
Abstract
SARS-CoV-2 infection poses a major threat to the lungs and multiple other organs, occasionally causing death. Until effective vaccines are developed to curb the pandemic, it is paramount to define the mechanisms and develop protective therapies to prevent organ dysfunction in patients with COVID-19. Individuals that develop severe manifestations have signs of dysregulated innate and adaptive immune responses. Emerging evidence implicates neutrophils and the disbalance between neutrophil extracellular trap (NET) formation and degradation plays a central role in the pathophysiology of inflammation, coagulopathy, organ damage, and immunothrombosis that characterize severe cases of COVID-19. Here, we discuss the evidence supporting a role for NETs in COVID-19 manifestations and present putative mechanisms, by which NETs promote tissue injury and immunothrombosis. We present therapeutic strategies, which have been successful in the treatment of immunο-inflammatory disorders and which target dysregulated NET formation or degradation, as potential approaches that may benefit patients with severe COVID-19.
Similar content being viewed by others


Facts
-
Patients with COVID-19 show signs of dysregulated innate and adaptive immune responses.
-
SARS-CoV-2-induces the formation of NETs through ACE2 and requires active TMPRSS2 and virus replication.
-
Immunothrombosis triggered by NETs mediates damage of distant organs.
Open questions
-
Would inhibition of neutrophil proteases ameliorate tissue injury in patients with COVID-19?
-
How are neutrophils and NETs influenced by a network of antibodies, complement proteins, clotting factors, CRP, nucleases, proteases, and anti-proteases?
-
Does the modulation of NET formation and its clearance complement current therapies?
-
Can the synergism of DNases and heparin in NET degradation be exploited as co-adjuvant therapy?
Basic aspects of neutrophil biology and their relevance for COVID-19
Neutrophils normally differentiate in the bone marrow and throughout this process start to express effector molecules that are stored in granules allowing them to mount inflammation and kill microbes [1, 2]. A distinctive feature of mature neutrophils is that they cannot proliferate and, thus stay for only short periods in the circulation [3]. Mature neutrophils transit from the bone marrow into the circulation and from the circulation into the tissues even under steady-state conditions. The trafficking and reactivity of neutrophils to pathogens follow circadian patterns [4,5,6]. The influx of neutrophils from the circulation into tissues happens in most organs, but in particular in highly vascularized ones such as lungs and kidneys, representing the prime targets in coronavirus disease 19 (COVID-19) [7]. Notably, although neutrophils homing into tissues have partially lost their prestored molecules [5], they remain active and can damage vessels and parenchyma. The neutrophil to lymphocyte ratio has been identified as the most important independent risk factor for severe COVID-19 [8].
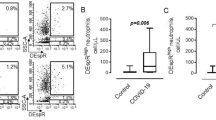
Mechanisms underlying neutrophil development have received increasing attention [9,10,11,12,13]. There is still no unified nomenclature for neutrophil developmental stages, which would be particularly useful to understand emerging observations in the context of COVID-19 and other disorders. For simplicity, we adopt here a naming system recently proposed [14], in which neutrophil development transits from proNeu1 via proNeu2, preNeu, and immature to mature neutrophils in the bone marrow. Immunophenotyping of COVID-19 blood samples revealed that the emergence of immature subsets of neutrophils (preNeu and immature) in the blood correlates with severe COVID-19, suggesting that precise delineation of neutrophil subsets could be used as a predictive marker for COVID-19 severity [15,16,17].
Mechanisms of neutrophil extracellular trap formation
Neutrophils are prompted to release neutrophil extracellular traps (NETs) upon encounter of danger signals (Supplementary Fig. 1), which in essence are structures composed of DNA decorated with histones and granule proteins such as lactoferrin, cathepsins, neutrophil elastase (NE), and myeloperoxidase (MPO) (Supplementary Fig. 2), as well as cytoplasmic and cytoskeletal proteins [18, 19]. Mitochondrial DNA is also found in NETs [20, 21]. NETs immobilize pathogens, limit their dissemination, and enable their killing by antimicrobial proteins. Beyond antimicrobial defense, there is growing evidence that NETs contribute to the pathogenesis of numerous diseases due to either excessive formation and/or impaired removal, which turns out to be toxic for the host [22].
Activation of neutrophils through Toll-like receptors, G protein-coupled receptors, Fc-, chemokine- and cytokine- receptors can stimulate NET formation (Fig. 1). Neutrophil activation by engagement of these receptors induces NET formation by various mechanisms, many of which are linked to the activation of the NADPH oxidase (NOX) complex. However, NOX-independent processes have also been described to lead to the NET formation [23]. Reactive oxygen species (ROS) produced in the context of NOX activation and mitochondrial dysfunction [21] are important in the rearrangement of the cytoskeleton [24] and glycolytic ATP production [25], which are required for NET formation. Early during NET formation, granular NE and MPO translocate to the nucleus and drive nuclear and chromatin decondensation [26]. Peptidylarginine deiminase 4 (PADI4) contributes to chromatin decondensation by histone hypercitrullination [27]. Citrullination licenses calpain to further decondensed nuclei before extracellular trap release [28]. However, like for NOX, PADI4- and NE-independent pathways have also been reported [29, 30]. In addition, necroptotic and pyroptotic pathways can be activated [31]. Cathepsin C (CatC) also plays an important role since it is required for the activation of NE and other serine proteases [32].

Viruses (SARS-CoV-2), ROS, calcium oxalate, co-infecting microorganisms, cytokines and chemokines, cationic antimicrobial peptides, nanodiamonds, monosodium urate (MSU), and platelets reportedly induce NET formation. See main text for references. Original illustration from the authors.
During NET formation, NE also cleaves gasdermin D (GSDMD), a molecule centrally involved in pyroptosis, and constituting a feed-forward loop to facilitate granule and plasma membrane permeabilization (Fig. 2) [33]. Conversely, NET formation is facilitated after cytosolic LPS sensing and caspase-11-dependent activation of GSDMD [34]. Disulfiram interferes with the papain-like proteases of the SARS-COVID-19 infection cycle [35] and was shown to modify Cys191–Cys192 in GSDMD to reduce pore [36] and possibly NET release.

Pathways that regulate NET formation (see body text for references). Pattern recognizing receptors (PRR) initiate NADPH oxidase activation and a spike of cytosolic calcium activating neutrophil peptidylarginine deiminase 4 (PADI4) causing histone citrullination (yellow circle) and DNA decondensation. Chromatin and/or mitochondrial DNA is expelled and form NETs. Several necrotic cell death pathways may contribute to NETosis. Necroptosis involves RIPK1/RIPK3-mediated activation of MLKL and plasma membrane permeabilization contributing to the release of NETs. Pyroptosis involves canonical or non-canonical inflammasome activation by the caspases-1 or 4, respectively. Caspase-1 and 4 as well as NE cleave GSDMD and generates the N-GSDMD fragment with a pore-forming activity that enables the release of NETs. In addition, autophagic processes contribute to the release of NETs. Original illustration from the authors.
Pro- and anti-inflammatory functions of NET
In general, NETs can exert both pro- and anti-inflammatory effects, which are context-dependent [37]. Proinflammatory effects include the induction of type I Interferons (IFNs) [38] and proinflammatory cytokines [21], induction of the NLRP3 inflammasome [39], promotion of adaptive immune responses [40], damage to the endothelium [41], and immunothrombosis [42]. In addition, NET aggregation can occlude ducts in various organs and promote organ damage [43, 44]. Indeed, pre-clinical strategies to interfere with the release of NETs or to promote the clearance of formed NETs can prevent organ injury in numerous models of inflammatory diseases [45, 46]. On the flip side of the coin, aggregation of NETs can promote the trapping and cleavage of proinflammatory mediators by NET-bound proteases (Fig. 3) [47,48,49], eventually leading to downregulation of inflammatory responses and resolution of inflammation.

Increased numbers of patrolling neutrophils in inflamed tissues form aggregated neutrophil extracellular traps (aggNETs). These are prone to occlude the ducts and glands of the pancreas, gall bladder, and ocular surface. The occlusions precipitate organ pathogeneses like pancreatitis and cholelithiasis. AggNETs also occlude blood vessels in particular the microvasculature of lungs, liver, kidney, heart, and thus cause pathogenesis. Original illustration from the authors.
