
Abstract
Alternative splicing (AS) is tightly regulated during cell differentiation and development. AS events are prevalent in the testis, but the splicing regulation in spermatogenesis remains unclear. Here we report that the spliceosome component Bud31 plays a crucial role during spermatogenesis in mice. Germ cell-specific knockout of Bud31 led to loss of spermatogonia and to male infertility. We further demonstrate that Bud31 is required for both spermatogonial stem cell pool maintenance and the initiation of spermatogenesis. SMART-seq revealed that deletion of Bud31 in germ cells causes widespread exon-skipping and intron retention. Particularly, we identified Cdk2 as one of the direct splicing targets of Bud31, knockout of Bud31 resulted in retention of the first intron of Cdk2, which led to a decrease in Cdk2 expression. Our findings suggest that Bud31-mediated AS within spermatogonial stem cells regulates the self-renewal and differentiation of male germ cells in mammals.
Similar content being viewed by others


Introduction
The process of male germ cell development is complex and highly coordinated. The primary spermatogonial stem cells (SSCs) originate from gonocytes in the postnatal testis, and they function by sustaining continuous sperm production [1]. Gonocytes originate from primordial germ cells during embryogenesis, proliferate until embryonic day (E)15–16, and then become quiescent [2]. After birth, the gonocytes resume proliferation and migrate to the basement membrane of the seminiferous tubules where they differentiate into SSCs [3]. SSCs either self-renew to maintain the stem cell pool or differentiate to produce spermatozoa [4]. PLZF, GFRα1, and ID4 have been identified as markers of undifferentiated SSCs [5]. RA (retinoic acid) and c-Kit are essential for SSC differentiation [6].
It is known that alternative splicing (AS) is regulated in a tissue-specific manner during development [7], and AS outcomes have been shown to regulate cell differentiation and lineage determination to support tissue identity specification [8]. AS occurs in about 95% of human genes and in about 60% of mouse genes [9], and AS is most prevalent in the testis and brain when compared to other tissues [10]. Defects in AS can lead to various diseases, and aberrant AS can result from mutations in RNA cis-regulatory elements or trans-acting regulatory factors [11]. The trans-acting factors include RNA binding proteins that can act as splicing enhancers or silencers [12], and several RNA binding proteins are differentially expressed in the testis and have been shown to play roles in spermatogenesis [13,14,15]. Despite these findings, understanding the splicing factors that control the AS of key genes in spermatogenesis remains a major challenge.
BUD31 belongs to the major Bact complex of the human minor spliceosome [16] and is required for spliceosome assembly and catalytic activity [17]. Bud31-null yeast cells show bud formation defects and have disorganized actin distribution [18], but the physiological roles of Bud31 remain unknown in mammals. Here, we generated germ cell conditional Bud31 knockout mice, which led to loss of spermatogonia and to infertility. Through integrative analysis of SMART-seq and RIP-seq data, we identified Cdk2 as a direct splicing target of Bud31. Our data suggest that Bud31-mediated AS is required for SSC pool maintenance and for the initiation of spermatogenesis.
Results
Germ cell-specific Bud31 knockout results in complete male infertility
The expression levels of splicing factors are tightly regulated during cell differentiation and development [8]. To characterize the splicing factors involved in spermatogenesis, we used publicly available RNA-seq data (E-MTAB-6798) [19] to obtain the levels of 134 known splicing factors [17] in testes at different stages of mouse spermatogenesis (E18.5, postnatal day (P)0, P3, P14, P28, and P63). We found that 10 splicing factors only began to be expressed after birth, which is when spermatogonia proliferation commences (Fig. 1A; Supplementary Table S1). Meanwhile, we analyzed AS events using RNA-seq data (E-MTAB-6798) and found that AS events were significantly increased in testes at P3 and reached a peak at P28 in comparison with P0 (Fig. 1B).

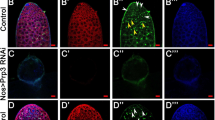
A Heatmap analysis of the expression of known splicing factors at E18 and at different postnatal day points (P0, P3, P14, P28, and P63) in mouse testes using the Evo-devo mammalian organs database (https://apps.kaessmannlab.org/evodevoapp/). B Histogram showing the occurrence of AS events at E18.5, P0, P3, P14, P28, and P63 in mouse testes using the Evo-devo mammalian organs database. C Immunofluorescence staining of Bud31 (red) and the germ cell marker Dazl (green) in the seminiferous tubules of P4 mice. The scale bar is 20 μm. D Immunofluorescence staining of Bud31 (red) and the germ cell marker Dazl (green) in seminiferous tubules of P4 testes of Bud31-vKO mice and in littermate controls. The scale bar is 10 μm. E Testes from P46 Bud31-vKO mice compared to their respective littermate controls. F Hematoxylin staining of the seminiferous tubules of the testes of P12 Bud31-vKO and littermate control mice. The scale bar is 20 μm. Red arrow: germ cells. G Immunofluorescence staining of the Sertoli cell marker Sox9 (red) and germ cell nuclear antigen Gcna (green) in the seminiferous tubules of P10 Bud31-vKO and their littermate controls. The scale bar is 10 μm.
Spliceosome component Bud31, which is required for spliceosome assembly and catalytic activity [17], caught our interest because its expression pattern was temporally coordinated with alternative splicing events (Fig. 1A, B). Bud31 is also known to regulate budding and cell cycle by facilitating G1/S transition in budding yeast [20], suggesting its role in spermatogenesis. We then performed immunoblotting of testes at different stages of mouse spermatogenesis. Consistent with the RNA-seq data, the Bud31 protein level in testes increased dramatically after birth (Supplementary Fig. S1A). We also determined the expression of Bud31 in different organs and found that Bud31 was highly expressed in testis, ovary, and spleen (Supplementary Fig. S1B). Additionally, co-immunofluorescence staining of Bud31 and Dazl, a marker for undifferentiated spermatogonia [21], showed that Bud31 was present in Dazl-positive cells in seminiferous tubules at P4 (Fig. 1C). We performed immunoblotting of purified germ and somatic cell fractions from P9 testes and found that Bud31 accumulation was more pronounced in germ cells than in somatic cells (Supplementary Fig. S1C), implying its potential role in spermatogenesis. Subcellular localization analysis revealed that Bud31 is mainly localized in the nuclei of spermatogenic cells (Supplementary Fig. S1D).
