
Abstract
Phenotypic modulation of vascular smooth muscle cells (VSMCs) plays critical roles in the pathogenesis of aortic aneurysm (AA). The function of nuclear receptor corepressor1 (NCOR1) in regulation of VSMC phenotype and AA is unclear. Herein, using smooth muscle NCOR1 knockout mice, we demonstrated that smooth muscle NCOR1 deficiency decreased both mRNA and protein levels of contractile genes, impaired stress fibers formation and RhoA pathway activation, reduced synthesis of elastin and collagens, and induced the expression and activity of MMPs, manifesting a switch from contractile to degradative phenotype of VSMCs. NCOR1 modulated VSMC phenotype through 3 different mechanisms. First, NCOR1 deficiency increased acetylated FOXO3a to inhibit the expression of Myocd, which downregulated contractile genes. Second, deletion of NCOR1 derepressed NFAT5 to induce the expression of Rgs1, thus impeding RhoA activation. Third, NCOR1 deficiency increased the expression of Mmp12 and Mmp13 by derepressing ATF3. Finally, a mouse model combined apoE knockout mice with angiotensin II was used to study the role of smooth muscle NCOR1 in the development of AA. The results showed that smooth muscle NCOR1 deficiency increased the incidence of aortic aneurysms and exacerbated medial degeneration in angiotensin II-induced AA mouse model. Collectively, our data illustrated that NCOR1 interacts with FOXO3a, NFAT5, and ATF3 to maintain contractile phenotype of VSMCs and suppress AA development. Manipulation of smooth muscle NCOR1 may be a potential approach for AA treatment.
Similar content being viewed by others


Introduction
Phenotypic modulation of vascular smooth muscle cells (VSMCs) is a critical process that are implicated in the pathogenesis of many vascular diseases, including atherosclerosis, aortic aneurysm (AA), and hypertension [1,2,3]. VSMCs are a group of highly specialized cells that are responsible for structure maintenance and contraction of blood vessels. In response to different environmental stimuli, VSMCs exhibit different phenotypes such as contractile, synthetic, and degradative phenotypes [4, 5]. Transcriptional or epigenetic reprogramming has been identified as a switch in governing VSMC plasticity [6] and targeting gene regulatory apparatus has emerged as an effective strategy in manipulating phenotypes of VSMCs. Although a multitude of transcriptional factors or epigenetic enzymes have been demonstrated to play essential roles in phenotypic switching [7,8,9], transcriptional regulation of phenotypic plasticity of VSMCs is still incompletely understood and more targets that effectively modulate phenotypes of VSMCs remain to be identified.
Nuclear receptor corepressor1 (NCOR1) is a master transcriptional coregulator through modifying the acetylation of histones or transcription factors in different cell types [10]. It has been shown that NCOR1 suppressed the activity of PPARβ/δ and ERRα in myotubes and is indispensable for modulating muscle function [11]. In macrophages, NCOR1 exacerbates insulin resistance of obese mice through repressing LXR [12] and protects against atherosclerosis by repressing PPARγ [13]. Moreover, recent studies have established that NCOR1 is strongly associated with cardiovascular diseases. Cardiomyocyte NCOR1 has been shown to interact with MEF2 to repress cardiac hypertrophy [14]. Macrophage NCOR1 deficiency attenuates myocardial infarction injury and neointimal hyperplasia [15]. However, the role of NCOR1 in VSMCs and vascular diseases such as AA have remained unknown.
To determine the function of NCOR1 in VSMCs, we first generated VSMC NCOR1 knockout (SNKO) mice and investigated the impact of NCOR1 deficiency on contractile genes, contractility, and extracellular matrix degradation of VSMCs. Subsequently, we unveiled the underlying mechanism how NCOR1 regulated the activities of transcription factors FOXO3a, NFAT5, and ATF3 in VSMCs. Finally, we investigated the role of smooth muscle NCOR1 in AngII-induced AA using SNKO mice.
Results
Smooth muscle NCOR1 deficiency downregulates contractile genes
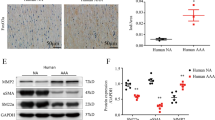
Western blotting analysis showed that the level of NCOR1 was significantly higher in aortas of AngII-infused mice than those of vehicle-infused mice (Supplemental Fig. 1A and 1B), suggesting the importance of NCOR1 in vascular pathology. To further understand the function of NCOR1 in the vasculature, we generated smooth muscle-specific NCOR1 knockout (SNKO) mice by crossing floxed Ncor1 mice [11] with SM22α-CreER mice [16]. Deletion of NCOR1 was induced in SNKO mice by administration of tamoxifen (Supplemental Fig. 1C). As expected, both mRNA level and protein level of NCOR1 decreased significantly in aortas of SNKO mice compared to littermate control (LC) mice (Supplemental Fig. 1D, E). Efficient deletion of NCOR1 in isolated vascular smooth muscle cells (VSMCs) was demonstrated at mRNA level by QRT-PCR (Supplemental Fig. 1F), and at protein level by western blotting analysis and immunofluorescence staining (Supplemental Fig. 1G, 1H).
We next investigated the effects of NCOR1 deficiency on gene expression in VSMCs. RNA sequencing (RNA-seq) detected 948 induced genes and 821 repressed genes in VSMCs isolated from SNKO mice compared to those from LC mice (Fig. 1A). Pathways related to smooth muscle contraction, cell adhesion, and regulation of actin cytoskeleton were highly enriched in the downregulated genes (Fig. 1B). Accordingly, Myocd and contractile genes including Acta2, Cnn1, Myl9, and Myh11 were downregulated in SNKO VSMCs (Fig. 1C). QRT-PCR results further revealed that expression of contractile genes was markedly suppressed in SNKO VSMCs compared to LC VSMCs (Fig. 1D). Consistently, results of immunofluorescence staining indicated that protein levels of α-SMA, Calponin1, MLC2, SMMHC, and SM22α were decreased in SNKO VSMCs (Fig. 1E and Supplemental Fig. 2). Western blotting analysis also demonstrated that SNKO VSMCs had less expression of contractile proteins compared to LC VSMCs (Fig. 1F, G). Moreover, QRT-PCR analysis revealed that expression of contractile genes and Myocd was downregulated in aortas from SNKO mice compared to LC mice (Fig. 1H). Consistent with the results in primary VSMCs, Ncor1 knockdown using siRNA (Supplemental Fig. 3A) strikingly decreased the level of contractile genes in human smooth muscle cells (hSMCs) (Supplemental Fig. 3B–D). Conversely, expression of contractile genes was markedly induced by NCOR1 overexpression using lentivirus in hSMCs (Supplemental Fig. 3E, F). Taken together, these data demonstrated a crucial role of NCOR1 in regulating contractile genes in VSMCs.

A Volcano plot of RNA-seq data showing significantly induced genes (948) or repressed genes (821) in VSMCs isolated from smooth muscle-specific NCOR1 knockout (SNKO) mice versus VSMCs from littermate control (LC) mice. The threshold for induced genes was fold change (FC) > 2 and that for repressed genes was FC < −2; false discovery rate (FDR) < 0.05 and P value < 0.05 for both induced and repressed genes. B Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of downregulated genes (SNKO vs LC). C Heatmap presentation of RNA-Seq results displaying relative expression of contractile genes. D QRT-PCR analysis of contractile genes and Myocd in VSMCs isolated from LC or SNKO mice. E Representative immunofluorescence staining of α-SMA, Calponin1, MLC2, and SMMHC in VSMCs. Scale bar: 50 μm. F Representative western blotting analysis of a-SMA, Calponin1, MLC2, SMMHC, and Myocardin in VSMCs. α-tubulin was used as a loading control. The samples were from the same experiment and gels/blots were processed in parallel. G Quantification of western blotting results. H QRT-PCR analysis of contractile genes and Myocd in aortas of LC and SNKO mice. n = 6:6 for Acta2, Cnn1, Myl9, Myh11; n = 6:5 for Myocd. The experiments were repeated for three times (D, E, and G). Values are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 determined by Student’s t-test. α-SMA indicates smooth muscle actin α2; MLC2 myosin light chain 2, SMMHC smooth muscle myosin heavy chain.
