
Abstract
Pancreatic β-cell failure by WFS1 deficiency is manifested in individuals with wolfram syndrome (WS). The lack of a suitable human model in WS has impeded progress in the development of new treatments. Here, human pluripotent stem cell derived pancreatic islets (SC-islets) harboring WFS1 deficiency and mouse model of β cell specific Wfs1 knockout were applied to model β-cell failure in WS. We charted a high-resolution roadmap with single-cell RNA-seq (scRNA-seq) to investigate pathogenesis for WS β-cell failure, revealing two distinct cellular fates along pseudotime trajectory: maturation and stress branches. WFS1 deficiency disrupted β-cell fate trajectory toward maturation and directed it towards stress trajectory, ultimately leading to β-cell failure. Notably, further investigation of the stress trajectory identified activated integrated stress response (ISR) as a crucial mechanism underlying WS β-cell failure, characterized by aberrant eIF2 signaling in WFS1-deficient SC-islets, along with elevated expression of genes in regulating stress granule formation. Significantly, we demonstrated that ISRIB, an ISR inhibitor, efficiently reversed β-cell failure in WFS1-deficient SC-islets. We further validated therapeutic efficacy in vivo with β-cell specific Wfs1 knockout mice. Altogether, our study provides novel insights into WS pathogenesis and offers a strategy targeting ISR to treat WS diabetes.
Similar content being viewed by others


Introduction
Wolfram syndrome (WS) is a rare autosomal recessive genetic disorder characterized by diabetes insipidus, diabetes mellitus, optic atrophy and deafness (also referred to as DIDMOAD) [1,2,3,4]. One of the hallmark features of WS is juvenile-onset diabetes, which presents at an average age of 6 years (range 3 weeks–16 years) [5]. The majority of WS patients require insulin treatment, resulted from selectively destroyed and functional failure of pancreatic β cells. However, due to the lack of understanding of the pathogenesis, there is currently no treatment available to reverse the progression of this disease other than provision of exogenous insulin.
The Wolframin gene (WFS1) encodes a protein with nine transmembrane domains across the endoplasmic reticulum (ER) membrane, which is highly expressed in pancreatic β cells and the brain [6,7,8]. WS is largely caused by pathogenic variants of WFS1 [9]. Analyses of pancreas from WS patients have demonstrated a selective loss of pancreatic β cells [10]. The ER is a central cell organelle responsible for protein folding and processing. It has been reported that WFS1 located in the ER membrane plays a role in protein trafficking from the ER to the Golgi, which directly interacts with a series of vesicular cargo proteins including proinsulin [11]. Wfs1-deficient mice exhibit reduced insulin processing and impaired insulin secretion in response to glucose stimulation [12, 13]. Loss-of-function of WFS1 in pancreatic β cells causes increased ER stress and activation of unfolded protein response (UPR), following pancreatic β-cell death [12, 14,15,16]. These findings indicate increased cell stress in WFS1-deficient pancreatic β cells.
Aberrant stress granule formation plays a pivotal role in various diseases, including diabetes, neurodegeneration and metabolic disorders. Stress granule formation is elicited by integrated stress response (ISR). Phosphorylation of eukaryotic initiation factor 2 (eIF2) on α subunit initiates ISR by four different kinases including protein kinase RNA-like endoplasmic reticulum kinase (PERK), which induces the activation of UPR under stress. Once ISR is triggered, the formation of the ternary complex (eIF2: GTP: methionyl-initiator-tRNA) is prevented, consequently limiting the AUG-initiated mRNA translation and inducing the assembly of stress granules and cell death [17,18,19,20]. WFS1 loss-of-function is known to up-regulate the PERK-signaling and trigger the apoptotic pathway in pancreatic β cells [8, 14, 21,22,23], leading us to hypothesize that WFS1 might control ISR and consequent stress granule formation.
Understanding of WS pathogenesis has been limited by the lack of proper human models. By modeling WS using human embryonic stem cell (hESCs) derived cerebral organoids and nerve cells, our recent study reveals Riluzole as a therapeutic molecule for WS neuropathy, demonstrating that human pluripotent stem cell derived tissues and cells could be applied as human models for WS pathogenesis investigation and drug discovery [21].
Aiming to elucidate the mechanism of pancreatic β-cell failure with WFS1 deficiency, with similar strategy, we characterize WFS1 deficiency through hESCs derived SC-islets as human WS β-cell failure model. Our results reveal that WFS1 deficiency drives SC-islets β cell fate to stressed trajectory, resulting in functional failure. Moreover, we identify the ISR as the key mechanism leading to the failure of SC-islets with WFS1 deficiency. Importantly, treatment with the ISR inhibitor, ISRIB, increases the proportion of SC-islets and mitigates apoptosis. Furthermore, ISRIB rescues impaired glucose homeostasis and increases insulin content in Wfs1 conditional knockout mice. Thus, our study provides mechanistic insights into pancreatic β-cell disorders of WS diabetes, and proposes a potential therapeutic approach with ISRIB.
Results
ScRNA-seq of SC-islets reveals subpopulations including main pancreatic endocrine cells
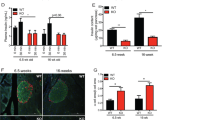
WFS1 deficiency causes childhood-onset insulin-dependent diabetes that is one of key features of WS [3, 5, 6, 24,25,26,27]. The impairment of pancreatic β-cell function and subsequent β-cell death are manifested as a result of WFS1 loss-of-function [8, 12, 13, 28, 29]. Previous studies illustrated the role of WFS1 associated with ER stress and UPR [8, 15, 16, 30], and WFS1 preserves β cell function by promoting insulin synthesis and mitigating ER stress [29]. However, deeper mechanism of pancreatic β-cell failure with WFS1 deficiency in WS diabetes remains unclear. To this end, we utilized SC-islets harboring WFS1 deficiency as a disease model from differentiation of hESCs by stepwise differentiation in vitro [31,32,33,34]. To recapitulate WS β-cell failure from a developmental perspective, pancreatic differentiation was initiated from hESCs reporter cell line (MEL1 Nkx6.1:linker2a:mCherry; INSGFP/w) that enable to precisely and dynamically trace SC-islet β cells during various stages of differentiation [35] (Fig. 1A). Meanwhile, WFS1 knockout hESCs reporter cell line (WFS1−/−) was established by using a CRISPR/Cas9 knockout strategy [31, 36]. WFS1 knockout caused a deletion of 49 bp and consequent early stop codon by transcript frameshift (deletion from cDNA 19–67 bp, p.Pro7 Arg fs Leu126*), and did not affect self-renewal and differentiation of pluripotency (Supplementary Fig. 1) [21]. Next, we performed qRT-PCR to examine WFS1 expression level from ES stage to SC-islet stage. We found that WFS1 was gradually expressed during the differentiation progress in wide type (WT) cell line, consistent with its high expression in human primary pancreatic β cells as reported (Fig. 1B) [7, 8, 11, 37]. To investigate the effects of WFS1 deficiency in SC-islets, we performed single-cell RNA sequencing (scRNA-Seq) for WT and WFS1−/− SC-islets (Fig. 1C). In total, we harvested single cells of SC-islets differentiated from WT and WFS1−/− hESCs for library construction, respectively. After quality control, 2654 and 3397 high-quality single-cell profiles from WT and WFS1−/− SC-islets were retained for downstream analysis (Supplementary Fig. 2). We found that the expression of WFS1 was largely diminished in WFS1−/− SC-islet, confirming the complete WFS1 knockout (Fig. 1D). Furthermore, dimensional reduction analysis using uniform manifold approximation and projection (UMAP) revealed a diversity of cell types. Unsupervised clustering analysis identified 8 distinct cell clusters in WT and WFS1−/− based on the top up-regulated genes in each cluster matched to cell types in terms of published scRNA-seq analysis (Fig. 1E and Supplementary Fig. 3A) [38,39,40,41]. Clusters consisted of mainly four types of endocrine cells including β cells (especially expressing INS, PCSK1 and G6PC2), α cells (especially expressing GCG), δ cells (especially expressing SST), ε cells (especially expressing GHRL), pancreatic progenitor cells (especially expressing SOX9), proliferation cells (especially expressing MKI67), EC cells (enterochromaffin cells, especially expressing FEV), and polyhormonal endocrine cells (co-expressing GCG and INS) (Fig. 1F, Supplementary Fig. 3B, C). Taken together, these results suggested that the utilization of SC-islet holds the promise to investigate the effect of WFS1 in WS diabetes in vitro.