To explore the potential functional impacts of Bud31 in spermatogenesis, we generated mice carrying a floxed Bud31 allele in which exons 4–5 was flanked by loxP sites (Supplementary Fig. S1E). Then Bud31 global knockout mice were generated by crossing Stra8-GFPCre deleter mice with Bud31-floxed mice (Supplementary Fig. S1F). We found that mice with global deletion of Bud31 were viable but had a decreased body size (Supplementary Fig. S1G). The testis size of Bud31 null mice was significantly smaller than littermate controls, with a severe germ cell loss in the tubules (Supplementary Fig. S1H, I). These findings implicate Bud31 in the development and spermatogenesis. To elucidate the potential role of Bud31 in spermatogenesis, we next created germ cell specific Bud31 knockout mice by crossing VASA-Cre transgenic mice with Bud31-floxed mice (Bud31-vKO). The genotype of the conditional knockout mice was confirmed by PCR of tail biopsy DNA (Supplementary Fig. S2A), and the knockout efficiency was assessed by qPCR and immunoblotting of testes at P4 (Supplementary Fig. S2B, C). Additionally, immunostaining of the germ cell marker DAZL and Bud31 in seminiferous tubules further confirmed the lack of Bud31 in germ cells of Bud31-vKO mice at P4 (Fig. 1D and Supplementary Fig. S2D).
Next, we analyzed the phenotypes and found that compared to their littermate controls (Bud31fl/fl or Bud31fl/+), Bud31-vKO mice (P46) had much smaller testes (Fig. 1E). Consistent with this phenotype, hematoxylin staining demonstrated a significant decrease in the number of germ cells in the testes of Bud31-vKO mice compared to their littermate controls (Fig. 1F). Furthermore, co-immunofluorescence staining of the Sertoli cell marker Sox9 and the germ cell marker Gcna revealed that Gcna-positive cells in Bud31-vKO testes were completely absent but were present in control testes at P10 (Fig. 1G). Moreover, no pups were obtained when adult Bud31-vKO male mice were housed with wild type fertile female mice for 3 months (Supplementary Table S2). These findings collectively suggest that Bud31 is essential for spermatogenesis.
Bud31 knockout leads to severe defects in SSC self-renewal
To determine the cause of germ cell loss observed in Bud31-vKO mice, we performed immunohistochemistry staining against Plzf (a known SSC marker) in Bud31-vKO and control mice at P8. Plzf-positive cells were lost in Bud31-vKO testes but were observed in control testes (Supplementary Fig. S2E). To investigate the time of onset of SSC loss in Bud31-vKO mice, we performed immunostaining against Bud31 and Plzf in seminiferous tubules at P1, P2, P3, P4, and P5 in Bud31-vKO and control mice. Compared to controls, the first point at which a significant reduction in the number of Plzf-positive cells became evident was P4 (Fig. 2A, B). Immunoblotting verified the reduced expression of Plzf in Bud31-vKO mice compared to controls at P4 (Supplementary Fig. S2F). Further, co-staining with an additional SSC marker (Lin28a) in seminiferous tubules of Bud31-vKO mice at P5 showed that the number of cells co-expressing Lin28a and Plzf was significantly lower in the Bud31-vKO tubules than in controls (Fig. 2C). Subsequently, we used qPCR to examine the expression of SSC markers (Plzf, Lin28a, Gfrα1, and Id4) in P4 testes, and the levels of these markers were significantly reduced in Bud31-vKO testes compared to controls (Fig. 2D). Further, flow cytometry analysis showed that cells expressing the SSC surface marker Thy1 but not expressing c-Kit decreased from 3.4% to 2.26% at P4 in Bud31-vKO testes compared with controls (Fig. 2E), confirming that the number of SSCs was reduced as early as P4.

A Immunofluorescence staining of Bud31 (red) and Plzf (green) in seminiferous tubules of P4 mice. The scale bar is 10 μm. B Quantification of Plzf-positive cells in seminiferous tubules from P1 to P5 mice assessed by immunofluorescence staining. Counts shown are Plzf-positive cells per tubule. At least 60 tubules were counted from at least three different mice. Student’s t test, error bars indicate the SEM. ***P < 0.001. C Immunofluorescence staining of SSC markers Plzf (red) and Lin28a (green) in seminiferous tubules of P5 mice. The scale bar is 10 μm. D qPCR to measure the expression of SSC self-renewal-related genes (Plzf, Lin28a, Gfrα1, and Id4) in P4 Bud31-vKO mice compared to littermate controls. Student’s t tests were performed, and data are shown as the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001. E The spermatogonial stem cell proportion was analyzed by assessing cells positive for the SSC marker Thy1 and negative for the differentiated spermatogonia marker c-Kit using flow cytometry in P3 and P4 Bud31-vKO mice (compared against littermate controls). F Gene Ontology analysis of differentially expressed genes (DEGs) in P4 mouse testes from SMART-seq analysis of SSCs. G Gene Set Enrichment Analysis of P13K-AKT-mTOR enrichment in P4 mouse testes from the SMART-seq dataset. H Western blotting analysis of mTOR pathway-related protein expression in testes from Bud31-vKO mice. Gapdh was used as the loading control.
We performed SMART-seq analysis of the sorted Thy1+/c-Kit– SSCs from Bud31-vKO and control mice at P4, Gene Ontology and Gene Set Enrichment Analysis (GSEA) of the differentially expressed genes revealed obvious enrichment for PI3K-AKT-mTOR signaling, an essential pathway for normal SSC self-renewal [22] (Fig. 2F, G). Immunoblotting verified the reduced levels of mTOR signaling components in sorted Thy1+/c-Kit– SSCs from the P4 Bud31-vKO animals (Fig. 2H). These results implicate Bud31 in the regulation of SSC self-renewal in newborn mice.
Bud31-deficient male germ cells fail to enter meiosis
Meiosis is a unique process for the differentiation of germ cells, and Stra8 (Stimulated by Retinoic Acid 8) functions as an essential gatekeeper of meiotic initiation [23]. To assess the potential role of Bud31 in male meiosis, we crossed Stra8-GFPCre mice with Bud31flox/flox (Bud31-sKO) mice. The knockout efficiency was assessed by qPCR, immunoblotting, and immunofluorescence staining of testes at P10 (Supplementary Fig. S3A–C). The testes size was also greatly reduced in Bud31-sKO mice compared to controls (Supplementary Fig. S3D). We then performed hematoxylin staining, which showed that meiotic cells were largely reduced in Bud31-sKO mice in comparison with their littermate controls (Bud31fl/fl or Bud31fl/+) at P10 and P12 (Fig. 3A and Supplementary Fig. S3E). Subsequently, we used immunofluorescence staining of the meiosis markers SYCP3 and γH2AX in the seminiferous tubules of P12 mice and found that SYCP3 and γH2AX double-positive cells were significantly decreased in Bud31-sKO testes compared with controls (Fig. 3B). Importantly, no spermatocytes later than those with preleptotene stage-like chromosomes were observed in Bud31-sKO testes at P12, a time when abundant zygotene chromosomes were already evident in control testes (Fig. 3C). Moreover, no meiotic cells were observed in Bud31-sKO testes at P17, a time when pachytene chromosomes were already evident in control testes (Supplementary Fig. S3G, H). This phenotype was similar to that of Stra8 knockout mice based on our data (Supplementary Fig. S3I, J) and previous study [24], indicating that Bud31 is required for entry into meiosis.