NETs activation
Viral infections induce the formation of NET
A wide array of pathogens triggers NET formation [18]. These include viruses such as a respiratory syncytial virus (RSV) [50] and influenza [51]. Initial studies showed that sera from COVID-19 patients triggered NET release by healthy control neutrophils in vitro [52] and more recent evidence suggests that viable SARS-CoV-2 can directly stimulate human neutrophils to release NETs in a dose-dependent manner (Fig. 4) [53]. SARS-CoV-2-mediated NET-induction requires the angiotensin converting enzyme 2 receptor (ACE2), expressed by neutrophils, the activity of the serine protease TMPRSS2, and virus replication. Similar to what was observed for RSV, the pan-PAD inhibitor Cl-Amidine abrogated SARS-CoV-2 induced NET formation, implying that inhibition of NET formation may represent a potential therapeutic option for COVID-19.

NET formation of human blood-derived neutrophils after treatment with SARS-CoV-2. Immunofluorescence staining of NETs was done using antibodies against elastase (red) and DNA-histone1-complexes (green), with a counterstain of DNA (blue). Yellow staining indicates colocalization of NETs (histone-DNA fibers) with elastase. The Bars represent 25 µm (left) and 50 µm (middle and right). Original illustration from the authors.
The role of citrullination in NETs
The physiological NET formation is typically associated with PADI4 activation (Fig. 2) [54]. PADI4 converts positively charged arginines to neutral citrullines in protein substrates, including core histones [55]. Citrullination unleashes the energy of coiled DNA, leading to the catapult-like ejection of NETs [56]. PADI4 retains enzymatic activity in the extracellular environment and modifies proteins, including those of the extracellular matrix [57] and coagulation factors [58]. Accumulation of citrullinated histones was found in COVID-19 and in influenza-infected mice [52, 59, 60]. Since pan-PADI and PADI4 inhibitors such as Cl-amidine, BB-CL-amidine, YW-56, or GSK484 have shown efficacy in the treatment of NET-mediated pathologies, such as lethal lung endotoxemia [61] and cellular damage due to hypoxia [62], the administration of such inhibitors may prove beneficial for the treatment of COVID-19.
The relation of platelet activation with NETs
Platelets are activated during COVID-19, forming aggregates with leukocytes, in particular in patients with severe disease [59, 63,64,65,66,67]. Platelets are well known to adhere to injured blood vessels, become activated, and express adhesion molecules, including P-selectin and ICAM-1, leading to neutrophil recruitment. Platelets—due to their number and privileged position in the blood—may represent major instigators of neutrophil activation [68] through direct contact [69]. The physiological importance of this interaction may be to trigger neutrophil-mediated repair [70, 71]. Circulating platelets do not spontaneously bind neutrophils, but do so in the context of bacterial [72] or viral infection [73]. This interaction relies on integrins [74] and may result in NET formation. Indeed, platelets can trigger NET formation (Fig. 2), but the platelet-derived molecules that induce NET release remain poorly characterized. While a consensus is lacking, HMGB1 [75] and inorganic polyphosphate (polyP) [76] are candidates to underlie this phenomenon. However, this mechanism is debated [77]. Thus, platelet activation may trigger the formation of intravascular NET aggregates in the pulmonary and renal microcirculation [59, 65], thereby contributing to the manifestations of COVID-19 [78].
Complement activation as a trigger for NETs
Complement activation fosters the cytokine storm and coagulopathy, both critical events in COVID-19 [79]. A history of macula degeneration, associated with complement-activation, predisposes to poor outcomes during COVID-19, while complement deficiencies appear to be protective [79]. SARS-CoV-2 activates compliment and complement regulators [79] and consistently C5a and C5b-9 accumulate in the blood of COVID-19 patients, indicating complement activation [80, 81]. Complement deposition is detected in the microvasculature, occasionally in proximity to SARS-CoV-2 glycoproteins [82]. Complement activation may thus represent an additional trigger for NETs also in COVID-19 [81].
NETs’ impact at the cellular level
The role of NET-bound enzymes
Neutrophil granules contain various serine proteases including NE, cathepsin G, and proteinase-3, lactoferrin, MPO, and lysozyme that can promote tissue damage [45]. These enzymes, which also appear in NETs, can modulate viral immune responses through modification of autoantigens and immune complexes [83]. NE can further cleave the spike protein and thus activate the fusogenic peptide of SARS-CoV-2 spike protein S2 [84]. These findings suggest that the proteolytic activity of neutrophil-derived enzymes may modulate membrane fusion of the virus [85]. The effects of NE may be modulated by protease inhibitors such as serum alpha-1-antitrypsin (serpinA1) to prevent tissue injury and virus activation. Increased serum NE activity was detected during severe COVID-19, despite the functional inhibitory activity of serpinA1 against exogenous soluble NE [59], thereby revealing a mechanism of resistance of NET-derived NE to serpinA1 that may be relevant during COVID-19 [86].
NET-induced thromboinflammation in COVID-19
Thrombotic complications contribute to morbidity and mortality in severe COVID-19 [87, 88]. Thrombosis in patients with COVID-19 affects both the arterial and venous circulation, leading to acute coronary syndrome, stroke, deep vein thrombosis, pulmonary embolism, and microvascular thrombosis (Fig. 3) [89,90,91,92]. The NET-remnants, including circulating cell-free DNA, citrullinated H3, or MPO-DNA complexes, are abundantly found in the circulation of patients with severe COVID-19 [52, 93]. Furthermore, neutrophil-platelet aggregates and neutrophil activation markers are also increased in patients with severe disease [65, 94]. Importantly, NETs from patients with COVID-19 are loaded with tissue factor (TF). Complement activation has been linked to the release of thrombogenic NETs decorated with TF [81]. The excessive NET formation may also cause direct vascular injury [41, 95] and indirectly support the formation of autoantibodies that determine the appearance of various forms of autoimmune vasculitis [96, 97]. Along this line, histopathology studies have shown that NET-based immunothrombosis is linked to organ damage in severe COVID-19 [98]. Lung autopsies from patients with COVID-19-related acute respiratory distress syndrome (ARDS) revealed widespread occlusion of small pulmonary vessels by aggregated NETs [93]. Neutrophils also infiltrate alveolar and interstitial areas of K18-hACE2 transgenic mice infected with SARS-CoV-2 leading to a comparable pulmonary pathology [32, 99]. NET-rich thrombi, platelets, and fibrin were also present in the lung, heart, and kidney [59, 65]. This clogging of microvessels by aggregated NETs (aggNETs) may contribute to fatal outcomes in COVID-19. Notably, in steady-state conditions, DNAses prevent vascular occlusions by non-canonical NET-driven thrombosis [100]. This observation indicates that NET-dissolving mediators can also be impaired or overwhelmed in the patients [101].
NET-induced endothelial activation and damage in COVID-19
Endothelial injury is considered an essential pathogenic process in COVID-19, leading to lung and kidney damage [102,103,104]. Organ- and microenvironment-associated endothelial heterogeneity likely contributes to different COVID-19 outcomes [105]. Similar to other SARS viruses, SARS-CoV-2 enters cells through ACE2, expressed on renal and pulmonary endothelial cells [106, 107]. In accordance, SARS-CoV-2 has been detected intracellularly in renal and pulmonary endothelial cells [107, 108]. Paracrine factors released from infected endothelial cells [109] may impact disease outcome by altering functions of epithelial or other neighboring cells, including neutrophils and pneumocytes. Furthermore, endothelial damage fosters perivascular T-cell recruitment and disrupts the alveolar-capillary barrier in the lungs [91]. Acute endothelial damage in COVID-19 is associated with structurally deformed capillaries and signs of compensatory neovascularization [91]. This compromised endothelial barrier triggers lung edema and proteinuria, which are common observations in severe lung and kidney diseases [91, 110, 111].
NETs directly activate endothelial cells, induce endothelial to mesenchymal transition, and apoptotic endothelial cell death. Thus, NETs compromise endothelial integrity and barrier function and promote endothelial dysfunction (Supplementary Fig. 3) [41, 112, 113]. Since NETs are abundant in the circulation and in lung and kidney tissues of patients with COVID-19 [65], their accumulation represents a key trigger to induce pulmonary and renal microvascular thrombosis, which triggers disease-related organ failure [59, 81, 93, 114]. The effect of classical anti-thrombotic treatments may be hampered as NETs have shown to be central components of vascular occlusion in COVID-19 [100].