A Schematic of pancreatic islets differentiation protocol from hESCs. The key markers of the main stages are described. The red and green colors represent expression of NKX6.1-mCherry and INS-GFP, respectively. B Expression profile of WFS1 (normalized to that of hESCs) during the differentiation process. n = 6 biological replicates for each stage. C Representative fluorescent images of INS-GFP and NKX6.1-mCherry in WT and WFS1−/− SC-islet. Scale bars, 25 μm. D Violin plot showing the expression pattern of WFS1 in WT and WFS1−/− SC-islets. E UMAP plot of 2654 and 3397 cells from WT and WFS1−/− SC-islets, respectively. Cells are colored according to their assigned type. F Violin plots showing the expression pattern of key marker genes of β cells, α cells, δ cells, ɛ cells, EC cells, pancreatic progenitor cells, proliferation cells and polyhormonal endocrine cells. Data are presented as mean ± SD. p values calculated by unpaired two-tailed Student’s t test were *p < 0.05, **p < 0.01, and ***p < 0.001.
Trajectory reconstruction reveals stressed and mature subpopulations of β cells
Pancreatic β cells are essential in maintaining glucose homeostasis by regulating insulin secretion in response to glucose stimulation [42]. Besides regulating cellular function, extensive researches have revealed significant heterogeneity within the pancreatic β-cell population [43,44,45]. To investigate the function of WFS1 in SC-islets β cell heterogeneity, we further re-clustered all merged SC-islet β cells using dimensionality reduction and unsupervised clustering into two main β cell subpopulations (Fig. 2A). We identified these two subpopulations according to top up-regulated genes and defined into mature β cells and stressed β cells. Mature β cells were characterized by high expression of mature pancreatic β cell markers such as INS, CHGA, G6PC2, and PCSK1, whereas stressed β cells showed high expression of stress-associated markers [19, 46] such as XBP1, ATF4, ATF6, DDIT3, HERPUD1, EIF2AK1, EIF2AK2, and EIF2AK4 (Fig. 2B). Together, we unveiled the heterogeneity with two subpopulations including mature β cells and stressed β cells.

A UMAP plot of scRNA-seq data of WT and WFS1−/− SC-islet β cells. SC-islet β cells are colored and annotated into two subtypes including mature β cells and stressed β cells. B Violin plots showing the expression patterns of β cell marker genes (labeled in blue) and cell stress marker genes (labeled in red) between mature β cells and stressed β cells. C, D Pseudotime analysis revealing the progression of two sub-clusters of SC-islet β cells as in (A). Trajectory reconstruction of all single cells revealing three branches: Pre-branch, Fate 1 (indicated as stress branch) and Fate 2 (indicated as maturation branch), defined by expression profiles. Cells are colored by pseudotime (C) and SC-islet β cell subtypes (D). E Heatmap showing the expression of the top 500 differently expressed genes in three branches as in (C), highlighting representative genes associated with pancreatic β cells specific genes and cell stress genes along the right margin. From the center to the left of the heatmap, the curve from the pre-branch along the trajectory to fate 1 branch. From the center to the right, the curve from pre-branch to fate 2 branch. F The expression dynamics of selected genes from pre-branch to stress and maturation branches, respectively. Each point represents one cell. Cells are colored by pseudotime.
Furthermore, to decipher the trajectory of SC-islets β cell fate, single-cell pseudotemporal analysis was performed to model the cell fates in SC-islet β cells. By utilizing Monocle [47,48,49] pseudotime analysis to determine the transcriptional fate, we found two different branches in SC-islet β cells (Fig. 2C). We attempted to elucidate the molecular dynamics to distinguish the two branches. The analysis of gene expression dynamics was focused on the top 500 differentially expressed genes (Fig. 2E and Supplementary Table 3). We found that stress-associated markers were highly expressed along fate 1 branch, such as ATF4 and JUN [50, 51], while SC-islets β cell identity and maturation markers were highly expressed along fate 2 branch, such as INS, INS-IGF2, CHGA and CDKN1C [52]. On the basis of the above features, we inferred fate 1 branch as stress state, and fate 2 branch as maturation state (Fig. 2C). Notably, total SC-islet β cells branched opposite divergent ends as two terminally differentiated cell types. Most of stressed β cells were found at the terminal end of fate 1 branch (stress branch), consistent with stressed β cell population in SC-islets (Fig. 2D). Meanwhile, we assessed the gene expression patterns associated with pancreatic β cell markers and stress-associated markers during two fate branches. The expression of pancreatic β cell markers, including INS and CHGA increased along the maturation branch. Notably, WFS1 was highly expressed along the maturation branch, indicating the function of WFS1 correlated to β-cell maturation. Whereas, the expression of ATF4, G3BP1 and ATF6 increased along the stress branch but notably decreased along the maturation branch (Fig. 2F). Taken together, through single-cell transcriptomic analysis, the composition of SC-islet β cells was identified into two different subpopulations including mature and stressed β cells, characterized with two distinct fate trajectories.
WFS1 deficiency directs β cells into the stress branch lack of functional maturation
To investigate the effect of WFS1 on cell fate, we performed Monocle pseudotime analysis for WT and WFS1−/− SC-islet β cells, respectively. Combining with defined two cell fate branches, we found that the pseudotime trajectory began with pre-branch and then mostly placed at maturation branch among WT SC-islet β cells. Of note, WFS1−/− SC-islet β cells mainly placed at the stress branch along the pseudotime trajectory, which were stagnated towards the maturation branch (Fig. 3A). Next, we assessed the proportions of WT and WFS1−/− SC-islet β cells in three cell fate branches (Fig. 3B). The percentage of WT and WFS1−/− SC-islet β cells exhibited about 58.6% and 41.4% of total cells in pre-branch, respectively. The proportion of WT cells reached 86.5% at the maturation branch, whereas the proportion of WFS1−/− cells accounted for 92.3% of total cells in the stress branch (Fig. 3B), which was consistent with the stagnant trajectory of WFS1−/− SC-islet β cells towards the maturation branch. Meanwhile, we performed a deeper analysis of transcriptomic changes in WT and WFS1−/− SC-islet β cells to detect the molecular changes. Differential expression analysis generated a large number of differential expression genes (DEGs) that comprised a distinctive gene expression profile with 2701 up-regulated and 446 down-regulated genes in WFS1−/− SC-islet β cells (Fig. 3C and Supplementary Table 4). We found that the expression of genes related to pancreatic β cell maturation and function such as INS, NKX6.1, PCSK1, and CDKN1C were down-regulated, and cell stress-associated genes such as ATF4, JUN, ROCK2 and HSP90B1 were highly up-regulated in WFS1−/− SC-islet β cells (Fig. 3C). A summary of the significant up-regulated or down-regulated DEGs within WFS1−/− SC-islet β cells was shown (Fig. 3D). Mature pancreatic β cells gain the capacity of glucose-stimulated insulin secretion (GSIS) [53].By contrast, our analysis of scRNA-seq data showed that WFS1−/− SC-islet β cells exhibited stressed cell fate lack of maturation. To validate this, we performed GSIS assays in WT and WFS1−/− SC-islets. Insulin secretion upon glucose stimulation was measured by scoring the ratio of insulin release in high glucose to that in low glucose [31]. Compared with WT SC-islets, WFS1−/− SC-islets showed significantly reduced insulin secretion (Fig. 3E, F). And the insulin content per cell was measured in WFS1−/− SC-islets and result revealed significantly lower intracellular insulin content compared with WT SC-islets (Fig. 3E, G). Collectively, our result revealed that WFS1 was required for advancing cell fate of β cells towards the maturation trajectory.

