
Abstract
Hypoxic-ischemic encephalopathy (HIE) is a main cause of mortality and severe neurologic impairment in the perinatal and neonatal period. However, few satisfactory therapeutic strategies are available. Here, we reported that a rapid nuclear translocation of phosphatase and tensin homolog deleted on chromosome TEN (PTEN) is an essential step in hypoxic-ischemic brain damage (HIBD)- and oxygen-glucose deprivation (OGD)-induced neuronal injures both in vivo and in vitro. In addition, we found that OGD-induced nuclear translocation of PTEN is dependent on PTEN mono-ubiquitination at the lysine 13 residue (K13) that is mediated by neural precursor cell expressed developmentally downregulated protein 4-1 (NEDD4-1). Importantly, we for the first time identified α- and γ-adaptin binding protein (Aagab) as a novel NEDD4-1 regulator to regulate the level of NEDD4-1, subsequently mediating Pten nuclear translocation. Finally, we demonstrated that genetic upregulation of Aagab or application of Tat-K13 peptide (a short interference peptide that flanks K13 residue of PTEN) not only reduced Pten nuclear translocation, but also significantly alleviated the deficits of myodynamia, motor and spatial learning and memory in HIBD model rats. These results suggest that Aagab may serve as a regulator of NEDD4-1-mediated Pten nuclear translocation to promote functional recovery following HIBD in neonatal rats, and provide a new potential therapeutic target to guide the clinical treatment for HIE.
Similar content being viewed by others


Introduction
Neonatal hypoxic-ischemic encephalopathy (HIE) caused by perinatal asphyxia, is one of the leading causes of mortality and neurological disability in infants. The incidence of HIE is about 1 to 3 per 1,000 term births [1, 2], and the survivor exhibits motor disability or a variety of serious neurological sequela, including cerebral palsy, severe learning impairment or intellectual deficiency [3, 4]. A large number of studies have demonstrated that the death of neurons following hypoxia-ischemia (HI) is caused, at least in part, by over-activation of N-methyl-D-aspartate receptors (NMDARs) [5,6,7,8,9,10]. However, the antagonists of NMDAR for clinical trials have thus far been unsuccessful after hypoxic-ischemic brain damage (HIBD), because these antagonists have the short therapeutic time-window and are able to block the normal functions of brain such as neuronal survival and learning and memory [11,12,13], when they exert neuroprotective roles after HIBD. Therefore, the research for a new potential therapeutic approach to guide the clinical treatment for HIE is particularly important.
Phosphatase and tensin homolog deleted on chromosome TEN (PTEN), the first discovered dual lipid/protein phosphatase, was previously identified as a candidate tumor suppressor gene [14,15,16,17], which plays a crucial role in cell growth, proliferation, survival, and death [18,19,20], by directly regulating phosphatidylinositol 3-kinase (PI3K) [21, 22]. More recent studies have suggested that PTEN is implicated in neuronal damage after HI [23,24,25]. Further studies from cancer have shown that point mutant of PTEN such as K13E and K289E, results in failure of PTEN nuclear translocation [26,27,28], thereby reducing the function of PTEN’s tumor suppression and cell apoptosis, suggesting that nuclear translocation of PTEN may be a crucial step for PTEN-mediated cell death during HI. Consistently, our recent study has found that increased Pten nuclear accumulation results in neuronal death in in vitro and in vivo models of excitotoxic/ischemic neuronal injuries, and prevention of Pten nuclear translocation by mutation of K13 or a short interfering peptide (Tat-K13) reduced the neuronal death induced by excitotoxicity/ischemia [29]. However, most of these researches have focused on mature brain injury, and much less is known about the effects of PTEN on the developing brain although two recent studies report that inhibition of PTEN is able to attenuate neuronal injury after HI [30, 31]. Moreover, the underlying mechanisms remain poorly characterized.
A growing body of evidence has shown that the enhanced nuclear PTEN accumulation is associated with increased PTEN mono-ubiquitination, particularly at K13 and K289 residues [26, 32,33,34]. Mono-ubiquitination enhances PTEN protein stability, whereas poly-ubiquitination results in PTEN degradation by proteasome pathway [35, 36]. Further studies have shown that neural precursor cell expressed developmentally downregulated protein 4 (NEDD4) family E3 ligases, NEDD4-1 and NEDD4-2 play critical roles in mediating PTEN nuclear translocation by PTEN mono-ubiquitination, and in mediating PTEN degradation by PTEN poly-ubiquitination [26, 33, 37]. However, whether NEDD4 is involved in and what mechanism mediates PTEN mono-ubiquitination and increases PTEN nuclear import in developing brain after HIBD remains poorly understood. Recent reports have revealed that Rab5 GTPase, which plays an important role in the endosomal trafficking pathways and the transport of proteins to subcellular locations [38,39,40,41], is able to mediate the ubiquitination of PTEN and the nuclear efficient translocation of ubiquitinated PTEN [42]. Since α- and γ-adaptin binding protein (Aagab), a cytosolic protein with a Rab-like GTPase domain, has also been shown to display a role in membrane traffic [43], it is reasoned that Aagab may regulate nuclear translocation of Pten, and regulating Aagab or inhibiting Pten nuclear translocation may alleviate neuronal damage after HIBD. Here, we tested this speculation using several well characterized in vitro and in vivo models of hypoxic-ischemic neuronal injuries.
Results
HI increases Pten nuclear translocation
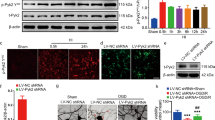
To determine whether HI results in an increase in Pten nuclear translocation, the brain tissues from rats subjected to HIBD were collected for preparation of cytoplasm/nuclei fractionations. The results showed that the nuclear Pten was increased in a time-dependent manner, peaking at 6 h after HI, and returning to control level within 24 h following HIBD (n = 5; Fig. 1A, C). Notably, there was no significant difference in the total level of Pten among these groups (n = 5; Fig. 1A, B). To determine the nuclear translocation of Pten directly in a cellular context, we next introduced primarily cultured neurons treated with OGD. The results from immunofluorescence assay showed that Pten was predominantly observed in the cytoplasm of neurons under normal circumstance, whereas the subcellular localization of Pten was increased in a time-dependent manner from the cytoplasm to the nucleus after OGD (Fig. 1D). Further western blot assay confirmed that nuclear Pten displayed an inverted-U curve and peaked at 6–9 h after OGD, and returned to control (CTR) levels within 24 h (n = 5; Fig. 1E, G). While no significant difference of total Pten levels was observed among these groups (n = 5; Fig. 1E, F). These results indicate that Pten nuclear translocation displays a time-dependent enhancement in HIBD rats and cultured neurons subjected to OGD treatment.

A–C The protein levels of cytoplasmic and nuclear Pten assessed by western blot in the brain tissues from HIBD rats. A The nuclear Pten protein levels increased in a time-dependent manner, but the total Pten protein levels among these groups were not changed in HIBD rats. B, C Bar graphs summarized the relative levels of total and nuclear Pten. D Immunofluorescence figure of Pten nuclear translocation induced by OGD at different time point in primarily cultured neurons. E–G The protein levels of cytoplasmic and nuclear Pten assessed by western blot in primarily cultured neurons after OGD. E The nuclear Pten levels increased in a time-dependent manner, but the global Pten protein levels were not changed in primarily cultured neurons after OGD. F, G Bar graphs summarized the relative levels of total and nuclear Pten. Data were expressed as mean ± SEM, **p < 0.01.