A Hematoxylin staining of the seminiferous tubules of the testes of P10 Bud31-sKO and littermate control mice. The scale bar is 50 μm. B Immunofluorescence staining of the meiotic marker γH2AX (green) and syndicate complex marker SYCP3 (red) in P12 testis tissue sections. The scale bar is 10 μm. C Immunofluorescence staining of SYCP3 (red) and γH2AX (green) was performed on chromosome spreads of Bud31-sKO mice. The scale bar is 5 μm. D Immunofluorescence staining of Stra8 (green) in the testicular tissue sections of P10 control and Bud31-sKO mice. The scale bar is 20 μm. E The expression of Stra8 and its downstream targets was assessed by qPCR in the testes of P10 Bud31-sKO and littermate control mice. The Stra8 expression by qPCR (F) and western blotting (G) in P4 testes of Bud31-vKO mice and littermate controls. *P < 0.05; **P < 0.01; ***P < 0.001.
The expression of Stra8 is necessary for germ cells to enter into meiosis [25], and we performed immunofluorescence staining of Stra8 in the seminiferous tubules of P10 mice and found that the Stra8 signal was significantly diminished in Bud31-sKO testes compared to controls (Fig. 3D). A subsequent qPCR assay revealed that the levels of Stra8 and its known downstream transcriptional activation targets Hoxa1 and Cyp26a1 [26] were decreased in P10 Bud31-sKO mice compared to controls (Fig. 3E). We further used both qPCR and immunoblotting to assess Stra8 and observed significantly reduced Stra8 levels in P4 Bud31-vKO testes compared to controls (Fig. 3F, G). We also noted that P10 Bud31-sKO testes had significantly reduced levels of genes involved in spermatogonia differentiation (c-kit, Stra8, Sohlh1, and Sohlh2) [27] and meiosis (Dmc1, Sycp1, Sycp3, and Rec8) [28] compared to controls (Supplementary Fig. S3F). These results again support that Bud31 functions to regulate meiotic initiation.
Bud31 regulates AS in spermatogenic cells
In order to explore the role of Bud31 in AS in spermatogenesis, we performed AS analysis on SMART-seq data using rMATS. Bud31 knockout resulted in a total of 1948 AS events (Supplementary Table S3), including skipped exons (57%), retained introns (20%), alternative 3′ splice sites (11%), alternative 5′ splice sites (5%), and mutually exclusive exons (7%) (Fig. 4A, B). We subsequently analyzed the reads mapping distribution and observed that Bud31 depletion significantly reduced the proportion of exonic reads and increased the proportion of intronic and intergenic reads (Fig. 4C). Furthermore, we calculated the splicing efficiency using the SMART-seq data and found that knockout of Bud31 resulted in significantly reduced efficiency of 5′ splice sites (D = 0.0885) and 3′ splice site (D = 0.0657) respectively (Fig. 4D), indicating impairment of AS upon loss of Bud31.

A Schematic diagram of AS types. B Pie chart depicting the proportions of different types of AS events in the SMART-seq data from spermatogenic cells in the testes of P4 Bud31-vKO mice and littermate controls. C Mapping reads coverage of the SMART-seq data in spermatogenic cells in the testes of P4 Bud31-vKO mice and littermate controls. The percentages of reads aligned to intergenic, intron, and exon regions are shown. D Global splicing efficiency at the 5′ splicing sites and 3′ splicing sites was analyzed with splicing efficiency analysis and annotation in spermatogenic cells in the testes of P4 Bud31-vKO mice and littermate controls. E Pie chart of the distribution of Bud31 binding peaks from RIP-seq data for primary cells in P14 wild-type mouse testes. F The normalized reads intensity distribution around the 3′ and 5′ splicing sites (±100 bp) was analyzed using RIP-seq data in primary cells of P14 wild-type mouse testes. G HOMER de novo motif analysis of Bud31 binding peaks based on the RIP-seq data. Two significant motifs with their corresponding E-values are shown. H Venn diagram of 392 genes common to both the Bud31-binding genes from the RIP-seq data and genes with AS events identified by SMART-seq. I Gene ontology (GO) enrichment analysis of biological processes for the 392 genes. J Volcano plot of differentially expressed genes (DEGs) from SMART-seq of SSCs in P4 Bud31-vKO mice compared to their littermate controls. |log2FC| > 0.7 and an adjusted p value <0.05 were considered significant. K Venn diagram of 12 genes common to both 392 genes and down regulated genes from SMART-seq of SSCs in P4 Bud31-vKO mice compared to their littermate controls.
To identify genome-wide binding sites and targets of Bud31 in spermatogenesis, RIP-seq (RNA immunoprecipitation sequencing) was conducted using spermatogenic cells from P14 wild-type mice. RIP peak distribution analysis revealed that Bud31 was predominantly mapped to intron regions (81.8%) of pre-mRNAs (Fig. 4E and Supplementary Table S4). We further analyzed the reads intensity and found that Bud31 was highly enriched in exon-intron regions around splicing sites (Fig. 4F). Moreover, we used the HOMER algorithm to identify the Bud31-recognizing RNA motif and found that the most abundant elements were UUUUAAAA and GAGGCAGG (Fig. 4G), which are useful for identifying potential binding sites. We also performed immunoprecipitation with Bud31 antibody coupled to mass spectrometry in P14 testis. Bubble map analysis revealed that the gene ratio related to the spliceosome and RNA binding was significantly enriched (Supplementary Fig. S4A). Importantly, we found that Bud31 was predominantly associated with hnRNP proteins, U2 snRNPs, and A-complexes (Supplementary Fig. S4B). Finally, coimmunoprecipitation and immunoblotting verified that Sf3b1, U2af2, and Ddx4 interact with Bud31 (Supplementary Fig. S4C). The interactions between Bud31 and spliceosome components indicate its role in the regulation of AS.
