
Abstract
Alveolar macrophages (AM) maintain airway immune balance; however, the regulation of heterogeneity of AMs is incompletely understood. We demonstrate that RGS1 coregulates the immunophenotype of AM subpopulations, including pro- and anti-inflammatory, injury- and repair-associated, and pro- and antifibrotic phenotypes, through the PLC-IP3R signal-dependent intracellular Ca2+ response. Flt3+ AMs and Tie2+ AMs had different immune properties, and RGS1 expression in the cells was targeted by exosomes (EXOs) containing miR-223 and miR-27b-3p that were derived from vascular endothelial cells (EnCs) and type II alveolar epithelial cells (EpCs-II), respectively. Imbalance of AMs was correlated with acute lung injury/acute respiratory distress syndrome (ALI/ARDS) and pulmonary fibrosis (PF) caused a lack of secretion of CD31+ and CD74+ EXOs derived from EnCs and EpCs-II. Timely treatment with EXOs significantly improved endotoxin-induced ALI/ARDS and bleomycin-induced PF in mice. Thus, EnC- and EpC-II-derived EXOs regulate the immune balance of AMs and can be used as potential therapeutic drugs.
Similar content being viewed by others


Introduction
Disorders of alveolar macrophage subpopulations (AMs) can cause various lung diseases, such as acute lung injury/acute respiratory distress syndrome (ALI/ARDS) [1], pulmonary fibrosis (PF) [2], asthma [3], and chronic obstructive pulmonary disease (COPD) [4]. ALI/ARDS involves the excessive recruitment of monocytes or neutrophils, which disrupts the vascular endothelial barrier and triggers blood-oxygen exchange disorders [5]. PF results from long-term chronic injury of pulmonary epithelial cells (such as long-term smoking or drug use), and the fibrous scar deposited by repeated repairs hinders ventilation function [6]. AMs have multiple phenotypes involved in the inflammatory response and regulation, tissue remodeling, and antifibrotic processes [7]; however, the molecular mechanism of the regulation of the immune balance by AMs in the lung microenvironment is poorly understood.
Increase in tissue macrophage numbers depends on chemokine-mediated monocyte recruitment and cytokine-mediated proliferation [8]. In the early stages of the pulmonary inflammatory response, AMs with a proinflammatory phenotype represent the major population, destroy the pathogens, and produce associated tissue damage. IL-4 activates the repair phenotype of AMs, which stimulate the fibroblasts to temporarily repair the damaged tissue [9]. Subsequently, AMs with anti-inflammatory and antifibrotic phenotypes become predominant; these AMs respond to IL-10 and IL-4, respectively [10, 11]. Failure to effectively control the AM balance can result in tissue injury or fibrosis.
Regulator of G protein signaling-1 (RGS1) is a regulatory member of the G protein signaling family that links G protein-coupled receptors (GPCRs) and calcium signaling [12]. RGS1 is specifically expressed in macrophages and B cells, and defects in RGS1 cause autoimmune diseases and macrophage-mediated atherosclerosis [13, 14]. However, the regulation of RGS1 in AMs is largely unclear. This study addresses the following issues: (i) characterization of immunophenotypic propensity for inflammatory injury and fibrosis repair in various AMs, (ii) clarification of the mechanism of RGS1-dependent regulation of immune response involving AMs in the lung, and (iii) identification of the effects of the intrapulmonary microenvironment on RGS1-mediated AM immunity in the lung. The resolution of these issues can illustrate the pathogenesis of airway diseases.
Results
RGS1 is related to the recruitment of AMs with different immunophenotypes during lung infection
Ly6C is upregulated in peripheral monocytes in the hematopoietic phase, and the abundance of Ly6C+ monocytes dynamically controls the numbers of tissue macrophages [15]. In the alveoli, Ly6Clow, Ly6Chi, and Ly6C− AMs can be sorted based on Ly6C (Fig. 1a). These AMs have different recruitment sequences during the infections induced by Gram-negative bacterium E. coli. Ly6C− AMs are the first to be recruited (0.5 days) (Fig. 1b) followed by Ly6Chi AMs (0.5–2 days) (Fig. 1c) and Ly6Clow AMs that start to increase at a later time (3–5 days) (Fig. 1d). Although the ratio of Ly6Chi AMs to Ly6Clow AMs is changed during the infection, Ly6C+, and Ly6C− AMs always maintained a ratio of approximately 1:1. Importantly, during the infection, the RGS1 expression in these AMs was downregulated. The role of the downregulation of RGS1 in these AMs is unknown. These subpopulations have similar transcriptional profiles [16]; however, changes in the alveolar immune microenvironment influence the heterogeneity and immunophenotypic properties of AMs. Ly6Chi and Ly6Clow AMs had a proinflammatory (high iNOS and IL-1β expression; Fig. 1e) or an antifibrotic phenotype (high MMP-9 and cathepsin K; Fig. 1g), respectively, and Ly6C− AMs had increased expression of anti-inflammatory cytokines (IL-10 and SOCS3; Fig. 1g) and tissue repair/fibrotic factors (Arg-1 and TGF-β1; Fig. 1h). In a model of ALI and PF induced by lipopolysaccharide (LPS) and bleomycin (BLM) (Supplementary Fig. 1), LPS mobilizes a large number of Ly6Chi and Ly6Clow AMs and downregulates the expression of RGS1; however, these changes do not influence the number of Ly6C− cells and the expression of RGS1 in these cells (Supplementary Fig. S1a–c). Although the recruitment time in the lung is slightly different, the immunophenotype of LY6Chi, LY6Clow, and LY6C− AMs does not change under LPS induction (Supplementary Fig. 1d). On the contrary, BLM induces an increase in LY6C− AMs and downregulates the expression of RGS1 (Supplementary Fig. 1e-g). Meanwhile, the immunophenotype of LY6Chi, LY6Clow, and LY6C− AMs does not change under BLM induction (Supplementary Fig. 1h).

Gating strategy for AMs (CD45+ CD11b-Siglec-F+ CD11c+ Ly6G-) subpopulation and RGS1 expression in Ly6C−, Ly6Clow, and Ly6Chi AMs (a). The frequency of mouse Ly6C− (b), Ly6Clow (c), and Ly6Chi (d) AMs in total lung cells and total AM changes and RGS1 expression within 2 weeks of tracheal administration of E. coli. At 5 days of E. coli infection, the expression of iNOS and IL-1β (e), activities of cathepsins and MMPs (f), expression of IL-10 and SOCS3 (g), and expression of ARG1 and TGFβ1 (h) in LY6Chi AMs (light green), LY6Clow AMs (dark blue), and LY6C− AMs (red) was assessed. Representative flow cytometry plots (left) and statistical graph (right) are shown; each small circle represents a mouse. The data are shown as the mean ± SD; *p < 0.05 and **p < 0.01 by ANOVA, and “n.s.” indicates not statistically significant. The results at each time point correspond to 3 mice.
Flt3 and Tie2 can be used as specific markers of Ly6C+ AMs and Ly6C− AMs
To assess the specificity of RGS1 expression, CD45+ myeloid cells and CD45− endothelial cells were sorted as shown in Fig. 2a, b; the results indicated that many myeloid and endothelial cells do not express RGS1 that is expressed in Ly6C+ monocytes. As shown in Supplementary Fig. 2a, Ly6Chi and Ly6Clow AMs were supplemented by peripheral Ly6C+ monocytes using chimeric mice that received sorted Ly6Chi monocytes from CD45.1+ mice. Ly6Chi monocytes migrated into the lung and were then transformed into Ly6Chi and LY6Clow AMs, and Ly6Clow monocytes were not a complementary source of Ly6C+ AMs (Supplementary Fig. 2b). This result indicates that RGS1 is specifically expressed in AMs regardless of the Ly6C+ or Ly6C− phenotype. Specific knockout of RGS1 in Ly6C+ and Ly6C− cells separately was needed to investigate whether RGS1 is involved in the maintenance of the proportional balance of AMs. However, efficient expression tools for generation of conditional knockout of RGS1 in various AMs are unavailable. Tie2 and Flt3 are expressed in macrophages originating from erythro-myeloid progenitors (EMPs) and hematopoietic stem cells (HSCs), respectively [17, 18]. Brain microglia is known to originate from EMPs [19], and intestinal macrophages are derived entirely from the supplementation of HSCs [20]. Therefore, we used Flt3 and Tie2 to distinguish Ly6C+ and Ly6C− AMs. As shown in Fig. 2c, d, mouse brain macrophages are only of the Ly6C-Tie2+ phenotype, and intestinal macrophages are only of the Ly6C + Flt3+ phenotype. In the lung, Ly6C− AMs are completely Tie2 + Flt3−, and Ly6C+ AMs are Flt3 + Tie2− (Fig. 2e). Therefore, Flt3 and Tie2 can be used as potential markers of Ly6C− AMs and Ly6C+ AMs. We crossed RGS1fl/fl mice (Fig. 2f) with Flt3-creERt2 or Tie2-creERt2 mice to obtain transgenic RGS1fl/fl;Flt3-creERt2(RGS1Flt3) and RGS1fl/fl;Tie2-creERt2(RGS1Tie2) mice with induced conditional knockout of RGS1 in Flt3+ or Tie2+ AMs after treatment with tamoxifen, respectively (Fig. 2g, h).