PTEN mono-ubiquitination at K13 residue enhances PTEN nuclear translocation
Previous findings from cancer research have indicated that the enhanced nuclear PTEN accumulation is associated with increased PTEN mono-ubiquitination [32], particularly at K13 and K289 residues [26, 33]. Here we wanted to evaluate whether Pten mono-ubiquitination contributed to Pten nuclear translocation after OGD in neurons. The results showed that mono-ubiquitinated PTEN increased in HEK293T cells and cultured neurons that were subjected to OGD, compared with CTR (Fig. 2A–C). Since our recent study has shown that only the K13 residue, but not the K289 residue, appears critically important for Pten nuclear accumulation in excitotoxic and ischemic neuronal injuries [16], we next examined whether the increase of Pten nuclear translocation induced by OGD was attributed to Pten mono-ubiquitination at K13 site. We transfected HEK293T cells with wild-type (PTENWT) or point mutant PTEN plasmid (PTENK13R or PTENK289R). As shown in Fig. 2D, nuclear PTEN significantly was decreased after OGD treatment in cells transfected with PTENK13R, but not PTENK289R, compared with PTENWT (n = 5; Fig. 2D). To further confirm this finding in neurons, we infected cultured neurons with lentivirus carrying wide-type (LVPTENWT) or mutant PTEN (LVPTENK13R or LVPTENK289R). Consistent with the findings in HEK293T cells, the level of nuclear Pten was also significantly decreased in neurons infected with LVPTENK13R, compared with that in LVPTENWT-infected cells (n = 5; Fig. 2E). Notably, the decrease of nuclear PTEN in HEK293T cells transfected with PTENK13R or neurons infected with LVPTENK13R was not attributed to different transfection rate or protein loading amount since the total PTEN displayed no difference among these groups (Fig. S1A, B). Furthermore, mono-ubiquitinated PTEN was reduced after OGD treatment in the case of K13R mutation, but not the case of K289R mutation (Fig. 2F–H). Thus, these results further confirm that OGD-induced Pten mono-ubiquitination leads to Pten nuclear import, and the K13 residue (but not the K289 residue) is critically important for OGD-induced Pten nuclear translocation.

PTEN mono-ubiquitination occurred in both HEK 293T cells (A) and primarily cultured neurons (B, C) after OGD treatment. D HEK293T cells transfected with wild-type (WT), K13R mutation or K289R mutation plasmid of PTEN for 24 h, were subjected to OGD for 1.5 h and harvested 6 h after hypoxia onset. PTEN was localized to the nucleus in WT or K289R plasmid transfection, while treatment with K13R plasmid inhibited the PTEN nuclear entry. E Primarily cultured neurons infected with lentivirus carrying WT, K13R or K289R of PTEN gene for 4 days, were subjected to OGD for 1.5 h and collected at 6 h after hypoxia onset. Nuclear Pten was decreased in K13R group, but not K289R group, compared with WT group. F PTEN mono-ubiquitination was decreased in K13R, but not K289R plasmid-transfected HEK293T cells, compared with WT transfection. G, H Pten mono-ubiquitination was decreased in primarily cultured neurons infected with lentivirus carrying K13R, but not K289R compared with WT. Data were expressed as mean ± SEM, **p < 0.01.
Over-expression of NEDD4-1 suppresses OGD-induced Pten nuclear translocation
Given previous reports that the NEDD4-family E3 ubiquitin ligases NEDD4-1 and NEDD4-2 may ubiquitinate PTEN and thereby regulate PTEN trafficking [26, 33, 37, 44], we next examined whether NEDD4-1 or NEDD4-2 had an effect on HIBD-induced Pten nuclear translocation. The results showed that NEDD4-1 expression was significantly decreased at 6–12 h (n = 5; Fig. 3A, B), while NEDD4-2 expression was significantly increased at 3–12 h (n = 6; Fig. 3A, C), and both of them returned to control levels within 24 h in rats subjected to HIBD. Similar to the findings in vivo, the protein level of NEDD4-1 was significantly decreased at 3–9 h (n = 5; Fig. 3D, E), but NEDD4-2 was significantly increased at 6–9 h (n = 4; Fig. 3D, F), and both of them returned to control levels within 12 h in primarily cultured neurons after OGD.

A–C Total protein levels of NEDD4-1 and NEDD4-2 assessed by western blot in the brain tissues from HIBD rats. A, B The expression of NEDD4-1 protein was decreased at 6–12 h, and returned to sham levels within 24 h after HIBD. A, C While NEDD4-2 protein expression was increased at 3–12 h, and returned to sham levels within 24 h after HIBD. D–F Total protein levels of NEDD4-1 and NEDD4-2 assessed by western blot in primarily cultured neurons subjected to OGD. D, E NEDD4-1 protein expression was decreased 3–9 h, and returned to control levels within 12 h after OGD. D, F While NEDD4-2 protein expression was increased at 6–9 h, and returned to control levels within 12 h after OGD. G–I The protein level of nuclear Pten assessed by western blot in primarily cultured neurons over-expressed or downregulated NEDD4-1 before OGD. G, H LVNEDD4-1 succeeded in rescuing the decrease of OGD-induced NEDD4-1 expression. G, I NEDD4-1 over-expression before OGD blocked Pten nuclear import induced by OGD. J–L The protein level of nuclear Pten assessed by western blot in primarily cultured neurons over-expressed or downregulated NEDD4-2 before OGD. J, K LVshNEDD4-2 succeeded in preventing the increase of OGD-induced NEDD4-2 expression. J, L Either knockdown or over-expression of NEDD4-2 before OGD failed to block Pten nuclear import induced by OGD. M–O The protein level of nuclear Pten assessed by western blot in primarily cultured neurons over-expressed or downregulated NEDD4-1. M, N LVshNEDD4-1 succeeded in downregulating NEDD4-1 expression in primarily cultured neurons. M, O Downregulation of NEDD4-1 expression failed to induce Pten nuclear translocation. P–R The protein level of nuclear Pten assessed by western blot in primarily cultured neurons over-expressed or downregulated NEDD4-2. P, Q LVNEDD4-2 succeeded in over-expressing NEDD4-2 in primarily cultured neurons. P, R Over-expression of NEDD4-2 failed to induce Pten nuclear translocation. Data were expressed as mean ± SEM, *p < 0.05, **p < 0.01.
To determine the role of NEDD4-1 and NEDD4-2 in HIBD-induced Pten nuclear localization, we infected cultured neurons with lentivirus to overexpress (LVNEDD4-1 and LVNEDD4-2) or knockdown (LVshNEDD4-1 and LVshNEDD4-2) NEDD4-1 and NEDD4-2. LVNEDD4-1 and LVNEDD4-2 infection succeeded in expressing exogenous NEDD4-1 and NEDD4-2 in cultured neurons (Fig. S2A, B), while LVshNEDD4-1 and LVshNEDD4-2 infection effectively knocked down endogenous NEDD4-1 and NEDD4-2 (Fig. S2C, D). Importantly, NEDD4-1 over-expression rather than knockdown (n = 6; Fig. 3G, H) before OGD treatment, significantly blocked OGD-induced Pten nuclear import in cultured neurons (n = 6; Fig. 3G, I). However, neither over-expression nor knockdown of NEDD4-2 (n = 4; Fig. 3J, K) affected OGD-induced Pten nuclear import (n = 4; Fig. 3J, L). To further clarify the different roles of NEDD4-1 and NEDD4-2 in OGD-induced Pten nuclear translocation, we directly examined the interaction between PTEN and NEDD4-1 or NEDD4-2. Results from co-IP assays showed that both NEDD4-1 and PTEN can pull down each other in HEK293T cells and primarily cultured neurons, while NEDD4-2 and PTEN cannot mutually co-precipitate (Fig. S3A-D). These data may explain why NEDD4-2 has no appreciate effect on PTEN nuclear import after OGD treatment.
Next, to determine whether NEDD4-1 or NEDD4-2 enabled to regulate Pten nuclear translocation without OGD treatment, we assayed the level of nuclear Pten in cultured neurons infected with lentivirus. We found that either over-expression or downregulation of NEDD4-1 (n = 5; Fig. 3M, N), did not affect Pten nuclear translocation (n = 5; Fig. 3M, O). Similarly, regulation of NEDD4-2 expression (n = 3; Fig. 3P, Q) had no effect on Pten nuclear translocation (n = 4; Fig. 3P, R). Together, these results demonstrate that over-expression of NEDD4-1 is able to suppress OGD-induced Pten nuclear translocation, but NEDD4-1 knockdown is insufficient to induce Pten nuclear import in primarily cultured neurons.
