
Abstract
Intratumor heterogeneity has been recognized in numerous cancers as a major source of metastatic dissemination. In uveal melanomas, the existence and identity of specific subpopulations, their biological function and their contribution to metastasis remain unknown. Here, in multiscale analyses using single-cell RNA sequencing of six different primary uveal melanomas, we uncover an intratumoral heterogeneity at the genomic and transcriptomic level. We identify distinct transcriptional cell states and diverse tumor-associated populations in a subset of the samples. We also decipher a gene regulatory network underlying an invasive and poor prognosis state driven in part by the transcription factor HES6. HES6 heterogenous expression has been validated by RNAscope assays within primary human uveal melanomas, which further unveils the existence of these cells conveying a dismal prognosis in tumors diagnosed with a favorable outcome using bulk analyses. Depletion of HES6 impairs proliferation, migration and metastatic dissemination in vitro and in vivo using the chick chorioallantoic membrane assay, demonstrating the essential role of HES6 in uveal melanomas. Thus, single-cell analysis offers an unprecedented view of primary uveal melanoma heterogeneity, identifies bona fide biomarkers for metastatic cells in the primary tumor, and reveals targetable modules driving growth and metastasis formation. Significantly, our findings demonstrate that HES6 is a valid target to stop uveal melanoma progression.
Similar content being viewed by others


Introduction
Uveal melanoma is an aggressive and deadly neoplasm, which develops from melanocytes in the choroid. At diagnosis, only 1–3% of the patients have detectable metastases. Rapid local treatments are crucial, as survival correlates with primary tumor size [1]. Despite successful treatment of the primary tumor, metastases, that display a pronounced liver tropism, develop in 50% of patients within a median time of 2.4 years [2]. Once it has spread, there are no approved systemic treatments for uveal melanoma. Overall, 90% of patients will die within 6 months after diagnosis of metastases (reviewed in [3, 4]).
The above observations imply that cell subpopulations responsible for metastases, and patient death, disseminate early from the primary tumor. The identity of these cell subpopulations and the identification of their specific markers are required to improve patient outcome. Supporting this idea, in skin melanomas, intratumoral heterogeneity has been shown to have a profound impact on tumor evolution, development of metastases and therapy resistance [5,6,7].
Previous studies have separated uveal melanoma into two classes according to their transcriptomic profile. Class 1 is predictive of poor metastatic risk and long-term survival, while class 2 is associated with a high risk of distant metastasis and a dismal prognosis [8]. However, to date none of the genes identified in these studies has been shown to be potential therapeutic targets for uveal melanoma treatment.
Very recently, single-cell RNA-seq analyses provided a glimpse into primary and metastatic uveal melanomas ecosystems, and disclosed a regulatory T-cell phenotype, highlighting LAG3 as a potential candidate for immune checkpoint blockade [9]. LAG3 has also been pointed out to be a potential regulator of uveal melanoma immunity in other studies [10,11,12].
Thus, assessing intratumoral heterogeneity and characterization of the different transcriptional states, might provide insights into the subpopulation of uveal melanoma cells, that favor the metastatic dissemination and may lead to the identification of biomarkers to prevent the metastatic disease.
Methods
Sample collection and processing
Single cells were isolated from tumor tissues (#A–F) of patients diagnosed with ocular melanoma, after written informed consent was obtained from the Nice CHU hospital. Samples were analyzed using the 10x Genomics’s protocol.
Cell cultures
Human uveal melanoma cell lines Mel270 (GNAQQ209P) [13], 92.1 (GNAQQ209L) [14], OMM2.5 (GNAQQ209P) [13] and OMM1 (GNA11Q209L) [15] were grown as previously described. They all express BAP1. Additional information about these cell lines may be found here [16]. MP46 (GNAQQ209L; no BAP1 expression) and MP65 (GNA11Q209L; BAP1 c1717del) cell lines were from ATCC. Cell lines are regularly tested for mycoplasma and are mycoplasma-free.
RNAscope
mRNAs for HES6 in sections from human uveal melanomas were detected with RNAscope assay (Advanced Cell Diagnostics, ACD) according to the manufacturer’s protocols. Images were captured with a confocal (Leica DMI6000) microscope.
Statistical analyses
No statistical methods were used to determine sample size. Sample size was determined to be adequate based on the magnitude and consistency of measurable differences between groups. The data are presented as the means + SD and analyzed using two-sided Student’s t-test with Prism or Microsoft Excel software (*p value ≤ 0.05; **p value ≤ 0.01; ***p value ≤ 0.001). For chick embryo chorioallantoic membrane (CAM) assay, a one-way ANOVA analysis with post-tests was done on the data.