To identify candidate AS targets regulated by Bud31 in spermatogenic cells, we compared the Bud31-bound genes from the RIP-seq data (3258 genes) with AS-related genes from our SMART-seq dataset (1591 genes) and identified 392 common genes (Fig. 4H). We further carried out pathway enrichment analysis on 392 genes and found pathways including meiotic cell cycle and cell division were highly enriched (Fig. 4I). To verify the reliability our analysis on RIP-seq and SMART-seq data, we subsequently examined candidate Bud31 targets, and visualization of alternatively spliced exons by sashimi plots revealed, for example, that exon 7 of Angel2 was skipped in the SMART-seq data (Supplementary Fig. S5A, top). Consistent with this, RIP-seq analysis showed that Bud31 binds to exon 7 of Angel2 (Supplementary Fig. S5A, bottom). The presence of the anticipated spliced isoforms of Angel2 was confirmed by semiquantitative RT-PCR in the testes of P10 Bud31-sKO and littermate control mice using isoform-specific primers (Supplementary Fig. S5B). We further performed qPCR to analyze long-to-short isoform ratio of Angel2 in the testes of P10 Bud31-sKO and littermate control mice. Consequently, long-to-short isoform ratio of Angel2 was significantly reduced upon loss of Bud31(Supplementary Fig. S5C). RIP-qPCR showed that Bud31 bound to Angel2 pre-mRNA (Supplementary Fig. S5D). We also validated Bud31 regulates AS of Hspa9 through intron 7 retention (Supplementary Fig. S5E–H).
Bud31 depletion leads to increased retention of the first intron of Cdk2
To identify the targets that account for the phenotype in Bud31 cKO mice, we interrogated 392 genes and 701 down-regulated genes (Fig. 4J and Supplementary Table S5) upon Bud31 deficiency. We found 12 genes, including Cdk2, Ccdc62 and Lbr were both differentially spliced and down-regulated (Fig. 4K). We next focus on Cdk2, which is not essential for mouse viability, but its deletion has been shown to cause male infertility [29]. SMART-seq analysis showed that disruption of Bud31 led to the retention of the first intron of the Cdk2 pre-mRNA (Fig. 5A, top), and our RIP-seq data support that Bud31 binds intron 1 of Cdk2 (Fig. 5A, bottom). We next validated the effect of Bud31 on the AS of Cdk2 by semiquantitative RT-PCR in the testes of P10 Bud31-sKO and littermate control mice using isoform-specific primers (Fig. 5B). To obtain further evidence, we constructed minigenes spanning intron 1 and performed splicing assays by transfecting GC-1 cells with minigenes in combination with Bud31 siRNAs. Bud31 depletion greatly increased the intron-containing transcripts of Cdk2 (Fig. 5C). We then analyzed long-to-short isoform ratio by qPCR in the testes of P10 and observed a significant increase long-to-short isoform ratio of Cdk2 upon loss of Bud31 (Fig. 5D). In addition, a splicing reporter assay was performed to confirm the effect of Bud31 on the AS of Cdk2. Knockdown of Bud31 using siRNAs significantly reduced the luciferase activity encoded by a Cdk2 intron 1-containing reporter fragment (Fig. 5E). To gain further evidence for the direct binding of Bud31 to Cdk2, a RIP assay was performed in P14 mouse testes using a Bud31 antibody, and, as expected, RIP-qPCR showed that Bud31 bound to intron 1 of Cdk2 pre-mRNA (Fig. 5F). Moreover, an RNA pull-down assay showed successful pull-down of Bud31 by a biotin-labeled Cdk2 probe (Fig. 5G).

A The AS pattern and Bud31 direct binding sites in the Cdk2 pre-mRNA were visualized with IGV using the SMART-seq and the RIP-seq data. The yellow region highlights the AS region and Bud31 binding sites. B Semiquantitative RT-PCR was performed to validate AS events in Cdk2. Percent spliced in (PSI) was quantified (n = 3). C Analysis of the splicing of Cdk2 minigene by semiquantitative RT-PCR. D qPCR was performed to analyze long-to-short isoform ratio of Cdk2 in the testes of P10 Bud31-sKO and littermate control mice. E A splicing reporter assay was performed to assess the splicing of Cdk2 intron 1 from the pre-mRNA molecule as regulated by Bud31 in GC-1 cells (n = 3 biologically independent samples). F RIP-qPCR was performed to validate the interaction between Bud31 and Cdk2 RNA in P14 mouse testes primary cells using the Bud31 antibody and IgG. G RNA pull-down assay showing the interaction between the Cdk2 intron 1 transcript and Bud31 protein. H qPCR was performed to measure Cdk2 expression in testes tissues of P10 Bud31-sKO and littermate control mice. I qPCR analysis of Cdk2 expression in GC-1 cells transfected with control and Bud31 siRNA. Student’s t test was performed, and data are shown as the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001. J Western blotting analysis of P-Cdk2, Cdk2 and Bud31 levels in testes from P4 Bud31-vKO and control mice. Tubulin was used as the loading control.
Intron retention can reduce gene expression at the post-transcriptional level [30]. We therefore performed qPCR in P10 mouse testes and found that Cdk2 expression was significantly down-regulated in Bud31-sKO mouse testes compared to controls (Fig. 5H). We also found that siRNA-mediated knockdown of Bud31 in GC-1 cells led to significantly reduced Cdk2 levels (Fig. 5I). Immunoblotting further demonstrated that both two protein isoforms were significantly reduced in Bud31-vKO mouse testes compared to controls (Fig. 5J). These findings indicate that deletion of Bud31 results in the retention of intron 1, which leads to decreased Cdk2 expression. Moreover, the expressions of Ccdc62 and Lbr, which have been reported to be involved in fertility [31, 32], were also compromised in the absence of Bud31 (Supplementary Fig. S5I–P). Taken together, these data suggest that Bud31-mediated alternative splicing is involved in the regulation of spermatogenesis.
Discussion
Regulation of AS is important for proper spermatogenesis [8], and splicing factors and AS are regulated in a stage-specific manner during spermatogenesis [33]. For example, Rbm5 is an essential splicing factor in round spermatids, and it regulates the splicing of several spermatid-expressed mRNAs that are critical for producing spermatozoa [34]. Ptbp2 regulates AS in the meiotic and post-meiotic stages of spermatogenesis, and Ptbp2 loss results in the absence of spermatozoa [35]. Also, Bcas2 is involved in pre-mRNA splicing, which is essential for the initiation of meiosis but has little effect on spermatogonia [14]. Nanos2 and Ddx5 are required for the splicing of key genes in the maintenance of spermatogonia [13, 15]. Our findings demonstrated that germ cell-specific Bud31 knockout results in spermatogonia loss and complete male infertility. We further observed significant loss of SSCs as early as P4 in Bud31-vKO mouse testes compared to controls (Fig. 2B, E). Our work thus highlights the importance of spliceosome components in SSC pool maintenance.