Aagab regulates NEDD4-1-mediated Pten nuclear translocation
Since Rab5 GTPase plays an important role in ubiquitinated PTEN nuclear translocation and Aagab holds on a Rab-like GTPase domain to mediate membrane traffic [38, 42, 43], we next wanted to determine whether Aagab was involved in NEDD4-1-mediated Pten nuclear translocation in HIBD model. We found that the protein level of Aagab was decreased in a time-dependent manner in the brain tissues of HIBD rats (n = 4; Fig. 4A) and in cultured neurons subjected to OGD (n = 4; Fig. 4B). To determine the role of Aagab in OGD-induced Pten nuclear translocation, lentivirus carrying Aagab gene (LVAagab) or Aagab small hairpin RNA (LVshAagab) was used to treat primarily cultured neurons. LVshAagab infection efficiently reduced Aagab expression, whereas LVAagab infection successfully expressed exogenously Aagab (Fig. S4A, B). Interestingly, infection with LVAagab, but not LVmNeonGreen, before OGD treatment significantly blocked Pten nuclear import in primarily cultured neurons (n = 5; Fig. 4C–E). To further determine whether Aagab was sufficient to regulate Pten nuclear translocation, we assayed the level of nuclear Pten in cultured neurons infected with lentivirus, and found that Pten nuclear translocation displayed a significant increase in cultured neurons infected with LVshAagab, but not LVAagab, compared with control lentivirus infection (n = 4; Fig. 4F–H).

A Aagab expression was decreased at 3–6 h, and returned to sham levels within 12 h in the brain tissues from HIBD rats. B Aagab expression was decreased at 3–9 h, and returned to control levels within 12 h after OGD. C–E The protein level of nuclear Pten assessed by western blot in primarily cultured neurons over-expressed Aagab before OGD. C, D LVAagab succeeded in rescuing the decrease of OGD-induced Aagab expression. C, E Aagab over-expression before OGD blocked Pten nuclear import induced by OGD. F–H The protein level of nuclear Pten assessed by western blot in primarily cultured neurons downregulated Aagab. F, G LVshAagab succeeded in downregulating Aagab expression in primarily cultured neurons. F, H Downregulation of Aagab expression enabled to induce Pten nuclear translocation. I Neither over-expression nor knockdown NEDD4-1 affected Aagab expression in primarily cultured neurons. J Downregulation of Aagab reduced NEDD4-1 expression in primarily cultured neurons. K Over-expression of NEDD4-1 partially rescued Aagab knockdown-induced Pten nuclear translocation in primarily cultured neurons. Data were expressed as mean ± SEM, *p < 0.05, **p < 0.01.
We next determined whether Aagab-regulated Pten nuclear localization was dependent on NEDD4-1. The results showed that knockdown (LVshNEDD4-1) or over-expression (LVNEDD4-1) of NEDD4-1 had no effects on Aagab expression (Fig. S5). However, infection with LVshAagab leaded to a significant decrease in NEDD4-1 (n = 5; Fig. 4I, J). More importantly, the increase of Pten nuclear translocation induced by Aagab knockdown can be partially suppressed by NEDD4-1 over-expression (n = 5; Fig. 4K). To further clarify the role of Aagab in NEDD4-1-mediated Pten nuclear translocation, we examined the interaction between Aagab and NEDD4-1. Results from co-IP assays showed that both Aagab and NEDD4-1 can pull down each other in primarily cultured neurons (Fig. 5A). We further found that the poly-ubiquitination of Aagab (Fig. 5B, C) and NEDD4-1 (Fig. 5D, E) were increased in primarily cultured neurons subjected to OGD. Taken together, these results indicate that OGD induces Aagab downregulation, and subsequently increases NEDD4-1-mediated Pten nuclear translocation.

A Co-IP assays showed that Aagab interacted with NEDD4-1 in primarily cultured neurons. B, C Aagab poly-ubiquitination was increased in primarily cultured neurons after OGD treatment. D, E NEDD4-1 poly-ubiquitination was increased in primarily cultured neurons after OGD treatment.
Upregulation of Aagab ameliorates behavioral defects in HIBD model rats
Since our recent study has found that inhibition of Pten nuclear translocation could promote behavioral recovery in rat stroke model [29], and our aforementioned results (Fig. 4) showed that over-expression of Aagab before OGD treatment significantly blocked Pten nuclear import in primarily cultured neurons, we next wanted to determine whether upregulation of Aagab can reverse HIBD-induced behavioral defects. Newborn rats were infected with LVAagab or LVshAagab, and grip strength and Morris water maze tests were introduced to evaluate myodynamia and cognitive functions in HIBD model rats (Fig. 6A).

A Diagram illustrating the experimental protocols for HIBD model establishment and lentivirus injection as well as behavioral tests. B The left and right forelimb myodynamia during the grip strength test. C–F Upregulation of Aagab ameliorates the deficit of spatial learning and memory in HIBD model rats in the Morris water maze paradigm. C The escape latency to the hidden platform during spatial learning period. The number of entries into the platform zone (D), the escape latency of the first entry into the platform zone (E), and time spent in the hidden platform-located quadrant (F) during the probe test with absence of the hidden platform. Data were expressed as mean ± SEM, *p < 0.05, **p < 0.01.
Grip strength test revealed that grip strength of the right forelimb was significantly decreased compared to that of the left in HIBD rats (sham: n = 14; HIBD: n = 10; Fig. 6B), indicating a significant impairment of myodynamia. As expected, rats infected with LVAagab (HIBD + LVAagab: n = 12), but not its control virus LVmNeonGreen (HIBD + LVmNeonGreen: n = 16; Fig. 6B), exhibited significant recovery in grip strength of the right forelimb. In addition, downregulation of Aagab by LVshAagab infection dramatically decreased the grip strength of the right forelimb compared with control virus LVshRNA infection in sham rats (sham + LVshRNA: n = 16; sham + LVshAagab: n = 15; Fig. 6B).
Since the impairment of learning and memory is a major sequelae of HIBD both in human and in a variety of animal models [45, 46], we next evaluated the influence of Aagab upregulation on spatial learning and memory in HIBD rats. The results showed that the escape latency for searching for the hidden platform in rats subjected to HIBD was much longer than that in the sham group during spatial training period (sham: n = 15; HIBD: n = 9; Fig. 6C). While upregulation of Aagab significantly ameliorated the spatial learning impairment, as reflected by a decrease in the escape latency (HIBD + LVAagab: n = 9; Fig. 6C). Similar to the findings in grip strength test, rats subjected to LVshAagab (sham + LVshAagab: n = 12), but not LVshRNA infection (sham + LVshRNA: n = 12; Fig. 6C), dramatically increased the escape latency, compared with sham rats. Furthermore, the probe test data also showed that upregulation of Aagab obviously prevented HIBD-induced the impairment of spatial memory retrieval, as the LVAagab-treated rats had a shorter escape latency to cross the location of hidden platform (Fig. 6E), although the number of entries into the platform zone (Fig. 6D) and time spent in the quadrant where the hidden platform was previously located (Fig. 6F) remained unchanged. In contrast, downregulation of Aagab by LVshAagab infection in sham rats induced significant impairment of spatial memory retrieval, as reflected by a decrease in the number of entries into the platform zone (Fig. 6D) and an increase in the escape latency to cross the location of hidden platform (Fig. 6E), although the time spent in the target quadrant remained unchanged (Fig. 6F).