Primers and siRNAs used
HES6 forward, TGA CCA CAG CCC AAA TTG C; reverse, CTA CCC CAC CAC ATC TGA AC; RPLP0 forward, AAG GTG TAA TCC GTC TCC ACA GA; reverse, CTA CCC CAC CAC ATC TGA AC. siRNA were obtained from Sigma (#EHU036431) and Horizon Discovery (#L-008408-00-0005).
Results
A subset of primary uveal melanomas displays intratumoral heterogeneity
To inspect intratumoral heterogeneity, we isolated individual cells from six freshly resected human primary uveal melanomas (#A–F) and generated single-cell transcriptomes using 10x genomics (Fig. 1A). The clinical, histopathologic and cytogenetic features are presented (Table 1). Histological examination showed epithelioid cells in tumors B and C (Supplementary Fig. 1), and a high mitotic index in tumor B (not shown). Examination of copy number variations (CNVs) by array comparative genomic hybridization (a-CGH) showed complete or partial loss of chromosome 3 in tumors A, C, E and partial gain of chromosome 8 in tumors A, C, D and F (Supplementary Fig. 2). These CNVs are associated with high metastatic risk [17]. Finally, whole exome sequencing of bulk lesions indicated that all, except tumor C, carried activating mutation in GNAQ or GNA11, two frequent driver mutations in uveal melanomas (Table 1). Tumor C neither harbors mutations in CYSLTR2 or PLCB4 which mutations are also considered as driver events in uveal melanomas [18,19,20].

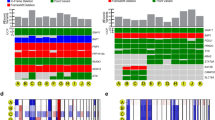
A Experimental workflow indicating the dissociation and isolation of individual cells from primary uveal melanomas for generating single-cell RNA-seq profiles. cDNA from the individual cells were synthesized, followed by library construction and massively parallel sequencing using the 10x genomic approach. B Heatmaps of the first two principal components from the principal component analysis (PCA) based on highly variable genes in the dataset. Both cells and genes are sorted by their PC scores allowing easy exploration of the principal sources of heterogeneity in the dataset. The first ten genes with the highest or lowest absolute PC values are shown for PC1 and PC2 (yellow). C Kaplan–Meier survival plot of the ten genes with the highest PC1 values (lower left quadrant of B). D Kaplan–Meier survival plot of ten genes with the lowest PC1 values (upper right quadrant of B). E Histograms showing PC1 score, based on the top ten up and down PC1 genes, of all the single cells in each tumor. The Youden index was used as the cutoff point (red line). The percentage of cells with high and low PC1 score as well as the percentage of “poor prognosis” cells per tumors are indicated below the histogram.
We first used the principal component analysis of which the two first principal components (PC) constituted the majority of the variance within the dataset (Fig. 1B and Supplementary Table 1). Among the ten genes with the highest PC1 and PC2 values was HTR2B, a gene previously associated with high metastatic risk and poor overall survival [8]. Cellular function or disease analysis using Ingenuity® Pathway Analysis (IPA) indicated that the PC1 signature (z score −1/+1; 258 genes up; 15 genes down) correlated with cell movement of tumor cell lines, migration of tumor cell lines, cell viability, cell survival, neoplasia of cells (Table 2). Interestingly, liver tumor function was also predicted and is consistent with a strong liver tropism of uveal melanomas. Conversely, cellular functions or diseases related to apoptosis or necrosis were inhibited. Analysis of upstream regulators with IPA highlighted activation of transcription regulators and cytokines, with role in inflammation, and cellular stress, including STAT, NFKB, ATF6, XBP1, HIF1 and TNF proteins (Supplementary Table 2). IPA revealed that PC2 was also linked to proliferation of tumor cells and invasion of tumor (Supplementary Fig. 3A).
Kaplan–Meier analysis of uveal melanoma patients (The Cancer Genome Atlas Program (TCGA) set) showed that expression of the top ten genes with the highest PC1 values was associated with shortened survival (Fig. 1C), whereas expression of the top ten genes with the lowest PC1 values correlated with an increased survival (Fig. 1D). Expression of the top ten genes with the highest values in PC2 was also predictive of a poor prognosis but that of the top ten genes with the lowest PC2 values did not correlate with survival (Supplementary Fig. S3B, C).
To gain insights into the prognosis sensitivity and specificity of the PC1 signature, we used the top ten up and down genes to calculate a “PC1 score” for each patient of the uveal melanoma TCGA cohort and plotted a ROC curve (Supplementary Fig. 4). The AUROC was 0.84 and the Youden index 0.63, thereby indicating that this “PC1 score” might be of interest to estimate patients’ prognosis. If we extrapolate this “score” to our single-cell analysis, cells with a “score” above the Youden index should be of “poor prognosis” i.e., expressed a gene signature associated with poor patient survival, while those with a “score” under the Youden index should be of “good prognosis”, because they expressed genes associated with low metastatic risk and long-term survival.