Deficiency of NCOR1 impairs RhoA signaling pathway in VSMCs
Activation of RhoA facilitates phosphorylation of myosin light chain (MLC2), which directly leads to the contraction of VSMCs [17], and then increases actin polymerization and stress fiber formation in VSMCs [18]. To evaluate the impact of NCOR1 deficiency on RhoA activation, VSMCs isolated from LC and SNKO mice were first treated with angiotensinII (AngII) to induce the formation of stress fibers. Immunofluorescence staining of F-actin showed that less elongated cells with well-defined stress fibers were observed among SNKO VSMCs compared to LC VSMCs (Fig. 2A, B). Western blotting analysis illustrated that deletion of NCOR1 significantly inhibited activation of RhoA in VSMCs with or without AngII stimulation (Fig. 2C, D). Similar results were obtained in VSMCs treated with lysophosphatidic acid (LPA) (Supplemental Fig. 4A, B). Consistently, markedly suppressed phosphorylation of MLC2 was detected in SNKO VSMCs compared to LC VSMCs within a 10-minute time course of stimulation by AngII (Fig. 2E, F) or phenylephrine (PE) (Supplemental Fig. 4C, D). Similar suppression on MLC2 phosphorylation was observed in hSMCs transfected with Ncor1 siRNA (Supplemental Fig. 4E, F). Furthermore, we examined phosphorylation of MLC2 in LC and SNKO aortas incubated with AngII or PE ex-vivo. Results of western blotting indicated that SNKO aortas had less phosphorylation of MLC2 compared to LC aortas (Fig. 2G, H and Supplemental Fig. 4G–I). Collectively, these data suggested that NCOR1 deficiency inhibited the contractility and stress fiber formation of VSMCs by impairing RhoA signaling pathway.

A Representative immunofluorescence staining of F-actin to detect stress fibers in VSMCs treated with vehicle (0.9% sodium chloride) or angiotensin II (AngII, 1 μM) for 2 h. Boxed areas are shown in higher magnification by inset images. Scale bar: 100 μm. B Quantification of cell roundness factor of VSMCs. Each circle represents a single cell. C Representative western blotting analysis of active RhoA in lysate pulldowns of VSMCs treated with vehicle or AngII (1 μM) for 10 min. Rhotekin-binding domain was used as a bait for the pulldowns. Total RhoA was used as a loading control. D Quantification of western blotting results as Active RhoA/Total RhoA. E Representative western blotting analysis of phosphorylation and total protein levels of myosin light chain 2 (MLC2) in VSMCs treated with AngII (1 μM) for indicated time periods. α-tubulin was used as a loading control. The samples were from the same experiment and gels/blots were processed in parallel. F Quantification of western blotting results as p-MLC2(ser19)/MLC2, p-MLC2(ser19)/α-tubulin, and MLC2/α-tubulin. G Representative western blotting analysis of phosphorylation and total protein levels of MLC2 in thoracic aortas taken from LC or SNKO mice and treated with vehicle or AngII (1 μM) for 20 min ex-vivo. GAPDH was used as a loading control. The samples were from the same experiment and gels/blots were processed in parallel. Thoracic aortas from 2 to 3 mice were pooled as one sample. H Quantification of western blotting results as p-MLC2(ser19)/MLC2 and p-MLC2(ser19)/GAPDH. n = 4:4:6:6. Data from three independent experiments were presented in B, D, and F. Values are expressed as mean ± SEM. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001 determined by 2-way ANOVA with Bonferroni post-tests.
Smooth muscle NCOR1 deficiency promotes degradation of extracellular matrix
In addition to contractile capacity, VSMCs acquire important secretory properties to ensure the synthesis of extracellular matrix and maintain the structure of vascular wall [19]. Gene ontology term enrichment analysis of RNA-Seq results revealed that NCOR1 deficiency in VSMCs downregulated the expression of genes highly enriched in terms related to extracellular matrix organization and cell adhesion (Fig. 3A). Synthetic genes such as Eln, Mfap4 and Col1a1 were dramatically suppressed in SNKO VSMCs (Fig. 3B). It has been shown that matrix metalloproteinases (MMPs) play critical roles in degradation of extracellular matrix [20]. Interestingly, the expression of Mmp3, Mmp12, Mmp13, and Mmp27 were markedly increased in SNKO VSMCs (Fig. 3B). Consistently, QRT-PCR analysis demonstrated that NCOR1 deletion markedly decreased the expression of Eln, Col1a1, Col6a1, and Col16a1, and significantly increased the expression of Mmp3, Mmp12, and Mmp13 in VSMCs (Fig. 3C). Results of western blotting further revealed that the protein level of elastin was reduced and the levels of MMP3, MMP12, and MMP13 were elevated in SNKO VSMCs compared to LC VSMCs with or without AngII stimulation (Fig. 3D, E). Immunofluorescence staining illustrated that SNKO VSMCs had less expression of elastin compared to LC VSMCs with or without AngII treatment (Fig. 3F, G). MMP activities, evaluated by in situ zymography (DQ gelatin), were induced in SNKO VSMCs compared to LC VSMCs with or without AngII stimulation (Fig. 3H, I). Accordingly, QRT-PCR results illustrated suppressed expression of Eln, Col6a1, and Col16a1, and increased expression of Mmp3, Mmp12 and Mmp13 in aortas of SNKO compared to LC mice (Fig. 3J). These results indicated that NCOR1 deficiency promoted degradation of extracellular matrix by suppressing synthetic genes and inducing MMPs. In addition, gene ontology term enrichment analysis of RNA-Seq results revealed upregulation of genes associated with inflammatory response and innate immune response in SNKO VSMCs (Supplemental Fig. 5A). Expression of genes related to immune response was substantially increased in SNKO compared to LC VSMCs (Supplemental Fig. 5B) and most of these genes were involved in macrophage activation, such as Lgals3, Tlr1, and JAK3, indicating that NCOR1 deficiency promoted VSMCs to acquire macrophage markers and exhibit proinflammatory phenotype. Therefore, NCOR1-deficient VSMCs shared similar characteristics with Tsc1-deficient VSMCs that were termed as degradative VSMCs [21].

A Gene ontology analysis of downregulated genes in primary VSMCs (SNKO vs LC) determined by RNA-seq. MF indicates molecular function; CC, cellular component; BP, biological process. B Heatmap presentation of RNA-Seq results displaying relative expression of matrix metalloproteinases and synthetic genes identified in Gene Ontology analysis. C QRT-PCR analysis of Eln, Col1a1, Col6a1, Col16a1, Mmp3, Mmp12, and Mmp13 in VSMCs. D Representative western blotting analysis of elastin, MMP3, MMP12, and MMP13 in VSMCs treated with vehicle or AngII (1 μM) for 24 h. GAPDH was used as a loading control. The samples were from the same experiment and gels/blots were processed in parallel. E Quantification of western blotting results. F Representative immunofluorescence staining of elastin in VSMCs treated with vehicle or AngII (1 μM) for 24 h. Boxed areas are shown in higher magnification by inset images. Scale bar: 200 μm. G Quantification of elastin expression as mean fluorescence intensity (MFI). H Representative immunofluorescence images of in situ zymography (DQ gelatin) assessing MMP activities in VSMCs treated with vehicle (0.9% sodium chloride) or AngII (1 μM) for 24 h. Boxed areas are shown in higher magnification by inset images. Scale bar: 200 μm. I Quantification of MMP activities using relative fluorescence units (RFU) in VSMCs. J QRT-PCR analysis of Eln, Col6a1, Col16a1, Mmp3, Mmp12, and Mmp13 in aortas of LC and SNKO mice. n = 4:5 for Eln, n = 5:5 for Col6a1 and Col16a1, n = 5:6 for Mmp3, n = 4:4 for Mmp12, n = 6:6 for Mmp13. The experiments were repeated for three times (C, E, G, and I). Values are expressed as mean ± SEM. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001 determined by Student’s t-test (C and J) or 2-way ANOVA with Bonferroni post-tests (E, G and I). Eln indicates elastin; Col1a1 indicates collagen type I alpha 1; Col6a1 indicates collagen type VI alpha 1; Col16a1 indicates collagen type XVI alpha 1.