Inhibition of Pten nuclear translocation ameliorates behavioral defects in HIBD model rats
Although over-expression of Aagab before OGD treatment significantly blocked Pten nuclear import, thereby ameliorating HIBD-induced myodynamic and cognitive impairments, genetic upregulation of Aagab is difficult to be used in the clinical treatment. We next wanted to determine whether inhibition of PTEN nuclear translocation directly by Tat-K13 peptide is able to reverse HIBD-induced behavior defects. We first confirmed that Tat-K13 peptide effectively blocked Pten nuclear translocation both in vitro (Fig. S6A–C) and in vivo (Fig. S7A, B), and exerted neuroprotective effect against OGD damage (Fig. S6D–F). The results from grip strength test revealed that grip strength of the right forelimb (sham: n = 14, HIBD + saline: n = 14; Fig. 7B) was significantly decreased compared to that of the left in HIBD rats. Importantly, rats treated with Tat-K13 (HIBD + Tat-K13: n = 16), but not its control peptide Tat-K13R (HIBD + Tat-K13R: n = 12; Fig. 7B), exhibited an obvious recovery in grip strength of the right forelimb. Since HIE survivor exhibits motor disability, rotarod test was sued to evaluate the motor function of HIBD rats. Results showed that the rats in HIBD group spent much less time on the rod compared with those in sham group (sham: n = 19; HIBD + saline: n = 13; Fig. 7C), indicating a significant impairment of motor balance and coordination. Tat-K13 peptide treatment (HIBD + Tat-K13: n = 18), but not Tat-K13R (HIBD + Tat-K13R: n = 15; Fig. 7C), dramatically increased the time spent on the rod during test. These results indicate that treatment with Tat-K13 rather than Tat-K13R ameliorates the deficits of myodynamia and motor function in the neonatal rats after HIBD.

A Diagram illustrating the experimental protocols for HIBD model establishment and peptide treatment as well as behavioral tests. B The left and right forelimb myodynamia during the grip strength test. C The latency to fall off the rod during the rotarod test. D–G Inhibition of Pten nuclear translocation by Tat-K13 peptide ameliorates the deficit of spatial learning and memory in HIBD model rats in the Morris water maze paradigm. D The escape latency to the hidden platform during spatial learning. The number of entries into the platform zone (E), the escape latency of the first entry into the platform zone (F), and time spent in the hidden platform-located quadrant (G) during the probe test with absence of the hidden platform. Data were expressed as mean ± SEM, *p < 0.05, **p < 0.01.
We next examined the effects of Tat-K13 on spatial learning and memory deficits in HIBD rats in the Morris water maze paradigm. The results showed that the escape latency for searching for the hidden platform in HIBD rats was much longer than that in the sham group during spatial training period (sham: n = 16; HIBD + saline: n = 14; Fig. 7D), indicating an impairment of learning after HIBD. As expected, treatment with Tat-K13 significantly ameliorated the impairment compared with saline group, as reflected by a dramatic decrease in the escape latency, although the escape latency was still longer than sham group (HIBD + Tat-K13: n = 16; Fig. 7D). However, treatment with Tat-K13R exhibited no protective effect on the impairment of spatial learning (HIBD + Tat-K13R: n = 15; Fig. 7D). Furthermore, the probe test data also showed that spatial memory retrieval was obviously impaired in HIBD rats, as reflected by a reduced number of entries into the platform zone (Fig. 7E), a longer escape latency to the first entry into the platform zone (Fig. 7F), and much less time spent in the quadrant where the hidden platform was previously located (Fig. 7G). Administration of Tat-K13, but not Tat-K13R, significantly increased the number of entries into the platform zone (Fig. 7E), shortened the escape latency to the first entry into the platform zone (Fig. 7F), and increased the time spent in the target quadrant (Fig. 7G). These results suggest that the inhibition of Pten nuclear translocation by systemic administration of Tat-K13 immediately after HIBD is able to prevent the HIBD-induced impairment of spatial learning and memory.
Discussion
Although PTEN is a well-established tumor suppressor, accumulating evidence implicates that PTEN is important for multiple aspects of neuronal function and development in the brain, containing cell proliferation, neuronal growth and synaptic plasticity [47]. For example, loss of PTEN function is sufficient to disrupt programmed interneuron cell death, which may in turn alter the cellular balance of excitation and inhibition in the cerebral cortex [48]. Deletion of PTEN in limited differentiated neuronal populations results in macrocephaly and neuron hypertrophy, subsequently contributing to human autism spectrum disorders (ASD) [49, 50]. Inhibition of PTEN is able to rescue normal synaptic function and cognition in cellular and animal models of Alzheimer’s disease (AD), whereas over-expression of PTEN leads to synaptic depression that can mimic and occlude Aβ-induced depression [51]. Our recent study has indicated that Pten nuclear accumulation induced by excitotoxia/ischemia resulted in increased neuronal death, and the mutation at K13 residue of Pten significantly inhibited Pten nuclear import and significantly reduced neuronal damage induced by excitotoxia/ischemia, thereby improved behavioral function in mature brain [29]. Here, we found that Pten nuclear translocation was obviously increased both in neonatal rats after HIBD and in primarily cultured neurons after OGD (Fig. 1). We further demonstrated that the mutation at K13 residue of Pten significantly inhibited Pten nuclear import (Fig. 2) and reduced neuronal damage (Fig. S6), and thereby ameliorated behavioral defects induced by HIBD in neonatal rats (Fig. 7). These findings are fully in agreement with recent reports that reduction in nuclear PTEN promotes axonal regeneration and neuronal survival [52, 53]. Thus, Pten nuclear translocation plays a critical role in excitotoxic and ischemic neuronal injuries, and subsequent motor and cognitive deficits in both mature and developing animals.
Previous reports have shown that NEDD4-1 mediated PTEN nuclear translocation [26, 33, 37]. However, the potential molecular mechanism remains poorly understood. Here, we reported that HIBD and OGD induced a significant decrease in NEDD4-1 expression in vivo and in vitro, respectively. Over-expression of NEDD4-1 dramatically inhibited OGD-induced Pten nuclear import, while downregulation of NEDD4-1 alone exhibited no effect on Pten nuclear import in cultured neurons (Fig. 3). We further found that Pten nuclear translocation medicated by NEDD4-1 was dependent on mono-ubiquitination of PTEN at K13 residue (Fig. 2). Notably, previous studies have revealed that nuclear PTEN accumulation are associated with increased PTEN mono-ubiquitination at lysine residues K13 and K289 [26, 32, 33]. However, the mutation at K289 residue of PTEN had no effect on the PTEN nuclear import induced by OGD in HEK 293T cells and in primarily cultured neurons in the present study (Fig. 2). One possible explanation is due to different residues of PTEN mono-ubiquitination observed in different cells or different diseases. Although the detailed mechanisms by which PTEN nuclear translocation increases neuronal vulnerability remain largely unclear, recent studies have pointed out that nuclear PTEN may antagonize nuclear PIP3/Akt survival signaling pathways [54,55,56,57,58], thereby mediates excitotoxic neuronal death. Alternatively, nuclear PTEN can form a physical complex with p53 in the nucleus [59, 60], thereby protecting p53 from ubiquitination-mediated degradation and facilitating p53-dependent apoptosis. In addition, recent study from cancer cells has revealed that nuclear PTEN accumulation activates autophagy by activating p-JUN-SESN2/AMPK pathway, leading to DNA damage and cell death [61]. Thus, further study to detect the effect of PTEN nuclear translocation on autophagy in neurons is necessary.
Recent developments of cancer research have indicated that P34SEI-1 (CDK4-binding protein p34SEI) controls switching between NEDD4-1-mediated PTEN mono- and poly-ubiquitination via regulating the auto-ubiquitination of NEDD4-1, thereby regulates nuclear import and degradation of PTEN [36]. However, in the present study, we did not observe any difference of the expression of P34SEI-1 in the brain tissues of rats subjected to HIBD and in the primarily cultured neurons subjected to OGD (Fig. S8). Discrepancies between the previous study and the present work still need to be resolved, but may be at least in part accounted for by differences in cell types and experimental conditions. According to recent reports that nuclear translocation of ubiquitinated PTEN can be mediated by Rab5 GTPase, which plays an important role in the endosomal trafficking pathways and the transport of proteins to subcellular locations [38, 42], and cytosolic protein Aagab, containing a Rab-like GTPase domain, is shown to bind both clathrin adapter protein complexes and participate in membrane traffic [43]. Consistently, we here found that downregulation of Aagab reduced NEDD4-1 expression, and subsequently induced Pten nuclear translocation (Fig. 4). In contrast, over-expression of Aagab is able to prevent OGD-induced Pten nuclear translocation (Fig. 4), thereby reducing HIBD-induced myodynamic and cognitive deficits (Fig. 6). Notably, we here found that the poly-ubiquitination of Aagab and NEDD4-1 were increased and the corresponding protein levels were decreased in primarily cultured neurons subjected to OGD (Fig. 5). One possible explanation is that the reduced expression of NEDD4-1 is not a downregulation event, but due to NEDD4-1 activation in response to HI/OGD or downregulated Aagab, which is accompanied with increased ubiquitination and degradation of NEDD4-1 itself. Therefore, future work should be necessary to determine the exact molecular mechanism underlying the downregulation of Aagab and NEDD4-1 following OGD.