A Pseudotime analysis showing the progression of SC-islet β cells in WT (left panel) and WFS1−/− SC-islet β cells (right panel), respectively. Cells are colored by pseudotime. B Bar plots presenting the percentage of stressed β cells or mature β cells in three trajectory branches. Blue represents WT and red represents WFS1−/−. C Volcano plot of differential expression analysis between WT and WFS1−/− SC-islet β cells. Each dot represents a gene, and selected DEGs are highlighted in red. D Particular up-regulated and down-regulated genes by log-fold change in WFS1−/− SC-islet β cells with adjusted p < 0.05. E Schematic of in vitro function assays and insulin content test in WT and WFS1−/− SC-islets. F GSIS in vitro of WT and WFS1−/− SC-islets, n = 8 per group. G Insulin content of WT and WFS1−/− SC-islets, n = 7. Data are presented as the mean ± SD. p values calculated by unpaired two-tailed Student’s t test for (F) and (G) were *p < 0.05 and **p < 0.01.
WFS1 deficiency induces ISR in β cells
To determine the signaling pathways regulating WFS1−/− SC-islets β cell functional failure, we performed gene set enrichment analysis (GSEA) on the SC-islet β cells and the top enriched pathways were identified (Supplementary Table 5). We found that Reactome pathways related to translation such as eukaryotic translation elongation, peptide chain elongation, eukaryotic translation termination, formation of a pool of free 40 S subunits, GTP hydrolysis and joining of the 60S ribosomal subunit, cap-dependent translation initiation and eukaryotic translation initiation were significantly enriched in WFS1−/− SC-islet β cells among top 25 down-regulated pathways collected from the pathway enrichment analysis. Meanwhile, Reactome pathways related to cellular response to stress such as PERK regulates gene expression and HSP90 chaperone cycle for steroid hormone receptors in the presence of ligand were significantly enriched in WFS1−/− SC-islet β cells among top 25 up-regulated pathways in the pathway enrichment analysis (Fig. 4A, Supplementary Fig. 4). To further verify the down-regulated translation in WFS1−/− SC-islet β cells, we compared genes expression involved in the eIF2 signaling pathway [50, 54, 55] between WT and WFS1−/− SC-islet β cells. 54 shared genes associated with eIF2 signaling pathway among DEGs within WFS1−/− SC-islet β cells were significantly enriched (Fig. 4B). The comparative heatmap of relative expression changes in eIF2 signaling pathway associated genes showed that 75% (42 genes in 54 shared genes) genes were down-regulated, indicating activation of the ISR (Fig. 4C). Stress granules are formed when translation initiation is inhibited by stress responses [56, 57]. Furthermore, we evaluated the core stress granule genes [58] between WT and WFS1−/− SC-islet β cells. 21 shared genes associated with core stress granule genes among DEGs within WFS1−/− SC-islet β cells were significantly enriched (Fig. 4B). Notably, 20 genes in total shared 21 genes associated with core stress granule genes were significantly up-regulated, including all seven genes that have been described as “essential” to stress granule assembly [58] (Fig. 4D). Previous study had shown that PERK phosphorylates eIF2α to activate the ISR, leading to a decrease in global protein translation [19, 59]. Consistently, we found that the expression of PERK pathway regulated genes was significantly up-regulated and the process of intracellular translation was significantly down-regulated in WFS1−/− SC-islet β cells. Meanwhile, we also examined the gene expression levels associated with the UPR, and found the elevated expression pattern of the UPR associated genes (Supplementary Fig. 5). Taken together, our results revealed that WFS1 deficiency activates the ISR in SC-islet β cells with global attenuation of translation. To further verify the activation of ISR in WFS1−/− SC-islet β cells, we tested the expression of ISR associated genes. We found that the expression of ISR associated genes was highly increased in WFS1−/− SC-islet β cells compared with WT (Fig. 4E). The expression levels of key factors of PERK/eIF2 signaling were examined by western blot, revealing significantly increased phosphorylation of eIF2α, and enhanced protein levels of PERK and ATF4 in WFS1−/− SC-islets compared with WT (Fig. 4F, G, Supplementary Fig. 6). Meanwhile, we applied immunostaining to examine the expression of G3BP1, which is an essential component of stress granules. The mean intensity of stress granules in INS+ cells was significantly higher in WFS1−/− SC-islets compared with WT (Fig. 4H, I).

A Reactome pathway enrichment analysis of differentially expressed genes from WFS1−/− SC-islet β cells using GSEA. Enrichment analysis showed that protein translation processes were enriched in down-regulated genes, whereas the stress pathways were enriched in up-regulated genes. B Venn diagrams showing the overlapping genes between WFS1−/− SC-islet β cell DEGs and eIF2-pathway-associated genes, between WFS1−/− SC-islet β cell DEGs and stress granule-associated genes, respectively. Up-regulated and down-regulated genes in overlapping genes were highlighted. C Average expression of signature genes in eIF2-pathway-associated genes overlapped with DEGs as in (B). D Average expression of signature genes in stress granule-associated genes overlapped with DEGs as in (B). Genes described as “essential” to SG assembly were labeled in red. E Violin plots showing the expression patterns of the ISR associated genes in WT and WFS1−/− SC-islet β cells. F, G PERK, Phosphorylation of eIF2a and ATF4 pattern were determined by western blot analysis in WT and WFS1−/− SC-islets. H, I Immunostaining of G3BP1 in WT and WFS1−/− SC-islets. Scale bars, 25 μm (H) and mean intensity measurements for G3BP1, n = 9 (I). Data are presented as the mean ± SD. p values calculated by unpaired two-tailed Student’s t test were *p < 0.05, **p < 0.01, and ***p < 0.001.
ISR inhibitor reverses pancreatic β-cell failure
As shown above, ISR is activated by WFS1 deficiency in β cells. To investigate whether inhibition of ISR could be applied to treat pancreatic β-cell failure in WS, we tested the ability of ISRIB (an ISR inhibitor [60, 61]) both in WFS1-deficient SC-islets and pancreatic Wfs1-deficient mice.
First, from PP cell stage to SC-islets stage, WFS1-deficient SC-islets were treated with ISRIB at the concentration of 100 nM (Fig. 5A). The mean intensity of G3BP1 was significantly decreased in WFS1-deficient SC-islets treated with ISRIB as compared to vehicle (Fig. 5B, C). Since activated ISR inhibits protein synthesis, we performed nascent polypeptide synthesis assay via O-propargyl-puromycin (OPP) labeling to assess total protein synthesis [62,63,64,65]. As a result, the decreased total protein synthesis was significantly restored in ISRIB-treated WFS1-deficient SC-islets as compared to control (Fig. 5D, E). Furthermore, we found that the percentage of INS-GFP NKX6.1-mCherry double positive SC-islet β cells in WFS1−/− SC-islet treated with ISRIB was significantly increased as compared to vehicle (Fig. 5F, G). Next, we treated WFS1-deficient SC-islet with 100 nM ISRIB or vehicle from SC-islets stage for 2 days (Fig. 5H). As compared to vehicle, the mean intensity of G3BP1 in WFS1-deficient SC-islets treated with ISRIB was significantly decreased, indicating the reduced formation of stress granules (Fig. 5I, J). Consistently, total protein synthesis was also restored with ISRIB treatment (Fig. 5K, L). Meanwhile, the apoptosis of SC-islet β cells was significantly reduced in WFS1-deficient SC-islet treated with ISRIB as compared to vehicle (Fig. 5M, N). These results suggested that ISR inhibition reverses pancreatic β-cell loss.