FOXO3a mediates the suppressive effects of NCOR1 deficiency on contractile genes
We next explored the mechanism how NCOR1 regulated the expression of contractile genes. Serum response factor (SRF) and its coactivators Myocd, Mrtfa, and Mrtfb, are the main regulators of contractile genes in VSMCs [7, 22]. QRT-PCR analysis indicated that deletion of NCOR1 did not affect the expression of SRF in VSMCs (Supplemental Fig. 6A). In addition, no interaction was observed between NCOR1 and SRF in VSMCs according to Co-immunoprecipitation (Co-IP) analysis (Supplemental Fig. 6B). Interestingly, NCOR1 deficiency significantly suppressed the expression of Myocd (Fig. 1D) and did not affect the expression of Mrtfa and Mrtfb (Supplemental Fig. 7). Thus, it was more likely that NCOR1 deletion inhibited expression of contractile genes by downregulating Myocd. It has been demonstrated that Myocd is a transcriptional target of MEF2, TEAD and FOXO in VSMCs [23]. Previous study has revealed that FOXO3a is recruited and deacetylated by HDAC3/NCOR1 complex to activate gene expression [24]. Acetylation of FOXO neutralizes the positive charges of lysine residues of forkead DNA-binding domain, interferes the interaction between the positively charged basic residues of FOXO and negatively charged residues of DNA chain, and thus reduces the DNA-binding affinity and transcriptional activity of FOXO [25]. Therefore, we hypothesized that NCOR1 deficiency increased acetylation of FOXO3 to inhibit Myocd and consequently contractile gene expression. We first identified a putative FOXO3 binding site on the promoter of Myocd using FIMO software (Fig. 4A) and then performed chromatin immunoprecipitation (ChIP) using antibodies against NCOR1 and FOXO3a in VSMCs. The results revealed that both NCOR1 and FOXO3a were enriched within the binding region on the promoter of Myocd (Fig. 4B). Co-IP analysis demonstrated that NCOR1 interacted with FOXO3a (Fig. 4C). Increased acetylation of FOXO3a was observed in SNKO VSMCs compared to LC VSMCs (Fig. 4D), as well as in VSMCs treated with acetylase inhibitors—nicotinamide and trichostatin A (Supplemental fig. 8). In line with this, results of ChIP indicated that NCOR1 deficiency reduced the enrichment of FOXO3a binding at the promoter of Myocd (Fig. 4E). These results together suggested that NCOR1 interacted with FOXO3a to regulate expression of Myocd in VSMCs. Moreover, QRT-PCR analysis revealed that FOXO3a knockdown (Fig. 4F) significantly suppressed expression of contractile genes in hSMCs (Fig. 4G). Results of western blotting showed that FOXO3a knockdown decreased levels of a-SMA, Calponin1, MLC2, SMMHC, and Myocardin in hSMCs (Fig. 4H, I). Taken together, these data implied that NCOR1 deficiency inhibited expression of contractile genes through impairing the transcriptional activity of FOXO3a.

A Schematic illustration of putative FOXO3 binding region on the promoter of mouse Myocd. Forward primer (fwd) and reverse primer (rev) for ChIP assay were indicated by arrows. B ChIP analysis showing enrichment of NCOR1 and FOXO3a on the promoter of Myocd in VSMCs. ChIP products were amplified using regular PCR and analyzed by gel electrophoresis. C Co-immunoprecipitation analysis of NCOR1 and FOXO3a in VSMCs. D Representative analysis of FOXO3a acetylation in VSMCs isolated from LC or SNKO mice. Antibodies against Ac-Lys (ac-K) were used for immunoprecipitation. E ChIP analysis of enrichment of FOXO3a on the promoter of Myocd in VSMCs isolated from LC or SNKO mice. ChIP products were analyzed using qPCR. QRT-PCR analysis of Foxo3a (F) and contractile genes (G) in human smooth muscle cells (hSMCs) transfected with control siRNA (siCTL) or Foxo3a siRNA (siFoxo3a) for 48 h. H Representative western blotting analysis of FOXO3a, α-SMA, Calponin1, MLC2, SMMHC, and Myocardin in hSMCs transfected with control siRNA or Foxo3a siRNA for 72 h. α-tubulin was used as a loading control. The samples were from the same experiment and gels/blots were processed in parallel. I Quantification of western blotting results. The experiments were repeated for three times (B–D, F, G, and I). Data from three independent experiments were presented in E. Values are expressed as mean ± SEM. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001 determined by Student’s t-test.
NFAT5 mediates the impacts of NCOR1 deficiency on RhoA activation in VSMCs
We went on to investigate how NCOR1 regulated RhoA. QRT-PCR analysis demonstrated that NCOR1 deficiency did not affect the expression of angiotensin II type 1a receptor (Agtr1a), the receptor for AngII, or α-adrenergic receptor (Adra1a), Adra1b and Adra1d, the receptors for phenylephrine, in VSMCs (Supplemental Fig. 9). Further analysis of RNA-seq data revealed that NCOR1 deficiency induced a group of genes enriched in a term related to GTPase activator activity (Fig. 5A), including Rgs1 and Rgs2 that were reported to inhibit vascular contraction in mice [26, 27]. Knockdown of Rgs1 in VSMCs (Fig. 5B) blocked the impairment of active-RhoA in SNKO VSMCs (Fig. 5C). Analysis of transcription factor-binding motif revealed a TGGAAA motif for NFAT that was highly enriched on promoters of Rgs1, Rgs2. There are five members in the NFAT family, NFAT1-NFAT5 [28]. Each of these five members was knocked down in LC and SNKO VSMCs (Supplemental Fig. 10A and Fig. 5D) to examine which member mediated the impacts of NCOR1 deficiency. Results of QRT-PCR analysis demonstrated that knockdown of NFATs had no effect on Rgs2 expression (Supplemental Fig. 10B). However, knockdown of NFAT5 or NFAT2 hindered the upregulation of Rgs1 in SNKO VSMCs (Fig. 5E and Supplemental Fig. 10C). Co-IP analysis illustrated interaction between NCOR1 and NFAT5 but not NFAT2 (Fig. 5F and Supplemental Fig. 10D). Thus, we next explored the possibility that NFAT5 mediated the effect of NCOR1 on Rgs1. As expected, a putative NFAT5 binding site was identified on the promoter of Rgs1 (Fig. 5G). ChIP analysis demonstrated enrichment of both NCOR1 and NFAT5 in the binding region on the Rgs1 promoter (Fig. 5H). Consistently, ChIP results showed that NCOR1 deficiency increased the acetylation of Histone 3 and Histone 4 of the Rgs1 promoter in VSMCs (Figs. 5I, 5J), indicating chromatin opening and accessibility. These results together suggested that NCOR1 deficiency induced Rgs1 expression and inhibited activation of RhoA through NFAT5 in VSMCs.

A Heatmap presentation of RNA-Seq results displaying relative expression of genes regulating GTPase activator activity in VSMCs isolated from LC or SNKO mice. B QRT-PCR analysis of Rgs1 in VSMCs transfected with control siRNA (siCTL) or Rgs1 siRNA (siRgs1) for 48 h. C Representative western blotting analysis of active RhoA in lysate pulldowns of LC or SNKO VSMCs transfected with control siRNA or Rgs1 siRNA for 48 h and then treated with AngII (1 μM) for 10 min. Rhotekin-binding domain was used as a bait for the pulldowns. Total RhoA was used as a loading control. QRT-PCR analysis of NFAT5 (D) and Rgs1 (E) in VSMCs transfected with control siRNA (siCTL) or NFAT5 siRNA (siNFAT5) for 48 h. F Co-immunoprecipitation analysis of NCOR1 and NFAT5 in VSMCs. G Schematic illustration of putative NFAT5 binding region on the promoter of mouse Rgs1. Forward primer (fwd) and reverse primer (rev) for ChIP assay were indicated by arrows. H ChIP analysis showing enrichment of NCOR1 and NFAT5 on the promoter of Rgs1 in VSMCs. ChIP products were amplified using regular PCR and analyzed by gel electrophoresis. ChIP analysis of enrichment of acetylated-Histone 3 (Ac-H3) (I) and acetylated-Histone 4 (Ac-H4) (J) on the promoter of Rgs1 in VSMCs isolated from LC or SNKO mice. ChIP products were analyzed using qPCR. The experiments were repeated for three times (B–H). Data from two independent experiments were presented in I and data from three independent experiments were presented in J. Values are expressed as mean ± SEM. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001 determined by Student’s t-test (B, I and J) or 2-way ANOVA with Bonferroni post-tests (D and E).
ATF3 mediates the impacts of NCOR1 deficiency on expression of Mmp12 and Mmp13 in VSMCs
It has been reported that ATF3 binds to AP-1 binding motif to regulate the transcription of Mmp13 directly [29]. We next examined whether ATF3 mediated the impact of NCOR1 deficiency on the expression of MMPs in VSMCs. QRT-PCR analysis showed that knockdown of ATF3 in VSMCs (Fig. 6A) attenuated the upregulation of Mmp12 and Mmp13, resulting in significantly lower expression of these two genes in SNKO VSMCs (Fig. 6B), but did not affect the expression of Mmp3 in either LC or SNKO VSMCs (Supplemental Fig. 11). Putative AP-1 binding sites were identified on the promoter of both Mmp12 and Mmp13 (Fig. 6C). ChIP assays verified the enrichment of NCOR1 and ATF3 in the AP-1 binding regions of Mmp12 and Mmp13 (Fig. 6D). Co-IP analysis demonstrated that NCOR1 interacted with ATF3 in VSMCs (Fig. 6E). Increased acetylation of Histone 3 and Histone 4 were also detected by ChIP assays on the promoters of Mmp12 and Mmp13 in SNKO VSMCs compared to LC VSMCs (Fig. 6F, G). Collectively, these data indicated that NCOR1 deficiency induced expression of Mmp12 and Mmp13 through ATF3 in VSMCs.