The peptide (Tat-K13) used in the current study do not affect the global protein level of Pten, but it obviously prevents Pten nuclear translocation, a delayed step in ischemia-induced cell death signaling cascade. Tat-K13 significantly inhibits OGD-mediated excitotoxic damage in the cultured neurons, and significantly alleviates the deficits of myodynamia, motor functions and spatial learning and memory in a rat model of HIBD (Fig. 7), suggesting that inhibition of Pten nuclear translocation plays a neuroprotective role in ischemic brain damage. Nonetheless, contradictory results challenge these findings. Two studies using Ndfip 1 (NEDD4 family-interacting protein 1) deficient mice have reported that without Ndfip 1 in vivo, PTEN fails to translocate to nuclei, resulting in an increase in neuronal vulnerability following ischemic insults [62, 63]. One possible explanation for these discrepancies is that Ndfip 1, as an adapter and activator of protein ubiquitination by Nedd4 E3 ligases, whose deficiency would affect not only PTEN but also numerous substrates of the E3-ligase NEDD4. Thereby, it is most likely that the increased neuronal vulnerability induced by ischemia in Ndfip1-deficient mice is due to its impact on one or more cell survival signaling pathways.
Overall, our present data provide the first evidence that Aagab, as a novel regulator for NEDD4-1, regulates the expression of NEDD4-1, and subsequently mediates Pten nuclear translocation. And we also demonstrate that systemic application of Tat-K13 peptide promotes behavioral recovery in neonatal HIBD rats. Thus, these findings provide scientific basis for the development of PTEN nuclear translocation blockers, such as Tat-K13, as potential therapeutics for treating the motor and cognitive deficits associated with patients with acute brain insults, such as HIE and brain trauma.
Materials and methods
Animals
Unsexed 7-day-old SD rats were used to establish the HIBD model as described previously with modification [64, 65]. Briefly, the 7-day-old SD rats were randomly divided into two groups, sham group and HIBD group, and then the HIBD rats were randomly divided into subgroups for different treatment. The left common carotid artery of the rats in HIBD group was isolated and ligated. Two hours after surgery, the pups were exposed to hypoxic conditions (8% O2 + 92% N2) for 2.5 h at 37 °C. The sham animals were only separated out the left common carotid artery without ligation and no exposure to hypoxic conditions. Pups were returned to their nest with their dam until weaning on P21. All procedures involving animals were housed in plastic cages with unlimited access to food and water and maintained in a temperature-controlled colony room (21 °C) under a cycle of 12-h light/12-h dark (8:00 am–8:00 pm). All procedures were performed in accordance with Chongqing Science and Technology Commission guidelines for animal research and approved by the Chongqing Medical University Animal Care Committee, and every effort was made to minimize both the animal suffering and the number of animals used. The mortality of rats was about 3% during and after HIBD, and around another 3–5% animals were excluded due to underweight after surgery. All experiments were performed double-blind.
Antibodies and reagents
Rabbit anti-lamin B1 (#ab133741), rabbit anti-HA tag (#ab9110) and rabbit anti-PTEN (#ab267787) for western blot were obtained from Abcam Inc (Cambridge, MA), and mouse anti-PTEN (#sc-7974) for immunocytochemistry was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-Aagab (#NBP1-87905) for western blot and rabbit anti-Aagab (#NBP2-59084) for co-immunoprecipitation were supplied by Novus (Novus Biologicals, USA). Rabbit anti-Ubiquitin (#10201-2-AP), rabbit anti-Histone-H3 (#17168-1-AP), rabbit anti-Tubulin (#11224-1-AP) and rabbit anti-NEDD4L (#13690-1-AP) were obtained from Proteintech (Rosemont, USA). Rabbit anti-NEDD4-1 (#MAB07-049) and rabbit anti-p34-Arc/ARPC2 (#07-227) were purchased from Millipore Corporation (Billerica, MA). Mouse anti-β-actin (#026M4780V) was obtained from Sigma-Aldrich (St. Louis, MO). Rabbit anti-GFP (#AG279) was obtained from Beyotime Biotechnology (Shanghai, China). Anti-Mouse IgG (Goat) HRP-Labeled (#NEF822001EA) was purchased from PerkinElmer (PerkinElmer, USA).
MinuteTM Cytoplasmic and Nuclear Extraction kit (#SC-003) was purchased from Invent Biotechnologies, Inc. (Beijing, China). Complete Protease Inhibitor Cocktail Tablets (#04693116001) was obtained from Roche Applied Science. PierceTM BCA Protein Assay Kit (#23225) was purchased from Thermoscientific. Protein A/G Magnetic Beads (#B23202) was obtained from Bimake (Hoston, TX). NeuroCultTM SM1 Neuronal Supplement (#05711) was obtained from STEMCELL (STEMCELL technologies, Canada). MG132 was purchased from MedChem Express (Monmouth Junction, NJ, USA). Tat-fused peptides (Tat-K13 and Tat-K13R) were synthesized by GL Biochem Ltd (Shanghai, China).
Buffers
All buffers were sterilized by autoclaving or vacuum filtration (Corning, Polyethersulfone with 0.22 µm pore size). Phosphate Buffered Saline (PBS) contains 137 mM NaCl, 8.1 mM Na2HPO4, 1.76 mM KH2PO4, 2.7 mM KCl, pH 7.4. Four times (4×) sample buffer contains 50% Glycerol, 125 mM Tris-HCl (pH 6.8), 4% SDS and 0.08% Bromophenol Blue. Running buffer contains 25 mM Tris-HCL, 0.25 mM glycine and 0.1% SDS. Transfer buffer contains 48 mM Tris-HCL, 39 mM glycine, 0.037% SDS and 20% methanol. Poly-D-Lysine coating solution contains 0.01% poly-D-lysine. Tris-buffered saline (TBS) contains 150 mM NaCl, 50 mM Tris-HCL, pH 7.6.
Primary culture of cortical/hippocampal neurons
Pregnant SD rats (E19) were used to culture cortical/hippocampal neurons as described previously with modification [29]. Briefly, rats were anaesthetized deeply with Urethane solution (1.5 g/kg, i.p.). The cortex or hippocampi were isolated in HBSS dissection buffer (HyClone, USA), then digested with 2 mL of pre-warmed 0.25% Trypsin-EDTA for 10 min at 37 °C. Then, the dissociated cells were washed with DMEM (with 10% FBS) for three times and pipetted gently to ensure a single cell suspension. Next, the cells were centrifuged at 600 rpm for 3 min, re-suspended in DMEM (with 10% FBS) media, counted using a hemocytometer, and plated onto poly-D-lysine-coated culture dishes at a density of 1.0 × 107 per 10 cm dish, 6.0 × 106 per 6 cm dish and 2.0 × 106 per 6 well plate. The cells were cultured in an incubator (Thermo Forma 3111, Thermo Scientific) containing 5% CO2 at 37 °C. Four hours later, the original media was replaced by Neurobasal Feeding Media (NF media, containing 500 mL of Neurobasal Media, 2% SM1 supplement and 0.5 mM GlutaMAXTM-I Supplement). Half of NF media was replaced every 3 days until cells were used for experiments.
OGD treatment
Primary culture of cortical/hippocampal neurons (7 DIV) and HEK293T cells were used in this study. The cell conditioned medium (taken out and saved for further use) was replaced by Earle’s balanced salt solution (EBSS) (Hyclone, USA). Followed up, the cells were transferred to an incubator (Thermo Scientific) with 5% O2 and 95% N2 at 37 °C. Ninety minutes later, the cells were returned to the previously saved conditional medium. Neurons were allowed to recover for different periods of time, ranging from 0 h to 24 h until further experiments and HEK293T cells were allowed to recover for 6 h.