NETs’ impact at the organ level
NETs in COVID-19-associated acute lung disease
Histopathological studies revealed that respiratory symptoms and shortness of breath in COVID-19 occur secondary to alveolar-capillary damage, hemorrhage, immune cell infiltration, fibrin deposition, and fluid-filled alveoli [115,116,117,118]. Detailed analysis of lungs revealed abnormal extracellular matrix remodeling, denuded alveolar epithelia, and proliferation of epithelial cells and fibroblasts. Importantly, neutrophilia directly correlates with disease severity in COVID-19 [102]. Increased serum levels of neutrophil-derived MPO-DNA and citrullinated histone H3, both NET degradation products, closely parallel lung distress and predict COVID-19 severity [52]. Furthermore, circulating nucleosomes were identified as potential markers to monitor COVID-19 disease progression [119]. Immature and low-density neutrophils predominate in severe COVID-19 [15, 16, 120]. Neutrophils that recently emigrate from the bone marrow have higher granule contents and enhanced NET release, which aggravates pulmonary injury in murine models [5]. It is therefore conceivable that immature neutrophils in the circulation of COVID-19 patients actively promote susceptibility to ARDS [5]. Likewise, hypogranular neutrophils produced during emergency myelopoiesis have a higher propensity to release NETs and may be causally related to COVID-19 severity [93]. NETs released by SARS-CoV-2–activated neutrophils promote lung epithelial cell death in vitro [53, 121]. In this line, COVID-19 goes along with massive infiltration of neutrophils into the lungs, including the formation of NETs as potential drivers of ARDS [122] and the associated immunothrombosis of patients with COVID-19 (Fig. 5) [59, 93].

Occlusion of small and intermediate-sized pulmonary vessels in COVID-19 published by Leppkes et al 2020 [59]. The former is marked by asterisks and the latter by a white frame. Note, that large fields of the (micro)-vasculature are occluded by NETs identified by extracellular neutrophil elastase (green). The nuclei of the cells were stained with propidium iodide (red). The bar represents 1000 µm. Original illustration from the authors.
NETs in kidneys and liver
NET formation contributes to numerous forms of acute and subacute kidney injury with proteinuria [123]. NOX-independent NETs directly induce kidney endothelial dysfunction, thereby offering a potential explanation for the proteinuria observed in most patients with COVID-19 [112]. NET-rich microvascular thrombi were also detected in the autopsy material of the kidneys in severe COVID-19 with renal failure [65]. It is still elusive whether involvement relates to the renal tropism of SARS-CoV-2 or to a systemic tendency for immunothrombosis and cytokine storm (Supplementary Fig. 4) [124, 125].
Liver injury emerges as a co-existing symptom in COVID-19 [126] and it might result from direct viral toxicity, but also from an overproduction of cytokines and/or NETs [127]. In patients with COVID-19 injured liver displays patchy necrosis alike in experimental models of the net-damaged liver [128, 129].
Targeting NETs in COVID-19 treatment
Glucocorticoids, hydroxychloroquine, and heparin in NET formation
COVID-19 patients frequently receive dexamethasone [130], heparin [131], and until recently also hydroxychloroquine, the latter emerged as highly controversial and not beneficial for the course of the disease [132,133,134]. The anti-inflammatory action of hydroxychloroquine relies on the inhibition of lysosomal activity and cytokine production. In vitro, neutrophils are more prone to release NETs when exposed to chloroquine [135]. The effect of hydroxychloroquine administered in vivo regarding NET formation has not been systematically studied yet [136,137,138]. In contrast, glucocorticoids including dexamethasone have been reported to reduce NET formation [139] most likely by suppressing the expression of inflammatory mediators that activate neutrophils. As mentioned above, activated neutrophils and platelets play key roles in thrombosis associated with severe COVID-19 [65]. Excessive NET formation harbors the risk of vascular occlusion [59], while heparin reduced NET formation in an experimental in vivo model of lung injury [140]. Heparin and low molecular weight heparins neutralize extracellular cytotoxic histones [141, 142], accelerate DNaseI-mediated degradation of NET mediated clots [59], and prevent NET aggregation by nano- and microparticles [143] in COVID-19. The therapeutic value of heparin in COVID-19 has been demonstrated recently [144], though some patients may develop heparin resistance [145].
Cytokine inhibitors and NET formation
Given the reduced incidence of COVID-19 in individuals treated with cytokine inhibitors [146] and the promising results with IL-6 and IL-1-blockade and immunosuppressants [130], inflammatory cytokines are likely important players in mediating inflammatory tissue damage in response to SARS-CoV-2. Stimulating results have been obtained with tocilizumab in a randomized clinical, double-blind, placebo-controlled, phase III study (preprint) [147] dampening the late IL-6-driven hyper-inflammatory phase [148] and reducing the need for mechanical and non-invasive ventilation. However, mortality after 28 days was not affected in another randomized fully peer-reviewed clinical trial [149]. The IL-1 receptor antagonist Anakinra is also currently under evaluation in RCTs, following early encouraging results in observational studies [150, 151]. Clinical studies with JAK-STAT inhibitors, also inhibiting IL-6, in addition, also IFNs, are still ongoing (NCT04320277). JAK inhibitors and direct blockade of IL-6 inhibit NET formation [152]. Previous studies in murine models have shown that the JAK inhibitor tofacitinib impairs NET formation in vitro and in vivo [153].
Complement-based therapies
Thrombogenic NETs elicited by the activated complement present in the blood of patients with COVID-19 [81] are candidates to amplify inflammation and thrombosis [154, 155]. Anti-complement strategies based on eculizumab or AMY-101 have successfully been used in small numbers of severe and critical/intubated patients [156,157,158,159,160]. Eculizumab is an anti-C5 humanized monoclonal antibody clinically approved for selected rare complement-mediated disorders. AMY-101 is clinically developed for various complement-mediated disorders and belongs to compstatin, a group of small-sized peptides that bind C3 and prevent its activation [160]. Early clinical data indicates that both inhibitors resulted in the resolution of SARS-CoV-2-associated ARDS; however, AMY-101 was associated with a more robust reduction in circulating neutrophils and NETs, highlighting the role of C3 in NET-driven thromboinflammation [160]. Several compassionate use programs or Phase II RCTs with complement inhibitors are in progress (NCT04346797, NCT04355494, NCT04288713, NCT04395456, EudraCT2020-004408-32).
Together with the complement components C1q-C4, C-reactive protein (CRP) functions in the disposal of bacteria and apoptotic or necrotic host cells [161, 162]. As COVID-19 is characterized by high CRP levels, it was proposed that reduction of the CRP levels by therapeutic apheresis, might reduce the pathological process in early disease [163]. Agarose bead-based CRP adsorption from the blood additionally depleted cell-free chromatin co-aggregates with C3 fragments [164]. The role of this approach in COVID-19 remains to be determined. In addition to CRP, calprotectin was identified as another acute-phase protein in severe pulmonary disease in COVID-19 [165].
Modulation of purinergic signaling
Injured cells release ATP that signals “danger” to neighboring tissues [166]. As a counterpoint, ectonucleotidases hydrolyze ATP to generate adenosine that supports local homeostasis. Activation of specific surface adenosine receptors suppresses NET formation via cyclic AMP-dependent signaling [167, 168]. Dipyridamole is an inexpensive, FDA-approved drug with a favorable safety profile. Dipyridamole potentiates adenosine receptor signaling by (i) inhibition of ectonucleoside reuptake, and (ii) stabilization of intracellular cyclic AMP. Dipyridamole tempers NET release in vitro while preventing NET-dependent thrombosis in mice [168]. In a small study, dipyridamole suppressed D-dimer levels in patients with COVID-19 [169]. Larger studies are required to evaluate clinical outcomes (NCT04391179) [170].