QRT-PCR analysis of Atf3 (A), Mmp12 and Mmp13 (B) in VSMCs transfected with control siRNA (siCTL) or Atf3 siRNA (siAtf3) for 48 h. C Schematic illustration of putative AP-1 binding regions on the promoters of mouse Mmp12 and Mmp13. Forward primer (fwd) and reverse primer (rev) for ChIP assay were indicated by arrows. D ChIP analysis showing enrichment of NCOR1 and ATF3 in putative AP-1 binding regions on the promoters of Mmp12 and Mmp13 in VSMCs. ChIP products were amplified using regular PCR and analyzed by gel electrophoresis. E Co-immunoprecipitation analysis of NCOR1 and ATF3 in VSMCs. The samples from the same experiment were loaded onto 2 parallel gels for detecting NCOR1 and ATF3. ChIP analysis of enrichment of acetylated-Histone 3 (Ac-H3) (F) and acetylated-Histone 4 (Ac-H4) (G) on the promoters of Mmp12 and Mmp13 in VSMCs isolated from LC or SNKO mice. ChIP products were analyzed using qPCR. The experiments were repeated for three times (A, B, D, and E). Data from two independent experiments were presented in F and data from three independent experiments were presented in G. Values are expressed as mean ± SEM. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001 determined by 2-way ANOVA with Bonferroni post-tests (A and B) or Student’s t-test (F and G).
So far, we demonstrated that FOXO3a, NFAT5 and ATF3 all mediated functions of NCOR1 in VSMCs. We further determined whether they had direct relationships with each other. QRT-PCR analysis revealed that knockdown of any one of these three transcription factors did not affect the expression of the other two (Supplemental Fig. 12A–C). Co-IP analysis did not detect any interaction among them in VSMCs (Supplemental Fig. 12D–F). These results together indicated that there was no direct relationship among FOXO3a, NFAT5, and ATF3 in VSMCs. Therefore, we proposed a working model to illustrate the mechanisms how NCOR1 regulated phenotypic switch of VSMCs (Supplemental Fig. 13).
Deficiency of smooth muscle NCOR1 exacerbates AngII-induced AA formation in mice
We next investigated the impacts of smooth muscle NCOR1 deficiency on the development of aortic aneurysm. SNKO mice were crossed with hyperlipidemic ApoE−/− mice to obtain double knockout mice. The incidence of abdominal AA was markedly increased in SNKO ApoE−/− mice compared to LC ApoE−/− mice after infusion of AngII (1200 ng/kg/min/) for 4 weeks (Fig. 7A, B). Consistently, ultrasound analysis showed that maximal diameters of suprarenal abdominal aortas were significant larger in SNKO ApoE−/− mice compared to LC ApoE−/− mice after AngII infusion (Fig. 7C, D). Noninvasive tail-cuff blood pressure (BP) measurement demonstrated that NCOR1 deficiency did not affect the systolic BP and slightly decreased the diastolic BP of ApoE−/− mice infused with AngII (Supplemental Fig. 14A–C), demonstrating that NCOR1 deficiency did not exacerbate AA by increasing blood pressure. Additionally, both the right ventricular systolic blood pressure (RVSP) and aortic media wall thickness were comparable between LC and SNKO ApoE−/− mice infused with AngII (Supplemental Fig. 14D–F). Nevertheless, NCOR1 deficiency exacerbated AngII-induced elastin degradation in abdominal aortas, which was demonstrated by van-Gieson staining (Fig. 7E–F). Immunofluorescence staining illustrated that the levels of α-SMA, Calponin1, and SMMHC were drastically lower in abdominal aortas of SNKO ApoE−/− mice compared to LC ApoE−/− mice after AngII infusion (Fig. 7G). In addition, higher MMP activities were observed in abdominal aortas of SNKO ApoE−/− mice than those of LC ApoE−/− mice (Fig. 7H, I). Similar results were obtained in thoracic aortas of LC ApoE−/− mice and SNKO ApoE−/− mice (Supplemental Fig. 15). Western blotting analysis demonstrated that smooth muscle NCOR1 deletion decreased the level of elastin and increased the levels of MMP3, MMP12, and MMP13 in aortas after AngII infusion (Fig. 7J, K). QRT-PCR results revealed that expression of Col1a1, Col3a1, Col6a1, Col8a1 and Col16a1 was markedly suppressed in aortas of SNKO ApoE−/− mice (Fig. 7L). Interestingly, expression of inflammatory genes, including Tnf-α, Il1β, Il6, Ccl2, Nos2, and Cxcl1, was upregulated in aortas of SNKO ApoE−/− mice compared to LC ApoE−/− mice (Supplemental Fig. 16A). Accordingly, media of abdominal aortas of SNKO ApoE−/− mice manifested more expression of Mac-2, a marker of macrophage, compared to those of LC ApoE−/− mice after AngII infusion (Supplemental Fig. 16B, C). Furthermore, QRT-PCR analysis revealed that smooth muscle NCOR1 deficiency decreased the expression of Myocd and increased the expression of Rgs1, Mmp12 and Mmp13 in aortas from ApoE−/− mice infused with AngII (Supplemental Fig. 17A). Results of ChIP assays using aortas of ApoE−/− mice infused with AngII illustrated that NCOR1 and FOXO3a were enriched within the binding region on the promoter of Myocd, NCOR1 and NFAT5 were enriched within the binding region on the promoter of Rgs1, and NCOR1 and ATF3 were enriched within the AP-1 binding regions on the promoters of Mmp12 and Mmp13 (Supplemental Fig. 17B–E). These in vivo data further supported the mechanism that NCOR1 regulated VSMC phenotype through FOXO3a, NFAT5 and ATF3. Collectively, these results indicated that smooth muscle NCOR1 plays a protective role against the development of aortic aneurysm.

A Representative images of dissected aorta of LC ApoE−/− and SNKO ApoE−/− mice infused with vehicle (0.9% sodium chloride) or AngII (1200 ng kg−1 min−1) for 4 weeks. Scale bar: 1 mm. B Incidence of aortic aneurysm (AA) in mice. n = 7:7:16:18. C Representative ultrasound images assessing maximal diameters of suprarenal abdominal aortas after infusion with vehicle or AngII for 4 weeks. The yellow arrows indicate the edges of the aorta. D Quantification of maximal diameters of suprarenal abdominal aortas. n = 7:7:16:18. E Representative van Gieson staining of elastin in cross-sections of suprarenal abdominal aortas from LC ApoE−/− and SNKO ApoE−/− mice infused with AngII for 4 weeks. The lower two panels are the larger magnification of the upper two corresponding panels. Scale bar: 200 μm. F Quantification of elastin degradation grade. n = 17:16. G Representative immunofluorescence staining of α-SMA, calponin1, and SMMHC in cross-sections of suprarenal abdominal aortas from LC ApoE−/− and SNKO ApoE−/− mice infused with AngII. Boxed areas are shown in higher magnification by inset images. Scale bar: 200 μm. H Representative immunofluorescence images of in situ zymography (DQ gelatin) assessing MMP activities in aortas of LC ApoE−/− and SNKO ApoE−/− mice infused with AngII for 4 weeks. Scale bar: 200 μm. I Quantification of MMP activities using relative fluorescence units (RFU) in aortas. n = 7:5. J Representative western blotting analysis of Elastin, MMP3, MMP12, and MMP13 in aortas of LC ApoE−/− and SNKO ApoE−/− mice infused with AngII for 4 weeks. GAPDH was used as a loading control. The samples were from the same experiment and gels/blots were processed in parallel. K Quantification of western blotting results. n = 4:4 for MMP3, n = 5:5 for Elastin and MMP12, n = 6:6 for MMP13. L QRT-PCR analysis of Col1a1, Col3a1, Col6a1, Col8a1, and Col16a1 in aortas of LC ApoE−/− and SNKO ApoE−/− mice infused with AngII for 4 weeks. n = 4:4. Values are expressed as mean ± SEM. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001 determined by 2-way ANOVA with Bonferroni post-tests (D) or Student’s t-test (F, I, K and L).