A Schematic diagram of ISRIB treatment starting from pancreatic progenitor stage. Immunostaining of G3BP1 in WFS1−/− SC-islets treated with ISRIB or vehicle. Scale bars, 10 μm (B) and mean intensity measurements for G3BP1, n = 9 (C). OPP protein synthesis assay of WT and WFS1−/− SC-islets treated with ISRIB or vehicle, Scale bars, 100 μm (D) and mean intensity measurements for OPP in INS+ β cells, n = 10 (E). Representative FACS plots (F) and quantifications (G) of INS-GFP+/NKX6.1-mCherry+ populations in WFS1−/− SC-islets treated with ISRIB or vehicle starting from pancreatic progenitor stage, n = 13. H Schematic diagram of ISRIB treatment in SC-islets. Immunostaining of G3BP1 in WFS1−/− SC-islets treated with ISRIB or vehicle. Scale bars, 10 μm (I) and mean intensity measurements for G3BP1, n = 9 (J). OPP protein synthesis assay of WT and WFS1−/− SC-islets treated with ISRIB or vehicle, Scale bars, 100 μm (K) and mean intensity measurements for OPP in INS+ β cells, n = 10 (L). Representative FACS plots (M) and quantifications (N) of Annexin V+/INS-GFP+ populations in WT and WFS1−/− SC-islets treated with ISRIB or vehicle, n = 5. Data are presented as the mean ± SD. p values calculated by unpaired two-tailed Student’s t test were *p < 0.05, **p < 0.01 and ***p < 0.001.
ISR inhibitor improves glucose homeostasis in Wfs1 pancreatic conditional knockout mice
To test the in vivo efficacy of ISRIB, we generated pancreatic Wfs1 conditional knockout mice by crossing the Wfs1-flox mice with Pdx1-Cre mice (Wfs1fl/fl, Pdx1-Cre mice; CKO mice) (Supplementary Fig. 7A) [21, 37]. We administered 2.5 mg/kg ISRIB or vehicle in CKO mice through intraperitoneal (i.p.) injections from 3 to 8 weeks old (Fig. 6A). We observed a significantly decreased fasting glucose level in Wfs1-deficient mice treated with ISRIB as compared to vehicle at 8 weeks old (Fig. 6B). To investigate the intracellular changes of pancreatic β cells, we performed the immunostaining of insulin in mouse pancreas. We found that insulin intensity and insulin content of pancreatic β cells in ISRIB-treated CKO mice were significantly restored as compared to vehicle, similar to that in WT mice (Fig. 6C–E). Consistently, OPP labeling showed that ISRIB treatment significantly restored the impaired total protein synthesis, and reduced stress granule formation in CKO mice (Fig. 6F–H).

A Schematic of the strategy of generating pancreatic Wfs1 conditional knockout mice for ISRIB treatment. B Fasting glucose levels in WT and CKO mice after ISRIB treatment, n ≥ 8. Immunostaining of Ins (C) and quantifications (D) of Ins intensity in WT and CKO mice after ISRIB treatment, n = 18 islets from n = 3 mice. Scale bar, 25 μm. E Insulin content in WT and CKO mice after ISRIB treatment, n = 9. OPP protein synthesis assay of WT and CKO mice after ISRIB treatment, Scale bars, 25 μm (F, left) and 5 μm (F, right) and mean intensity measurements for OPP (G) and G3BP1 (H) in β cells, n = 9 from n = 4 mice (G, H). IPGTT (I) and AUC analysis (J) in WT and CKO mice after ISRIB treatment, n ≥ 7. K GSIS in WT and CKO mice after ISRIB treatment, n ≥ 4. L Schematic diagram showing activated ISR in WFS1-deficient β cells and reversed β cell function treated with ISRIB in vitro and in vivo. Data are presented as the mean ± SD. p values calculated by unpaired two-tailed Student’s t test for (B), (D), (E), (G), (H), and (I); two-way ANOVA with Sidak’s test for multiple comparisons for (H) and (J). n.s. not significant, *p < 0.05, **p < 0.01 and ***p < 0.001.
Furthermore, intraperitoneal glucose tolerance test (IPGTT) showed that ISRIB treatment significantly improved glucose tolerance in CKO as compared to vehicle-treated mice (Fig. 6I, J). Meanwhile, we examined the insulin secretion by GSIS in vivo. We found that ISRIB-treated CKO mice showed significantly improved insulin secretion upon high glucose stimulation as compared to vehicle (Fig. 6K). Notably, there was no significant difference of insulin secretion upon high glucose stimulation between WT and ISRIB-treated CKO mice (Fig. 6K). To explore the potential toxicity of ISRIB in the body, we detected the histological images of major organs including heart, liver, spleen, lung and kidney. No inflammatory cell infiltration and histopathological changes were observed with ISRIB treatment (Supplementary Fig. 7B). These results demonstrated that ISRIB could reverse β-cell failure and improve glucose homeostasis in vivo. Overall, our results suggested that ISRIB could be applied to treat pancreatic β-cell failure and function in WS diabetes (Fig. 6L).
Discussion
The clinical progression of WS diabetes is characterized by severe β-cell loss during juvenile stage, which ultimately becomes resistant to treatment [2, 5]. Unfortunately, the lack of understanding regarding its pathogenesis has hindered the development of effective therapies for this disease. The investigation of WS diabetes has been limited due to ethical concerns and the scarcity of human samples. The effect of WFS1 deficiency on pancreatic β-cell function has been modeled in rodents [12, 13, 29, 66, 67]. Meanwhile, the phenotypes resulted from WFS1 loss-of-function have also been analyzed in cell lines and SC-islets [16, 28, 68]. However, the underlying mechanisms of WFS1 deficiency causing β-cell functional failure and loss are elusive, remaining as a challenge for WS drug discovery. To achieve a deeper understanding of WS diabetes, we applied SC-islets differentiated from WFS1 knockout hESCs as human model. Our investigation revealed the presence of two distinct cell fate trajectories in β cells: the maturation branch and the stress branch. Notably, WFS1 deficiency blocked β-cell fate trajectory to maturation and directed them towards stress trajectory. Further analysis identified the activated ISR as a crucial pathogenic mechanism for WS β-cell failure.
WFS1 is a protein with nine transmembrane domains across the ER membrane, which is highly expressed in pancreatic β cells and brain [6,7,8]. Previous studies using rodent models mainly focused on the relationship between the ER stress and pancreatic β cell function. It has been reported that loss-of-function of WFS1 results in pancreatic β-cell failure by eliciting chronic ER stress [12, 13, 16, 28, 66, 67]. To date, significant efforts have been devoted to treat WS by decongesting ER stress [28]. In rodent and SC-islet models, ER stress mitigators, such as valproic acid and GLP-1 receptor agonists have been proposed as potential drug candidates [69,70,71,72]. Several studies have suggested WFS1 deficiency causes calcium dyshomeostasis, thus compounds targeting calcium signaling such as dantrolene sodium (an ER Ca2+ stabilizer), ibudilast and calpain inhibitor were also identified as drug candidates to treat with WS [73,74,75]. Nevertheless, given the fact that calcium is a universal second messenger ubiquitously existing in various cell types, application of calcium stabilizers is limited due to their potential for systemic side effects [76]. Besides targeting ER stress and calcium, it is of substantial importance to identify alternative therapeutic targets and molecules to decongest stress in WFS1-deficient β cells. Here, by targeting the activated ISR in WFS1-deficient SC-islets, we found ISRIB, an ISR inhibitor [60, 61, 77,78,79], efficiently reduced stress granule formation and cell death in SC-islet β cells. Moreover, we validated in vivo efficacy of ISRIB by observing its effects in pancreatic Wfs1 conditional knockout mice. ISRIB treatment led to a significant restoration of insulin content and β cell mass. Wfs1-deficient CKO mice treated with ISRIB exhibited improved glucose homeostasis. These results, obtained from both in vitro human model and in vivo animal model, suggested that ISRIB could be a potential drug candidate for the treatment of WS diabetes.
The mechanism of stress granules assembly mediated by phase separation is not fully understood [80]. Our results demonstrated that WFS1 deficiency up-regulated stress granule-associated gene such as G3BP1, indicating that WFS1 deficiency enhances stress granule formation. Previous studies have shown ISR leads to a decreased global protein translation and the induction of specific gene expression, including ATF4 [19, 59]. ATF4 acts as an effector of the ISR, regulating stress-responsive gene expression [81,82,83,84,85,86,87]. In WFS1-deficient SC-islet β cells, we observed increased expression of G3BP1, which is a crucial component for stress granule. This finding suggested that ATF4, up-regulated by WFS1 deficiency mediated ISR, might activate G3BP1 transcriptional expression. Further investigation is needed to confirm this. Moreover, we also discovered that overexpression of WFS1 alleviated the phosphorylation of eIF2α and stress granule assembly in β cells (data not shown). Our discoveries indicate a critical role of WFS1 in stress granule formation and emphasize the need for future research in this area.
