. 68, 1236–1242 (2013).
Inose, H. et al. Risk factors of nonunion after acute osteoporotic vertebral fractures: a prospective multicenter cohort study. Spine (Philos. Pa 1976) 45, 895–902 (2020).
Aitola, M., Carlsson, P., Mahlapuu, M., Enerbäck, S. & Pelto-Huikko, M. Forkhead transcription factor FoxF2 is expressed in mesodermal tissues involved in epithelio-mesenchymal interactions. Dev. Dyn. 218, 136–149 (2000).
Wang, S. et al. FOXF2 reprograms breast cancer cells into bone metastasis seeds. Nat. Commun. 10, 2707 (2019).
Xu, J., Liu, H., Lan, Y., Park, J. S. & Jiang, R. Genome-wide Identification of Foxf2 Target Genes in Palate Development. J. Dent. Res. 99, 463–471 (2020).
Reyahi, A. et al. Foxf2 is required for brain pericyte differentiation and development and maintenance of the blood-brain barrier. Dev. Cell 34, 19–32 (2015).
Logan, M. et al. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77–80 (2002).
Maridas, D. E., Rendina-Ruedy, E., Le, P. T. & Rosen, C. J. Isolation, Culture, and Differentiation of Bone Marrow Stromal Cells and Osteoclast Progenitors from Mice. J. Vis. Exp. https://doi.org/10.3791/56750 (2018).
Dempster, D. W. et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 28, 2–17 (2013).
Mulati, M. et al. The long noncoding RNA Crnde regulates osteoblast proliferation through the Wnt/β-catenin signaling pathway in mice. Bone 130, 115076 (2020).
Yamada, T. et al. After repeated division, bone marrow stromal cells express inhibitory factors with osteogenic capabilities, and EphA5 is a primary candidate. Bone 57, 343–354 (2013).
Fukuda, T. et al. MicroRNA-145 regulates osteoblastic differentiation by targeting the transcription factor Cbfb. FEBS Lett. 589, 3302–3308 (2015).
Yamaguchi, A. et al. Effects of BMP-2, BMP-4, and BMP-6 on osteoblastic differentiation of bone marrow-derived stromal cell lines, ST2 and MC3T3-G2/PA6. Biochem. Biophys. Res. Commun. 220, 366–371 (1996).
Wu, M., Chen, G. & Li, Y.-P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 4, 16009 (2016).
Uppal, S. et al. Mutations in 15-hydroxyprostaglandin dehydrogenase cause primary hypertrophic osteoarthropathy. Nat. Genet. 40, 789–793 (2008).
Shibata, Y., Tsukazaki, T., Hirata, K., Xin, C. & Yamaguchi, A. Role of a new member of IGFBP superfamily, IGFBP-rP10, in proliferation and differentiation of osteoblastic cells. Biochem. Biophys. Res. Commun. 325, 1194–1200 (2004).
Wang, T. et al. Forkhead transcription factor Foxf2 (LUN)-deficient mice exhibit abnormal development of secondary palate. Dev. Biol. 259, 83–94 (2003).
Cserjesi, P. et al. MHox: a mesodermally restricted homeodomain protein that binds an essential site in the muscle creatine kinase enhancer. Development 115, 1087–1101 (1992).
Wang, C. et al. NOTCH signaling in skeletal progenitors is critical for fracture repair. J. Clin. Invest 126, 1471–1481 (2016).
Greenbaum, A. et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495, 227–230 (2013).
Moore, E. R., Zhu, Y. X., Ryu, H. S. & Jacobs, C. R. Periosteal progenitors contribute to load-induced bone formation in adult mice and require primary cilia to sense mechanical stimulation. Stem Cell Res. Ther. 9, 190 (2018).
Kuo, T.-R. & Chen, C.-H. Bone biomarker for the clinical assessment of osteoporosis: recent developments and future perspectives. Biomark. Res. 5, 18 (2017).
Oryan, A., Kamali, A., Moshiri, A. & Baghaban Eslaminejad, M. Role of mesenchymal stem cells in bone regenerative medicine: what is the evidence? Cells Tissues Organs 204, 59–83 (2017).
Inose, H. et al. Bone turnover markers as a new predicting factor for nonunion after spinal fusion surgery. Spine (Philos. Pa 1976) 43, E29–E34 (2018).
Takahashi, A. et al. Loss of cyclin-dependent kinase 1 impairs bone formation, but does not affect the bone-anabolic effects of parathyroid hormone. J. Biol. Chem. 293, 19387–19399 (2018).