Gating strategy for AMs (CD45 + Siglec-F + CD11b-CD11c + CD64 + ), CD11b + DCs (CD11b + MHC-II + CD11c + CD24 + CD64-), CD103 + DCs (CD11c + CD103 + CD24 + ), eosinophils (Siglec-F + CD11b + CD11c-), interstitial macrophages (CD11b + MHC-II + CD11c + CD64 + CD24-), Ly6C+ monocytes and macrophages, Ly6C− monocytes and macrophages (CD11b + MHC-II-CD64+/−), and neutrophils (CD11b+ Ly6G+) in the mouse lung and the RGS1 expression in the cells were evaluated (a). Gating strategy for endovascular progenitor (EVP, CD45-CD31lowVEGFRlow), transit-amplifying (TA, CD45-CD31intVEGFRint), and definitive differentiated cells (D, CD45-CD31hiVEGFRhi) and their RGS1 expression were evaluated (b). The expression of Flt3 and Tie2 was evaluated in Ly6C+ or Ly6C− brain macrophages (c), intestinal macrophages (d), and lung macrophages (e). Construction mode diagrams of RGS1fl/fl;Flt3-CreERt2 and RGS1fl/fl;Tie2-CreERt2 (f). One week after tamoxifen treatment, the expression of RGS1 in RGS1Flt3 and RGS1Tie2 mice in Flt3 + Ly6Chi AMs, Flt3 + Ly6Clow AMs, and Tie2 + Ly6C− AMs was detected (g, h). The data are shown as the mean ± SD; *p < 0.05 and **p < 0.01 by ANOVA, and “n.s.” indicates not statistically significant. The data in a-e were derived from 3 independent experiments. The data in g and h are based on 3 mice in each group.
RGS1 influenced the Flt3+/Tie2+ AM ratio and lung immune response
The defects in RGS1 influenced the number and proportion of Flt3+ and Tie2+ AMs during the E. coli infection (Fig. 3a, b and Supplementary Fig. 3a, b). An increase in the proportion of Flt3+ AMs resulted in a decrease in blood oxygenation (Fig. 3c). The defects in Tie2+ AMs apparently improved blood oxygenation (Fig. 3d); however, similar to RGS1Flt3 mice, RGS1Tie2 mice had a reduced survival rate after E. coli infection (Fig. 3e). RGS1Flt3 mice manifested endogenous L-lactic acidosis, and RGS1Tie2 mice manifested exogenous D-lactic acidosis (Fig. 3f, g) suggesting that RGS1Flt3 mice die from hypoxia and RGS1Tie2 mice die from bacterial lung invasion. The proinflammatory and antifibrotic molecules, including iNOS, CaK, and MMP9, were overexpressed in the lung of RGS1Flt3 mice (Fig. 3h) antagonizing the formation of fibroblasts (ɑ-smooth muscle actin (ɑSMA) was used as a marker) and collagen precipitation (Fig. 3h) and aggravating the release of damage-associated molecular patterns (DAMPs) (e.g., mtDNA, cellular proteome, HMGB1, and GAPDH) into BALF (Fig. 3i). In contrast, the lack of proinflammatory and antifibrotic molecules in the lung of RGS1Tie2 mice resulted in extensive fibroblast production and collagen deposition (Fig. 3h). In LPS-induced ALI and BLM-induced PF mouse models, RGS1Flt3 mice manifested more severe lung injury and respiratory distress than that in the wild-type (WT) mice (Fig. 3j), and RGS1Tie2 mice manifested more severe respiratory dysfunction and fibrotic deposition (Fig. 3k) than that in the WT mice. These data indicate that RGS1 maintains the ratio of AMs and that imbalance in Flt3+ or Tie2+ AMs leads to the development of ALI/ARDS and PF, respectively.

The ratios of Flt3+/Tie2+ AMs (a) and PaO2/FiO2 (b) in RGS1Flt3 and RGS1Tie2 mice within 2 weeks of E. coli infection; values were compared to the corresponding Flt3-creERt2 or Tie2-creERt2 control mice. Survival curves (e) for RGS1Flt3 and RGS1Tie2 mice within 4 weeks of E. coli infection. Serum L-lactic acid (f) and serum D-lactic acid (g) levels in RGS1Flt3 and RGS1Tie2 mice at 14 days of E. coli infection. The expression of cathepsin K, MMP9, iNOS, α-SMA, collagen I, and collagen IV in the lung of RGS1Flt3 and RGS1Tie2 mice was compared with that in WT mice at 14 days of E. coli infection (h). The levels of released mtDNA, proteome, HMGB1, and GAPDH in BALF of RGS1Flt3 and RGS1Tie2 mice at 14 days of E. coli infection were determined by dot blotting, Coomassie blue staining, and western blot (i). j Lung H&E staining, lung Masson’s trichrome staining, tissue damage scores, and the blood PaO2/FiO2 ratio in RGS1Flt3 and RGS1Tie2 mice in the LPS-induced ALI model. k Lung H&E staining, lung Masson’s trichrome staining, lung collagen deposition area, and assessment of blood Rsc in RGS1Flt3 and RGS1Tie2 mice in the BLM-induced PF model. The data are shown as the mean ± SD; *p < 0.05 and **p < 0.01 by ANOVA. The data in a–d, j, k are based on 3 mice at each time point; the data in e are based on 8–11 mice used to assess the survival rate; the data in h, i were derived from 3 independent experiments using 3 mice.
RGS1 regulates the Ca2+ signaling-dependent immune response of AMs
Stable primary Flt3+ AMs and Tie2+ AMs lines were generated in vitro to confirm the effect of RGS1 on the immune response of AMs. As shown in Supplementary Fig. 4a, RGS1fl/fl;Flt3-creERt2YFP and RGS1fl/fl;Tie2-creERt2YFP mice were obtained. Primary Flt3+ AMs and Tie2+ AMs isolated from these mice were immortalized using the pGMLV-SV40T lentivirus, and these AMs have reduced RGS1 and associated fluorescent YFP expression after induction with tamoxifen (Supplementary Fig. 4b, c). In vitro, RGS1-deficient Flt3+ AMs and Tie2+ AMs exhibited highly responsive Ca2+ influx (Fig. 4a) and activation of PLC-IP3 phosphorylation signals (Fig. 4b, c) in response to ATP (PLC-IP3-coupled receptor agonist) induction. RGS1 deficiency influenced the immune response of each type of AMs; in Flt3+ AMs, the expression of iNOS and IL-1β in response to LPS induction was enhanced (Fig. 4d), and IL-4 induced an increase in the cathepsin and MMP activities (Fig. 4e). In Tie2+ AMs, RGS1 deficiency enhanced IL-10 and SOCS3 expression (Fig. 4f), and IL-4 induced Arg-1 and TGFβ1 expression (Fig. 4g). These proinflammatory/injury and repair/fibrotic expression patterns and cell proliferation activity depended on the level of calcium ions in the cell culture medium (Supplementary Fig. 5a–f). Moreover, calcium influx and PLC-IP3 signals in peripheral monocytes from RGS1Flt3-deficient mice were highly responsive (Supplementary Fig. 6a, b). In a transwell model of simulation of monocyte transendothelial-epithelial migration (Supplementary Fig. 6c), RGS1-deficient Flt3+ monocytes had higher transendothelial-epithelial migration than that of WT monocytes (Supplementary Fig. 6d). The levels of several calcium-dependent adhesion molecules [21, 22] associated with monocyte migration were also upregulated (Supplementary Fig. 6e). These data indicate that RGS1 regulates the calcium-dependent immune response of AMs.

Oligomycin A was used to block ATP production in YFP+Flt3+ and YFP+Tie2+ AMs in vitro, and 1 μM ATP was used to induce intracellular Ca2+ signaling. YFP images (first column) were captured prior to data acquisition to identify YFP-positive cells (defined by dashed circles), and 340/380 ratio images were captured 30 s before the application of ATP (second column) and 10, 50, 150, and 250 s after stimulation. The right side shows the ATP-induced Ca2+ 340/380 ratio (recorded every 10 s) and the statistical chart of the area under the curve (n = 7–9). Western blot analysis of the phosphorylation levels (b, c) of PLCβ2 and IP3R stimulated by 1 μM ATP at 250 s. Expression of iNOS and IL-1β in YFP+Flt3+ AMs induced by LPS in vitro (d). The activity of cathepsins and MMPs in YFP+Flt3+ AMs induced by rmIL-4 (e). Expression of IL-10 and SOCS3 in YFP+Tie2+ AMs induced by rmIL-10 (f). Expression of ARG-1 and TGFβ1 in YFP+Tie2+ AMs induced by rmIL-4 (g). The data are shown as the mean ± SD; *p < 0.05 and **p < 0.01 by ANOVA. The data in a are based on 3 independent fields of view with 7–9 cells; the data in b–g are based on 3 replicate wells of culture plates for each group.