Peptide administration
To examine the effects of Tat-K13 against Pten nuclear translocation induced by OGD, cultured neurons received pretreatment of Tat-K13 peptide (50 μM) as previously described [29], with modifications. To illustrate the effects of Tat-K13 against HIBD, experimental pups were subdivided into four groups (sham, HIBD + saline, HIBD + Tat-K13, HIBD + Tat-K13R), and received different treatments of interfering peptides (10 mg/kg, i.p.) or saline at 0 h and 3 h after hypoxia onset. To achieve the optimal outcome, another two doses of peptide were conducted on the second and third days, respectively.
Lentivirus
To regulate the expression of genes in cultured neurons and in vivo, lentivirus (LV) carrying corresponding genes or their siRNAs was constructed. LVPTEN (PTENWT, PTENK13R or PTENK289R, PTEN expressed in LVPTEN is EGFP-fusion protein), LVNEDD4-1, LVNEDD4-2, LVAagab and LVshAagab were constructed by OBiO Technology (Shanghai, China). LVshNEDD4-1 was constructed by Genechem Technology (Shanghai, China). LVshNEDD4-2 was constructed by GenePharma Technology (Shanghai, China). The sequences of all plasmids were confirmed by DNA sequencing. LVshRNA oligonucleotides were as follows: LVshNEDD4-1-1, 5’-AGCCACAAATCAAGAGTTAAA-3’; LVshNEDD4-1-2, 5’- TTGGAAGGACCTACTACGTAA -3’; LVshNEDD4-1-3, 5’- CTGGATTGAGTTTGATGGTGA -3’; LVshNEDD4-2-1, 5’-GCTCGCCAACAGTAACCTTAT-3’; LVshNEDD4-2-2, 5’-GGGAAGATCCACGGCTGAAAT-3’; LVshNEDD4-2-3, 5’-GCAGAAATACGACTACTTTAG-3’; LVshAagab-1, 5’-GCTGTTGTGGAAGCGACTT-3’; LVshAagab-2, 5’-GGACGATGACTTCCCTGAA-3’; LVshAagab-3, 5’-GGATTGTACAAGCATTGAA-3’; LVshRNA, 5’-TTCTCCGAACGTGTCACGT-3’. Titers were 3.0E + 8–2.5E + 9 TU/ml. Through preliminary experiments, we screened LVshNEDD4-1-1, LVshNEDD4-2-1 and LVshAagab-2 with the best knockdown effects for subsequent experiments.
Stereotaxic viral injection
P1 rat pups were received microinjection of lentivirus (LVshRNA, LVshAagab, LVmNeonGreen or LVAagab) into left hippocampus (2.6 mm lateral to midline, 2.6 mm anterior to bregma, 1.2 mm below skull) and motor cortex (1.0 mm lateral to midline, 1.9 mm posterior to bregma, 1.2 mm below skull), using a 30-gauge needle on a Hamilton syringe. The syringe was attached to a nanoliter infusion pump (Legato130, KD Scientific, USA) and a volume of 2 μl of lentivirus was infused at a rate of 0.5 μl/min for each brain region. Following injection, the pups were returned to homecage with their dam until P7 to establish HIBD model.
Nuclei fractionation
MinuteTM cytoplasmic and nuclear extraction kits were used to fractionate cytoplasm/nuclei. Briefly, cultured cortical/ hippocampal neurons or brain tissues of hippocampus were homogenized with cytoplasmic extraction buffer on ice, and then centrifuged at 14,000 rpm for 5 min at 4 °C. The supernatant (cytoplasmic fraction) was transferred to a new tube. The cell deposit was re-suspended in nuclear extraction buffer by vortex and then rested on the ice for 1 min, and this operation was repeated for 4 times. Followed by, the sample was pipetted into filter cartridge and centrifuged at 16,000 rpm for 30 s at 4 °C. Both cytoplasmic and nuclei fractions were saved at −80 °C for further use.
Western blotting
Total protein was extracted using ice-cold whole cell lysis buffer. Approximately 30 μg of total protein or 15 μg nuclear protein was boiled with 4× sample buffer at 96 °C for 5 min. Followed by, the samples were separated on 10% (total protein) or 15% (nuclear protein) SDS-PAGE gels and transferred onto Immobilon-PTM polyvinylidene fluoride (PVDF) membranes (Bio-Rad, Hercules, CA, USA). Then, the membranes were incubated with 5% fat-free milk in TBS contains 0.1% Tween-20 for 90 min at room temperature to block non-specific binding site. The target proteins were immunoblotted with primary antibody overnight at 4 °C.
After incubation with corresponding horseradish peroxidase (HRP)-conjugated secondary antibody (1: 5000, Perkin Elmer) at room temperature for 90 min, the protein was visualized in the Bio-Rad Imager using ECL western blotting substrate (Pierce). The band intensity of each protein was quantified by the Bio-Rad Quantity One software as described previously [66]. The relative level of target protein was expressed as the percentage between intensity of target protein and that of marker protein on the same blot, such as cytoplasmic marker β-actin, and nuclear marker H3.
Immunocytochemistry
Primary culture of hippocampal neurons (7–8 DIV) were fixed with 4% paraformaldehyde in 100 mM PBS for 20 min, and then permeabilized by 1% Triton X-100 in PBS for 20 min at room temperature. To block non-specific background, the neurons were incubated with 2% Bovine Serum Albumin (BSA) in PBS for 90 min at room temperature. Thereafter, the neurons were incubated with mouse anti-PTEN (1:50) for overnight at 4 °C. On the next day, the neurons were incubated with fluorophore-conjugated secondary antibodies (1:200) for 90 min at room temperature, and then nuclei were stained with fluorescent dye Hoechst 33342 (1 μg/ml) for 20 min at room temperature, avoiding bright light. After sealing, images of Pten and nuclei were taken with a Nikon Eclipse Ti inverted fluorescence microscope.
Co-immunoprecipitation
Cortical/hippocampal neurons and HEK293T cells were lysed with ice-cold cell lysis buffer for immunoprecipitation (IP) with protease inhibitors. The lysates were pre-cleared with protein A/G Magnetic Beads. Followed by, the pre-cleared lysates were incubated with anti-PTEN, anti-NEDD4-1, anti-Aagab or anti-NEDD4-2 antibodies for 2 h at 4 °C with continuous agitation, and then protein A/G magnetic beads was added. On the next day, the lysate-antibody-magnetic beads complex was collected with magnetic frame for 2 min on the ice, and then washed three times with PBS. Finally, proteins were eluted from the beads by boiling with 1 X sample buffer, and western blot was used to analyze the corresponding proteins.
Ubiquitination assay
EGFP-PTENWT, EGFP-PTENK13R or EGFP-PTENK289R plasmids were transiently transfected into HEK293T cells for 24 h by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s recommendation protocol. Primary culture of cortical/hippocampal neurons were infected with LVPTEN (LVPTENWT, LVPTENK13R or LVPTENK289R) and returned to incubator for another 4 days. For PTEN mono-ubiquitination assay, cultured neurons or HEK293T cells were lysed with IP buffer with protease inhibitors for 30 min on the ice, and then centrifuged at 16000 rpm for 30 min at 4 °C to obtain cytosolic protein. The extract was subjected to IP with anti-ubiquitin, anti-PTEN or anti-EGFP primary antibody, followed by immunobloting with anti-PTEN, anti-EGFP or anti-ubiquitin primary antibody. For Aagab and NEDD4-1 poly-ubiquitination assay, the protocol is similar to PTEN mono-ubiquitination assay. The only difference is that primarily cultured neurons (7 DIV) were treated with MG132 (15 μM for 6 h) when they were subjected to OGD, as previously described with modifications [67].
Grip strength test
Grip strength test was used to evaluate myodynamia as described previously [65]. Briefly, 3 weeks after HIBD model established, the left and right forelimbs of each rat were placed on the grasping force sensor, respectively. The animals grasped a horizontal bar of the Grip Strength Meter (Chatillon, USA), and then they were gently pulled away by an experienced handler until their grasp was broken. Each rat was given 5 trials at 10-min intervals, and the mean value was calculated to illustrate the grip strength. The Grip Strength Meter was cleaned with 70% ethanol and water between tests.