Discussion
The function of NCOR1 in VSMCs and vascular diseases has been largely unknown. In the current study, we discovered a central role of NCOR1 in maintaining the homeostasis of VSMCs. NCOR1 deficiency decreased the contractility of VSMCs through downregulation of contractile genes and upregulation of Rgs1 that inhibited RhoA activation. NCOR1 deficiency also reduced the production of elastin and collagens and induced the secretion of MMPs, which promoted extracellular matrix degradation. Additionally, absence of NCOR1 increased the expression of innate immunity markers in VSMCs. Therefore, NCOR1 deficiency induced VSMC to switch from a contractile phenotype to a degradative phenotype. Mechanistically, NCOR1 interacted with FOXO3, NFAT5, and ATF3 to regulate contractile genes, Rgs1, and MMPs, respectively. Finally, NCOR1 deficiency in VSMCs increased the incidence of AA in mice through impairing vascular contractility and worsening medial degeneration.
This study has identified NCOR1 as a crucial regulator of VSMC phenotypic switch. VSMCs adopt different phenotypes to adapt to different environments. Traditionally, quiescent VSMCs acquire a contractile phenotype that expresses a repertoire of contractile genes such as Acta2, Myh11, and Cnn1 and is responsible for vascular contraction [30]. Upon pathological stimuli, VSMCs transdifferentiate to a synthetic phenotype that exhibits high rates of proliferation and migration, and secrets more extracellular matrix, to participate in vascular repair [4]. A recent study has described a degradative phenotype of VSMCs induced by chronic mTOR activation, and characterized by decreased contractile and synthetic functions, as well as increased proteolysis, endocytosis, phagocytosis, and lysosomal clearance [21]. Herein, we demonstrated that NCOR1 was a novel regulator that controlled the phenotypic switch between contractile and degradative VSMCs. NCOR1 deficiency resulted in a degradative phenotype of VSMCs with suppression of contractile genes and impairment of contractility, degradation of extracellular matrix, as well as induction of innate immune response. Therefore, NCOR1 is required for maintaining the homeostasis of VSMCs.
NCOR1 regulates phenotypic switch of VSMCs through modifying activities of FOXO3, NFAT5, and ATF3. Previous studies have reported that the FOXO family is associated with contraction of VSMCs. For instance, FOXO4 represses contractile genes via interacting with Myocardin and inhibiting its activity [31]. Phosphorylated FOXO3 inhibits Myocd expression in VSMCs [32]. In the current study, we identified a new regulatory mechanism that NCOR1 deacetylated FOXO3 to increase its transcriptional activity and promote Myocd expression, emphasizing the crucial role of NCOR1 in regulating FOXO3 activity. In addition, we unveiled that NFAT5 mediated suppressive effect of NCOR1 deficiency on contractility of VSMCs. NFAT5 is a hypertonic stress-sensitive transcription factor and has been demonstrated to play an important role in phenotypic modulation of VSMCs through upregulating AngII-induced Acta2 expression and promoting PDGF-BB-stimulated migration of VSMCs [33]. In this study, we revealed the importance of the NFAT5/NCOR1 complex in regulating contractility of VSMCs through the Rgs1/RhoA axis. Moreover, we identified ATF3 as a transcription factor that mediated the impacts of NCOR1 deficiency on expression of MMPs in VSMCs. ATF3 belongs to AP-1 family [34] and has been reported to regulate Mmp13 in chondrocytes [29]. It has been demonstrated that NCOR1 is recruited by cJun, another member in the AP-1 family, to the promoter of Mmp12 in macrophages [35]. Similarly, our data indicated that NCOR1 interacted with ATF3 to repress the expression of Mmp12 and Mmp13 in VSMCs. Collectively, we demonstrated that NCOR1 is a core regulator in VSMCs through modulating activities of multiple transcription factors.
Our results have provided new evidence that NCOR1 not only has suppressive effects but also exhibits transcriptional activation on gene expression. NCOR1 has been demonstrated to be a corepressor and repress gene expression in many cell types, including myocytes, adipocytes, macrophages, T cells, and cardiomyocytes [11,12,13,14, 36, 37]. It has been reported that NCOR1 exerts its function via deacetylase [10, 38]. Our data demonstrated that NCOR1 not only regulated acetylation of histone but also modified acetylation of transcription factors in VSMCs. NCOR1 suppressed Rgs1, Mmp12, and Mmp13 by deacetylating histone 3 and histone 4 on their promoters. Conversely, NCOR1 activates Myocd expression through deacetylating FOXO3a. These results have painted a new layer to the mechanism of NCOR1 in transcriptional regulation, adding to its central role as a multifaceted master regulator of gene transcription.
Finally, we have demonstrated that smooth muscle NCOR1 plays a crucial role in the development of AA. AA is a fatal disease that accounts for about 200,000 deaths per year worldwide [39]. Since there is no efficient pharmacological treatment to AA, it is essential to identify novel targets and strategies to restrain this deadly disease. Although thoracic AA and abdominal AA exhibit distinct pathologies, phenotypic switching of VSMCs is an initiating factor for both AAs [2]. Therefore, modulating VSMC phenotypes is a promising strategy for intervention of AA. It is traditionally considered that synthetic and proinflammatory phenotypes contribute to AA formation [40, 41]. The present study demonstrated that deletion of smooth muscle NCOR1 exacerbated AA formation in a classical mouse model. Smooth muscle NCOR1 deficiency decreased levels of contractile proteins, suppressed the expression of elastin and collagen, increased MMP expression and activity, and elevated inflammation in aortas of mice with AA, reflecting the degradative phenotype observed in NCOR1-deficient VSMCs. Therefore, it is plausible to intervene AA by targeting NCOR1 in VSMCs.
In conclusion, NCOR1 is a master regulator in modulating VSMC phenotypes and AA development. Mechanistically, NCOR1 controls phenotypic switch of VSMCs through interactions with transcription factors FOXO3a, NFAT5, and ATF3. These data have revealed novel functions of smooth muscle NCOR1 and provided mechanistic insights on how smooth muscle NCOR1 regulates AA. These findings suggest that smooth muscle NCOR1 may be a potential therapeutic target for AA by modulating phenotypes of VSMCs.
Methods
Animals
Smooth muscle-specific Ncor1 knockout (SNKO) and littermate control (LC) mice were generated by crossing Ncor1 floxed mice [11] with SM-creERT2 mice [16]. Double knockout (SNKO ApoE−/−) mice were obtained by breeding SNKO mice to ApoE−/− mice (Shanghai Model Organisms). Five-week-old SNKO and SNKO ApoE−/− mice were intraperitoneally injected with tamoxifen (1 mg per mouse per day, Sigma-Aldrich) for five consecutive days to induce Ncor1 deletion. Tamoxifen-treated LC and LC ApoE−/− mice were used as controls. All mice were kept in a C57BL6 background. Eight- to twelve-week-old male mice were used for experiments. Mice with the same genotype were allocated randomly into different experimental groups. Investigators were blinded to the group allocation when assessing the results of animal studies.
Cell culture
Mouse primary vascular smooth muscle cells (VSCMs) were isolated as previously described [42]. Briefly, 8-week-old male mice were sacrificed and aortas were excised. After the adventitia and intima were removed from the medial, the aortas were scissored into small pieces and digested with collagenase II (1 mg/ml, Worthington) and elastase (1 mg/ml, Worthington) at 37 °C for 1 h. Digestion was stopped by adding culture media containing Dulbecco’s Modified Eagle Medium/Nutrient Mixture F12 (DMEM/F-12) (Thermo Fisher Scientific) supplemented with 20% fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Gibco). After centrifugation, VSMCs were maintained in culture media supplemented with gentamicin/amphotericin (Gibco) for 7 days. VSMCs were used for experiment at passage 4–6. VSMCs were identified by positive for α-SMA (Supplemental Fig. 1H) and negative for CD90 (Supplemental Fig. 18). Ncor1 deletion was induced by adding 4-hydroxytamoxifen (4-OHT) (1 μM, Sigma-Aldrich) into the media.
Human vascular smooth muscle cells (hSMCs) were obtained from ATCC (PCS-100-012). NIH/3T3 fibroblast cell line was obtained from ATCC (CRL-1658). hSMCs and NIH/3T3 cells were cultured in DMEM/F-12 (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Gibco).
All cell cultures were maintained at 37 °C in a humidified, 5% CO2 atmosphere.