Besides diabetes, neuropathy is also a major manifestation for WS. As recently reported, dysregulated ISR signaling contributes to the pathogenesis of several neurodegenerative diseases [19, 88, 89], and the effect of ISRIB on the neurodegeneration has been tested in amyotrophic lateral sclerosis, brain injury and age-related memory decline [61, 78, 79, 89,90,91]. Combined with our result, it will be valuable for further study to test therapeutic effect of ISRIB on WS manifestations of both diabetes and neurodegeneration in clinical trial. Moreover, with similar strategy to model WS neuropathy by application hESCs derived cerebral organoids and neural cells, we demonstrate that WFS1 deficiency delays neuronal differentiation with disrupted expression of genes associated with psychiatric disorders, impairs synapse formation, and renders astrocytes toxic to neurons by reducing EAAT2 (Glutamate transporter). On the basis of this, we demonstrate Riluzole as a therapeutic molecule to reverse neuronal loss and abnormality of synapse formation associated with WS neuropathy and further validate its efficacy in vivo in animal model [21]. Altogether, in future study, it would be intriguing to explore the synergistic curative effect of combining Riluzole with ISRIB for WS treatment.
Materials and methods
Generation of WFS1 knockout hESCs with CRISPR/Cas9
One or two sgRNAs inserts targeting WFS1 gene were cloned into vector containing U6 promoter and CMV promoter drove hCas9 (vector as a gift from Prof. Zhili Rong, Southern Medical University, Guangzhou, China). MEL1 Nkx6.1:linker2a:mCherry; INSGFP/w gene-edited cell line was provided by Dr. Xin Cheng. All cell lines were checked to be mycoplasma-free once a month by PCR assay. Mel1-Reporter line was transfected with above-mentioned plasmid carrying sgRNAs and hCas9. After 24–48 h of electroporation, puromycin was added into hESCs culture medium for selection as concentration of 400 ng/ml, and hESCs culture medium with puromycin was replaced after 2 days selection. Cell clones were picked and cultured for 5–7 days later. PCR was used to validate the WFS1 knockout cell line.
In vitro differentiation of SC-islets derived from hESCs
hESCs were differentiated into SC-islets as described previously [31, 33, 92]. In brief, hESCs were cultured on the Matrigel-coated plate to start differentiation. Then the differentiation process contained planar culture through stage 1 to stage 4 to generate NKX6.1+ progenitor cells and 3D culture from stage 5 to stage 7 to yield mature SC-islets. SC-islets differentiation that did not pass a minimum criteria of 30% INS+/mCherry+ double positive cells at the final stage were excluded from further analysis. No data were excluded for other analyses of this study, and the randomization was not applicable. The in vitro experiments, mean intensity measurement, cell type and apoptosis quantification were blinded to investigator to analyze.
Library construction and NGS sequencing
DNBelab C Series High-throughput Single-Cell RNA Library (MGI, #940-000047-00) was utilized for scRNA-seq library preparation. In brief, the single-cell suspensions were converted to barcoded scRNA-seq libraries through steps including droplet encapsulation, emulsion breakage, mRNA captured beads collection, reverse transcription, cDNA amplification and purification. cDNA production was sheared to short fragments with 250-400 bp, and indexed sequencing libraries were constructed according to the manufacturer’s protocol. Qualification was performed using Qubit ssDNA Assay Kit (Thermo Fisher Scientific) and Agilent Bioanalyzer 2100. All libraries were further sequenced by the MGISEQ-2000RS with pair-end sequencing. The sequencing reads contained 30-bp read 1 (including the 10-bp cell barcode 1, 10-bp cell barcode 2 and 10-bp unique molecular identifiers (UMI)), 100-bp read 2 for gene sequences and 10-bp barcodes read for sample index. Generation of FASTQ files were carried out using DNBelab_C4_scRNA_v2.3.
ScRNA-seq data processing (alignment, barcode assignment, and UMI counting)
The sequencing data were processed using an open-source pipeline (https://github.com/MGI-tech-bioinformatics/DNBelab_C_Series_scRNA-analysis-software). Briefly, all samples were performed sample de-multiplexing, barcode processing, and single-cell 3’ unique molecular identifier (UMI) counting with default parameters. Processed reads were then aligned to GRCh38 genome reference using STAR (2.5.1b). Valid cells were automatically identified based on the UMI number distribution of each cell by using the “barcodeRanks()” function of the DropletUtils tool to remove background beads and the beads that had UMI counts less than the threshold value. Finally, we used PISA to calculate the gene expression of cells and create a gene x cell matrix for each library.
ScRNA-seq data analysis
Raw sequencing data were aligned to the GRCh38 human reference genome. We processed the resulting data using Seurat [93] and removed low-quality cells with less than 200 or more than 9000 detected genes, or mitochondria gene content was more than 10%. Genes were filtered with less than 3 cells detected in each sample. SCTransform-v2 was used for normalization. SelectIntegrationFeatures and PrepSCTIntegration functions were used to perform integration. RunPCA function in Seurat was used for principal component analysis and selected 40 PCs for cell clustering and downstream analysis. FindNeighbors and FindClusters (the resolution was adjusted to 0.8) functions were run to get 11 clusters. Dimensionality reduction was performed by RunUMAP function in Seurat. Marker genes were identified of each cluster to define cell types using FindAllMarkers in Seurat. According to known gene markers including GCG, INS, SST, CHGA, GHRL, FEV, we defined those different clusters. Finally, we merged 11 clusters to 8 cell types including α cells, β cells, δ cells, ε cells, polyhormonal cells, pancreatic progenitor, EC cells and proliferation cells. We re-identified marker genes for 8 cell types and highlighted top 10 positive marker genes per cell type. We compared the differential expression pattern in different cell types between WT and WFS1−/−. Differential expression genes (DEGs) were identified using FindMarkers function in Seurat with adjusted p < 0.05 and |log2-fold change| > 0.1 as cutoffs. β cells were further classified into 3 sub-clusters by re-running steps above. 3 sub-clusters were merged to 2 subtypes according to β cell identity markers and stress markers. DEGs between 2 β cell types were identified using Findmarkers function in Seurat. Visualizations were generated using Seurat, ggplot2 (https://ggplot2.tidyverse.org) and pheatmap (https://github.com/raivokolde/pheatmap).
Pathway analysis
Pathway analysis was based on Reactome pathway database using R package clusterProfiler [94, 95]. All genes ranked by the fold change calculated above between WT and WFS1−/− were considered.
Pseudotime analysis
R package Monocle2 was used to order β cells and progenitor cells for pseudotime analysis. SetOrderingFilter function was used to get a list of gene ids to be used for ordering. Reducing the Dimensionality of data was performed by ReduceDimension function. DDRTree was used to learn tree-like trajectories. OrderCells function was used to ordering cells along the trajectory. The roots of these trajectories were determined by the expression of unique genes. The trajectory plots were generated by plot_cell_trajectory function. Beam function was used to figure out genes differentially expressed between the branches. Genes were selected with q < 1e-4 as cutoff. The heatmap were generated by plot_genes_branched_heatmap function with top 500 genes selected.
Quantitative real-time PCR
Total RNA was extracted using the TRNzol Universal kit and reverse transcribed into cDNA using Quantscript RT Kit (TianGen). Quantitative real-time PCR was carried out using SuperReal SYBR Green kit on LightCycler 96 (Roche). The amplification efficiency for each primer and the cycle threshold were determined automatically by Lightcycler software (Roche). The fold-change was calculated by the comparative CT (2−ΔΔCT) method against GAPDH. Primer sequences were shown in Supplementary Table 1.