Rotarod test
Rotarod test was used to evaluate motor performance as described previously with modifications [65]. In brief, one day after grasping test, rats received 2 rounds of pre-training on the rotarod (Stoelting Co.). During pre-training, the rotarod was maintained at constant speed of 10 rpm and 20 rpm for 3 min, respectively. Twenty-four hours after the pre-training, rats received formal rotarod testing in 11 consecutive trails (20-min intervals) with the continuous increasing rotation from 10 rpm to 60 rpm (increasing by 5 rpm each time). During each test, the time that the animals remained on the rotarod was monitored. Maximum test time (cut-off limit) was 180 s. The motor performance of the rat was expressed as the latency to fall off the rotarod. The rotarod was cleaned with 70% ethanol and water between tests.
Morris water maze test
Four weeks after HIBD, rats were subjected to the Morris water maze test to measure their spatial learning and memory functions as previously described [66, 68]. In brief, on the first day of adaptation, the rats were allowed to adapt to the water maze for a 60-s free swim, which consists of a circular pool (180-cm diameter) filled with water (25 °C ± 1 °C) made opaque with nontoxic black paint. On the following 5 days, the rats were continuously trained in the pool over four trails per day to find an invisible platform (1-cm below the surface of the water). During each trial, the rats were guided to the hidden platform to remain 30 s when they failed to find the platform within 60 s. Twenty-four hours after the final training trial, a 60-s probe test was performed without an escape platform to assess the memory retrieval. All trials were recorded by using Any-maze tracking system (Stoelting, USA).
Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) is released from cells into medium when the plasma membrane integrity is compromised. The released LDH can convert oxidized nicotinamide adenine dinucleotide (NAD+) into NADH (the reduced form). Therefore, the amount of release represents the degree of cell death. In the present study, LDH cytotoxicity assay kit (Beyotime Biotechnology, Shanghai, China) was used to measure the extracellular LDH level. Briefly, hippocampal neurons were plated onto poly-D-lysine-coated culture 96-well plate at a density of 1.0 × 105 per well. Seven day later, the cell conditioned medium was replaced with 200 μL new medium and continue cultured for 24 h. Then, the cells were centrifuged at 400 rpm for 5 min. Followed by, 120 μl supernatant of each well was transferred into a new 96-well plate, added 60 μL LDH working solution, then mixed gently and incubated at room temperature for 30 min, avoiding bright light. The absorbance was determined at 490 nm by microplate reader (Bio Tek Insruments). Finally, the cell death rate was expressed as a ratio (%) between the absorbance of the treated group and that of the control group.
MTT assay
The 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay was performed to test cell viability. MTT cell proliferation and cytotoxicity assay kit (Beyotime Biotechnology, Shanghai, China) was used in the present study. In brief, hippocampal neurons (7 DIV) in 96-well were incubated with 100 μl MTT (0.5 mg/ml, dissolved in NF media) for 4 h, and added 100 μl dimethyl sulfoxide (DMSO) to dissolve the dark blue formazan crystals. The absorbance was determined at 570 nm by microplate reader. Finally, the cell viability rate was expressed as a ratio (%) between the absorbance of the treated group and that of the control group.
CCK-8 assay
Cell counting kit-8 (CCK-8, DOJINDO, Japan) was used to further test cell viability in the present study. Briefly, hippocampal neurons (7 DIV) in 96-well were washed with PBS, then added 10% CCK-8 solution (dissolved in NF media) and cultured in an incubator containing 5% CO2 at 37 °C for 1–4 h. Then, the absorbance was determined at 450 nm by microplate reader. Finally, the cell viability rate was expressed as a ratio (%) between the absorbance of the treated group and that of the control group.
Statistical analysis
All data were expressed as means ± standard error (mean ± SEM). The differences of rotarod test and spatial learning in water maze test among different groups were analyzed by two-way AVOVA with treatment (group) as the between-subjects factor and trials as the within- subjects factor. The data of all other experiments were analyzed by one-way ANOVA followed by Tukey’s post hoc test. Statistical significance was set as at *p < 0.05, **p < 0.01, ***p < 0.001.
References
Kurinczuk JJ, White-Koning M, Badawi N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev. 2010;86:329–38.
Graham EM, Ruis KA, Hartman AL, Northington FJ, Fox HE. A systematic review of the role of intrapartum hypoxia-ischemia in the causation of neonatal encephalopathy. Am J Obstet Gynecol. 2008;199:587–95.
Martinez-Biarge M, Diez-Sebastian J, Kapellou O, Gindner D, Allsop JM, Rutherford MA, et al. Predicting motor outcome and death in term hypoxic-ischemic encephalopathy. Neurology. 2011;76:2055–61.
Shankaran S, Pappas A, McDonald SA, Vohr BR, Hintz SR, Yolton K, et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N Engl J Med. 2012;366:2085–92.
Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, et al. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science. 2002;298:846–50.
Lai TW, Shyu WC, Wang YT. Stroke intervention pathways: NMDA receptors and beyond. Trends Mol Med. 2011;17:266–75.
Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–22.
Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164:719–21.
Rothman SM. Synaptic activity mediates death of hypoxic neurons. Science. 1983;220:536–7.
Simon RP, Swan JH, Griffiths T, Meldrum BS. Blockade of N-methyl-D-aspartate receptors may protect against ischemic damage in the brain. Science. 1984;226:850–2.
Gladstone DJ, Black SE, Hakim AM, Heart and Stroke Foundation of Ontario Centre of Excellence in Stroke R. Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke. 2002;33:2123–36.
Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1:383–6.
Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500.
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7.
Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–8.
Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39.
Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–62.
Tamura M, Gu J, Tran H, Yamada KM. PTEN gene and integrin signaling in cancer. J Natl Cancer Inst. 1999;91:1820–8.
Weng L, Brown J, Eng C. PTEN induces apoptosis and cell cycle arrest through phosphoinositol-3-kinase/Akt-dependent and -independent pathways. Hum Mol Genet. 2001;10:237–42.
Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M, et al. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell. 2011;144:187–99.
Parsons R. Human cancer, PTEN and the PI-3 kinase pathway. Semin Cell Dev Biol. 2004;15:171–6.
Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–96.
Ning K, Pei L, Liao M, Liu B, Zhang Y, Jiang W, et al. Dual neuroprotective signaling mediated by downregulating two distinct phosphatase activities of PTEN. J Neurosci. 2004;24:4052–60.
Chen X, Du YM, Xu F, Liu D, Wang YL. Propofol prevents hippocampal neuronal loss and memory impairment in cerebral ischemia injury through promoting PTEN degradation. J Mol Neurosci. 2016;60:63–70.
Gary DS, Mattson MP. PTEN regulates Akt kinase activity in hippocampal neurons and increases their sensitivity to glutamate and apoptosis. Neuromolecular Med. 2002;2:261–9.
Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–56.
Georgescu MM, Kirsch KH, Kaloudis P, Yang H, Pavletich NP, Hanafusa H. Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res. 2000;60:7033–8.
Walker SM, Leslie NR, Perera NM, Batty IH, Downes CP. The tumour-suppressor function of PTEN requires an N-terminal lipid-binding motif. Biochem J. 2004;379:301–7.
Zhang S, Taghibiglou C, Girling K, Dong Z, Lin SZ, Lee W, et al. Critical role of increased PTEN nuclear translocation in excitotoxic and ischemic neuronal injuries. J Neurosci. 2013;33:7997–8008.
Zhao J, Qu Y, Wu J, Cao M, Ferriero DM, Zhang L, et al. PTEN inhibition prevents rat cortical neuron injury after hypoxia-ischemia. Neuroscience. 2013;238:242–51.
Li D, Qu Y, Mao M, Zhang X, Li J, Ferriero D, et al. Involvement of the PTEN-AKT-FOXO3a pathway in neuronal apoptosis in developing rat brain after hypoxia-ischemia. J Cereb Blood Flow Metab. 2009;29:1903–13.
Baker SJ. PTEN enters the nuclear age. Cell. 2007;128:25–8.
Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129–39.