Gerin, I. et al. On the role of FOX transcription factors in adipocyte differentiation and insulin-stimulated glucose uptake. J. Biol. Chem. 284, 10755–10763 (2009).
Ormestad, M. et al. Foxf1 and Foxf2 control murine gut development by limiting mesenchymal Wnt signaling and promoting extracellular matrix production. Development 133, 833–843 (2006).
Jia, B. et al. Long noncoding RNA LINC00707 sponges miR-370-3p to promote osteogenesis of human bone marrow-derived mesenchymal stem cells through upregulating WNT2B. Stem Cell Res. Ther. 10, 67 (2019).
Hellqvist, M., Mahlapuu, M., Samuelsson, L., Enerbäck, S. & Carlsson, P. Differential activation of lung-specific genes by two forkhead proteins, FREAC-1 and FREAC-2. J. Biol. Chem. 271, 4482–4490 (1996).
Meyer-Schaller, N., Heck, C., Tiede, S., Yilmaz, M. & Christofori, G. Foxf2 plays a dual role during transforming growth factor beta-induced epithelial to mesenchymal transition by promoting apoptosis yet enabling cell junction dissolution and migration. Breast Cancer Res. 20, 118 (2018).
Kang, L. J. et al. Reciprocal transrepression between FOXF2 and FOXQ1 controls basal-like breast cancer aggressiveness. Faseb J. 33, 6564–6573 (2019).
Lam, E. W., Brosens, J. J., Gomes, A. R. & Koo, C. Y. Forkhead box proteins: tuning forks for transcriptional harmony. Nat. Rev. Cancer 13, 482–495 (2013).
Reynolds, K. et al. Wnt signaling in orofacial clefts: crosstalk, pathogenesis and models. Dis. Model. Mech. 12, dmm037051 (2019).
Medio, M. et al. Wnt/β-catenin signaling and Msx1 promote outgrowth of the maxillary prominences. Front. Physiol. 3, 375 (2012).
Yan, J., Sun, W., Zhang, J., Goltzman, D. & Miao, D. Bone marrow ablation demonstrates that excess endogenous parathyroid hormone plays distinct roles in trabecular and cortical bone. Am. J. Pathol. 181, 234–244 (2012).
Kósa, J. P. et al. Effect of menopause on gene expression pattern in bone tissue of nonosteoporotic women. Menopause 16, 367–377 (2009).
Saad, M. A., Aboelwafa, R. A. & Elsayed, E. H. Could procollagen type I N-terminal propeptide (PINP) and bone alkaline phosphatase (B-ALP) be valid alternative diagnostic markers to dual X-ray absorptiometry (DEXA) in elderly females with osteoporosis? An Egyptian radiological and laboratory monocentric study. Egypt. Rheumatol. Rehabil. 48, 20 (2021).
Tehrani, S. S. et al. Status of circulating bone turnover markers in elderly osteoporosis/osteopenia patients in comparison with healthy subjects. Asian Biomed. 14, 97–106 (2020).
Finnes, T. E. et al. Procollagen type 1 amino-terminal propeptide (P1NP) and risk of hip fractures in elderly Norwegian men and women. A NOREPOS study. Bone 64, 1–7 (2014).
Acknowledgements
The authors would like to thank Dr. Philipp Kaldis and Dr. Daisuke Kajimura for the discussion and Nobuko Nakajima for patient recruitment. The authors would also like to thank Drs. Takashi Hirai, Motonori Hashimoto, Yu Matsukura, and Hisato Higashikawa for collecting human samples. This work was supported by the Japan Society for the Promotion of Science grants (to T.K., A.O., and H.I.) and a grant from the Japanese Society for Bone and Mineral Research (to H.I.).
Author information
Authors and Affiliations
Contributions
A.O., P.C., and H.I. designed the research and wrote the paper. T.T., A.T., S.X., and C.J. performed the in vivo experiments. T.T., Y.K., M.S., T.K., and T.Y. performed the cell culture. T.T. and Y.I. performed the ChIP PCR analysis. T.T., H.O., and S.S. performed the micro-CT analysis. T.T. prepared the figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tanaka, T., Takahashi, A., Kobayashi, Y. et al. Foxf2 represses bone formation via Wnt2b/β-catenin signaling. Exp Mol Med 54, 753–764 (2022). https://doi.org/10.1038/s12276-022-00779-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-022-00779-z
This article is cited by
-
Network regulatory mechanism of ncRNA on the Wnt signaling pathway in osteoporosis
Cell Division (2023)