Exosomes (EXOs) in BALF regulate RGS1 in AMs
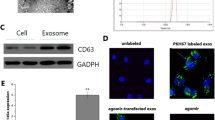
The mechanism of downregulation of RGS1 expression in AMs is unknown. A number of inflammatory mediators and cytokines are present in the pulmonary microenvironment during lung infection. Pathogen-associated molecular patterns (PAMPs) (e.g., LPS), DAMPs (e.g., mtDNA and HMGB1), chemokines (e.g., CX3CL1), proinflammatory cytokines (e.g., TNF-α), and ILs (e.g., IL-4 and IL-10) did not directly downregulate RGS1 expression in Flt3+ and Tie2+ AMs in vitro (Supplementary Fig. 7a, b). However, BALF harvested from infected mice induced downregulation of RGS1 in AMs in vitro (Fig. 5a, b). BALF samples treated with an exosomal uptake inhibitor Annexin V [23] lost the ability to downregulate RGS1 (Fig. 5a, b). This finding suggests that EXOs, which may be mediators in BALF, downregulate RGS1 in AMs. EXOs in BALF were isolated by differential centrifugation (Fig. 5c) and identified and characterized by transmission electron microscopy (TEM) (Fig. 5d) and nanoparticle tracking analysis (NTA) (Fig. 5e). Specific molecules carried by the vesicles isolated by differential centrifugation were assayed (Fig. 5f) (extracellular vesicles (EVs) were pan labeled with ACTN, apoptotic bodies (ABs) were labeled with THBS1, microvesicles (MVs) were labeled with Arf6, and EXOs were labeled with TSG-101 and CD63; Erp72 and LAMP1 were used as negative controls for endoplasmic reticulum (ER) and lysosome contamination). The isolated EVs, including ABs, MVs, and EXOs with variable particle sizes [24], were individually added to cultured Flt3+ and Tie2+ AMs (Fig. 5g). BALF-derived EVs downregulated the expression of RGS1 in Flt3+ and Tie2+ AMs in vitro (Fig. 5h); however, only EXOs downregulated RGS1 in AMs (Fig. 5i), and ABs and MVs did not impact the expression of RGS1 (Fig. 5j, k). EVs often carry protein markers from the cells that secreted them [25, 26], such as CD31 from endothelial cells (EnCs), CD74 from type II alveolar epithelial cells (EpCs-II), PDPN from EpCs-I, and β-tubulin from ciliated cells (CC), as shown in Fig. 5f. EXOs that can affect the expression of RGS1 in AMs may be mainly derived from CD74+ EpCs-II and CD31+ EnCs. Comparison of the number and size of EVs in BALF of RGS1flt3 and RGS1Tie2 mice did not demonstrate significant differences with the characteristics of EVs from the isotype control negative mice (Supplementary Fig. 3c–f).

BALF samples were collected from mice after 24 h of E. coli infection, centrifuged to remove cells and debris, and added to in vitro culture of YFP+Flt3+ AMs and YFP+Tie2+ AMs; additionally, BALF samples were treated with an exosome uptake inhibitor, Annexin V. Flow cytometry was used to detect the expression of RGS1 in YFP+Flt3+ AMs and YFP+Tie2+ AMs (b). Differential centrifugation was used to isolate EVs (c) from the BALF samples, and 8 centrifugation gradients were used. EVs obtained at 12,000–100,000 × g were combined and evaluated by TEM (d), and the particle size of the EVs was measured by NTA. Western blot was used to analyze the surface markers of EVs (f) obtained by differential centrifugation. BALF samples were collected from mice after 0, 5, or 14 days of E. coli infection, and EVs isolated by differential centrifugation were separated into the total EV group (h), EXO group (i), AB group (j) and MV group (k). The separated groups were added to YFP+Flt3+ and YFP+Tie2+ AMs in vitro, and flow cytometry was used to detect the expression of RGS1 (g). The data are shown as the mean ± SD; *p < 0.05 and **p < 0.01 by ANOVA. All data were derived from 3 independent experiments using 3 mice.
EXOs derived from EpCs-II and EnCs inhibit expression of RGS1 in Flt3+ and Tie2+ AMs
EpCs-II and EnCs are important non-professional immune cells in the lung that are activated by soluble CD14 (sCD14) [27, 28]. EpCs-II and EnCs can directly respond to induction with sCD14 by secreting a large number of EVs with particle sizes ranging from 100 to 400 nm (Fig. 6a, b). EXOs isolated from EVs derived from EpCs-II and EnCs carry specific markers, CD74 and CD31, respectively (Fig. 6c, d). We used ELISA [29] (Fig. 6e) to indirectly detect the levels of exosomal CD74 and CD31 (Fig. 6f, g). ELISA was also used to detect specific EXOs in BALF samples as shown in Fig. 6h. The levels of CD73+ and CD31+ EXOs were increased in BALF of E. coli-infected mice. To determine whether CD73+ and CD31+ EXOs have uptake specificity, PKH67-labeled EXOs (Fig. 6i) were added to the culture medium of Flt3+ or Tie2+ AMs. As shown in Fig. 6j, k, Flt3+ and Tie2+ AMs were able to specifically uptake CD74+ and CD31+ EXOs secreted by EpCs-II and EnCs, respectively. Importantly, these specific EXOs inhibited the expression of RGS1 in Flt3+ and Tie2+ AMs in vitro (Fig. 6l, m). As expected, Flt3+ and Tie2+ AMs treated with CD74+ or CD31+ EXOs exhibited highly responsive Ca2+ influx and high expression of inflammatory and fibrotic cytokines (Supplementary Fig. 8a–e). Thus, EXOs in BALF are important regulators of the AM immune balance. EnC-derived CD31+ EXOs enhanced the anti-inflammatory and profibrotic effects of Tie2+ AMs, and EpC-II-derived CD74+ EXOs enhanced the proinflammatory and antifibrotic effects of Flt3+ AMs. Interestingly, loss of EpCs-II and EnCs is a known pathological feature of PF and ALI [30, 31], respectively. Whether these losses lead to disordered EXO regulation in the microenvironment requires further verification.

After sCD14 treatment of EpCs-II (a) or EnCs (b) in vitro for 12 h, EVs were isolated from the medium by differential centrifugation, and the size of EVs obtained at 12,000–100,000 × g was evaluated by NTA; EVs isolated at 100,000 × g were evaluated using TEM. Western blot was used to analyze sCD14-treated EpCs-II (c) and EnCs (d) at 0, 1, 4, 8, 12, and 24 h for EV surface markers, including ACTN, EXO markers TSG101 and CD63, EnC marker CD31, EpC-II marker CD74, EpC-I marker PDPN, ER marker Erp72, and lysosomal marker LAMP1. A schematic diagram (e) shows the method of detection of CD31+ EXOs and CD74+ EXOs by ELISA. EXOs were isolated from the medium of sCD14-induced EpC-II or EnC at 12 h and quantified based on protein content; linear relationships between the content of CD31 (f) or CD74 (g) and the total protein content were determined by ELISA. The content of CD31+ EXOs and CD74+ EXOs in BALF at 0, 1, 3, 5, and 7 days in E. coli-infected mice was determined by ELISA (h). EpC-II and EnC EXOs induced by sCD14 treatment for 12 h were fluorescently labeled with PKH67, added to Flt3+ or Tie2+ AMs, and defined as the Annexin V rescue group. After 24 h, flow cytometry was used to detect the fluorescence intensity at 504 nm (i–k). Western blot was used to analyze the effects of EXOs from EpCs-II (l) and EnCs (m) induced by sCD14 on the RGS1 expression in Flt3+ and Tie2+ AMs at 0, 1, 4, 8, 12, and 24 h. The data are shown as the mean ± SD *p < 0.05 and **p < 0.01 by ANOVA. All data were derived from 3 independent experiments with 3 replicate wells of culture plates.
Imbalance of CD31+ and CD74+ EXOs causes ALI/ARDS and PF
In ALI/ARDS and PF, EnCs and EpCs, respectively, are often severely damaged. Drugs or treatments that improve EnC or EpC functions can improve the symptoms of ALI and PF, respectively [32, 33]. As shown in Fig. 7a, b, LPS and BLM induce EnC and EpC-II proliferation abnormalities, respectively. As expected, high concentrations of LPS and BLM resulted in a lack of CD31+ EXOs and CD74+ EXOs in BALF of ALI or PF mice (Fig. 7c, d), respectively.