RNA sequencing
Primary VSMCs were isolated from 3 LC mice and 3 SNKO mice and treated with 4-OHT to induce Ncor1 deletion. VSMCs from 3 technical replicates per group were harvested and subjected to total RNA extraction using TRIzol reagent (Thermo Fisher Scientific). RNA sequencing was performed by WuXi NextCODE (Shanghai, China) using HiSeq X Ten (Illumina). Clean reads were mapped to mouse genome. Differentially expressed genes (DEGs) were analyzed using DESeq and defined by the p-value < 0.05, FDR < 0.05 and fold change >2 or <−2. Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were applied to identify enriched functional annotations and pathways for DEGs. The Molecular Signatures Database (MSigDB, Broad Institute) was used to assess the enrichment of common conserved putative cis-regulatory elements of DEGs and predict transcription factors according to a previously described protocol [24]. FIMO (Find Individual Motif Occurences) (https://meme-suite.org/meme/tools/fimo) was used to predict binding site of transcription factors on target genes [43].
Immunofluorescence staining
For cell morphology analysis, VSMCs plated on glass coverslips were incubated with 4-OHT dissolved in serum-free media for 72 h, and then treated with vehicle (0.9% sodium chloride) or AngII (1 μM, Sigma-Aldrich) for 2 h. Subsequently, VSMCs were fixed in 4% formaldehyde for 15 min and rinsed in PBS for 3 times (5 min each time). After being incubated in blocking buffer containing 5% goat serum and 0.3% TritonX-100 in PBS at 37 °C for 1 h, VSMCs were stained with phalloidin coupled to Alexa Fluor 488 (8878, Cell Signaling Technology) at room temperature for 15 min to detect F-actin, then rinsed once with PBS. The coverslips were finally mounted with ProLongTM Gold Antifade Mountant with DAPI (P36931, Thermo Fisher Scientific) and cell images were captured using a fluorescent microscope (Leica). For quantification of the cell roundness, formula (4 x Area)/ [π x (Major axis)2] was used according to a protocol previously described [44].
For immunofluorescence staining of VSMCs, after being treated with 4-OHT, VSMCs grown on coverslips were fixed in 4% formaldehyde for 15 min and washed with PBS for 3 times, and then incubated in blocking buffer at 37 °C for 1 h. Subsequently, VSMCs were stained with primary antibodies at 4 °C overnight and fluorochrome-conjugated secondary antibodies (Thermo Fisher Scientific) at room temperature for 2 h the next day. The coverslips were mounted with ProLongTM Gold Antifade Mountant with DAPI (P36931, Thermo Fisher Scientific) and images were acquired using a fluorescent microscope (Leica).
For immunofluorescence staining of aortas, paraformaldehyde-fixed thoracic or abdominal aortas were embedded in paraffin and sectioned at a thickness of 5 μm. Cross-sections of aortas were subsequently subjected to antigen retrieval treatment after being deparaffinized and rehydrated. After being incubated in blocking buffer at 37 °C for 1 h, the aortic sections were stained with primary antibodies at 4 °C overnight and then with fluorochrome-conjugated secondary antibodies (Thermo Fisher Scientific) at room temperature for 2 h. The nuclei were stained with ProLongTM Gold Antifade Mountant with DAPI (P36931, Thermo Fisher Scientific) and the sections were viewed with a fluorescent microscope (Leica).
The following primary antibodies were used: anti-α-SMA (BM0002, Boster bio-tech), anti-Calponin 1 (17819, Cell Signaling Technology), anti-MLC2 (3672, Cell Signaling Technology), anti-SMMHC (for VSMCs, sc-6956, Santa Cruz Biotechnology), anti-SMMHC (for aortic tissue, ab53219, Abcam), anti-SM22α (ab14106, Abcam), anti-Elastin (sc-58756, Santa Cruz Biotechnology), anti-NCOR1 (5948, Cell Signaling Technology), anti-Mac-2 (14-5301-81, eBioscience) and anti-CD90 (sc-53456, Santa Cruz Biotechnology).
Quantitative RT-PCR
Total RNA was isolated from mouse VSMCs, hSMCs, or aortas using TRIzol reagent (Thermo Fisher Scientific), and complementary DNA (cDNA) was synthesized using reverse transcription kits (Takara) according to the manufacturer’s instruction. Quantitative RT-PCR was performed on a Light Cycler 480 II (Roche) using cDNA, TB Green Premix (Takara), and specific primers. Expression of target genes was normalized to that of gapdh. Specific primer sequences are available upon request.
Plasmid and Lentivirus Infection
Mouse NCOR1-flag plasmid was kindly provided by Dr. Johan Auwerx. The full-length of the coding region with flag tag was subcloned into pHAGE-fEF1a-IRES-ZsGreen vector to make pHAGE-NCOR1-flag plasmids, which were co-transfected with lentivirus packaging plasmids into HEK-293FT cells using lipofectamine 2000 (Thermo Fisher Scientific) to produce NCOR1 overexpression lentivirus. Empty pHAGE plasmids were used to produce control lentivirus. Supernatants were harvested 48 h after transfection and passed through a 0.22 μm filter. hSMCs were infected with lentivirus for 72 h.
RhoA activity
VSMCs isolated from LC and SNKO mice were incubated with 4-OHT in serum-free media for 48 h. After treatment, active-RhoA in VSMCs was examined using RhoA Pull-Down Activation Assay Biochem Kit (BK036, Cytoskeleton) according to the manufacturer’s instruction. Briefly, Rhotekin beads were used to pull down active-RhoA as Rhotekin is a Rho effector protein that comprises the rho binding domain (RBD), which has a high affinity for active GTP-bound form of RhoA [45]. The amount of active RhoA was then determined by western blot using RhoA specific antibodies.
Matrix Metalloproteinase (MMP) activity
MMP activity was assessed using in situ zymography as previously described [41]. Briefly, freshly frozen sections of aortas or VSMCs cultured on coverslips were incubated with quenched fluorescein-conjugated gelatin substrate (DQ gelatin, D12054, Thermo Fisher Scientific) in zymography buffer (50 mmol/L Tris-HCl, 150 mmol/L NaCl, 5 mM CaCl2, pH 7.6) for one hour according to the manufacturer’s instruction. The gelatin colocalized with active MMP yields green fluorescence after being degraded. The nuclei were stained with ProLongTM Gold Antifade Mountant with DAPI (P36931, Thermo Fisher Scientific) and images were captured using a fluorescent microscope (Leica). Active MMPs were assessed by quantifying the green fluorescence intensity.
Western blotting analysis
Total protein was exacted from mouse VSMCs, hSMCs or aortas using RIPA buffer supplemented with protease inhibitor cocktail (Med Chem Express) and phenylmethylsulfonylfluoride (PMSF, Sigma-Aldrich). Protein samples were separated by SDS-PAGE and transferred onto PVDF membranes. After being blocked with 5% non-fat dry milk, the membrane was sequentially incubated with primary antibodies and HRP-conjugated secondary antibodies. Immunoblots were visualized using ECL Western Blotting Substrates (Thermo Fisher Scientific). For western blots presented in the same figure panel, protein samples from the same experiment were loaded onto parallel gels when indicated in the figure legends. Full-length uncropped original western blots were provided in Supplementary file.
For assessment of phosphorylation level of MLC2 in aortic tissues, thoracic aortas or abdominal aortas were excised from LC and SNKO mice, and connective tissues were removed from the aortas. After being equilibrated in Krebs solution (in mM: NaCl 118, KCl 4.6, NaHCO3 25, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, glucose 12, pH 7.4) for 30 min, the aortas were incubated with vehicle (0.9% sodium chloride), AngII (1 μM, Sigma-Aldrich) or phenylephrine (PE) (1 μM, Sigma-Aldrich) for 20 min. Subsequently, the aortas were snap frozen in liquid nitrogen, and protein was exacted for western blotting analysis.
The following primary antibodies used for western blotting: anti-α-SMA (19245, Cell Signaling Technology), anti-Calponin 1 (17819, Cell Signaling Technology), anti-MLC2 (3672, Cell Signaling Technology), anti-SMMHC (ab53219, Abcam), anti-Myocardin (bs-9472R, Biosynthesis Biotech), antiphospho-MLC2 (3671, Cell Signaling Technology), anti-Elastin (sc-58756, Santa Cruz Biotechnology), anti-MMP3 (ab52915, Abcam), anti-MMP12 (sc-390863, Santa Cruz Biotechnology), anti-MMP13 (ab39012, Abcam), anti-GAPDH (KC-5G4, KangChen Bio-tech), anti-α-tubulin (T6199, Sigma-Aldrich), anti-FOXO3a (2497, Cell Signaling Technology), and anti-NCOR1 (5948, Cell Signaling Technology).