Immunofluorescence staining and quantification of immunofluorescence image
SC-islets were rinsed once with precooled PBS and fixed with 4% paraformaldehyde overnight at 4 °C and washed with DPBS for three times. SC-islets were then embedded in optimum cutting temperature (O.C.T) for cryosectioning at 8 µm with Leica CM1950. Mouse pancreases were collected in PBS, fixed in 4% paraformaldehyde on ice for 1 h, washed in PBS for three times, and dehydrated in 30% sucrose at 4 °C overnight. Mouse pancreases were embedded in O.C.T for cryosectioning at 10 µm with Leica CM1950. Next, for SC-islets or mouse pancreases, sections were permeabilized with PBST (PBS with 0.1% Triton X-100) for 15 min and then blocked with PBS containing 5% donkey serum for 2 h at RT. Primary antibodies were added at appropriate dilutions at 4 °C overnight. The next day, cells were washed with PBS for three times, secondary antibodies were added at appropriate dilutions for 1 h at RT. Cell nuclei were stained with DAPI. Primary antibodies used in this study included: WFS1 (Abcam, ab259362, 1:50), INSULIN (Sigma, I2018, 1:500), G3BP1 (Proteintech, 13057-2-AP, 1:500). Secondary antibodies were available from Jackson ImmunoResearch Laboratories. Images were captured with Leica SP8 confocal microscope. We processed the quantification using the National Institutes of Health ImageJ software to analyze the fluorescence intensity.
Flow cytometry and apoptosis analysis
SC-islets were dissociated into single cells with 0.25% trypsin and washed with precooled PBS. Then cells were subjected to be labeled Annexin V using the Annexin V-647/PI apoptosis detection kit (YEASEN). The death of SC-islet β cells was measured by flow cytometry with Annexin V+/INS-GFP+ double labeling.
Insulin secretion assays
The protocol was adapted from a previous study [31]. For GSIS in vitro, SC-islets needed to be fasted in Krebs-Ringer buffer [33] supplemented with 2 mM glucose for 2 h in a 37 °C, 5% CO2 incubator. Then the SC-islets were stimulated alternately by Krebs-Ringer buffer with low (2 mM) or high (20 mM) glucose. Supernatants were collected after 30 min of each stimulation and the pellets were lysed overnight in acidified alcohol (75% alcohol, 1.5% HCl) at −20 °C, for insulin content measurement. Secreted insulin or total insulin content was measured by a human insulin ELISA kit (ALPCO).
Western blot
The protocol was performed as described previously [31], using primary antibodies and secondary antibodies. Primary antibodies were listed as below: β-actin (Beyotime, AF5001, 1:5000), PERK (CellSignalingTechnology, 3192S, 1:1000), eIF2α (Beyotime, AF6771, 1:1000), P-eIF2α (Beyotime, AF1237, 1:1000), ATF4 (Proteintech, 10835-1-AP, 1:500). Secondary antibodies were available from Cell Signaling Technology.
ISRIB administration
ISRIB solution was made by dissolving 5 mg ISRIB in 2 ml dimethylsulfoxide (DMSO). The solution was kept at RT throughout the experiment. The vehicle solution consisted of DPBS containing of 10% DMSO. ISRIB was given at 100 nM as final concentration in cell culture medium. And ISRIB was given at 2.5 mg/kg/day through i.p. injections for mice from 3 weeks old for 5 weeks. Animals were randomized using randomized number table to be treated with DMSO or ISRIB. All animals received food and water ad libitum.
Total protein synthesis assay
Cells were plated and recovered overnight, and cell medium was replaced with OPP working solution (1:1000, 20 μM in cell culture medium). After 30 min, cells were washed once with DPBS and fixed with 4% PFA for 30 min at room temperature (RT), and then stained with Click-iT™ Plus OPP Alexa Fluor™ 647 protein synthesis kit (Invitrogen, C10458). Additional immunostaining could be processed following OPP staining.
Animal studies
All experiments were performed in accordance with the University of Health Guide for the Care and Use of Laboratory Animals and approved by the Biological Research Ethics Committee of Tongji University. The in vivo experiments were blinded to data collectors.
Generation of conditional Wfs1 knockout mice
Mice were obtained as described [21, 37]. Briefly, Wfs1fl/fl mice were generated by Shanghai Model Organisms Center, Inc (Shanghai, China) using the CRISPR-Cas9 technology. Pdx1-Cre mice were ordered from Shanghai Model Organisms Center, Inc (Shanghai, China). All mice were maintained in the animal facility of Tongji University, Shanghai, China. Mice were maintained on the C57BL6 background, housed in standard cages, fed a normal diet and maintained in a 12-h light/dark cycle. PCR primers were designed to verify the correctly targeted allele (Supplementary Table 1).
Intraperitoneal glucose tolerance (IPGTT) Test
Mice were fasted for 16 h before the experiment. 20% (w/v) Glucose solution were administered intraperitoneally at a dose of 2 g/kg body weight. Blood glucose levels were measured at 0, 15, 30, 60, and 120 min, respectively, from the tail vein.
Glucose-stimulated insulin secretion (GSIS) in vivo
Mice were fasted for 16 h before the experiment. 20% (w/v) Glucose solution were administered intraperitoneally at a dose of 2 g/kg body weight. Blood samples were collected from the venous sinus immediately before glucose administration and after 20 min to measure blood insulin levels. Blood was put on ice for 1 h, and centrifuged for 15 min to collect serum. Serum insulin levels were measured using a mouse insulin ELISA kit (ALPCO) according to the manufacturer’s instructions.
Statistical analyses
All statistical analyses were performed using GraphPad Prism® 8.0/9.0 (GraphPad Software, San Diego, CA). Western blots were quantified by densitometric analyses using ImageJ software and standardized to β-actin. Sample sizes were chosen based on availability of experimental samples. The sample sizes were sufficient since we used many experimental techniques to confirm the results. Statistical methods were not used to determine sample size on animal studies. Results represent mean ± SD. All experiments were repeated at least three independent biological replicates. Comparisons between groups were calculated using unpaired, two-tailed, Student’s t test (two groups) and one-way ANOVA or two-way ANOVA (multiple groups). *p < 0.05, **p < 0.01, ***p < 0.001, n.s. not significant.
Data availability
The raw and processed data used for this study have been deposited on GEO with accession numbers GSE235331.
Code availability
Code for the analysis used in this study have been deposited on GitHub at https://github.com/xychen24/Code_for_ISRIB
References
Khanim F, Kirk J, Latif F, Barrett TG. WFS1/wolframin mutations, Wolfram syndrome, and associated diseases. Hum Mutat. 2001;17:357–67.
Urano F. Wolfram syndrome: diagnosis, management, and treatment. Curr Diab Rep. 2016;16:6.
Rigoli L, Bramanti P, Di Bella C, De Luca F. Genetic and clinical aspects of Wolfram syndrome 1, a severe neurodegenerative disease. Pediatr Res. 2018;83:921–9.
Barrett TG, Bundey SE. Wolfram (DIDMOAD) syndrome. J Med Genet. 1997;34:838–41.
Rigoli L, Lombardo F, Di Bella C. Wolfram syndrome and WFS1 gene. Clin Genet. 2011;79:103–17.
Inoue H, Tanizawa Y, Wasson J, Behn P, Kalidas K, Bernal-Mizrachi E, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet. 1998;20:143–8.
Takeda K, Inoue H, Tanizawa Y, Matsuzaki Y, Oba J, Watanabe Y, et al. WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet. 2001;10:477–84.
Yamada T, Ishihara H, Tamura A, Takahashi R, Yamaguchi S, Takei D, et al. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Hum Mol Genet. 2006;15:1600–9.
Kitamura RA, Maxwell KG, Ye W, Kries K, Brown CM, Augsornworawat P, et al. Multidimensional analysis and therapeutic development using patient iPSC-derived disease models of Wolfram syndrome. JCI Insight. 2022;7:e156549.
Karasik A, O’Hara C, Srikanta S, Swift M, Soeldner JS, Kahn CR, et al. Genetically programmed selective islet beta-cell loss in diabetic subjects with Wolfram’s syndrome. Diabetes Care. 1989;12:135–8.
Wang L, Liu H, Zhang X, Song E, Wang Y, Xu T, et al. WFS1 functions in ER export of vesicular cargo proteins in pancreatic β-cells. Nat Commun. 2021;12:6996.
Riggs AC, Bernal-Mizrachi E, Ohsugi M, Wasson J, Fatrai S, Welling C, et al. Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia. 2005;48:2313–21.