Low (0.01 mg/kg) and high doses of LPS (1.5 mg/kg) were administered intratracheally, and 1 mg of BrdU was injected intraperitoneally 20 h later. After 1 day, the cell cycle disruption mediated by LPS in sorted EnCs was observed by flow cytometry (a). Low (0.125 U/kg) and high doses of BLM (4 U/kg) were administered intratracheally, and BrdU was injected. After 14 days, flow cytometry was used to observe the effects of BLM on the cell cycle in sorted EpC-II to evaluate toxicity (b). The levels of CD31+ EXOs and CD74+ EXOs in BALF were measured 1 day after the tracheal administration of various doses of LPS (c). The levels of CD31+ EXOs and CD74+ EXOs in BALF (d) were measured 14 days after the tracheal administration of various doses of BLM. BALF samples were collected from 72 ARDS/ALI patients who were defined according to the Berlin criteria, 17 PF patients who were defined according to the ERS criteria, and 20 healthy controls. The RGS1 expression in Flt3+ AMs (e) and Tie2+ AMs (g) in BALF was detected by flow cytometry. Correlations between CD31+ EXOs (f) and CD74+ EXOs (h) in BALF with PaO2/FiO2 were compared in ARDS/ALI patients, PF patients, and healthy controls. The ratio of Flt3+ AMs to Tie2+ AMs in BALF of ARDS/ALI patients, PF patients, and healthy controls was detected (i). CD31+ EXOs (500 μg/kg) and CD74+ EXOs (1 mg/kg) were administered by tracheal instillation in mice, and the expression of RGS1 in Tie2+ AMs and Flt3+ AMs was examined (j). CD31+ EXOs were administered to high-dose LPS-induced ALI mice. The Tie2+/Flt3+ AM ratio (k) was measured at specified time points to evaluate the blood oxygenation index (l) and lung tissue damage (m, n). CD74+ EXOs were administered in the high-dose BLM-induced PF model mice, and the Tie2+/Flt3+ AM ratio (o) was measured at specified time points. Respiratory system compliance (p), pulmonary hydroxyproline content (q), and H&E and Masson’s trichrome staining (r) were assessed. The data are shown as the mean ± SD; *p < 0.05 and **p < 0.01 by ANOVA or Pearson correlation analysis. All data of the mouse experiments included 3 mice at each time point.
In collected BALF samples from 72 patients with ALI/ARDS, extensive epithelial injury could have induced a significant reduction in the RGS1 expression in Flt3+ AMs (Fig. 7e). Moreover, the number of CD31+ EXOs in BALF was proportional to the PaO2/FiO2 ratio (Fig. 7f), and the ratio of Flt3+ AMs was significantly upregulated (Fig. 7i) compared with that in the healthy subjects. On the other hand, apparently due to endothelial cell injury, the expression of RGS1 in Tie2+ AMs in BALF from 17 patients with PF was significantly decreased (Fig. 7g), and the number of CD74+ EXOs was proportional to the PaO2/FiO2 ratio (Fig. 7h). All these changes could have resulted in significant upregulation of the ratio of Tie2+ AMs in PF patients (Fig. 7i) compared to that in the healthy subjects. These results indicate that ALI/ARDS and PF patients lack CD31+ EXOs and CD74+ EXOs in the pulmonary microenvironment, respectively, leading to obstructed blood-oxygen exchange and respiratory failure.
To assess whether EXOs have potential therapeutic effects, CD31+ or CD74+ EXOs were administered by tracheal instillation in mice. As shown in Fig. 7j, CD31+ or CD74+ EXOs downregulated the expression of RGS1 in Tie2+ AMs or Flt3+ AMs in vivo, respectively. Due to severe endothelial damage, a high dose of LPS induced a gradual downregulation of the ratio of Tie2+ AMs to Flt3+ AMs (Fig. 7k) that resulted in a decrease in the PaO2/FiO2 ratio (Fig. 7l) and severe lung injury (Fig. 7m, n). The treatment with CD31+ EXOs increased the Tie2+ AM proportion, reduced lung injury, and improved blood oxygenation. In the BLM-induced PF model, supplementation with CD74+ EXOs resulted in a normal Tie2+ AM proportion and a significant improvement in fibrosis progression (Fig. 7o–r). To detect the effect of a reduction in the number of CD74+ EXO or CD31+ EXOs on ALI or PF, mice were treated with CD74 or CD31 antibodies. The treatments resulted in the inhibition of the release of CD74+ EXOs or CD31+ EXOs in BALF induced by low doses of LPS or BLM (Supplementary Fig. 9a, b) and subsequent reduction in the number of Flt3+ AMs and Tie2+ AMs in BALF in mice (Supplementary Fig. 9c) thereby aggravating high-dose LPS- or BLM-induced ALI (Supplementary Fig. 9f) or PF (Supplementary Fig. 9g–i), respectively. Moreover, the administration of Flt3 and Tie2 antibodies to mice only slightly reduced the number of Flt3+ AMs or Tie2+ AMs (Supplementary Fig. 9l). An increase in the number of CD74+ EXOs or CD31+ EXOs was detected (Supplementary Fig. 9j, k); however, these changes had little effect on the severity of ALI and PF induced by high doses of LPS or BLM (Supplementary Fig. 9m–r).
miR-223 and miR-27b-3p inhibit the expression of RGS1
BALF-EVs contain a large number of microRNAs (miRNAs) [34]. Thus, we identified miRNAs that target the inhibition of RGS1 expression. Acridine orange (AO) was used to label RNA in EXOs (Fig. 8a). Flt3+ and Tie2+ AMs were specifically able to uptake AO-labeled RNA from EXOs secreted by EpCs-II and EnCs, respectively (Fig. 8b). Dicer is an enzyme necessary for miRNA synthesis [35]. Interference with Dicer expression in EpCs-II or EnCs (Fig. 8c) resulted in the loss of the ability of secreted EXOs to inhibit the expression of RGS1 in Flt3+ and Tie2+ AMs, respectively (Fig. 8d). This result indicated that miRNAs carried by EXOs inhibit the expression of RGS1 in AMs and suggests that the expression of miRNAs that inhibit RGS1 expression can be targeted as shown in Fig. 8e and g. miR-27b-3p and miR-223 are potential RGS1 inhibitors. The presence of miR-223 was detected in CD31+ EXOs secreted by EnCs (Fig. 8f), and the presence of miR-27b-3p was detected in CD74+ EXOs secreted by EpCs-II (Fig. 8h). These findings suggest that miR-27b and miR-223 are targeted to inhibit the expression of RGS1. To validate this hypothesis, miR-223 or miR-27b-3p mimics were cotransfected with an RGS1 promoter-bearing luciferase plasmid into 293T cells (Fig. 8i), and both miR-223 and miR-27b-3p significantly inhibited the initiation of RGS1 expression; these effects were rescued by miR-223 and miR-27b-3p inhibitors, respectively (Fig. 8j–k). In addition, the PAR-CLIP assay was used to detect the binding of miRNA-223/27b to the RGS1 3′-UTR (Fig. 8l). As shown in Fig. 8m, n, miR-27b and miR-223 significantly increased the recruitment of RGS1 to RNA-induced silencing complex (RISC). Importantly, miRNA-223 and miRNA-27b influenced RGS1 expression in Flt3+ and Tie2+ AMs in vitro (Fig. 8r, s) and in vivo (Fig. 8q, r). Moreover, miRNA-223 and miRNA-27b-3p were able to influence the expression of Ca2+-dependent inflammatory/fibrotic molecules in Flt3+ AMs and Tie2+ AMs (Supplementary Fig. 10a–e).

EpC-II-derived CD74+ EXOs and EnC-derived CD31+ EXOs were labeled with acridine orange and added to YFP+Flt3+ or YFP+Tie2+ AMs. After 24 h, quantification of the acridine orange content (650 nm) in the cells was performed by flow cytometry (a, b). shRNA that interfered with Dicer expression was transfected into EpCs-II or EnCs. Secreted CD31+ EXOs and CD74+ EXOs were collected and added to YFP+Flt3+ or YFP+Tie2+ AMs, and the effects on RGS1 expression were detected (c, d). The TargetScan database was used to predict the target sites in the RGS1 3′-UTR (e, g). qPCR was used to detect the copy number of miR-223 (f) and miR-27b-3p (h) in CD31+ EXOs and CD74+ EXOs. The design of the mutated luciferase reporter gene for the predicted target in the 3′-UTR of the RGS1 mRNA transcript is shown (i). Luciferase reporter genes were cotransfected with miR-223 (j) or miR-27b-3p (k) mimics/inhibitors into 293 T cells, and the luciferase activity of the cells was measured by a microplate reader. The PAR-CLIP method (l) was used to detect the enrichment in miR-223 (m) or miR-27b-3p (n) of the RGS1 3′-UTR associated with RISC. miR-223 or miR-27b-3p mimics/inhibitors were transfected into YFP+Flt3+ or YFP+Tie2+ AMs, and the effects on the expression of RGS1 were examined (o, p). The NLE nanoparticle delivery system was used to deliver miR-223 or miR-27b-3p mimics/inhibitors to mice. After 48 h, flow cytometry was used to detect the expression level of RGS1 in mouse lung Flt3+ AMs and Tie2+ AMs (q, r). The data are shown as the mean ± SD; *p < 0.05 and **p < 0.01 by ANOVA. The data in a–d, o, p correspond to 3 replicate wells of culture plates in each group; the data in e–m were derived from 3 independent experiments. The data in q, r are based on 3 mice in each group.