The mitosis-meiosis transition is a crucial step for spermatogenesis [36]. Retinoic acid (RA) is an essential extrinsic inducer of meiotic initiation by inducing Stra8 in mammalian germ cells [37]. Dazl is a key intrinsic factor for initiating meiosis, which is required for Stra8 expression [37]. Meiosin together with Stra8 acts as an upstream regulator for cell-cycle switching from mitosis to meiosis [38]. Dmrt1 blocks entry intro meiosis via suppresses RA-dependent transcriptional activity [39]. RNA binding protein Bcas2 is required for the initiation of meiosis via regulating Dazl splicing [14], whereas Nanos2 prevents meiosis initiation by repressing Stra8 expression, which is required for premeiotic DNA replication [40]. Here we crossed Stra8-GFPCre mice with Bud31flox/flox (Bud31-sKO) mice to study the roles of Bud31 in meiosis. We found that SYCP3 and γH2AX positive cells were significantly decreased in Bud31-sKO testes compared with controls (Fig. 3B). No spermatocytes later than preleptotene-like stage chromosomes were observed in Bud31-sKO testes at P12, a time when abundant zygotene chromosomes were already evident in control testes (Fig. 3C). Importantly, levels of Stra8 and its known downstream transcriptional activation targets Hoxa1 and Cyp26a1 were decreased in P10 Bud31-sKO mice compared to controls (Fig. 3E). These observations support Bud31 functions to regulate meiotic initiation in male germ cells.
One of our key findings was that we identified the binding motif and splicing targets of Bud31 during spermatogenesis through combined analysis of RIP-seq and SMART-seq data, which are high sensitivity and low variability RNA sequencing technologies [41]. We observed that the predominant AS events regulated by Bud31 were skipped exons (57%) and retained introns (20%). Importantly, we identified Cdk2 as a direct splicing target of Bud31. Cdk2 is a member of the CDK family that controls the G1/S and S/G2 transitions [42]. Cdk2 knockout mice are viable and survive for up to two years, yet both male and female Cdk2−/− are sterile [43, 44]. Germ cell-specific Cdk2 knockout mice by crossing Hspa2-cre with Cdk2flox/flox mice in our study further confirmed that loss of Cdk2 causes meiotic failure in spermatocytes (Supplementary Fig. S6A–D). Mice bearing a deregulated allele (Cdk2Y15S) are defective in spermatogonial self-renewal and differentiation, resulting in sertoli cell only syndrome [29], suggesting that Cdk2 is one of the crucial splicing targets of Bud31. Mouse Cdk2 is known to produce two protein isoforms, Cdk2-beta (a full-length isoform) and Cdk2-alpha (an exon 6-skipped isoform) [45]. We also identified the exon 6-skipped isoform of Cdk2 in our SMART-seq data, but we failed to validate the skipped exon 6 in the mini-gene assay. Furthermore, both Cdk2-beta and Cdk2-alpha were significantly reduced when Bud31 was depleted (Fig. 5J). Very recently, it was reported that an Mt2b2 retrotransposon promoter generates a truncated isoform of Cdk2, which is essential for preimplantation embryo development [46]. In this report, we found that disruption of Bud31 induced the retention of intron 1 of the Cdk2 pre-mRNA, which led to decreased Cdk2 expression. Thus, AS appears to be a crucial regulatory mechanism for the function of Cdk2.
In summary, our findings suggest that the spliceosome component Bud31 is a critical splicing regulator in SSC self-renewal and meiotic initiation. We also provide strong evidence that Bud31 predominantly regulates exon-skipping and intron retention during spermatogenesis. Specifically, we verified Cdk2 as one of the splicing targets of Bud31. Our study demonstrates that Bud31-mediated AS is essential for early spermatogenesis.
Materials and methods
Mice
Bud31flox/+ mice were established at the Nanjing Biomedical Research Institute of Nanjing University. Cdk2flox/+ mice and Stra8 conventional knockout were from Cyagen Biosciences. Vasa-Cre mice were obtained from the Jackson Laboratory, and Stra8-GFPCre mice were a generous gift from Prof. Ming-han Tong at the Chinese Academy of Sciences [47, 48]. Hspa2-Cre were a generous gift from Prof. Yujun Xu at Nanjing Medical University. Germ cell-specific Bud31-knockout male mice were generated by crossing Bud31-floxed mice with Vasa-Cre (vKO) or Stra8-GFPCre (sKO) transgenic mice. All animals used in our study were housed under a controlled lighting regime (12 h light; 12 h darkness) at 21 °C–22 °C with freely available food and water. The Animal Ethics Committee approved all experimental protocols of the School of Medicine of Shandong University.
Cell culture
Mouse spermatogonia cell line GC-1 was purchased from Wuhan Procell Technology (Wuhan, China). The GC-1 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS, Gibco), 100 units/mL penicillin, and 100 μg/mL streptomycin. All cells were cultured in a humidified atmosphere of 5% CO2 at 37 °C.
Enrichment of spermatogenic cells
The spermatogenic and somatic cells were enriched using a two-step enzymatic digestion process followed by a differential adhesion method [14]. Briefly, after disrupting the tunica albuginea, testes from P9 wild-type mice were transferred into 120 U/ml collagenase type I (Gibco, 17100–017) and incubated at 37 °C for 10 min with gentle shaking every 3–5 min. The cell suspension was then filtered through a 70 μm filter into a new tube, and the lower sediment was digested with 0.25% trypsin-EDTA (Gibco, 25200–072) at 37 °C for 10 min to dissociate the seminiferous tubules into single cells. The suspension was neutralized with FBS and filtered and mixed with collagenase-digested suspension and centrifuged at 1000 g for 5 min. The cell pellet was suspended in 8 ml of DMEM medium and seeded in a 10 cm culture dish. After 2–3 h of incubation at 37 °C, the floating and weakly adhering cells were transferred to a new 10 cm dish. The fraction of spermatogenic cells comprised the floating and weakly adhering cells, while the attached cells on the bottom of the dish were collected as the fraction of somatic cells. The efficiency of the separation was determined using RT-PCR and western blotting analysis.
RNA extraction and qPCR
Total RNA was extracted from cells or whole testes using TRIzol reagent (Invitrogen, 15596–026) following the manufacturer’s instructions. After removing the residual genomic DNA, 1 μg of total RNA was reverse-transcribed into cDNA using the HiScript III RT SuperMix for qPCR (+gDNA wiper) (Vazyme, R223–01) according to the manufacturer’s protocol. qPCR was performed using ChamQ SYBR Colour qPCR Master Mix (Vazyme, Q411–02) on a Quant Studio 3 system (Thermo Fisher). The primers for the qPCR assay are listed in Supplementary Table S6.