Yang JM, Schiapparelli P, Nguyen HN, Igarashi A, Zhang Q, Abbadi S, et al. Characterization of PTEN mutations in brain cancer reveals that pten mono-ubiquitination promotes protein stability and nuclear localization. Oncogene. 2017;36:3673–85.
Wang X, Shi Y, Wang J, Huang G, Jiang X. Crucial role of the C-terminus of PTEN in antagonizing NEDD4-1-mediated PTEN ubiquitination and degradation. Biochem J. 2008;414:221–9.
Hong SW, Moon JH, Kim JS, Shin JS, Jung KA, Lee WK, et al. p34 is a novel regulator of the oncogenic behavior of NEDD4-1 and PTEN. Cell Death Differ. 2014;21:146–60.
Drinjakovic J, Jung H, Campbell DS, Strochlic L, Dwivedy A, Holt CE. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron. 2010;65:341–57.
Miaczynska M, Pelkmans L, Zerial M. Not just a sink: endosomes in control of signal transduction. Curr Opin Cell Biol. 2004;16:400–6.
Bucci C, Parton RG, Mather IH, Stunnenberg H, Simons K, Hoflack B, et al. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 1992;70:715–28.
Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–25.
Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–17.
Li Y, Low LH, Putz U, Goh CP, Tan SS, Howitt J. Rab5 and Ndfip1 are involved in Pten ubiquitination and nuclear trafficking. Traffic. 2014;15:749–61.
Pohler E, Mamai O, Hirst J, Zamiri M, Horn H, Nomura T, et al. Haploinsufficiency for AAGAB causes clinically heterogeneous forms of punctate palmoplantar keratoderma. Nat Genet. 2012;44:1272–6.
Putz U, Howitt J, Doan A, Goh CP, Low LH, Silke J, et al. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci Signal. 2012;5:ra70.
Dilenge ME, Majnemer A, Shevell MI. Long-term developmental outcome of asphyxiated term neonates. J Child Neurol. 2001;16:781–92.
Golan H, Huleihel M. The effect of prenatal hypoxia on brain development: short- and long-term consequences demonstrated in rodent models. Dev Sci. 2006;9:338–49.
Knafo S, Esteban JA. PTEN: local and global modulation of neuronal function in health and disease. Trends Neurosci. 2017;40:83–91.
Wong FK, Bercsenyi K, Sreenivasan V, Portales A, Fernandez-Otero M, Marin O. Pyramidal cell regulation of interneuron survival sculpts cortical networks. Nature. 2018;557:668–73.
Frazier TW, Embacher R, Tilot AK, Koenig K, Mester J, Eng C. Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol Psychiatry. 2015;20:1132–8.
Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, et al. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–88.
Knafo S, Sanchez-Puelles C, Palomer E, Delgado I, Draffin JE, Mingo J, et al. PTEN recruitment controls synaptic and cognitive function in Alzheimer’s models. Nat Neurosci. 2016;19:443–53.
Sun F, Park KK, Belin S, Wang D, Lu T, Chen G, et al. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature. 2011;480:372–5.
Zheng M, Liao M, Cui T, Tian H, Fan DS, Wan Q. Regulation of nuclear TDP-43 by NR2A-containing NMDA receptors and PTEN. J Cell Sci. 2012;125:1556–67.
Vasko V, Saji M, Hardy E, Kruhlak M, Larin A, Savchenko V, et al. Akt activation and localisation correlate with tumour invasion and oncogene expression in thyroid cancer. J Med Genet. 2004;41:161–70.
Gimm O, Perren A, Weng LP, Marsh DJ, Yeh JJ, Ziebold U, et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am J Pathol. 2000;156:1693–700.
Ahn JY, Liu X, Cheng D, Peng J, Chan PK, Wade PA, et al. Nucleophosmin/B23, a nuclear PI(3,4,5)P(3) receptor, mediates the antiapoptotic actions of NGF by inhibiting CAD. Mol Cell. 2005;18:435–45.
Ahn JY, Rong R, Liu X, Ye K. PIKE/nuclear PI 3-kinase signaling mediates the antiapoptotic actions of NGF in the nucleus. EMBO J. 2004;23:3995–4006.
Deleris P, Bacqueville D, Gayral S, Carrez L, Salles JP, Perret B, et al. SHIP-2 and PTEN are expressed and active in vascular smooth muscle cell nuclei, but only SHIP-2 is associated with nuclear speckles. J Biol Chem. 2003;278:38884–91.
Tang Y, Eng C. PTEN autoregulates its expression by stabilization of p53 in a phosphatase-independent manner. Cancer Res. 2006;66:736–42.
Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117–30.
Chen JH, Zhang P, Chen WD, Li DD, Wu XQ, Deng R, et al. ATM-mediated PTEN phosphorylation promotes PTEN nuclear translocation and autophagy in response to DNA-damaging agents in cancer cells. Autophagy. 2015;11:239–52.
Howitt J, Lackovic J, Low LH, Naguib A, Macintyre A, Goh CP, et al. Ndfip1 regulates nuclear Pten import in vivo to promote neuronal survival following cerebral ischemia. J Cell Biol. 2012;196:29–36.
Goh CP, Putz U, Howitt J, Low LH, Gunnersen J, Bye N, et al. Nuclear trafficking of Pten after brain injury leads to neuron survival not death. Exp Neurol. 2014;252:37–46.
Rice JE 3rd, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981;9:131–41.
Dai C, Liu Y, Dong Z. Tanshinone I alleviates motor and cognitive impairments via suppressing oxidative stress in the neonatal rats after hypoxic-ischemic brain damage. Mol Brain. 2017;10:52.
Dong Z, Han H, Li H, Bai Y, Wang W, Tu M, et al. Long-term potentiation decay and memory loss are mediated by AMPAR endocytosis. J Clin Invest. 2015;125:234–47.
Ren G, Zhang X, Xiao Y, Zhang W, Wang Y, Ma W, et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 2019;38:e100376.
Ge Y, Dong Z, Bagot RC, Howland JG, Phillips AG, Wong TP, et al. Hippocampal long-term depression is required for the consolidation of spatial memory. Proc Natl Acad Sci USA. 2010;107:16697–702.
Acknowledgements
We thank all other members in the Wang and Dong laboratories for the helpful discussions and support.
Funding
This work was supported by grants from the National Natural Science Foundation of China (NSFC) 81571042, 82071395 and 91749116 to ZD, the Chongqing Science and Technology Commission cstc2020jcyj-zdxmX0004 to ZD, the Science and Technology Research Program of Chongqing Municipal Education Commission KJZD-K201900403 to ZD, Innovation Research Group at Institutions of Higher Education in Chongqing CXQTP19034 to ZD, Doctoral Initiation Fund of Women and Children’s Medical Center of Guangzhou Medical University 1600037-04 to CD, Graduate Student Innovation Project of Chongqing CYS17139 to YC, and the Canadian Institutes of Health Research (CIHR) to YTW. YTW is the holder of Heart and Stroke Foundation of British Columbia and Yukon Chair in Stroke Research.
Author information
Authors and Affiliations
Contributions
CD, YTW and ZD conceived the study and CD, YD and ZD wrote the manuscript. CD, BW, YC, YB and YP performed behavioral studies. CD, BW and XL performed biochemical assays. All authors have contributed to and approved the final paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
All procedures were performed in accordance with Chongqing Science and Technology Commission guidelines for animal research and approved by the Chongqing Medical University Animal Care Committee.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by P. Salomoni
Rights and permissions
About this article

Cite this article
Dai, C., Wu, B., Chen, Y. et al. Aagab acts as a novel regulator of NEDD4-1-mediated Pten nuclear translocation to promote neurological recovery following hypoxic-ischemic brain damage. Cell Death Differ 28, 2367–2384 (2021). https://doi.org/10.1038/s41418-021-00757-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-021-00757-4
This article is cited by
-
Inhibiting NLRP3 inflammasome signaling pathway promotes neurological recovery following hypoxic-ischemic brain damage by increasing p97-mediated surface GluA1-containing AMPA receptors
Journal of Translational Medicine (2023)
-
Dihydroartemisinin attenuates hypoxic-ischemic brain damage in neonatal rats by inhibiting oxidative stress
Molecular Brain (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}