Discussion
Fate-mapping studies have shown that tissue-resident macrophages originate from at least three macrophage precursors, including yolk sac-derived macrophages (YS-Macs) [36], fetal liver monocytes (FL-MOs) [37], and bone marrow monocytes (BM-MOs). Brain microglia, liver Kupffer cells, and skin Langerhans cells are exclusively derived from YS-Macs [20, 38, 39]. Intestinal FL-MO-derived macrophages are completely replaced by BM-MOs after birth [20]. Pulmonary- [40] and cardiac-resident macrophages [41] originate from FL-MOs and are long-lived cells that are gradually replaced by BM-MOs over time. The impact of macrophages from various sources on local immunity has been a matter of controversy because the transcriptional profiles of these macrophages are similar [16]. The results of our study indicate that Ly6C can be used to classify macrophages into three types of AMs with different immune predispositions in the lung of adult mice unlike the standard macrophage classification method [42]. Tie2 can label almost all brain microglia derived from FL-MOs and Ly6C+ AMs, and Flt3 labels BM-MO-derived intestinal macrophages and Ly6C− AMs. Therefore, in the lung, Flt3 and Tie2 can effectively distinguish between AMs originating from BM-MOs and FL-MOs.
In WT mice, the ratio of Flt3+ AMs to Tie2+ AMs remains essentially constant even during infection. Although Flt3+ AMs with a LY6Chi or LY6Clow phenotype are different, both subpopulations are supplemented by peripheral Ly6C+ monocytes [43]. Survival of mice with lethal pneumonia depends on whether Flt3+ and Tie2+ AMs are balanced and whether RGS1 expression can be continuously downregulated in both types of AMs. The roles of RGS1 in the macrophage immune response and regulation remain unclear. A single defect in RGS1 in Flt3+ or Tie2+ AMs can cause severe lung disease in response to E. coli infection. In the lung of Rgs1flt3 mice, the proportion of Flt3+ AMs with a proinflammatory phenotype is increased resulting in an acute decline in blood oxygenation and death from respiratory acidosis. The proportion of Tie2+ AMs in Rgs1Tie2 mice is abnormally high, and high immunosuppressive activity and fibrosis resulted in low resistance against bacterial infection leading to death of the animals from sepsis. Rgs1flt3 or Rgs1Tie2 mice developed severe lung injury or fibrosis in response to treatment with relatively low doses of LPS or BLM, respectively. Defects in RGS1 resulted in high responsiveness of calcium signaling in AMs, which influenced a calcium-dependent increase in AM numbers and the expression of inflammatory/fibrotic molecules.
During E. coli lung infection, RGS1 expression in Tie2+ and Flt3+ AMs was downregulated, which maintained the balance between Tie2+ and Flt3+ AMs. Our results indicate that this balance is maintained by EXOs in the pulmonary microenvironment. EVs have been shown to play a role in the immune environment in the lungs, including vesicles secreted by AMs carrying SOCS1 and SOCS3 that are taken up by pulmonary epithelial cells to inhibit inflammatory signals [44]. AMs uptake leukotrienes (LTs) from EVs secreted by epithelial cells to induce chemotaxis and proliferation that aggravate asthma-type lung injury. miRNAs, such as miR-223, are active in the EVs in the pulmonary microenvironment and are involved in the immune regulation in the lung [45, 46]. Our study showed that EXOs isolated from BALF derived from mouse lung during the infection inhibit RGS1 expression in Flt3+ and Tie2+ AMs; this effect is mediated by miR-27b-3p and miR-223 carried by EXOs, respectively, to regulate the RGS1-mediated calcium signaling response confirming that pulmonary cells exchange information using EXOs. Importantly, this extracellular regulation is precise and specific. EpCs-II and EnCs release CD74+ and CD31+ EXOs, respectively, targeting Flt3+ and Tie2+ AM RGS1 expression. The number of CD74+ and CD31+ EXOs in the pulmonary environment are associated with two difficult to treat pulmonary diseases, ALI/ARDS and PF.
Extreme conditions, such as ischemia-reperfusion injury [47] and ALI/ARDS [5], damage endothelial barrier, which is responsible for the excessive infiltration of a number of inflammatory cells [48]. A lack of EnC-derived CD31+ EXOs reduces the levels of anti-inflammatory mediators and repair Tie2+ cells thus increasing the severity of lung damage. EpCs-II remains proliferating in response to low doses of BLM to complement damaged EpCs-I. High doses of BLM induce extensive damage to mouse pulmonary EpCs, and a lack of CD74+ EXOs originating from EpCs-II inhibits the supplementation of Flt3+ AMs thus suppressing expedient elimination of bacterial infection and fibrotic tissue. Patients with PF often manifest high immunosuppression in the pulmonary environment [49]. There is increasing evidence that EXOs can be used as carriers for therapeutic drugs, such as small RNA or DNA molecules [50]. EXOs endogenously produced by the host cells are less immunogenic and/or less toxic than exogenous nanoparticles [51]. In the present study, we administered EnC-derived CD31+ EXOs or CD74+ EXOs by tracheal instillation to alleviate LPS- or BLM-induced ALI/ARDS or PF, respectively. This study suggests that EXOs can be used as potential drugs for the treatment of immune imbalance in pulmonary diseases.
Materials and methods
Subjects
BALF samples from patients with ALI/ARDS or PF were obtained from the intensive care unit of the YiJishan Hospital affiliated with Wannan Medical College. Screening for ALI/ARDS was performed according to the Berlin definition criteria [52]. PF patients were screened according to the American Thoracic Society (ATS) and the European Respiratory Society (ERS) criteria [53]. The clinical characteristics of all patients are listed in Supplementary Tables S1 and S2. This experimental protocol was approved by the Medical Ethics Review Committee of Wennan Medical College. Informed consent was obtained from all subjects. The study conformed to the principles outlined in the Helsinki Declaration. A bronchial fiberscope was used to repeatedly perfuse the lung segments of the ALI/ARDS and PF patients with sterile saline to obtain 25 ml of BALF, which was centrifuged at 1500 × g for 5 min; the supernatant was collected and stored at −80 °C before isolation of BALF-EXOs. The cell pellet was resuspended in PBS containing 1% BSA and passed through a filter to prepare a single-cell suspension, and flow cytometry was used to detect the ratio of Flt3+ AMs to Tie2+ AMs. In addition, 10 ml of peripheral blood was collected from all subjects, and the oxygenation index (PaO2/FiO2) in the whole blood was measured within 30 min using a blood gas analyzer (Radiometer, Denmark).
Mice
Exon 2 of the RGS1 gene was deleted via Cre-mediated recombination using the Cre/loxP system to generate a conditional RGS1-mutated allele mouse. Exon 2 encodes a binding domain that recognizes the B/R4 domain; thus, its deletion results in defective expression and function of RGS1. Two RGS1flox/+ mice were bred to obtain RGS1fl/fl homozygous mice. To obtain a conditional RGS1-deficient mouse, RGS1fl/fl mice were bred with Flt3-creERt2 or Tie2-creERt2 mice to produce RGS1flox/+;Flt3-creERt2 and RGS1flox/+;Tie2-creERt2 mice, respectively. Further backcrossing resulted in RGS1fl/fl; Flt3-creERt2 (RGS1Flt3) and RGS1fl/fl;Tie2-creERt2 (RGS1Tie2) mice. To induce genetic recombination in Flt3-creERt2 or Tie2-creERt2 mice, tamoxifen was dissolved in warm corn oil and administered subcutaneously twice at 48 h intervals, and RGS1 expression was detected after one week. To sort Flt3+ and Tie2+ AMs for in vitro culture and functional experiments, RGS1Flt3 or RGS1Tie2 mice were crossed with Rosa26-LSL-eYFP mice to obtain tamoxifen-induced RGS1 knockout YFP-expressing mice, Rosa26-LSL-eYFP;RGS1fl/fl;Flt3-creERt2 (RGS1fl/fl;Flt3-creERt2YFP) or Rosa26-LSL-eYFP;RGS1fl/fl;Tie2-creERt2 (RGS1fl/fl;Tie2-creERt2YFP) mice, respectively. All mice in experimental were 6–8 weeks of age, and their sexes and weight were randomization. Pregnant mice and lactating mice were excluded. Flt3-creERt2 and Tie2-creERt2 mice were a gift from Dr. Xiaai Zhang (Central South University Xiangya School of Medicine) and Prof. Tao Song (the Affiliated Honghui Hospital of Xi’an Jiaotong University), respectively. Rosa26-LSL-eYFP mice were purchased from The Jackson Laboratory. C57BL/6J-CD45.1 mice were obtained from Vital River Laboratory (Beijing, China). All protocols of this study involving animals were approved by the institutional animal care and use committee (IACUC) and performed in accordance with approved guidelines.