Immunoblotting
Protein samples were prepared for western blotting and immunoprecipitation using tissue lysis buffer with a protease inhibitor cocktail (Beyotime, P011) and were quantified using a BCA reagent kit (Beyotime, P0012–1). Equal amounts of total protein were separated on a 12% SDS–PAGE gel (Servicebio, G2063–50T) and transferred onto PVDF membranes (Millipore, ISEQ00010). The membranes were blocked with 5% nonfat milk for 1 h at room temperature and incubated with diluted primary antibodies overnight at 4 °C. After washing with TBST three times, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:10000 dilution, Jackson ImmunoResearch) for 1 h at room temperature. The signals were developed with Pierce ECL Substrate (Vazyme, E412–02). The details of the primary antibodies used in this study are shown in Supplementary Table S7. Full and uncropped western blots are shown in Supplementary Figs. S7 and S8.
Histological analysis
Mouse testes were collected immediately after euthanasia and were fixed in 4% paraformaldehyde (Servicebio, G1101) overnight. Following dehydration, the samples were embedded in paraffin and sectioned at a thickness of 4 μm. After deparaffinization, the slides were stained with hematoxylin, immunohistochemical staining, or immunofluorescence staining. The hematoxylin-stained slides were imaged using a fluorescence microscope (BX53, Olympus), and images were processed using Image J.
Chromosome spread immunofluorescence analysis
Chromosome spread immunofluorescence was performed as described previously [49]. Briefly, seminiferous tubules were incubated in hypotonic solution for 1 h and then transferred to a 100 mM sucrose solution. The samples were minced and spread on slides dipped in 1% PFA and 0.15% Triton X100 (pH 9.0). Slides were incubated in a humidified chamber for 4–5 h, dried, and washed in PBS followed by immunofluorescence staining. Primary antibodies were detected with Alexa Fluor 488- and 594-conjugated secondary antibodies (1:500 dilution, Thermo Fisher Scientific A-11070 and A-11012) for 1 h at room temperature. The slides were washed with PBS three times and mounted with mounting medium with DAPI (Abcam, ab104139). Immunolabeled chromosome spreads were imaged by confocal microscopy using a Dragonfly 200 system (Andor Technology). Projection images were then prepared using Image J software. Primary antibodies used for immunofluorescence are shown in Supplementary Table S7.
SMART-seq
SMART-seq provides sensitive full-length transcriptome profiling in single or few cells [50]. In this study, THY1+/c-Kit– SSCs were sorted by flow cytometry in Bud31-vKO and littermate control mice at P4. About 1000 cells were collected in tubes with lysis buffer and ribonuclease inhibitor. Oligo dT primers were introduced to the reverse transcription reaction for first strand cDNA synthesis, followed by PCR amplification to enrich the cDNA and magnetic bead purification to clean up the products. The cDNA product was checked with a Qubit 3.0 Fluorometer and Agilent 2100 Bioanalyzer to ensure the expected production with a length of around 1000–2000 bp. The cDNA was then sheared into random lengths by ultrasonication for use in the Illumina library preparation protocol, including DNA fragmentation, end-repair, 3′ end A-tailing, adapter ligation, PCR amplification, and library validation. After library preparation, a PerkinElmer LabChip GX Touch and Step OnePlus Real-Time PCR System were used for library quality inspection. Qualified libraries were then loaded onto the Illumina Hiseq platform for PE150 sequencing by Annoroad Gene Tech. After obtaining paired-end reads, clean reads were aligned to the mm10 genome with HISAT2 (version 2.2.0) and sorted with samtools (version 1.9). Mapped reads were visualized with the Integrative Genomics Viewer (IGV). Differential expression was determined with DESeq2. The cutoff was set as p value < 0.05 and |log2FC| > 0.7. The differentially expressed genes were used for Gene Set Enrichment Analysis. The RNA-seq data generated in this study has been deposited in the NCBI GEO database under the accession number GSE189714.
AS and splicing efficiency analysis
Aligned mapped reads of SMART-Seq data were used for further analysis. AS events were identified by rMATS (version 4.1.0) [51] and were classified into skipped exons, retained introns, alternative 5′ splice sites, alternative 3′ splice sites, and mutually exclusive exons. Significant events were filtered out with p < 0.05 and |IncLevelDifference| > 0.1. AS events over the course of testis development were identified with ASprofile [52] based on the RNA-seq data.
Splicing efficiency was determined separately for the 5′ and 3′ splice sites. We used Kolmogorov-Smirnov test to determine the difference and uniformity of the SMART-seq samples. Splicing efficiency was analyzed using the SMART-seq data with an R package SEAA. The source code used to generate the results has been deposited at Github (https://github.com/PrinceWang2018/SEAA). The D statistic indicates the absolute max distance between the CDFs of the two samples. The D values for two CDFs were 0.0885 for 5′ splice sites and 0.0657 for 3′ splice sites.
RNA immunoprecipitation sequencing (RIP-seq)
RIP was performed using the EZ-Nuclear RIP (Cross-Linked) Kit (Merck Millipore) following the manufacturer’s instructions. In brief, the P14 mouse testis tissues were digested into single cells through a two-step enzymatic digestion method. The cells were cross-linked with 0.3% formaldehyde and then lysed and ultrasonicated, and the nucleic acids were sheared into fragments of about 500–1000 bp. The cell lysates were incubated with magnetic beads coated with Bud31 antibody (Proteintech). RNA was extracted from the immune complex while removing DNA and protein, and RNA quality was checked with Quibit and an Agilent 2100. The library was constructed by the RiboBio Biotechnology Company and loaded onto the Illumina Hiseq platform for PE150 sequencing. Clean reads were aligned to the mm10 genome by Tophat (v2.0.13) and sorted with samtools (1.9). RIP peaks were identified with the R package RIPSeeker (v1.26.0) [53] and annotated with HOMER [54]. De novo Bud31 binding motif analysis was conducted using MEME program (v4.11.1) [55]. Reads intensity distribution among the exon and intron genome elements was visualized with Deeptools. The RIP-seq data generated in this study has been deposited in the NCBI GEO database under the accession number GSE189715.
RNA pull-down
Cdk2 transcripts were obtained using the RNAMAX-T7 in vitro transcription kit (RiboBio) and then biotin-labeled using the Pierce™ RNA 3′ End Desthiobiotinylation Kit (Thermo Fisher Scientific). According to the manufacturer’s protocol, RNA pull-down was performed using the Magnetic RNA-Protein Pull-Down Kit (Thermo Fisher Scientific). The proteins were detected by western blot analysis.