Treatment with DNases
Recombinant DNAse1 efficiently breaks down the chromatin of NETs that contributes to immunothrombosis and luminal obstructions of airways and vessels [59, 171]. NET-driven mucus accumulation, rigidity, and airway occlusion in severe COVID-19 may benefit from the same treatment [172, 173]. A small single-center case series (preprint) suggested that nebulized endotracheal DNAse1 (Dornase) reduced supplemental oxygen requirements in the patients [174]. COVIDornase (NCT04355364) and COVASE (NCT04359654) are two current initiatives that evaluated nebulized dornase α in prospective randomized controlled multicentre trials [175]. Since DNase1L3, which degrades extracellular DNA, works in a tandem with DNase1 to prevent immunothrombosis in an animal model of leukophilia [100], they are both candidates for the treatment of vascular occlusions in COVID-19. However, it is important to highlight that digestion of extracellular DNA with DNase1 and/or DNase1L3, while potentially reducing the occlusive capacity of aggregated NETs, may not successfully remove remnants that retain pro-inflammatory activities (Fig. 2) [176].
Other interventions to inhibit the NET formation
Treatment options targeting the pro-inflammatory action of NETs such as PADI4 inhibitors, or antibodies that block extrusion of NETs [46], or R406, a potent SYK inhibitor and the metabolically active component of fostamatinib [177], are potential new classes of drugs to tackle NET formation and to alleviate NET toxicity and in patients with severe COVID-19 [178]. In addition, the pharmacological inhibition of CatC to counterbalance the unwanted effects of neutrophil serine proteases in severe COVID-19 is considered a potential therapeutic target [179]. Some of these mediators are already in the development pipelines of pharmaceutical companies awaiting clinical trials. After the successful implementation of glucocorticoids [130], and the positive data on routine heparin use [180], future therapies will have to show how they perform compared to this standard of care. The studies of tocilizumab in severe COVID had heterogeneous outcomes [181, 182]. The patient cohort that had the most benefits from tocilizumab was mostly cotreated with glucocorticoids. This points to possible combination therapy as a future strategy. Combination therapy may also be useful for NET-targeted therapies: blocking new NET formation and improving degradation of preformed NETs. Given that NET degradation is negatively affected in severe COVID, the synergistic effect of DNase1 and heparin in NET degradation [59] may have identified these agents as suitable combination partners to effectively improve NET degradation in severe COVID. This needs to be proven by future clinical studies.
Conclusion
Here, we have highlighted the multifaceted functions that NETs play in the pathogenesis of COVID-19. The role of NETs in COVID-19 is increasingly supported by multiple lines of evidence and in fact, explains the wide range of manifestations seen in the most severe and critical cases. In the pathophysiology of COVID-19, there appears to be an important role for neutrophil dysregulation, oxidative stress, and aberrant NET formation as well as clearance. Neutrophils and NETs are at the crossroads of innate immune responses like pathogen killing, thrombosis, and activation of the adaptive immune system. This cardinal position helps to understand why a dysregulated neutrophil response upon SARS-COV-2 infection leads to such severe and uncontrolled disease manifestations. The pleiotropic complications caused by deposition of NETs in vessels and tissues in fact match disease manifestations in patients with COVID-19, and demand urgent actions to set trials with NET inhibitors. Identifying subgroups of individuals at risk for neutrophil dysregulation following SARS-CoV-2 exposure may help further refine individualized therapies. This strategy aims to prevent devastating complications including lung injury, kidney damage, endotheliitis, and immunothrombosis in severe COVID-19.
Search strategy and selection criteria
The data for this review were identified through searches of MEDLINE, PubMed, and references from relevant articles using the search terms “neutrophils” and “COVID-19” or “immunothrombosis” and “neutrophils”. Abstracts and reports from meetings were excluded. It uses 683 articles published in English from 2014 to April 2020. The publication date of additional articles was unlimited.
References
Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev. 2016;273:11–28.
Rubio-Ponce A, Hidalgo A, Ballesteros I. How to bridle a neutrophil. Curr Opin Immunol. 2021;68:41–47.
Hidalgo A, Chilvers ER, Summers C, Koenderman L. The neutrophil life cycle. Trends Immunol. 2019;40:584–97.
Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chevre R, AG N, et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell. 2013;153:1025–35.
Adrover JM, Aroca-Crevillen A, Crainiciuc G, Ostos F, Rojas-Vega Y, Rubio-Ponce A, et al. Programmed ‘disarming’ of the neutrophil proteome reduces the magnitude of inflammation. Nat Immunol. 2020;21:135–44.
Adrover JM, Del Fresno C, Crainiciuc G, Cuartero MI, Casanova-Acebes M, Weiss LA, et al. A neutrophil timer coordinates immune defense and vascular protection. Immunity. 2019;51:966–7.
Libby P, Luscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. 2020;41:3038–44.
Liu Y, Du X, Chen J, Jin Y, Peng L, Wang HHX, et al. Neutrophil-to-lymphocyte ratio as an independent risk factor for mortality in hospitalized patients with COVID-19. J Infect. 2020;81:e6–e12.
Evrard M, Kwok IWH, Chong SZ, Teng KWW, Becht E, Chen J, et al. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions. Immunity. 2018;48:364–79. e368
Muench DE, Olsson A, Ferchen K, Pham G, Serafin RA, Chutipongtanate S, et al. Mouse models of neutropenia reveal progenitor-stage-specific defects. Nature. 2020;582:109–14.
Kwok I, Becht E, Xia Y, Ng M, Teh YC, Tan L, et al. Combinatorial single-cell analyses of granulocyte-monocyte progenitor heterogeneity reveals an early uni-potent neutrophil progenitor. Immunity. 2020;53:303–18. e305
Xie X, Shi Q, Wu P, Zhang X, Kambara H, Su J, et al. Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat Immunol. 2020;21:1119–33.
Dinh HQ, Eggert T, Meyer MA, Zhu YP, Olingy CE, Llewellyn R, et al. Coexpression of CD71 and CD117 identifies an early unipotent neutrophil progenitor population in human bone marrow. Immunity. 2020;53:319–34. e316
Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol. 2019;19:255–65.
Schulte-Schrepping J, Reusch N, Paclik D, Bassler K, Schlickeiser S, Zhang B, et al. Severe COVID-19 Is marked by a dysregulated myeloid cell compartment. Cell. 2020;182:1419–40. e1423
Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martinez-Colon GJ, McKechnie JL, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med. 2020;26:1070–6.
Corley MJ, Pang APS, Dody K, Mudd PA, Patterson BK, Seethamraju H, et al. Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J Leukoc Biol. 2021. https://doi.org/10.1002/JLB.5HI0720-466R.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5.
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5:e1000639.
Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16:1438–44.
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22:146–53.
Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18:134–47.
Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci USA. 2015;112:2817–22.
Stojkov D, Amini P, Oberson K, Sokollik C, Duppenthaler A, Simon HU, et al. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J Cell Biol. 2017;216:4073–90.
Amini P, Stojkov D, Felser A, Jackson CB, Courage C, Schaller A, et al. Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat Commun. 2018;9:2958.
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–91.
Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207:1853–62.
Gosswein S, Lindemann A, Mahajan A, Maueroder C, Martini E, Patankar J, et al. Citrullination licenses calpain to decondense nuclei in neutrophil extracellular trap formation. Front Immunol. 2019;10:2481.
Diaz-Godinez C, Fonseca Z, Nequiz M, Laclette JP, Rosales C, Carrero JC. Entamoeba histolytica trophozoites induce a rapid non-classical NETosis mechanism independent of NOX2-derived reactive oxygen species and PAD4 activity. Front Cell Infect Microbiol. 2018;8:184.
Martinod K, Witsch T, Farley K, Gallant M, Remold-O’Donnell E, Wagner DD. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J Thromb Haemost. 2016;14:551–8.
Desai J, Kumar SV, Mulay SR, Konrad L, Romoli S, Schauer C, et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur J Immunol. 2016;46:223–9.
McGuire MJ, Lipsky PE, Thiele DL. Generation of active myeloid and lymphoid granule serine proteases requires processing by the granule thiol protease dipeptidyl peptidase I. J Biol Chem. 1993;268:2458–67.
Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3:eaar6689.
Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol. 2018;3:eaar6676.
Lin MH, Moses DC, Hsieh CH, Cheng SC, Chen YH, Sun CY, et al. Disulfiram can inhibit MERS and SARS coronavirus papain-like proteases via different modes. Antivir Res. 2018;150:155–63.
Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21:736–45.
Gupta S, Kaplan MJ. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol. 2016;12:402–13.
Rosa BA, Ahmed M, Singh DK, Choreno-Parra JA, Cole J, Jimenez-Alvarez LA, et al. IFN signaling and neutrophil degranulation transcriptional signatures are induced during SARS-CoV-2 infection. Commun Biol. 2021;4:290.
Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol. 2013;190:1217–26.
Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol. 2017;2:eaag3358.
Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis. 2015;74:1417–24.
von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–35.
Munoz LE, Boeltz S, Bilyy R, Schauer C, Mahajan A, Widulin N, et al. Neutrophil extracellular traps initiate gallstone formation. Immunity. 2019;51:443–50. e444
Leppkes M, Maueroder C, Hirth S, Nowecki S, Gunther C, Billmeier U, et al. Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis. Nat Commun. 2016;7:10973.
Carmona-Rivera C, Carlucci PM, Goel RR, James E, Brooks SR, Rims C, et al. Neutrophil extracellular traps mediate articular cartilage damage and enhance cartilage component immunogenicity in rheumatoid arthritis. JCI Insight. 2020;5:e139388.
Chirivi RGS, van Rosmalen JWG, van der Linden M, Euler M, Schmets G, Bogatkevich G, et al. Therapeutic ACPA inhibits NET formation: a potential therapy for neutrophil-mediated inflammatory diseases. Cell Mol Immunol. 2020. https://doi.org/10.1038/s41423-020-0381-3.
Euler M, Hoffmann MH. The double-edged role of neutrophil extracellular traps in inflammation. Biochem Soc Trans. 2019;47:1921–30.
Schauer C, Janko C, Munoz LE, Zhao Y, Kienhofer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. 2014;20:511–7.
Hahn J, Schauer C, Czegley C, Kling L, Petru L, Schmid B, et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019;33:1401–14.
Muraro SP, De Souza GF, Gallo SW, Da Silva BK, De Oliveira SD, Vinolo MAR, et al. Respiratory syncytial virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci Rep. 2018;8:14166.
Short KR, von Kockritz-Blickwede M, Langereis JD, Chew KY, Job ER, Armitage CW, et al. Antibodies mediate formation of neutrophil extracellular traps in the middle ear and facilitate secondary pneumococcal otitis media. Infect Immunol. 2014;82:364–70.
Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5:e138999.
Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. 2020;217:e20201129.
Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE, et al. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci USA. 2020;117:7326–37.
Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol. 2008;180:1895–902.
Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14:949–53.
Stefanelli VL, Choudhury S, Hu P, Liu Y, Schwenzer A, Yeh CR, et al. Citrullination of fibronectin alters integrin clustering and focal adhesion stability promoting stromal cell invasion. Matrix Biol. 2019;82:86–104.
Tilvawala R, Nguyen SH, Maurais AJ, Nemmara VV, Nagar M, Salinger AJ, et al. The rheumatoid arthritis-associated citrullinome. Cell Chem Biol. 2018;25:691–704. e696
Leppkes M, Knopf J, Naschberger E, Lindemann A, Singh J, Herrmann I, et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine. 2020;58:102925.
Ashar HK, Mueller NC, Rudd JM, Snider TA, Achanta M, Prasanthi M, et al. The role of extracellular histones in influenza virus pathogenesis. Am J Pathol. 2018;188:135–48.
Liang Y, Pan B, Alam HB, Deng Q, Wang Y, Chen E, et al. Inhibition of peptidylarginine deiminase alleviates LPS-induced pulmonary dysfunction and improves survival in a mouse model of lethal endotoxemia. Eur J Pharmcol. 2018;833:432–40.
Lange S, Rocha-Ferreira E, Thei L, Mawjee P, Bennett K, Thompson PR, et al. Peptidylarginine deiminases: novel drug targets for prevention of neuronal damage following hypoxic ischemic insult (HI) in neonates. J Neurochem. 2014;130:555–62.
Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136:1317–29.
Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pao CRR, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood. 2020;136:1330–41.
Nicolai L, Leunig A, Brambs S, Kaiser R, Weinberger T, Weigand M, et al. Immunothrombotic dysregulation in covid-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation. 2020;142:1176–89.
Taus F, Salvagno G, Cane S, Fava C, Mazzaferri F, Carrara E, et al. Platelets promote thromboinflammation in SARS-CoV-2 pneumonia. Arterioscler Thromb Vasc Biol. 2020;40:2975–89.
Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, et al. Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ Res. 2020:127:1404–18.
Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. 2014;346:1234–8.
Martinod K, Deppermann C. Immunothrombosis and thromboinflammation in host defense and disease. Platelets. 2021;32:314–24.
McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–6.
Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science. 2017;358:111–6.
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–9.
Jenne CN, Wong CH, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, et al. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe. 2013;13:169–80.
McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe. 2012;12:324–33.
Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12:2074–88.
Chrysanthopoulou A, Kambas K, Stakos D, Mitroulis I, Mitsios A, Vidali V, et al. Interferon lambda1/IL-29 and inorganic polyphosphate are novel regulators of neutrophil-driven thromboinflammation. J Pathol. 2017;243:111–22.
McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. 2017;129:1357–67.
Lou M, Yuan D, Liao S, Tong L, Li J. Potential mechanisms of cerebrovascular diseases in COVID-19 patients. J Neurovirol. 2021;27:35–51.
Lo MW, Kemper C, Woodruff TM. COVID-19: complement, coagulation, and collateral damage. J Immunol. 2020;205:1488–95.
Cugno M, Meroni PL, Gualtierotti R, Griffini S, Grovetti E, Torri A, et al. Complement activation in patients with COVID-19: a novel therapeutic target. J Allergy Clin Immunol. 2020;146:215–7.
Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Investig. 2020;130:6151–7.
Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1–13.
Paryzhak S, Dumych T, Mahorivska I, Boichuk M, Bila G, Peshkova S, et al. Neutrophil-released enzymes can influence composition of circulating immune complexes in multiple sclerosis. Autoimmunity. 2018;51:297–303.
Hu J, He C-L, Gao Q-Z, Zhang G-J, Cao X-X, Long Q-X, et al. D614G mutation of SARS-CoV-2 spike protein enhances viral infectivity. bioRxiv. 2020: 2020.2006.2020.161323.
Belouzard S, Madu I, Whittaker GR. Elastase-mediated activation of the severe acute respiratory syndrome coronavirus spike protein at discrete sites within the S2 domain. J Biol Chem. 2010;285:22758–63.
Bai X, Hippensteel J, Leavitt A, Maloney JP, Beckham D, Garcia C, et al. Hypothesis: alpha-1-antitrypsin is a promising treatment option for COVID-19. Med Hypotheses. 2021;146:110394.
Levi M, Thachil J, Iba T, Levy JH. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020;7:e438–e440.
Seitz R, Gurtler L, Schramm W. Thromboinflammation in COVID-19: can alpha2 -macroglobulin help to control the fire? J Thromb Haemost. 2021;19:351–4.
Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, et al. Extrapulmonary manifestations of COVID-19. Nat Med. 2020;26:1017–32.
Shahjouei S, Naderi S, Li J, Khan A, Chaudhary D, Farahmand G, et al. Risk of stroke in hospitalized SARS-CoV-2 infected patients: a multinational study. EBioMedicine. 2020;59:102939.
Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383:120–8.
Pramitasuri TI, Laksmidewi A, Putra IBK, Dalimartha FA. Neutrophil extracellular traps in coronavirus disease-19-associated ischemic stroke: a novel avenue in neuroscience. Exp Neurobiol. 2021;30:1–12.
Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136:1169–79.
Petito E, Falcinelli E, Paliani U, Cesari E, Vaudo G, Sebastiano M, et al. Association of neutrophil activation, more than platelet activation, with thrombotic complications in coronavirus disease 2019. J Infect Dis. 2021;223:933–44.
Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15:623–5.
Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. 2010;107:9813–8.
O’Sullivan KM, Holdsworth SR. Neutrophil extracellular traps: a potential therapeutic target in MPO-ANCA associated vasculitis? Front Immunol. 2021;12:635188.