Inducible mouse model
E. coli, LPS, and BLM were administered by tracheal instillation. E. coli (ATCC 25922) was cultured in TSB medium (Merck) and grown to an exponential phase (OD600 = 1) at 37 °C, and each mouse was intratracheally inoculated with 50 μl of E. coli bacterial solution. LPS and BLM were purchased from Sigma-Aldrich (USA) and administered at the indicated concentrations. Mice were anesthetized with a mixture of 100 mg/kg ketamine (Ford Dodge) and 10 mg/kg xylazine (Shenandoah). Mice were euthanized at the indicated time points after E. coli, LPS, or BLM administration, and lung tissue, peripheral blood, and BALF samples were collected for the detection of pulmonary inflammatory injury and fibrosis. For respiratory function assessment in acute injury, the mice were anesthetized, and 200 μl of the blood was collected by intracardiac puncture; the PaO2/FiO2 ratio was determined in the blood by a blood gas analyzer within 30 min. Respiratory function testing in mice with PF was performed using a flexiVent pulmonary system (SCIREQ Inc.) to evaluate respiratory system compliance (Crs) by Snapshot-150 perturbation. Pathological assessment of pulmonary tissue damage involved tracheal intubation after anesthesia and lung perfusion using 1 ml of 10% formalin. The lung tissue was removed, fixed overnight, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) or Masson’s trichrome. Inflammation and injury in the lung tissue sections were scored separately by three experienced pathologists according to the following range: normal (0–4), mild (5–8), moderate (9–12), and severe (13–15), and a single-blind method was used to read the pathological results. The calculation of the collagen deposition area assessed by Masson’s trichrome staining was performed using ImageJ software. For the detection of hydroxyproline, lung tissue was mixed with alkaline hydrolysate, boiled, adjusted to pH 6.5, and centrifuged at 4000 × g. The supernatant was collected and tested using a kit according to the manufacturer’s instructions (JianCheng Bioengineering Institute, Nanjing). Detection of L-lactic acid and D-lactic acid levels in the mouse serum was performed using a kit according to the manufacturer’s instructions (AAT Bioquest, USA). For BALF acquisition, the trachea was rinsed three times with 0.8 ml of sterile saline, the sample was centrifuged at 1500 rpm for 5 min, and the supernatant was collected for extraction and identification of EVs.
AM detection and primary culture
After mice were anesthetized, intracardiac perfusion was performed using 20 ml of PBS-heparin. Lung tracheal intubation was performed, 0.6 ml of 1% low-melting-point agarose heated to 42–45 °C was injected to fill the lungs, and the entire lung tissue was carefully dissected. The lungs were cut into the fragments of <1 mm3, which were homogenized in 10 ml of Dulbecco’s modified Eagle medium (DMEM) (Thermo Fisher Scientific, Inc.) containing 2 mg/ml collagenase A (Roche) and 1X DNase I (Sigma) for 30 min at 37 °C. The digestion was terminated by adding 2 ml of fetal bovine serum (FBS), and the digested solution was passed through 70, 40, and 20 μm nylon mesh filter membranes and centrifuged at 2000 rpm at 4 °C for 5 min to obtain cell pellet, which was resuspended in staining buffer containing 1% BSA. The single-cell suspensions were blocked with anti-mouse CD16/32 Fc (eBioscience) for 10 min. For AM detection, 100 μL of fluorophore-conjugated anti-mouse antibodies at the recommended dilution were incubated in the dark for 30 min, and a flow cytometer (BD Biosciences) was used to sort AMs (CD45+ CD11b- Siglec-F + CD11c + Ly6G-) for further testing. The sorted AMs were incubated for 24 h at 37 °C and 5% CO2 in RPMI 1640 medium supplement with 10% FBS to detach the labeled antibodies. The same method was used for the detection of macrophages in the brain and intestine. Information on the antibodies used for flow cytometry analyses and other experiments is shown in Supplementary Table S3.
EpC-II and EnC primary cultures
Primary EpCs-II were sorted and cultured according to the protocol of Sinha et al. [54]. Briefly, mouse lung tissue was perfused, exfoliated, digested, and filtered to prepare a single-cell suspension, and magnetic purification and FACS cell sorting were used to obtain high-purity EpCs-II (Lin-MHCII+EpCAM+). EpCs-II were cultured in the keratinocyte-SFM medium containing 1 mM isoproterenol, 10 μg/ml fibronectin, 30 μg/ml vitamins, and 10 μg/ml BSA. For EnC sorting, preprocessed tissue and single-cell procedures were performed as indicated for EpCs-II. CD45 and CD34 were used to sort all subpopulations of vascular EnCs [55]. Isolated vascular EnCs were cultured in the EnC-specific medium (PriCells). To obtain a large number of secreted EXOs from stable EpCs-II and EnCs, sorted cells were subjected to an immortalization treatment using the pGMLV-SV40T lentivirus (Genomeditech, USA).
Detection of MMP and cathepsin activities in AMs
To evaluate the activities of cathepsins (ProSsense-680; excitation wavelength: 680 ± 10 nm, emission wavelength: 700 ± 10 nm; VisEn Medical) and MMPs (MMPSense-680; excitation wavelength: 680 ± 10 nm; emission wavelength: 700 ± 10 nm; VisEn Medical), ProSense-680 and MMPsense-680 probes were dissolved in 150 μl of PBS and injected into mice via the tail vein at the doses of 5 nmol 24 h before the animals were sacrificed. Flow cytometry was used to detect the fluorescence intensity at indicated emission wavelengths.
EXO isolation from BALF and culture medium
EVs were obtained by differential centrifugation [56]. Human BALF, mouse BALF, and serum-free cell culture media were collected and centrifuged at 300 × g for 5 min to remove exfoliated inflammatory cells and dead cells. To isolate three subpopulations of EVs, including ABs, MVs, and EXOs, for subsequent experiments, the supernatant after the removal of the cells and cell debris was centrifuged at 3000 × g for 10 min to precipitate ABs. The AB-depleted supernatant was centrifuged at 12,000 × g for 1 h to obtain MVs. Finally, the supernatant was ultracentrifuged at 100,000 × g for 12 h to precipitate EXOs. Purified EXOs were added dropwise onto a formvar carbon-coated nickel grid and stained with 2% uranyl acetate; the grid was air-dried and observed using a JEM-1230 transmission electron microscope (Joel). The sizes and concentrations of EVs purified from BALF samples or cell culture supernatants were determined using a NanoSight NS300 instrument (Malvern Instruments). EVs were quantified using the BCA assay (Thermo Fisher) and used in the subsequent experiments as a reference control.
EXO uptake analysis
Isolated EXOs were labeled with PKH67 (Sigma) and AO (Thermo Fisher) according to the manufacturer’s recommendations. Prior to EXO labeling, PKH67 was centrifuged at 16,000 × g for 5 min to remove PKH67 aggregates. Primary AMs were exposed to PKH67- or AO-labeled EXOs overnight. Then, EXOs attached to the cell surface were thoroughly washed with an acidic buffer (pH 2.2); the cells were fixed, and the fluorescence intensity of PKH67 (504 nm) and AO (650 nm) in AMs was measured by flow cytometry.
ELISA
To detect CD31 and CD74 on the surface of EXOs, an adsorption immunoassay ELISA plate (96 wells) (Thermo Fisher) was coated with 0.25 μg/well of monoclonal anti-mouse or anti-human CD31 or CD74 antibodies overnight at 4 °C. Free binding sites were blocked for 1 h at room temperature with 200 μl of blocking buffer (Thermo Fisher). Then, 100 μl of EXOs samples with various protein concentrations (determined by the BCA method) was added to the wells. After overnight incubation at 4 °C, a monoclonal anti-CD31 or anti-CD74 antibody was added to each well and incubated for 1 h at room temperature. After washing, an HRP-labeled goat anti-rabbit secondary antibody was added for 2 h, a TBM substrate was used for color development, and the plate was read at 450 nm using a BioTek plate reader. Standard curves were prepared using recombinant CD31 (R&D Systems, cat # 3628-PC-050 and ADP6-200) and CD74 proteins (R&D Systems, cat # AF7478 and AF3590). A recombinant CD68 protein (R&D Systems, cat # MAB20401) was used as a negative control to verify detection specificity.