Ishihara H, Takeda S, Tamura A, Takahashi R, Yamaguchi S, Takei D, et al. Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet. 2004;13:1159–70.
Fonseca SG, Fukuma M, Lipson KL, Nguyen LX, Allen JR, Oka Y, et al. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J Biol Chem. 2005;280:39609–15.
Fonseca SG, Ishigaki S, Oslowski CM, Lu S, Lipson KL, Ghosh R, et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest. 2010;120:744–55.
Shang L, Hua H, Foo K, Martinez H, Watanabe K, Zimmer M, et al. β-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes. 2014;63:923–33.
Jain C, Ansarullah, Bilekova S, Lickert H. Targeting pancreatic beta cells for diabetes treatment. Nat Metab. 2022;4:1097–108.
Baumann K. mRNA translation in stress granules is not uncommon. Nat Rev Mol Cell Biol. 2021;22:164.
Costa-Mattioli M, Walter P. The integrated stress response: from mechanism to disease. Science. 2020;368:eaat5314.
Hinnebusch AG, Ivanov IP, Sonenberg N. Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science. 2016;352:1413–6.
Yuan F, Li Y, Hu R, Gong M, Chai M, Ma X, et al. Modeling disrupted synapse formation in wolfram syndrome using hESCs-derived neural cells and cerebral organoids identifies Riluzole as a therapeutic molecule. Mol Psychiatry. 2023;28:1557–70.
Chen D, Xu T, Li Y, Xu J, Peng B, Xu W, et al. Stress regulation of WFS1 and PERK-p-eIF2alpha-ATF4 signaling pathway in placental tissue cells of intrahepatic cholestasis of pregnancy. Placenta. 2023;139:1–11.
Smedley GD, Walker KE, Yuan SH. The role of PERK in understanding development of neurodegenerative diseases. Int J Mol Sci. 2021;22:8146.
de Heredia ML, Clèries R, Nunes V. Genotypic classification of patients with Wolfram syndrome: insights into the natural history of the disease and correlation with phenotype. Genet Med. 2013;15:497–506.
Medlej R, Wasson J, Baz P, Azar S, Salti I, Loiselet J, et al. Diabetes mellitus and optic atrophy: a study of Wolfram syndrome in the Lebanese population. J Clin Endocrinol Metab. 2004;89:1656–61.
Barrett TG, Bundey SE, Macleod AF. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet. 1995;346:1458–63.
Sandhu MS, Weedon MN, Fawcett KA, Wasson J, Debenham SL, Daly A, et al. Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet. 2007;39:951–3.
Maxwell KG, Augsornworawat P, Velazco-Cruz L, Kim MH, Asada R, Hogrebe NJ, et al. Gene-edited human stem cell-derived β cells from a patient with monogenic diabetes reverse preexisting diabetes in mice. Sci Transl Med. 2020;12:eaax9106.
Abreu D, Asada R, Revilla JMP, Lavagnino Z, Kries K, Piston DW, et al. Wolfram syndrome 1 gene regulates pathways maintaining beta-cell health and survival. Lab Invest. 2020;100:849–62.
Morikawa S, Tajima T, Nakamura A, Ishizu K, Ariga T. A novel heterozygous mutation of the WFS1 gene leading to constitutive endoplasmic reticulum stress is the cause of Wolfram syndrome. Pediatr Diabetes. 2017;18:934–41.
Ma Q, Xiao Y, Xu W, Wang M, Li S, Yang Z, et al. ZnT8 loss-of-function accelerates functional maturation of hESC-derived β cells and resists metabolic stress in diabetes. Nat Commun. 2022;13:4142.
Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH, et al. Generation of functional human pancreatic β cells in vitro. Cell. 2014;159:428–39.
Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. 2014;32:1121–33.
McGaugh EC, Nostro MC. Efficient differentiation of pluripotent stem cells to NKX6-1+ pancreatic progenitors. J Vis Exp. 2017;7:55265.
Micallef SJ, Li X, Schiesser JV, Hirst CE, Yu QC, Lim SM, et al. INS(GFP/w) human embryonic stem cells facilitate isolation of in vitro derived insulin-producing cells. Diabetologia. 2012;55:694–706.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308.
Su Q, Yuan F, Li X, Wang X, Yang K, Shao L, et al. Wfs1 loss-of-function disrupts the composition of mouse pancreatic endocrine cells from birth and impairs Glut2 localization to cytomembrane in pancreatic β cells. Biochem Biophys Res Commun. 2023;658:80–87.
Balboa D, Barsby T, Lithovius V, Saarimaki-Vire J, Omar-Hmeadi M, Dyachok O, et al. Functional, metabolic and transcriptional maturation of human pancreatic islets derived from stem cells. Nat Biotechnol. 2022;40:1042–55.
Peng J, Sun BF, Chen CY, Zhou JY, Chen YS, Chen H, et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019;29:725–38.
Veres A, Faust AL, Bushnell HL, Engquist EN, Kenty JH, Harb G, et al. Charting cellular identity during human in vitro β-cell differentiation. Nature. 2019;569:368–73.
Olaniru OE, Kadolsky U, Kannambath S, Vaikkinen H, Fung K, Dhami P, et al. Single-cell transcriptomic and spatial landscapes of the developing human pancreas. Cell Metab. 2023;35:184–99.e5.
Bartolomé A. The pancreatic beta cell: editorial. Biomolecules. 2023;13:495.
Dror E, Fagnocchi L, Wegert V, Apostle S, Grimaldi B, Gruber T, et al. Epigenetic dosage identifies two major and functionally distinct β cell subtypes. Cell Metab. 2023;35:821-836.e7.
Dorrell C, Schug J, Canaday PS, Russ HA, Tarlow BD, Grompe MT, et al. Human islets contain four distinct subtypes of β cells. Nat Commun. 2016;7:11756.
Benninger RKP, Hodson DJ. New Understanding of β-Cell Heterogeneity and In Situ Islet Function. Diabetes. 2018;67:537–47.
Tritschler S, Theis FJ, Lickert H, Böttcher A. Systematic single-cell analysis provides new insights into heterogeneity and plasticity of the pancreas. Mol Metab. 2017;6:974–90.
Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017;14:979–82.
Qiu X, Hill A, Packer J, Lin D, Ma YA, Trapnell C. Single-cell mRNA quantification and differential analysis with Census. Nat Methods. 2017;14:309–15.
Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–6.
Tang X, Uhl S, Zhang T, Xue D, Li B, Vandana JJ, et al. SARS-CoV-2 infection induces beta cell transdifferentiation. Cell Metab. 2021;33:1577–91.e7.
Nayak RR, Bernal WE, Lee JW, Kearns MJ, Cheung VG. Stress-induced changes in gene interactions in human cells. Nucleic Acids Res. 2014;42:1757–71.
Muraro MJ, Dharmadhikari G, Grün D, Groen N, Dielen T, Jansen E, et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016;3:385–94.e3.
Blum B, Hrvatin S, Schuetz C, Bonal C, Rezania A, Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol. 2012;30:261–4.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29.
Aleksander SA, Balhoff J, Carbon S, Cherry JM, Drabkin HJ, Ebert D, et al. The gene ontology knowledgebase in 2023. Genetics. 2023;224:iyad031.
Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol. 2009;10:430–6.
Protter DSW, Parker R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016;26:668–79.
Yang P, Mathieu C, Kolaitis RM, Zhang P, Messing J, Yurtsever U, et al. G3BP1 Is a Tunable Switch that Triggers Phase Separation to Assemble Stress Granules. Cell. 2020;181:325–45.e28.
Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17:1374–95.
Zyryanova AF, Kashiwagi K, Rato C, Harding HP, Crespillo-Casado A, Perera LA, et al. ISRIB Blunts the Integrated Stress Response by Allosterically Antagonising the Inhibitory Effect of Phosphorylated eIF2 on eIF2B. Mol Cell. 2021;81:88–103.e106.
Sidrauski C, McGeachy AM, Ingolia NT, Walter P. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife. 2015;4:e05033.