Morris G, Bortolasci CC, Puri BK, Olive L, Marx W, O’Neil A, et al. Preventing the development of severe COVID-19 by modifying immunothrombosis. Life Sci. 2021;264:118617.
Winkler ES, Bailey AL, Kafai NM, Nair S, McCune BT, Yu J, et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat Immunol. 2020;21:1327–35.
Jimenez-Alcazar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science. 2017;358:1202–6.
Gustine JN, Jones D. Immunopathology of hyperinflammation in COVID-19. Am J Pathol. 2021;191:4–17.
Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180:934–43.
Hirsch JS, Ng JH, Ross DW, Sharma P, Shah HH, Barnett RL, et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020;98:209–18.
Dupont A, Rauch A, Staessens S, Moussa M, Rosa M, Corseaux D, et al. Vascular endothelial damage in the pathogenesis of organ injury in severe COVID-19. Arterioscler Thromb Vasc Biol. 2021: ATVBAHA120315595.
Monteil V, Kwon H, Prado P, Hagelkruys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181:905–13. e907
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–8.
Oudit GY, Crackower MA, Backx PH, Penninger JM. The role of ACE2 in cardiovascular physiology. Trends Cardiovasc Med. 2003;13:93–101.
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–8.
Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature. 2016;529:316–25.
Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18:1217–23.
Garsen M, Rops AL, Rabelink TJ, Berden JH, van der Vlag J. The role of heparanase and the endothelial glycocalyx in the development of proteinuria. Nephrol Dial Transpl. 2014;29:49–55.
Pieterse E, Rother N, Yanginlar C, Gerretsen J, Boeltz S, Munoz LE, et al. Cleaved N-terminal histone tails distinguish between NADPH oxidase (NOX)-dependent and NOX-independent pathways of neutrophil extracellular trap formation. Ann Rheum Dis. 2018;77:1790–8.
Pieterse E, Rother N, Garsen M, Hofstra JM, Satchell SC, Hoffmann M, et al. Neutrophil extracellular traps drive endothelial-to-mesenchymal transition. Arterioscler Thromb Vasc Biol. 2017;37:1371–9.
Dolhnikoff M, Duarte-Neto AN, de Almeida Monteiro RA, da Silva LFF, de Oliveira EP, Saldiva PHN, et al. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J Thromb Haemost. 2020;18:1517–9.
Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med. 2020;217:e20200652.
Barton LM, Duval EJ, Stroberg E, Ghosh S, Mukhopadhyay S. COVID-19 autopsies, Oklahoma, USA. Am J Clin Pathol. 2020;153:725–33.
Zhang H, Zhou P, Wei Y, Yue H, Wang Y, Hu M, et al. Histopathologic changes and SARS-CoV-2 immunostaining in the lung of a patient with COVID-19. Ann Intern Med. 2020;172:629–32.
Ouwendijk WJD, Raadsen MP, van Kampen JJA, Verdijk RM, von der Thusen JH, Guo L, et al. Neutrophil extracellular traps persist at high levels in the lower respiratory tract of critically ill COVID-19 patients. J Infect Dis. 2021;223:1503–15.
Cavalier E, Guiot J, Lechner K, Dutsch A, Eccleston M, Herzog M, et al. Circulating nucleosomes as potential markers to monitor COVID-19 disease progression. Front Mol Biosci. 2021;8:600881.
Silvin A, Chapuis N, Dunsmore G, Goubet AG, Dubuisson A, Derosa L, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell. 2020;182:1401–18. e1418
Becker K, Beythien G, de Buhr N, Stanelle-Bertram S, Tuku B, Kouassi NM, et al. Vasculitis and neutrophil extracellular traps in lungs of golden Syrian hamsters with SARS-CoV-2. Front Immunol. 2021;12:640842.
Wang J, Jiang M, Chen X, Montaner LJ. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J Leukoc Biol. 2020;108:17–41.
Nakazawa D, Marschner JA, Platen L, Anders HJ. Extracellular traps in kidney disease. Kidney Int. 2018;94:1087–98.
Puelles VG, Lutgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med. 2020;383:590–2.
Braun F, Lutgehetmann M, Pfefferle S, Wong MN, Carsten A, Lindenmeyer MT, et al. SARS-CoV-2 renal tropism associates with acute kidney injury. Lancet. 2020;396:597–8.
Schurink B, Roos E, Radonic T, Barbe E, Bouman CSC, de Boer HH, et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe. 2020;1:e290–e299.
Li D, Ding X, Xie M, Tian D, Xia L. COVID-19-associated liver injury: from bedside to bench. J Gastroenterol. 2021;56:218–30.
Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M, et al. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol. 2020;33:1007–14.
Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673.
Group RC, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, et al. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med. 2021;384:693–704.
Wang T, Chen R, Liu C, Liang W, Guan W, Tang R, et al. Attention should be paid to venous thromboembolism prophylaxis in the management of COVID-19. Lancet Haematol. 2020;7:e362–e363.
Boulware DR, Pullen MF, Bangdiwala AS, Pastick KA, Lofgren SM, Okafor EC, et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for Covid-19. N Engl J Med. 2020;383:517–25.
Geleris J, Sun Y, Platt J, Zucker J, Baldwin M, Hripcsak G, et al. Observational study of hydroxychloroquine in hospitalized patients with Covid-19. N Engl J Med. 2020;382:2411–8.
Arshad S, Kilgore P, Chaudhry ZS, Jacobsen G, Wang DD, Huitsing K, et al. Treatment with hydroxychloroquine, azithromycin, and combination in patients hospitalized with COVID-19. Int J Infect Dis. 2020;97:396–403.
Munoz LE, Bilyy R, Biermann MH, Kienhofer D, Maueroder C, Hahn J, et al. Nanoparticles size-dependently initiate self-limiting NETosis-driven inflammation. Proc Natl Acad Sci USA. 2016;113:E5856–E5865.
Smith CK, Vivekanandan-Giri A, Tang C, Knight JS, Mathew A, Padilla RL, et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: an additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. 2014;66:2532–44.
Germic N, Stojkov D, Oberson K, Yousefi S, Simon HU. Neither eosinophils nor neutrophils require ATG5-dependent autophagy for extracellular DNA trap formation. Immunology. 2017;152:517–25.
Murthy P, Singhi AD, Ross MA, Loughran P, Paragomi P, Papachristou GI, et al. Enhanced neutrophil extracellular trap formation in acute pancreatitis contributes to disease severity and is reduced by chloroquine. Front Immunol. 2019;10:28.
Vargas A, Boivin R, Cano P, Murcia Y, Bazin I, Lavoie JP. Neutrophil extracellular traps are downregulated by glucocorticosteroids in lungs in an equine model of asthma. Respir Res. 2017;18:207.
Sun Y, Chen C, Zhang X, Wang S, Zhu R, Zhou A, et al. Heparin improves alveolarization and vascular development in hyperoxia-induced bronchopulmonary dysplasia by inhibiting neutrophil extracellular traps. Biochem Biophys Res Commun. 2020;522:33–39.
Buijsers B, Yanginlar C, Maciej-Hulme ML, de Mast Q, van der Vlag J. Beneficial non-anticoagulant mechanisms underlying heparin treatment of COVID-19 patients. EBioMedicine. 2020;59:102969.
Niu C, Du Y, Kaltashov IA. Towards better understanding of the heparin role in NETosis: feasibility of using native mass spectrometry to monitor interactions of neutrophil elastase with heparin oligomers. Int J Mass Spectrom. 2021;463. https://doi.org/10.1016/j.ijms.2021.116550.
Bilyy R, Bila G, Vishchur O, Vovk V, Herrmann M. Neutrophils as main players of immune response towards nondegradable nanoparticles. Nanomaterials. 2020;10:1273.
Liu J, Li J, Arnold K, Pawlinski R, Key NS. Using heparin molecules to manage COVID-2019. Res Pract Thromb Haemost. 2020;4:518–23.
White D, MacDonald S, Bull T, Hayman M, de Monteverde-Robb R, Sapsford D, et al. Heparin resistance in COVID-19 patients in the intensive care unit. J Thromb Thrombolysis. 2020;50:287–91.
Simon D, Tascilar K, Kronke G, Kleyer A, Zaiss MM, Heppt F, et al. Patients with immune-mediated inflammatory diseases receiving cytokine inhibitors have low prevalence of SARS-CoV-2 seroconversion. Nat Commun. 2020;11:3774.