Detection of abnormal proliferation of EnC and EpC-ll
5-Bromo-2′-deoxyuridine (BrdU) (Sigma-Aldrich) labeling was used to detect irreversible damage to mouse EnCs and EpCs-ll induced by LPS or BLM. Various doses of LPS or BLM were administered by bronchial instillation. The treated mice were sacrificed on day 1 and day 14 for injury detection, and 1 mg of BrdU was injected intraperitoneally 3 h before sacrifice. EnCs and EpCs-ll were isolated according to established methods, and a BrdU staining kit (eBioscience), a flow cytometry FITC kit, and propidium iodide (PI) (Sigma-Aldrich) were used to analyze the cell cycle according to the manufacturer’s instructions.
Tracheal instillation of EXOs
AMs can efficiently uptake EXOs administered by tracheal instillation [57]. EnCs and EpCs-II were treated with 10 ng/ml recombinant mouse sCD14 (Abcam, ab207103), and EXOs were extracted after the supernatant was collected and quantified by the BCA method. For the LPS-induced ALI model, 100 μg of CD31+ EXOs were resuspended in 200 μl of physiological saline, and mice were treated with tracheal instillation at a dose of 25 μl/25 g. For the BLM-induced PF model, 200 μg of CD74+ EXOs were resuspended in 200 μl of physiological saline, and mice were treated with tracheal instillation at a dose of 25 μl/25 g.
qPCR
Total RNA was extracted from isolated AMs or monocytes using TRIzol (Qiagen). The concentration of the purified RNA was measured using a NanoDrop Lite spectrophotometer (Thermo Scientific); cDNA was generated via reverse transcription (Thermo Scientific), and qPCR based on SYBR Green was performed to detect specific mRNAs. GAPDH was used as a reference gene. For the detection of miRNAs in EXOs, total miRNA was extracted using an RNeasy small RNA extraction kit (Qiagen); miRNAs were reverse transcribed using specific neck loop primers and quantified by qPCR. The copy number of miRNAs in EXOs was calculated based on the absolute quantification method [58], and the results were normalized to the number of vesicles determined using a reference control. All primer sequences are shown in Supplementary Table 4.
Western blotting
Protein expression assays were performed as described previously [59].
Luciferase reporter assays, overexpression/interference plasmids, and mimics/inhibitors
The binding site sequences of miR-27b-3p and miR-223 specific for the 3′-UTR of RGS1 were predicted using the miRNA-binding target prediction website (http://www.targetscan.org/). The luciferase expression plasmid pGL3-luc-RGS1 3′-UTR, the corresponding plasmid with a mutant sequence, and the Dicer shRNA interference plasmid were generated by Genepharm (Shanghai, China). miR-27b-3p and miR-223 mimics and inhibitor sequences were synthesized by Sangon (Shanghai, China). All sequence information is shown in Supplementary Table 4. The 3′-UTR-targeted assay evaluating the inhibition of RGS1 by miR-27b-3p or miR-223 was performed in 293 T cells (American Type Culture Collection). 293 T cells were cultured in DMEM containing 10% FBS and 2 mM L-glutamine at 37 °C in 5% CO2, and 2 × 104,293 T cells were seeded in the wells of 96-well plates. Scrambled miRNAs, mimics, or inhibitors were cotransfected into 293 T cells with the corresponding luciferase plasmids using Lipofectamine® 3000 (Thermo Fisher) according to the instruction manual. After 24 h, the fluorescence intensity corresponding to luciferase expression was detected using a Multiskan Sky microplate spectrophotometer (Thermo Fisher). miRNA-mimics or inhibitor were administered in vivo as described in our previous study [59].
PAR-CLIP
The PAR-CLIP experiment was performed according to a previously published method [59].
Imaging and measurement of the intracellular Ca2+ concentrations
Intracellular Ca2+ concentrations were estimated from the ratio of Fura-2 fluorescence intensities excited at 340 nm and 380 nm. Basal ATP production in YFP-positive AMs was blocked by 1 μM oligomycin A (Sigma). After 6 h, the cells were loaded with 4 μM Fura-2 (Beyotime, China) for 40 min at room temperature, and images were captured using a Nikon inverted microscope (400× oil immersion objective, N.A. = 1.4) for 30 s before 1 μM ATP stimulation of AMs. The cells were alternately excited at 340 and 380 nm using a single-frequency microscope filter set, and emission was monitored with 409 nm single frequency and 510/84 nm dichroic filters. The EM gain of the EMCCD camera was set to a linear dynamic range with exposure times ranging from 50 ms to 150 ms to acquire images at 10 s intervals. The area under the curve of the fluorescence intensity ratio excited at 340 nm/380 nm from 0 to 600 s was used as the total cumulative value of intracellular calcium responsiveness.
Transendothelial-epithelial transwell model
EpCs-II (7.5 × 104 cells/well) were cultured at the bottom of a transwell filter (Corning). After 5 days, the top surface of the filter was coated with 1 mg/ml fibronectin, and 1.5 × 105 EnCs were inoculated and allowed to adhere for 4 days to form a two-cell layered model. Monocytes (107 cells/ml) were placed in the upper transwell chamber in HBSS or Ca2+-free HBSS, and the chemokine rmCCL2 was added to the lower chamber. The fluorescence intensity in the upper and lower chambers was measured using a fluorescence spectrophotometer and compared with the fluorescence intensity of all monocytes (set to 100%).
Coomassie blue staining and dot blotting
BALF proteomes in the lung of mice were assayed using BALF from E. coli-infected mice that was mixed with 2× SDS loading buffer, heated at 100 °C for 15 min, and loaded onto an SDS-PAGE gel. After electrophoretic separation, the gel was stained using a Coomassie blue staining kit (Beyotime) according to the manufacturer’s instructions. For the detection of dsRNA, an equivalent amount of 10 µl of BALF was directly applied to a nitrocellulose membrane for dot blotting; the spots were air-dried and UV cross-linked, and anti-dsRNA (Scicons), anti-HMGB1, and anti-GAPDH antibodies were used for membrane treatment; ponceau S (Beyotime) staining was used as an internal reference.
Statistics and analysis
Data were analyzed using analysis of variance (ANOVA) followed by Tukey post hoc test for parametric data or using Kruskal–Wallis test followed by Dunn’s test for nonparametric data. Pearson correlation coefficient was used to estimate correlations between two sets of data. P < 0.05 was considered to indicate a statistically significant difference. Sample size was chosen according to previous observations, which perform similar experiments to see significant results, or the results from our preliminary experiments. Variance was similar between the groups that are being statistically compared. Statistical analyses and graphical representations of the data were performed using GraphPad Prism 6.0 (GraphPad Inc.) and SPSS version 19.0 (IBM Corp.).
Data availability
All data needed to evaluate the conclusions of the study are present in the paper and/or the Supplementary Materials. Additional data related to this study may be requested from the authors.
References
Dagvadorj J, Shimada K, Chen S, Jones HD, Tumurkhuu G, Zhang W, et al. Lipopolysaccharide induces alveolar macrophage necrosis via CD14 and the P2X7 receptor leading to interleukin-1alpha release. Immunity. 2015;42:640–53.
Liu SS, Lv XX, Liu C, Qi J, Li YX, Wei XP, et al. Targeting degradation of the transcription factor C/EBPbeta reduces lung fibrosis by restoring activity of the ubiquitin-editing enzyme A20 in macrophages. Immunity. 2019;51:522–34 e527.
Fricker M, Gibson PG. Macrophage dysfunction in the pathogenesis and treatment of asthma. Eur Respir J. 2017;50:1700196.
Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138:16–27.
Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–63.
Mora AL, Rojas M, Pardo A, Selman M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat Rev Drug Disco. 2017;16:810.
Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–62.
Gundra UM, Girgis NM, Ruckerl D, Jenkins S, Ward LN, Kurtz ZD, et al. Alternatively activated macrophages derived from monocytes and tissue macrophages are phenotypically and functionally distinct. Blood. 2014;123:e110–e122.
Jenkins SJ, Ruckerl D, Thomas GD, Hewitson JP, Duncan S, Brombacher F, et al. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med. 2013;210:2477–91.
Vannella KM, Barron L, Borthwick LA, Kindrachuk KN, Narasimhan PB, Hart KM, et al. Incomplete deletion of IL-4Ralpha by LysM(Cre) reveals distinct subsets of M2 macrophages controlling inflammation and fibrosis in chronic schistosomiasis. PLoS Pathog. 2014;10:e1004372.
Saraiva M, O’garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–81.