Yeomans A, Thirdborough SM, Valle-Argos B, Linley A, Krysov S, Hidalgo MS, et al. Engagement of the B-cell receptor of chronic lymphocytic leukemia cells drives global and MYC-specific mRNA translation. Blood. 2016;127:449–57.
Altman MK, Alshamrani AA, Jia W, Nguyen HT, Fambrough JM, Tran SK, et al. Suppression of the GTPase-activating protein RGS10 increases Rheb-GTP and mTOR signaling in ovarian cancer cells. Cancer Lett. 2015;369:175–83.
Slomnicki LP, Malinowska A, Kistowski M, Palusinski A, Zheng JJ, Sepp M, et al. Nucleolar Enrichment of Brain Proteins with Critical Roles in Human Neurodevelopment. Mol Cell Proteom. 2016;15:2055–75.
Morral C, Stanisavljevic J, Batlle E. Protocol for Efficient Protein Synthesis Detection by Click Chemistry in Colorectal Cancer Patient-Derived Organoids Grown In Vitro. STAR Protoc. 2020;1:100103.
Hatanaka M, Tanabe K, Yanai A, Ohta Y, Kondo M, Akiyama M, et al. Wolfram syndrome 1 gene (WFS1) product localizes to secretory granules and determines granule acidification in pancreatic beta-cells. Hum Mol Genet. 2011;20:1274–84.
Plaas M, Seppa K, Reimets R, Jagomäe T, Toots M, Koppel T, et al. Wfs1- deficient rats develop primary symptoms of Wolfram syndrome: insulin-dependent diabetes, optic nerve atrophy and medullary degeneration. Sci Rep. 2017;7:10220.
Gong Y, Xiong L, Li X, Su L, Xiao H. A novel mutation of WFS1 gene leading to increase ER stress and cell apoptosis is associated an autosomal dominant form of Wolfram syndrome type 1. BMC Endocr Disord. 2021;21:76.
Kakiuchi C, Ishigaki S, Oslowski CM, Fonseca SG, Kato T, Urano F. Valproate, a mood stabilizer, induces WFS1 expression and modulates its interaction with ER stress protein GRP94. PLoS One. 2009;4:e4134.
Toots M, Seppa K, Jagomäe T, Koppel T, Pallase M, Heinla I, et al. Preventive treatment with liraglutide protects against development of glucose intolerance in a rat model of Wolfram syndrome. Sci Rep. 2018;8:10183.
Kondo M, Tanabe K, Amo-Shiinoki K, Hatanaka M, Morii T, Takahashi H, et al. Activation of GLP-1 receptor signalling alleviates cellular stresses and improves beta cell function in a mouse model of Wolfram syndrome. Diabetologia. 2018;61:2189–201.
Gorgogietas V, Rajaei B, Heeyoung C, Santacreu BJ, Marín-Cañas S, Salpea P, et al. GLP-1R agonists demonstrate potential to treat Wolfram syndrome in human preclinical models. Diabetologia. 2023;66:1306–21.
Lu S, Kanekura K, Hara T, Mahadevan J, Spears LD, Oslowski CM, et al. A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome. Proc Natl Acad Sci USA. 2014;111:E5292–5301.
Abreu D, Stone SI, Pearson TS, Bucelli RC, Simpson AN, Hurst S, et al. A phase Ib/IIa clinical trial of dantrolene sodium in patients with Wolfram syndrome. JCI Insight. 2021;6:e145188.
Nguyen LD, Fischer TT, Abreu D, Arroyo A, Urano F, Ehrlich BE. Calpain inhibitor and ibudilast rescue β cell functions in a cellular model of Wolfram syndrome. Proc Natl Acad Sci USA. 2020;117:17389–98.
Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21.
Bugallo R, Marlin E, Baltanas A, Toledo E, Ferrero R, Vinueza-Gavilanes R, et al. Fine tuning of the unfolded protein response by ISRIB improves neuronal survival in a model of amyotrophic lateral sclerosis. Cell Death Dis. 2020;11:397.
Krukowski K, Nolan A, Frias ES, Boone M, Ureta G, Grue K, et al. Small molecule cognitive enhancer reverses age-related memory decline in mice. Elife. 2020;9:e62048.
Rabouw HH, Langereis MA, Anand AA, Visser LJ, de Groot RJ, Walter P, et al. Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc Natl Acad Sci USA. 2019;116:2097–102.
Hofmann S, Kedersha N, Anderson P, Ivanov P. Molecular mechanisms of stress granule assembly and disassembly. Biochim Biophys Acta Mol Cell Res. 2021;1868:118876.
Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. Embo j. 2005;24:1243–55.
Wang Q, Mora-Jensen H, Weniger MA, Perez-Galan P, Wolford C, Hai T, et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci USA. 2009;106:2200–5.
Hiwatashi Y, Kanno K, Takasaki C, Goryo K, Sato T, Torii S, et al. PHD1 interacts with ATF4 and negatively regulates its transcriptional activity without prolyl hydroxylation. Exp Cell Res. 2011;317:2789–99.
Köditz J, Nesper J, Wottawa M, Stiehl DP, Camenisch G, Franke C, et al. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood. 2007;110:3610–7.
Jousse C, Deval C, Maurin AC, Parry L, Chérasse Y, Chaveroux C, et al. TRB3 inhibits the transcriptional activation of stress-regulated genes by a negative feedback on the ATF4 pathway. J Biol Chem. 2007;282:15851–61.
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–33.
Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. 2008;40:14–21.
Bond S, Lopez-Lloreda C, Gannon PJ, Akay-Espinoza C, Jordan-Sciutto KL. The Integrated Stress Response and Phosphorylated Eukaryotic Initiation Factor 2alpha in Neurodegeneration. J Neuropathol Exp Neurol. 2020;79:123–43.
Wang S, Sun S. Translation dysregulation in neurodegenerative diseases: a focus on ALS. Mol Neurodegener. 2023;18:58.
Halliday M, Radford H, Sekine Y, Moreno J, Verity N, le Quesne J, et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 2015;6:e1672.
Frias ES, Hoseini MS, Krukowski K, Paladini MS, Grue K, Ureta G, et al. Aberrant cortical spine dynamics after concussive injury are reversed by integrated stress response inhibition. Proc Natl Acad Sci USA. 2022;119:e2209427119.
Nostro MC, Sarangi F, Yang C, Holland A, Elefanty AG, Stanley EG, et al. Efficient generation of NKX6-1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Rep. 2015;4:591–604.
Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–87.e29.
Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov (Camb). 2021;2:100141.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 32170740; 32370855; 82171166; 32122030; 31970751; 81971078); the National Key R&D Program of China (2020YFA0112500); the Key Project of the Science and Technology Commission of Shanghai Municipality (Grant No. 19JC1415300, 23J21901500); Natural Science Foundation of Shanghai (21ZR1452400); the Health System Leading Talents Training Program of Pudong New district, Shanghai (PWR12020-07); Shanghai Rising-Star Program (A type) of the Science and Technology Commission of Shanghai Municipality (21QA1409500, 21QA1409400); the Science and Technology Innovation Action Plan of the Science and Technology Commission of Shanghai Municipality (21JC1405500).
Author information
Authors and Affiliations
Contributions
RH, XC, QS, Z-NZ, LS and WL conceived and designed the experiments. RH performed SC-islets differentiation, immunostaining, ELISA, flow cytometry, total protein synthesis assay and other experiments. RH and YG performed single-cell RNA-seq library construction. XC performed single-cell RNA-seq profiling analyses. QS performed experiments associated with Wfs1 conditional knockout mice including IPGTT, GSIS in vivo, mouse pancreas immunostaining and other experiments of animals. RL and PD designed protein synthesis assay. RH, XC, QS, ZW, XW, MG, MX, LS, Z-NZ and WL wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Animal studies were performed in accordance with the University of Health Guide for the Care and Use of Laboratory Animals and approved by the Biological Research Ethics Committee of Tongji University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, R., Chen, X., Su, Q. et al. ISR inhibition reverses pancreatic β-cell failure in Wolfram syndrome models. Cell Death Differ 31, 322–334 (2024). https://doi.org/10.1038/s41418-024-01258-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-024-01258-w