Salama C, Han J, Yau L, Reiss WG, Kramer B, Neidhart JD, et al. Tocilizumab in nonventilated patients hospitalized with Covid-19 pneumonia. medRxiv. 2020: 2020.2010.2021.20210203.
Quartuccio L, Sonaglia A, McGonagle D, Fabris M, Peghin M, Pecori D, et al. Profiling COVID-19 pneumonia progressing into the cytokine storm syndrome: results from a single Italian Centre study on tocilizumab versus standard of care. J Clin Virol. 2020;129:104444.
Hermine O, Mariette X, Tharaux PL, Resche-Rigon M, Porcher R, Ravaud P, et al. Effect of tocilizumab vs usual care in adults hospitalized with COVID-19 and moderate or severe pneumonia: a randomized clinical trial. JAMA Intern Med. 2021;181:32–40.
Iglesias-Julian E, Lopez-Veloso M, de-la-Torre-Ferrera N, Barraza-Vengoechea JC, Delgado-Lopez PD, Colazo-Burlato M, et al. High dose subcutaneous Anakinra to treat acute respiratory distress syndrome secondary to cytokine storm syndrome among severely ill COVID-19 patients. J Autoimmun. 2020;115:102537.
Huet T, Beaussier H, Voisin O, Jouveshomme S, Dauriat G, Lazareth I, et al. Anakinra for severe forms of COVID-19: a cohort study. Lancet Rheumatol. 2020;2:e393–e400.
Perez-Sanchez C, Ruiz-Limon P, Aguirre MA, Jimenez-Gomez Y, Arias-de la Rosa I, Abalos-Aguilera MC, et al. Diagnostic potential of NETosis-derived products for disease activity, atherosclerosis and therapeutic effectiveness in Rheumatoid Arthritis patients. J Autoimmun. 2017;82:31–40.
Furumoto Y, Smith CK, Blanco L, Zhao W, Brooks SR, Thacker SG, et al. Tofacitinib ameliorates murine lupus and its associated vascular dysfunction. Arthritis Rheumatol. 2017;69:148–60.
de Bont CM, Boelens WC, Pruijn GJM. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol. 2019;16:19–27.
Zuo Y, Kanthi Y, Knight JS, Kim AHJ. The interplay between neutrophils, complement, and microthrombi in COVID-19. Best Pr Res Clin Rheumatol. 2021;35:101661.
Diurno F, Numis FG, Porta G, Cirillo F, Maddaluno S, Ragozzino A, et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharm Sci. 2020;24:4040–7.
Laurence J, Mulvey JJ, Seshadri M, Racanelli A, Harp J, Schenck EJ, et al. Anti-complement C5 therapy with eculizumab in three cases of critical COVID-19. Clin Immunol. 2020;219:108555.
Giudice V, Pagliano P, Vatrella A, Masullo A, Poto S, Polverino BM, et al. Combination of ruxolitinib and eculizumab for treatment of severe SARS-CoV-2-related acute respiratory distress syndrome: a controlled study. Front Pharm. 2020;11:857.
Mastaglio S, Ruggeri A, Risitano AM, Angelillo P, Yancopoulou D, Mastellos DC, et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol. 2020;215:108450.
Mastellos DC, Pires da Silva BGP, Fonseca BAL, Fonseca NP, Auxiliadora-Martins M, Mastaglio S, et al. Complement C3 vs C5 inhibition in severe COVID-19: early clinical findings reveal differential biological efficacy. Clin Immunol. 2020;220:108598.
Ries W, Heigl F, Garlichs C, Sheriff A, Torzewski J. Selective C-reactive protein-apheresis in patients. Ther Apher Dial. 2019;23:570–4.
Sheriff A, Kayser S, Brunner P, Vogt B. C-reactive protein triggers cell death in ischemic cells. Front Immunol. 2021;12:630430.
Kayser S, Kunze R, Sheriff A. Selective C-reactive protein apheresis for Covid-19 patients suffering from organ damage. Ther Apher Dial. 2021:25:251–2.
Torzewski J, Heigl F, Zimmermann O, Wagner F, Schumann C, Hettich R, et al. First-in-man: case report of selective C-reactive protein apheresis in a patient with SARS-CoV-2 infection. Am J Case Rep. 2020;21:e925020.
Shi H, Zuo Y, Yalavarthi S, Gockman K, Zuo M, Madison JA, et al. Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J Leukoc Biol. 2021;109:67–72.
Yadav V, Chi L, Zhao R, Tourdot BE, Yalavarthi S, Jacobs BN, et al. Ectonucleotidase tri(di)phosphohydrolase-1 (ENTPD-1) disrupts inflammasome/interleukin 1beta-driven venous thrombosis. J Clin Investig. 2019;129:2872–7.
Carmona-Rivera C, Khaznadar SS, Shwin KW, Irizarry-Caro JA, O’Neil LJ, Liu Y, et al. Deficiency of adenosine deaminase 2 triggers adenosine-mediated NETosis and TNF production in patients with DADA2. Blood. 2019;134:395–406.
Ali RA, Gandhi AA, Meng H, Yalavarthi S, Vreede AP, Estes SK, et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat Commun. 2019;10:1916.
Liu X, Li Z, Liu S, Sun J, Chen Z, Jiang M, et al. Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta Pharm Sin B. 2020;10:1205–15.
Kanthi Y, Knight JS, Zuo Y, Pinsky DJ. New (re)purpose for an old drug: purinergic modulation may extinguish the COVID-19 thromboinflammatory firestorm. JCI Insight. 2020;5:e140971.
Yang C, Montgomery M. Dornase alfa for cystic fibrosis. Cochrane Database Syst Rev. 2018;9:CD001127.
Earhart AP, Holliday ZM, Hofmann HV, Schrum AG. Consideration of dornase alfa for the treatment of severe COVID-19 acute respiratory distress syndrome. N Microbes N Infect. 2020;35:100689.
Lee YY, Park HH, Park W, Kim H, Jang JG, Hong KS, et al. Long-acting nanoparticulate DNase-1 for effective suppression of SARS-CoV-2-mediated neutrophil activities and cytokine storm. Biomaterials. 2021;267:120389.
Weber AG, Chau AS, Egeblad M, Barnes BJ, Janowitz T. Nebulized in-line endotracheal dornase alfa and albuterol administered to mechanically ventilated COVID-19 patients: a case series. Mol Med. 2020;26:91.
Desilles JP, Gregoire C, Le Cossec C, Lambert J, Mophawe O, Losser MR, et al. Efficacy and safety of aerosolized intra-tracheal dornase alfa administration in patients with SARS-CoV-2-induced acute respiratory distress syndrome (ARDS): a structured summary of a study protocol for a randomised controlled trial. Trials. 2020;21:548.
Swaidani S, McCrae KR. “HIT”ing back against NETs. Blood. 2020;135:706–7.
Strich JR, Ramos-Benitez MJ, Randazzo D, Stein SR, Babyak A, Davey RT, et al. Fostamatinib inhibits neutrophils extracellular traps induced by COVID-19 patient plasma: a potential therapeutic. J Infect Dis. 2021;223:981–4.
Zhang Y, Han K, Du C, Li R, Liu J, Zeng H, et al. Carboxypeptidase B blocks ex vivo activation of the anaphylatoxin-neutrophil extracellular trap axis in neutrophils from COVID-19 patients. Crit Care. 2021;25:51.
Korkmaz B, Lesner A, Marchand-Adam S, Moss C, Jenne DE. Lung protection by cathepsin C inhibition: a new hope for COVID-19 and ARDS? J Med Chem. 2020;63:13258–65.
Nadkarni GN, Lala A, Bagiella E, Chang HL, Moreno PR, Pujadas E, et al. Anticoagulation, Bleeding, Mortality, and Pathology in Hospitalized Patients With COVID-19. J Am Coll Cardiol. 2020;76:1815–26.
Investigators R-C, Gordon AC, Mouncey PR, Al-Beidh F, Rowan KM, Nichol AD, et al. Interleukin-6 receptor antagonists in critically Ill patients with Covid-19. N Engl J Med. 2021;384:1491–1502.