Squires KE, Montanez-Miranda C, Pandya RR, Torres MP, Hepler JR. Genetic analysis of rare human variants of regulators of G protein signaling proteins and their role in human physiology and disease. Pharm Rev. 2018;70:446–74.
Han SB, Moratz C, Huang NN, Kelsall B, Cho H, Shi CS, et al. Rgs1 and Gnai2 regulate the entrance of B lymphocytes into lymph nodes and B cell motility within lymph node follicles. Immunity. 2005;22:343–54.
Moratz C, Hayman JR, Gu H, Kehrl JH. Abnormal B-cell responses to chemokines, disturbed plasma cell localization, and distorted immune tissue architecture in Rgs1-/- mice. Mol Cell Biol. 2004;24:5767–75.
Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35.
Gibbings SL, Goyal R, Desch AN, Leach SM, Prabagar M, Atif SM, et al. Transcriptome analysis highlights the conserved difference between embryonic and postnatal-derived alveolar macrophages. Blood. 2015;126:1357–66.
Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547–51.
Boyer SW, Schroeder AV, Smith-Berdan S, Forsberg EC. All hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cells. Cell Stem Cell. 2011;9:64–73.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–5.
Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15:929–37.
Schenkel AR, Mamdouh Z, Muller WA. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol. 2004;5:393–400.
Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. 2006;203:2569–75.
Li J, Liu K, Liu Y, Xu Y, Zhang F, Yang H, et al. Exosomes mediate the cell-to-cell transmission of IFN-alpha-induced antiviral activity. Nat Immunol. 2013;14:793–803.
Lee H, Zhang D, Laskin DL, Jin Y. Functional evidence of pulmonary extracellular vesicles in infectious and noninfectious lung inflammation. J Immunol. 2018;201:1500–9.
Lee JH, Kim J, Gludish D, Roach RR, Saunders AH, Barrios J, et al. Surfactant protein-C chromatin-bound green fluorescence protein reporter mice reveal heterogeneity of surfactant protein C-expressing lung cells. Am J Respir Cell Mol Biol. 2013;48:288–98.
Marsh LM, Cakarova L, Kwapiszewska G, von Wulffen W, Herold S, Seeger W, et al. Surface expression of CD74 by type II alveolar epithelial cells: a potential mechanism for macrophage migration inhibitory factor-induced epithelial repair. Am J Physiol Lung Cell Mol Physiol. 2009;296:L442–452.
Pugin J, Heumann ID, Tomasz A, Kravchenko VV, Akamatsu Y, Nishijima M, et al. CD14 is a pattern recognition receptor. Immunity. 1994;1:509–16.
Nova Z, Skovierova H, Calkovska A. Alveolar-capillary membrane-related pulmonary cells as a target in endotoxin-induced acute lung injury. Int J Mol Sci. 2019;20:831.
Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–6.
Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367.
Mason RJ. Biology of alveolar type II cells. Respirology. 2006;11:S12–15.
Laterre PF. Clinical trials in severe sepsis with drotrecogin alfa (activated). Crit Care. 2007;11:S5.
Serrano-Mollar A, Gay-Jordi G, Guillamat-Prats R, Closa D, Hernandez-Gonzalez F, Marin P, et al. Safety and tolerability of alveolar type II Cell transplantation in idiopathic pulmonary fibrosis. Chest. 2016;150:533–43.
Lee H, Groot M, Pinilla-Vera M, Fredenburgh LE, Jin Y. Identification of miRNA-rich vesicles in bronchoalveolar lavage fluid: Insights into the function and heterogeneity of extracellular vesicles. J Control Release. 2019;294:43–52.
Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–4.
Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91.
Christensen JL, Wright DE, Wagers AJ, Weissman IL. Circulation and chemotaxis of fetal hematopoietic stem cells. PLoS Biol. 2004;2:E75.
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–43.
Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity. 2013;39:925–38.
Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 2013;210:1977–92.
Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, et al. Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med. 2014;211:2151–8.
Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61.
Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. 2010;115:e10–19.
Bourdonnay E, Zaslona Z, Penke LR, Speth JM, Schneider DJ, Przybranowski S, et al. Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J Exp Med. 2015;212:729–42.
Ismail N, Wang Y, Dakhlallah D, Moldovan L, Agarwal K, Batte K, et al. Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood. 2013;121:984–95.
Neudecker V, Brodsky KS, Clambey ET, Schmidt EP, Packard TA, Davenport B, et al. Neutrophil transfer of miR-223 to lung epithelial cells dampens acute lung injury in mice. Sci Transl Med. 2017;9:eaah5360.
de Perrot M, Liu M, Waddell TK, Keshavjee S. Ischemia-reperfusion-induced lung injury. Am J Respir Crit Care Med. 2003;167:490–511.
Chin AC, Parkos CA. Pathobiology of neutrophil transepithelial migration: implications in mediating epithelial injury. Annu Rev Pathol. 2007;2:111–43.
Molyneaux PL, Cox MJ, Willis-Owen SA, Mallia P, Russell KE, Russell AM, et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190:906–13.
ELA S, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12:347–57.
Ha D, Yang N, Nadithe V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm Sin B. 2016;6:287–96.
Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307:2526–33.
American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161:646–64.
Sinha M, Lowell CA. Isolation of highly pure primary mouse alveolar epithelial type II cells by flow cytometric cell sorting. Bio Protoc. 2016;6:e2013.
Patel J, Seppanen EJ, Rodero MP, Wong HY, Donovan P, Neufeld Z, et al. Functional definition of progenitors versus mature endothelial cells reveals key SoxF-dependent differentiation process. Circulation. 2017;135:786–805.
Greening DW, Xu R, Ji H, Tauro BJ, Simpson RJ. A protocol for exosome isolation and characterization: evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods Mol Biol. 2015;1295:179–209.
Zhang D, Lee H, Wang X, Rai A, Groot M, Jin Y. Exosome-mediated small RNA delivery: a novel therapeutic approach for inflammatory lung responses. Mol Ther. 2018;26:2119–30.
Chevillet JR, Kang Q, Ruf IK, Briggs HA, Vojtech LN, Hughes SM, et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc Natl Acad Sci USA. 2014;111:14888–93.
Feng Z, Qi S, Zhang Y, Qi Z, Yan L, Zhou J, et al. Ly6G+ neutrophil-derived miR-223 inhibits the NLRP3 inflammasome in mitochondrial DAMP-induced acute lung injury. Cell death Dis. 2017;8:e3170–e3170.
Acknowledgements
We thank the institutes’ platform support and all the patients who agreed for this study.
Funding
This study was supported by the National Natural Science Foundation of China (81800082, 81802651, 81172790, and 81671586) and the grants from The Recruitment Program of Overseas High-Level Young Talents, “Innovative and Entrepreneurial Team” (No.(2018)2015), Science and Technology Grant (BE2019758) and the Six Talent Peaks Project (TD-SWYY-007) of Jiangsu Province and High-Level Talents Program of Nanjing Medical University; Funding of Postdoctoral Funding of Nanjing Drum Tower Hospital Affiliated to Medical School of Nanjing University, and “Peak” Training Program for Scientific Research of Yijishan Hospital, Wannan Medical College (No.GF2019G19); The Medical Interdisciplinary Research Funding of Henan University (No. CJ1205A0240011).
Author information
Authors and Affiliations
Contributions
ZF and HX conceived the project. ZF, JZ, and RX performed the majority of the experiments. QL, LY, XC, and YJ helped in some experiments. YL and QC provide the clinical samples. GC and MW provided the transgenic mice. YZ and YW provided critical advice to the project and funding support. ZF wrote the manuscript and HX supervised the project.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics
All the sample collection and experimental protocols were approved by the Ethics Review Committee of the institutions of the authors.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Degterev
Supplementary information
Rights and permissions
About this article

Cite this article
Feng, Z., Zhou, J., Liu, Y. et al. Epithelium- and endothelium-derived exosomes regulate the alveolar macrophages by targeting RGS1 mediated calcium signaling-dependent immune response. Cell Death Differ 28, 2238–2256 (2021). https://doi.org/10.1038/s41418-021-00750-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-021-00750-x
This article is cited by
-
Endothelial cell dynamics in sepsis-induced acute lung injury and acute respiratory distress syndrome: pathogenesis and therapeutic implications
Cell Communication and Signaling (2024)
-
Exosomal mediators in sepsis and inflammatory organ injury: unraveling the role of exosomes in intercellular crosstalk and organ dysfunction
Military Medical Research (2024)
-
UTX deletion promotes M2 macrophage polarization by epigenetically regulating endothelial cell-macrophage crosstalk after spinal cord injury
Journal of Nanobiotechnology (2023)
-
Extracellular vesicles: emerging roles, biomarkers and therapeutic strategies in fibrotic diseases
Journal of Nanobiotechnology (2023)
-
Macrophage polarization and metabolism in atherosclerosis
Cell Death & Disease (2023)