Luciferase assay
The Cdk2 intron 1 sequence was cloned into CMV-LUC2CP/intron/ARE plasmids (Addgene). The plasmids encoding luciferase with or without Cdk2 intron 1 were co-transfected with Bud31 siRNAs into GC-1 cells. The pRL-TK vector was used as a control. Luciferase activity was measured 48 h after transfection using the Dual-Luciferase Reporter Assay System (Vazyme, DL101–01). The relative luciferase activity was calculated as the ratio between firefly and Renilla luciferase activity.
Minigene assay
The Cdk2 minigene was constructed by inserting an additional 708 bp from intron 1 (corresponding to chr10:128534147–128534854, mm10) into pcDNA3.1 vector between the hind111 and xho1 sites. Cdk2 intron 1 region was amplified and cloned into pcDNA3.1vector by ClonExpress II One Step Cloning Kit (Vazyme, C112–01) GC-1 cells were transfected with the si-Bud31 together with Cdk2 intron1 minigene plasmids using jetPRIME® DNA & siRNA Transfection Reagent (PolyPlus). The cells were harvested for RNA extraction 48 h after transfection. Semi-RT-PCR was performed for splicing analysis. The sequence of siRNAs and primers are shown in Supplementary Table S6.
Statistical analysis
The results of all quantitative experiments were based on at least three independent biological samples. All data are presented as means ± SEM. The statistical significance of the differences between the mean values for the different genotypes was measured by Student’s two-tailed t test with a paired t-test. The data were considered significant when the p value was less than 0.05.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Song HW, Bettegowda A, Lake BB, Zhao AH, Skarbrevik D, Babajanian E, et al. The homeobox transcription factor RHOX10 drives mouse spermatogonial stem cell establishment. Cell Rep. 2016;17:149–64.
Krawetz SA, De Rooij DG, Hedger MP. Molecular aspects of male fertility. International Workshop on Molecular Andrology. EMBO Rep. 2009;10:1087–92.
Feng LX, Chen Y, Dettin L, Pera RA, Herr JC, Goldberg E, et al. Generation and in vitro differentiation of a spermatogonial cell line. Science. 2002;297:392–5.
Oatley JM, Brinster RL. Regulation of spermatogonial stem cell self-renewal in mammals. Annu Rev Cell Dev Biol. 2008;24:263–86.
Song HW, Wilkinson MF. Transcriptional control of spermatogonial maintenance and differentiation. Semin Cell Dev Biol. 2014;30:14–26.
Jung H, Song H, Yoon M. The KIT is a putative marker for differentiating spermatogonia in stallions. Anim Reprod Sci. 2015;152:39–46.
Liu MM, Zack DJ. Alternative splicing and retinal degeneration. Clin Genet. 2013;84:142–9.
Baralle FE, Giudice J. Alternative splicing as a regulator of development and tissue identity. Nat Rev Mol Cell Biol. 2017;18:437–51.
Zhang X, Ameer FS, Azhar G, Wei JY. Alternative Splicing Increases Sirtuin Gene Family Diversity and Modulates Their Subcellular Localization and Function. Int J Mol Sci. 2021;22:E473.
de la Grange P, Gratadou L, Delord M, Dutertre M, Auboeuf D. Splicing factor and exon profiling across human tissues. Nucleic Acids Res. 2010;38:2825–38.
Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17:19–32.
Havens MA, Duelli DM, Hastings ML. Targeting RNA splicing for disease therapy. Wiley Interdiscip Rev RNA. 2013;4:247–66.
Legrand JMD, Chan AL, La HM, Rossello FJ, Anko ML, Fuller-Pace FV, et al. DDX5 plays essential transcriptional and post-transcriptional roles in the maintenance and function of spermatogonia. Nat Commun. 2019;10:2278.
Liu W, Wang F, Xu Q, Shi J, Zhang X, Lu X, et al. BCAS2 is involved in alternative mRNA splicing in spermatogonia and the transition to meiosis. Nat Commun. 2017;8:14182.
Sada A, Suzuki A, Suzuki H, Saga Y. The RNA-binding protein NANOS2 is required to maintain murine spermatogonial stem cells. Science. 2009;325:1394–8.
Bai R, Wan R, Wang L, Xu K, Zhang Q, Lei J, et al. Structure of the activated human minor spliceosome. Science. 2021;371:eabg0879.
Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015;525:384–8.
Masciadri B, Areces LB, Carpinelli P, Foiani M, Draetta G, Fiore F. Characterization of the BUD31 gene of Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2004;320:1342–50.
Cardoso-Moreira M, Halbert J, Valloton D, Velten B, Chen C, Shao Y, et al. Gene expression across mammalian organ development. Nature. 2019;571:505–9.
Saha D, Banerjee S, Bashir S, Vijayraghavan U. Context dependent splicing functions of Bud31/Ycr063w define its role in budding and cell cycle progression. Biochem Biophys Res Commun. 2012;424:579–85.
Costoya JA, Hobbs RM, Barna M, Cattoretti G, Manova K, Sukhwani M, et al. Essential role of Plzf in maintenance of spermatogonial stem cells. Nat Genet. 2004;36:653–9.
Kanatsu-Shinohara M, Shinohara T. Spermatogonial stem cell self-renewal and development. Annu Rev Cell Dev Biol. 2013;29:163–87.
Ferder IC, Fung L, Ohguchi Y, Zhang X, Lassen KG, Capen D, et al. Meiotic gatekeeper STRA8 suppresses autophagy by repressing Nr1d1 expression during spermatogenesis in mice. PLoS Genet. 2019;15:e1008084.
Anderson EL, Baltus AE, Roepers-Gajadien HL, Hassold TJ, de Rooij DG, van Pelt AM, et al. Stra8 and its inducer, retinoic acid, regulate meiotic initiation in both spermatogenesis and oogenesis in mice. Proc Natl Acad Sci USA. 2008;105:14976–80.
Hogarth CA, Griswold MD. The key role of vitamin A in spermatogenesis. J Clin Invest. 2010;120:956–62.
Nagaoka SI, Nakaki F, Miyauchi H, Nosaka Y, Ohta H, Yabuta Y, et al. ZGLP1 is a determinant for the oogenic fate in mice. Science. 2020;367:aaw4115.
Barrios F, Filipponi D, Campolo F, Gori M, Bramucci F, Pellegrini M, et al. SOHLH1 and SOHLH2 control Kit expression during postnatal male germ cell development. J Cell Sci. 2012;125:1455–64.