Chromatin Immunoprecipitation (ChIP) and Co-immunoprecipitation (Co-IP)
ChIP experiments were performed in aortas or VSMCs isolated from LC mice, SNKO mice, or wild-type mice using a ChIP-IT High Sensitivity Kit (53040, Active Motif) according to the manufacturer’s protocol. ChIP products were analyzed using quantitative PCR (qPCR). Enrichments of FOXO3a, acetyl-Histone H3, or acetyl-Histone H4 on target genes detected by ChIP was presented as ratios of qPCR products in proximal regions (normalized to input) to those in distal regions. The following antibodies were used: anti-NCOR1 (sc-515934X, Santa Cruz Biotechnology), anti-FOXO3a (720128, Thermo Fisher Scientific), anti-NFAT5 (ab3446, Thermo Fisher Scientific), anti-ATF3 (33593, Cell Signaling Technology), anti-acetyl-Histone H3 (06-599, Millipore), anti-acetyl-Histone H4 (39925, Active Motif), and IgG (sc-2025, sc-2027, Santa Cruz Biotechnology). The following primers for predicted binding regions (proximal regions) were used:
Myocd (proximal) F: 5′-TGGTTGGATCCTTCAGCTG-3′,
Myocd (proximal) R: 5′-CTCTGTGAAACATGAAAAAGAAAG-3′;
Rgs1 (proximal) F: 5′-GAAGTAGCATTAAAAAACAGAATG-3′,
Rgs1 (proximal) R: 5′- TGTCTACACCACTGTGGTGTC-3′;
Mmp12 (proximal) F: 5′-GAGTGATGAATTAGACAATGGT-3′,
Mmp12 (proximal) R: 5′-TTAAAGCCCCACTTTATATAAC-3′;
Mmp13 (proximal) F: 5′- ACAAACCACACTTAGGAAGAA-3′,
Mmp13 (proximal) R: 5′- AGCAGTGCCTGGAGTCTC-3′.
The following primers for distal regions were used:
Myocd (distal) F: 5′- CTATGCTCTGTCTCCAGAGACA-3′,
Myocd (distal) R: 5′- CAGAGATTTTGACCATGTAAATT-3′,
Rgs1 (distal) F: 5′- CGATGATAAGCACTTTCTTGG-3′,
Rgs1 (distal) R: 5′- CCTGTCAAGTTGACAGTTTAACTT-3′,
Mmp12 (distal) F: 5′- TGTTCTTGGATTCAGTTTGCA-3′,
Mmp12 (distal) R: 5′- AACACCACCTAACTTGTTCTATGA-3′,
Mmp13 (distal) F: 5′- CTGTAGATGTACATCCCTGGAG-3′,
Mmp13 (distal) R: 5′- CATATTGATAGAACACTGTTCACT-3′ .
Co-IP analyses were performed in VSMCs isolated from wild-type mice according to the protocol previously described [46] with small modifications. Briefly, whole-cell lysates were incubated with antibodies against NCOR1 (sc-515934X, Santa Cruz Biotechnology), FOXO3a (2497, Cell Signaling Technology), NFAT5 (sc-398171, Santa Cruz Biotechnology), ATF3 (33593, Cell Signaling Technology) or normal mouse IgG (sc-2025, Santa Cruz Biotechnology) with rotation at 4°C overnight, followed by immunoprecipitation with protein A/G magnetic beads (HY-K0202, Med Chem Express) at 4 °C for 4 h. After being washed with PBS containing 0.3% TritonX-100 for 5 times, the immunoprecipitates were eluted in 1 X loading buffer (Takara) and boiled at 95 °C for 10 min. The samples were used for western blotting analysis with the following antibodies: NCOR1 (sc-515934X, Santa Cruz Biotechnology), anti-FOXO3a (2497, Cell Signaling Technology), anti-NFAT5 (sc-398171, Santa Cruz Biotechnology), anti-ATF3 (33593, Cell Signaling Technology), anti-SRF (5147, Cell Signaling Technology), and anti-NFAT2 (sc-7294, Santa Cruz Biotechnology).
For evaluation of FOXO3a acetylation in VSMCs, total cell-lysates from VSMCs isolated from LC and SNKO mice or from VSMCs treated with or without trichostatin A (an inhibitor of HDAC class I/II) and nicotinamide (an inhibitor of Sirt1) were immunoprecipitated with antibodies against with acetylated- lysine (9441, Cell Signaling Technology) at 4 °C overnight, followed by incubation with protein A/G magnetic beads (HY-K0202, Med Chem Express) at 4 °C for 4 h. Samples were eluted in 1X loading buffer (Takara) and acetylation of FOXO3 were assessed using western blotting analysis with antibodies against FOXO3a (2497, Cell Signaling Technology).
siRNA transfection
VSMCs or hSMCs were transfected with small interfering RNA (siRNA) using lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instruction. siRNA targeting Ncor1, Foxo3a, Nfatc1, Nfatc2, Nfac3, Nfatc4, Nfat5, or Atf3 was synthesized by GenePharma (Shanghai, China) and the siRNA sequences were as follows:
siRNA for mouse Ncor1, GAAAUCCCACGGCAAGAUA;
siRNA for human Ncor1, GCTCTCAAAGTTCAGACTCTT;
siRNA for human Foxo3a, GACGAUGAUGCGCCUCUCU;
siRNA for mouse Nfatc1, TGAACCTCTCACGCTACAG;
siRNA for mouse Nfatc2, GAACCGATCGCACATAAGG;
siRNA for mouse Nfatc3, ATTGAGAAGTACAGTATCA
siRNA for mouse Nfatc4, ATTCAGACATTGAGCTACG
siRNA for mouse Nfat5, CCAGUUCCUACAAUGAUAACACU
siRNA for mouse Atf3, CACCCUUUGUCAAGGAAGATT
Commercial siRNA (sc-36409, Santa Cruz Biotechnology) was used to target mouse Rgs1.
AngII-induced Abdominal Aortic Aneurysm Model
Abdominal aortic aneurysm (AAA) mouse model was established as previously described [47]. Briefly, 12-week-old LC ApoE−/− mice and SNKO ApoE−/− mice were infused with AngII (1200 ng kg−1 min−1, Sigma-Aldrich) or vehicle (0.9% sodium chloride) via Alzet mini-osmotic pumps (model 2004, DURECT) for 4 weeks. The criterion for occurrence of AAA was that the external diameter of suprarenal aortas increased by 50% or greater compared with that of suprarenal aortas from vehicle-infused mice [48]. AAA incidence was quantified 4 weeks after implantations of pumps.
Ultrasound
B-mode ultrasound imaging was performed to measure the abdominal aortic diameters using a Vevo2100 Ultrasound system (Visual Sonics) according to the protocol previously described [49].
Measurements of Blood Pressure (BP) and Right Ventricular Systolic Pressure (RVSP)
Mouse BP was measured using tail-cuff BP-2000 Blood Pressure Analysis System (Visitech Systems) as previously described [50]. Briefly, mice were trained for 1 week to adapt the BP measurement condition. After training, BP was measured daily for 3 consecutive days per week, and data were presented as the mean of 3 BP measurements. For each measurement, twenty cycles were taken and at least ten consecutive readings per mouse were recorded and averaged.
Mouse RVSP was monitored using radiotelemetry according to the protocol described previously [51] with modifications. In brief, mouse was anesthetized with 2% isoflurane and the heart was exposed at the fourth intercostal space using two blunt forceps. The right ventricle was punctured with a 22G syringe needle and the pressure transmitter (HD-X10, Data Sciences International) was carefully inserted into the right ventricle using a vessel canulation forceps after removing the needle. Pressures were recorded every 10 seconds for 1–2 min.
Histological analysis
Paraformaldehyde-fixed and paraffined-embedded sections of thoracic aortas were stained with hematoxylin and eosin. Media thickness was measured using ImageJ software (National Institutes of Health).
Paraffin sections (5 μm) of abdominal aortas were used for elastin Van Gieson staining. Elastin degradation was graded as previously described [48]. Based on the standard for elastin degradation score, aortas with no degradation were scored as 1, those with mild degradation were scored as 2, those with severe degradation were scored as 3, and aortic ruptures were scored as 4.
Statistical analysis
All data were presented as mean ± SEM. Statistical analyses were performed using Prism (GraphPad Software). Unpaired Student’s t-test was used to make comparisons of parameters between two groups. Multiple comparisons were analyzed by 2-way ANOVA followed by Bonferroni post hoc test. A value of p ≤ 0.05 was considered statistically significant.
Data availability
Original data of RNA-sequencing are available from the NCBI Gene Expression Omnibus (GEO) database under accession number GSE185635.
References
Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156–64.
Ailawadi G, Moehle CW, Pei H, Walton SP, Yang Z, Kron IL, et al. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg. 2009;138:1392–9.
Brozovich FV, Nicholson CJ, Degen CV, Gao YZ, Aggarwal M, Morgan KG. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharm Rev. 2016;68:476–532.
Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801.
Hulsmans M, Nahrendorf M. Proliferative, degradative smooth muscle cells promote aortic disease. J Clin Invest. 2020;130:1096–8.
Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2012;74:13–40.
Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A, Olson EN. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 2004;428:185–9.
Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, et al. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation 2013;128:2047–57.
Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. 2019;25:1280–9.
Mottis A, Mouchiroud L, Auwerx J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013;27:819–35.
Yamamoto H, Williams EG, Mouchiroud L, Canto C, Fan W, Downes M, et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 2011;147:827–39.
Li P, Spann NJ, Kaikkonen MU, Lu M, Oh DY, Fox JN, et al. NCoR repression of LXRs restricts macrophage biosynthesis of insulin-sensitizing omega 3 fatty acids. Cell 2013;155:200–14.
Oppi S, Nusser-Stein S, Blyszczuk P, Wang X, Jomard A, Marzolla V, et al. Macrophage NCOR1 protects from atherosclerosis by repressing a pro-atherogenic PPARgamma signature. Eur Heart J. 2020;41:995–1005.
Li C, Sun XN, Chen BY, Zeng MR, Du LJ, Liu T, et al. Nuclear receptor corepressor 1 represses cardiac hypertrophy. EMBO Mol Med. 2019;11:e9127.
Du LJ, Sun JY, Zhang WC, Wang YL, Zhu H, Liu T, et al. Macrophage NCOR1 deficiency ameliorates myocardial infarction and neointimal hyperplasia in mice. J Am Heart Assoc. 2020;9:e015862.
Kuhbandner S, Brummer S, Metzger D, Chambon P, Hofmann F, Feil R. Temporally controlled somatic mutagenesis in smooth muscle. Genesis 2000;28:15–22.
Loirand G, Guerin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res. 2006;98:322–34.
Mack CP, Somlyo AV, Hautmann M, Somlyo AP, Owens GK. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J Biol Chem. 2001;276:341–7.
El-Hamamsy I, Yacoub MH. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol. 2009;6:771–86.
Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–39.
Li G, Wang M, Caulk AW, Cilfone NA, Gujja S, Qin L, et al. Chronic mTOR activation induces a degradative smooth muscle cell phenotype. J Clin Invest. 2020;130:1233–51.
Parmacek MS. Myocardin-related transcription factors: critical coactivators regulating cardiovascular development and adaptation. Circ Res. 2007;100:633–44.
Creemers EE, Sutherland LB, McAnally J, Richardson JA, Olson EN. Myocardin is a direct transcriptional target of Mef2, Tead and Foxo proteins during cardiovascular development. Development 2006;133:4245–56.
Nott A, Cheng J, Gao F, Lin YT, Gjoneska E, Ko T, et al. Histone deacetylase 3 associates with MeCP2 to regulate FOXO and social behavior. Nat Neurosci. 2016;19:1497–505.
Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci USA. 2005;102:11278–83.
Tang KM, Wang GR, Lu P, Karas RH, Aronovitz M, Heximer SP, et al. Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat Med. 2003;9:1506–12.
Patel J, Chuaiphichai S, Douglas G, Gorvin CM, Channon KM. Vascular wall regulator of G-protein signalling-1 (RGS-1) is required for angiotensin II-mediated blood pressure control. Vasc Pharm. 2018;108:15–22.
Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–84.
Chan CM, Macdonald CD, Litherland GJ, Wilkinson DJ, Skelton A, Europe-Finner GN, et al. Cytokine-induced MMP13 expression in human chondrocytes is dependent on activating Transcription Factor 3 (ATF3) Regulation. J Biol Chem. 2017;292:1625–36.
Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75:487–517.
Liu ZP, Wang Z, Yanagisawa H, Olson EN. Phenotypic modulation of smooth muscle cells through interaction of Foxo4 and myocardin. Dev Cell. 2005;9:261–70.
Yang X, Gong Y, Tang Y, Li H, He Q, Gower L, et al. Spry1 and Spry4 differentially regulate human aortic smooth muscle cell phenotype via Akt/FoxO/myocardin signaling. PLoS One. 2013;8:e58746.
Halterman JA, Kwon HM, Zargham R, Bortz PD, Wamhoff BR. Nuclear factor of activated T cells 5 regulates vascular smooth muscle cell phenotypic modulation. Arterioscler Thromb Vasc Biol. 2011;31:2287–96.
Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–6.
Ogawa S, Lozach J, Jepsen K, Sawka-Verhelle D, Perissi V, Sasik R, et al. A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proc Natl Acad Sci USA. 2004;101:14461–6.
Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J, et al. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell 2011;147:815–26.
Wang J, He N, Zhang N, Quan D, Zhang S, Zhang C, et al. NCoR1 restrains thymic negative selection by repressing Bim expression to spare thymocytes undergoing positive selection. Nat Commun. 2017;8:959.
Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet. 2010;11:109–23.
Liu B, Granville DJ, Golledge J, Kassiri Z. Pathogenic mechanisms and the potential of drug therapies for aortic aneurysm. Am J Physiol Heart Circ Physiol. 2020;318:H652–H70.
Liu R, Lo L, Lay AJ, Zhao Y, Ting KK, Robertson EN, et al. ARHGAP18 protects against thoracic aortic aneurysm formation by mitigating the synthetic and proinflammatory smooth muscle cell phenotype. Circ Res. 2017;121:512–24.
Nogi M, Satoh K, Sunamura S, Kikuchi N, Satoh T, Kurosawa R, et al. Small GTP-binding protein GDP dissociation stimulator prevents thoracic aortic aneurysm formation and rupture by phenotypic preservation of aortic smooth muscle cells. Circulation 2018;138:2413–33.
Guo X, Shi N, Cui XB, Wang JN, Fukui Y, Chen SY. Dedicator of cytokinesis 2, a novel regulator for smooth muscle phenotypic modulation and vascular remodeling. Circ Res. 2015;116:e71–80.
Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics 2011;27:1017–8.
de Carcer G, Wachowicz P, Martinez-Martinez S, Oller J, Mendez-Barbero N, Escobar B, et al. Plk1 regulates contraction of postmitotic smooth muscle cells and is required for vascular homeostasis. Nat Med. 2017;23:964–74.
Ito H, Morishita R, Nagata KI. Functions of Rhotekin, an effector of Rho GTPase, and its binding partners in mammals. Int J Mol Sci. 2018;19:2121.
Sun XN, Li C, Liu Y, Du LJ, Zeng MR, Zheng XJ, et al. T-cell mineralocorticoid receptor controls blood pressure by regulating interferon-gamma. Circ Res. 2017;120:1584–97.
Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–12.
Satoh K, Nigro P, Matoba T, O’Dell MR, Cui Z, Shi X, et al. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat Med. 2009;15:649–56.
Hadi T, Boytard L, Silvestro M, Alebrahim D, Jacob S, Feinstein J, et al. Macrophage-derived netrin-1 promotes abdominal aortic aneurysm formation by activating MMP3 in vascular smooth muscle cells. Nat Commun. 2018;9:5022.
Krege JH, Hodgin JB, Hagaman JR, Smithies O. A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension 1995;25:1111–5.
Handoko ML, Schalij I, Kramer K, Sebkhi A, Postmus PE, van der Laarse WJ, et al. A refined radio-telemetry technique to monitor right ventricle or pulmonary artery pressures in rats: a useful tool in pulmonary hypertension research. Pflug Arch. 2008;455:951–9.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81725003, 81991503, 81991500, 81921002, 31900810, 81900227, 82100446), the China Postdoctoral Science Foundation (2018M640402), and the Innovative Research Team of High-Level Local Universities in Shanghai (SHSMU-ZDCX20212500).
Author information
Authors and Affiliations
Contributions
L-JD, J-YS, W-CZ, YL, YL, W-ZL, TL, HZ, Y-LW, SS, L-JZ, and B-YC performed the experiments and acquisition of data; L-JD, J-YS and W-CZ designed the experiments and analyzed the data; L-JD, HL, R-GL, FJ, and S-ZD wrote and revised the manuscripts; R-GL, FJ, and S-ZD supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All animal procedures in this study were approved by the Institutional Review and Ethics Board of Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by H Ichijo
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Du, LJ., Sun, JY., Zhang, WC. et al. NCOR1 maintains the homeostasis of vascular smooth muscle cells and protects against aortic aneurysm. Cell Death Differ 30, 618–631 (2023). https://doi.org/10.1038/s41418-022-01065-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-022-01065-1
This article is cited by
-
Periodontitis exacerbates pulmonary hypertension by promoting IFNγ+ T cell infiltration in mice
International Journal of Oral Science (2024)