Rosas IO, Brau N, Waters M, Go RC, Hunter BD, Bhagani S, et al. Tocilizumab in hospitalized patients with severe Covid-19 pneumonia. N Engl J Med. 2021;384:1503–16.
Funding
This work was supported by the Swiss National Science Foundation (grant numbers 310030_184816 to H-US and 31003A_173215 to SY); by the CIHR Foundation to PK; by a Radboudumc Ph.D. fellowship to CY; by the National Science Center, Poland (NCN) grant no. 2018/29/B/NZ6/00713/ to EK; by the Pershing Square Foundation; by the Singapore Immunology Network (SIgN) core funding, A*STAR, Singapore to LGN; by the Greek General Secretariat for Research and Technology (GSRT), EYDE-ETAK Research & Innovation Programme CYTONET, Grant no: Τ1ΕDK-00617 to PS, IM, and KR; by the Intramural Research Program at NIAMS/NIH to MJK; by the H2020-FETOPEN 861878 NeutroCure to M Herrmann and M Hoffmann; by the Intramural Research Program of the NIH and NHLBI, Lasker Foundation, NIH (K08HL131993, R01HL150392), A. Alfred Taubman Medical Research Institute, Falk Medical Research Trust Catalyst Award, and the JOBST-American Venous Forum Award to YK; Methusalem (BOF16/MET_V/007) to PV; by the grants of the Ministry of Healthcare of Ukraine 0119U101338 and of National Research Foundation of Ukraine 2020.02/0131 to RB; by the grant RTI2018-095497-B-I00 to AH from Ministerio de Ciencia e Innovación (MICINN). the CNIC is supported by MICINN and the Pro-CNIC Foundation and is a Severo Ochoa Center of Excellence (MICINN award SEV-2015-0505) to AH; by the Ministerium für Wissenschaft und Kultur, Lower Saxony, Germany (14-76103-184 CORONA-15/20) to MvK-B; by the COVID-2020-12371617 grant from the Italian Ministry of Health to AAM, PR-Q, and NM.; by the the intramural support through the CORNET Collaborative Award of 2020 to MR and IN; by the Programm zur Förderung von Corona-Forschungsprojekten, StMWK, München to M Herrmann, ML, and M Stürzl; by the VW foundation (grant 97744) to M Herrmann, HJA and RB; and the Pershing Square Foundation to ME. We would further like to acknowledge the support of the German Research Foundation (DFG) FOR 2438 subproject 2 to and M Stürzl, SFB/TRR241 subproject A06 to M Stürzl AN372/14-3, and 24-1 to HJA; SCHA 2040/1-1 to CS; project number 387509280 and SFB 1350 to CD; MA 7770/1-1 to CM; SFB 914, SFB1123 and the DZHK to K.S.; TRR241: B04 to ML and M Herrmann; CRC1181: C03 to M Herrmann and M Hoffmann; IZKF and ELAN of FAU to CS; IZKF Münster Za2/001/18, KFO 342, ZA428/18-1, INST 211/984-1 and ZA428/20-1 to AZ, and the EU (ERC-Synergy grant 4D Nanoscope) LM and GS. None of the above-mentioned funders had any role in the paper design, data collection, data analysis, interpretation, or writing of the paper. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
All authors contributed equally in the design of the figures, edition, and proofreading of the manuscript. The literature search and writing of the abstract were performed by LEM, M Herrmann, and GS. The literature search and writing of the section “Basic aspects of neutrophil biology and their relevance for COVID-19” were performed by AH, MJK, HS, EK, LGN, AM, CM, and SY. The literature search and writing of the section “Mechanisms of neutrophil extracellular trap formation” were performed by AH, MJK, HS, EK, LGN, AM, CS, and SY. The literature search and writing of the section “Pro- and anti-inflammatory functions of neutrophil extracellular trap” were performed by MJK, M Hoffmann, and CS. The literature search and writing of the section “Viral infections induce the formation of neutrophil extracellular trap” was performed by MvK-B. The literature search and writing of the section “The role of neutrophil extracellular trap-bound enzymes” were performed by MJK, RB, JK, and ML. The literature search and writing of the section “The role of citrullination in neutrophil extracellular traps” were performed by MR, GLB, IN, JK, ML, and TN. The literature search and writing of the section “Neutrophil extracellular trap-induced thromboinflammation in COVID-19” were performed by ME, ML, M Herrmann, BH, KR, EK, IM, HJA, PS, CM, and KS. The literature search and writing of the section “Neutrophil extracellular trap induced endothelial activation and damage in COVID-19” were performed by JV, MJK, BH, MA, EN, and M Stürzl. The literature search and writing of the section “The relation of platelet activation with neutrophil extracellular traps” were performed by AZ, AAM, EK, NM, KS, and PK. The literature search and writing of the section “Complement activation as a trigger for neutrophil extracellular traps” were performed by AAM, PR-Q, KR, LGN, LV, and MS. The literature search and writing of the section “NETs in COVID-19-associated acute lung disease” were performed by AH, MR, TN, GLB, MvK, and KR. The literature search and writing of the section “NETs in kidneys and liver” were performed by EK, HJA, JV, and CD. The literature search and writing of the section “Targeting NETs in COVID-19 treatment-Glucocorticoids, hydroxychloroquine, and heparin in NET formation” were performed by ME, ML, and M Herrmann. The literature search and writing of the section “Targeting NETs in COVID-19 treatment-Cytokine inhibitors and NET formation” were performed by RdL, TH, JS, PV, and CY. The literature search and writing of the section “Targeting NETs in COVID-19 treatment-Complement-based therapies” were performed by KR, IM, PS, AAM, PR-Q, and M Herrmann. The literature search and writing of the section “Targeting NETs in COVID-19 treatment–modulation of purinergic signaling” were performed by JSK and YK. The literature search and writing of the section “Targeting NETs in COVID-19 treatment-treatment with DNase” were performed by HJA, EK, HS, JV, RB, SY, and ML. The literature search and writing of the section “Other interventions to inhibit NET formation” were performed by LEM, GS, and M Herrmann. The writing of the section “Outstanding questions” was performed by LEM, M Herrmann, and CS.
Corresponding author
Ethics declarations
Conflict of interest
MA, RB, GLB, CD, RdL, TH, AH, M Hoffmann, BH, MJK, JSK, JK, EK, PK, ML, AM, AAM, CM, NM, IM, LEM, TN, EN, IN, LGN, MZR, KR, PR-Q, M Schapher, CS, GS, H-US, JS, PS, KS, M Stürzl, PV, JvV, LV, MvK-B, CY, SY, and AZ have nothing to disclose. H-JA reports personal fees from AstraZeneca, personal fees from Boehringer, personal fees from Previpharma, personal fees from Secarna, personal fees from Inositec, personal fees from Novartis, personal fees from Bayer, personal fees from GSK, outside the submitted work. ME reports personal fees from CytoxM, personal fees from MPM Capital, non-financial support from Santhera, outside the submitted work. M Herrmann reports non-financial support from Neutrolis outside the submitted work; YK reports personal fees from Surface oncology, personal fees from Acer Therapeutics, outside the submitted work.
Ethics
The use of human material in some illustrations was in accordance with international and institutional guidelines and with the clearance of the Ethical Committees of each participating institution.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Melino
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ackermann, M., Anders, HJ., Bilyy, R. et al. Patients with COVID-19: in the dark-NETs of neutrophils. Cell Death Differ 28, 3125–3139 (2021). https://doi.org/10.1038/s41418-021-00805-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-021-00805-z
This article is cited by
-
Enhancement of NETosis by ACE2-cross-reactive anti-SARS-CoV-2 RBD antibodies in patients with COVID-19
Journal of Biomedical Science (2024)
-
What is the actual relationship between neutrophil extracellular traps and COVID-19 severity? A longitudinal study
Respiratory Research (2024)
-
The role of vasculature and angiogenesis in respiratory diseases
Angiogenesis (2024)
-
A review of different deep learning methods in processing the CT scan images of the COVID-19 patients’ lungs
Multiscale and Multidisciplinary Modeling, Experiments and Design (2024)
-
Risk factors for severe COVID-19 infection and the impact of COVID-19 infection on disease progression among patients with AAV
Clinical and Experimental Medicine (2024)