Xu K, Yang Y, Feng GH, Sun BF, Chen JQ, Li YF, et al. Mettl3-mediated m(6)A regulates spermatogonial differentiation and meiosis initiation. Cell Res. 2017;27:1100–14.
Singh P, Patel RK, Palmer N, Grenier JK, Paduch D, Kaldis P, et al. CDK2 kinase activity is a regulator of male germ cell fate. Development. 2019;146:dev180273.
Schmitz U, Pinello N, Jia F, Alasmari S, Ritchie W, Keightley MC, et al. Intron retention enhances gene regulatory complexity in vertebrates. Genome Biol. 2017;18:216.
Li Y, Li C, Lin S, Yang B, Huang W, Wu H, et al. A nonsense mutation in Ccdc62 gene is responsible for spermiogenesis defects and male infertility in repro29/repro29 mice. Biol Reprod. 2017;96:587–97.
Mylonis I, Drosou V, Brancorsini S, Nikolakaki E, Sassone-Corsi P, Giannakouros T. Temporal association of protamine 1 with the inner nuclear membrane protein lamin B receptor during spermiogenesis. J Biol Chem. 2004;279:11626–31.
Schmid R, Grellscheid SN, Ehrmann I, Dalgliesh C, Danilenko M, Paronetto MP, et al. The splicing landscape is globally reprogrammed during male meiosis. Nucleic Acids Res. 2013;41:10170–84.
O’Bryan MK, Clark BJ, McLaughlin EA, D’Sylva RJ, O’Donnell L, Wilce JA, et al. RBM5 is a male germ cell splicing factor and is required for spermatid differentiation and male fertility. PLoS Genet. 2013;9:e1003628.
Zagore LL, Grabinski SE, Sweet TJ, Hannigan MM, Sramkoski RM, Li Q, et al. RNA binding protein Ptbp2 is essential for male germ cell development. Mol Cell Biol. 2015;35:4030–42.
Nakagawa T, Zhang T, Kushi R, Nakano S, Endo T, Nakagawa M, et al. Regulation of mitosis-meiosis transition by the ubiquitin ligase beta-TrCP in male germ cells. Development. 2017;144:4137–47.
Lin Y, Gill ME, Koubova J, Page DC. Germ cell-intrinsic and -extrinsic factors govern meiotic initiation in mouse embryos. Science. 2008;322:1685–7.
Ishiguro KI, Matsuura K, Tani N, Takeda N, Usuki S, Yamane M, et al. MEIOSIN directs the switch from mitosis to meiosis in mammalian germ cells. Dev Cell. 2020;52:429–45.e410.
Matson CK, Murphy MW, Griswold MD, Yoshida S, Bardwell VJ, Zarkower D. The mammalian doublesex homolog DMRT1 is a transcriptional gatekeeper that controls the mitosis versus meiosis decision in male germ cells. Dev Cell. 2010;19:612–24.
Suzuki A, Saga Y. Nanos2 suppresses meiosis and promotes male germ cell differentiation. Genes Dev. 2008;22:430–5.
Picelli S. Full-length single-cell RNA sequencing with smart-seq2. Methods Mol Biol. 2019;1979:25–44.
Faber EB, Wang N, Georg GI. Review of rationale and progress toward targeting cyclin-dependent kinase 2 (CDK2) for male contraceptiondagger. Biol Reprod. 2020;103:357–67.
Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, et al. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet. 2003;35:25–31.
Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol. 2003;13:1775–85.
Kwon TK, Buchholz MA, Jun DY, Kim YH, Nordin AA. The differential catalytic activity of alternatively spliced cdk2 alpha and cdk2 beta in the G1/S transition and early S phase. Exp Cell Res. 1998;238:128–35.
Modzelewski AJ, Shao W, Chen J, Lee A, Qi X, Noon M, et al. A mouse-specific retrotransposon drives a conserved Cdk2ap1 isoform essential for development. Cell. 2021;184:5541–58.e5522.
Lin Z, Hsu PJ, Xing X, Fang J, Lu Z, Zou Q, et al. Mettl3-/Mettl14-mediated mRNA N(6)-methyladenosine modulates murine spermatogenesis. Cell Res. 2017;27:1216–30.
Chen Y, Lyu R, Rong B, Zheng Y, Lin Z, Dai R, et al. Refined spatial temporal epigenomic profiling reveals intrinsic connection between PRDM9-mediated H3K4me3 and the fate of double-stranded breaks. Cell Res. 2020;30:256–68.
Huang T, Yuan S, Gao L, Li M, Yu X, Zhan J, et al. The histone modification reader ZCWPW1 links histone methylation to PRDM9-induced double-strand break repair. Elife. 2020;9:e53459.
Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171–81.
Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci USA. 2014;111:E5593–601.
Florea L, Song L, Salzberg SL. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res. 2013;2:188.
Li Y, Zhao DY, Greenblatt JF, Zhang Z. RIPSeeker: a statistical package for identifying protein-associated transcripts from RIP-seq experiments. Nucleic Acids Res. 2013;41:e94.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89.
Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–7.
Acknowledgements
We thank Translational Medicine Core Facility of Shandong University for consultation and instrument availability that supported this work. We also thank the Laboratory Animal Center of Shandong University for mouse housing and care.
Funding
This work was supported by National Key R&D Program of China (2020YFA0804400). National Natural Science Foundation of China (81972437, 81672578, 82071854). This study is also supported by the Taishan Scholars Program of Shandong Province.
Author information
Authors and Affiliations
Contributions
Conception and design: JQ, HL, and ZL. Development and methodology: JQ, TH, JW, YZ, and QD. Acquisition of data: JQ, TH, ZW, JW, DC, and XW. Analysis and interpretation of data: JQ, TH, ZW, GL, XZ, SL, and ZL. Administrative, technical, or material support: GL, XZ, SL, HL, and ZL. Study supervision: HL and ZL. Writing, review, and/or revision of the manuscript: All authors. Final approval: All authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Our studies did not include human participants, human data, or human tissue.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Hardwick
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Qin, J., Huang, T., Wang, Z. et al. Bud31-mediated alternative splicing is required for spermatogonial stem cell self-renewal and differentiation. Cell Death Differ 30, 184–194 (2023). https://doi.org/10.1038/s41418-022-01057-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-022-01057-1
This article is cited by
-
Alternative splicing and related RNA binding proteins in human health and disease
Signal Transduction and Targeted Therapy (2024)
-
Intron detention tightly regulates the stemness/differentiation switch in the adult neurogenic niche
Nature Communications (2024)