Applying this concept, we found that tumors A, C and E classified in the poor prognosis class 2c group by the cytogenetic analysis (Table 1), contained between 80 and 100% of “poor prognosis cells”, while tumor B (class 2a, Table 1) contained only 20% (Fig. 1E). Among specimens with favorable predictable outcome (class 1b, Table 1), tumors D and F comprised only 0.8% and 3.5% of poor prognosis cells, respectively. This analysis, based on the “PC1 score” which reflects the ability of cells to metastasize and cause patient death, demonstrated an intratumoral transcriptomic and functional heterogeneity in uveal melanomas. On a clinical point of view, even a small number of cells with a high PC1 score might be sufficient to support distant metastasis development and impair patient survival.
To identify salient biological cell states, we next performed clustering of the individual cells with the Seurat analysis pipeline and used non-linear dimensionality reduction method [t-distributed stochastic neighbor embedding (t-SNE)], to visualize cell clusters. This analysis revealed that most cells grouped by tumor of origin, thereby indicating intertumor heterogeneity (Supplementary Fig. 5A). Further unbiased clustering of the individual cells identified 12 clusters (Fig. 2A). Tumors B, D and E each comprised a single cluster, while two clusters were identified in tumors A and F, and three clusters in tumor C, again emphasizing the existence of intratumor heterogeneity. Few non-malignant cells were detected in the tumors. Cluster 9 was annotated as immune cells since it was enriched in the expression of T-cells and monocytes/macrophages markers and cluster 11 as endothelial cells since it was enriched in the expression of PECAM1, CD34, FLT1 CDH5 (Supplementary Fig. 5B–D). These two latter clusters gathered by cell type and not by patient. Finally, representation of the cluster composition of each tumor, further demonstrated the transcriptomic heterogeneity of uveal melanoma cells within a tumor, and different cluster ratios in distinct tumors (Supplementary Fig. 6).

A Seurat analysis showing t-SNE plots of 7890 uveal melanoma cells colored by clusters. Each point represents a single cell. Red circles indicate non-malignant cells. B Kaplan–Meier survival plot for the top 20 genes of the indicated clusters. C Heatmap of inferred copy number variation (CNV) signal normalized against the topmost cluster composed by the pool of all putative non-malignant cells (no CNV variation). Cells (rows, n = 7890 cells) are ordered from non-malignant cells (NMC, n = 101 cells) to cancer cells (n = 7789 cells), from the six uveal melanomas. Chromosomal regions (columns) with amplifications (red) or deletions (blue) are shown. The additional tracks, on the right, show the associated cluster number from Seurat.
List of genes associated with each cluster (Supplementary Table 3) was used in IPA comparison analysis to address enrichment in canonical pathways. Clusters 2, 4, 7, 8 and 10 clustered together and disclosed clear activation of Rho GTPase-dependent signaling pathways, regulation of actin cytoskeleton and integrin signaling (Supplementary Fig. 7A). Equally important, in these clusters, Rho-GDI, a negative regulator of signaling through Rho GTPases, was downregulated. Other pathway more robustly expressed in these clusters included mitochondria oxidative phosphorylation [21]. In keeping with the recognized role of Rho GTPases and mitochondrial metabolism as markers of tumor invasion and metastasis, Kaplan–Meier survival plot generated from the top 20 genes in each cluster revealed that only clusters 2, 4, 7, 8 and 10 were associated with a poor prognosis (Fig. 2B and Supplementary Fig. 7B). In tumor C, whereas cluster 5 was not related to the prognosis, clusters 7 and 8 contained cells conveying a dismal prognosis, further supporting the existence of transcriptomic and functional intratumoral heterogeneity in primary uveal melanomas (Fig. 2B and Supplementary Fig. 7B).
In addition, as previously described [22], large-scale copy number aberrations for each cell by averaging relative expression levels over large genomic regions was used to infer CNVs from scRNA-seq data (Fig. 2C). Inferred-CNV profiles uncovered distinct chromosomal imbalance, including chromosome 3 loss and 8q gain, that are characteristic uveal melanoma alterations. However, tumors C and F appeared to contain more than one genetic clone. Cryptic alterations, in cell subsets of tumors A and D can also be observed in chromosomes 6 and 8, respectively. Globally, inferred-CNV analysis was in agreement with bulk array-based CGH (Supplementary Fig. 2).
Collectively, in addition to intertumor heterogeneity, this dataset discloses an intratumoral heterogeneity at both the genomic and transcriptomic level in a subset of tumors. Transcriptomic and genomic heterogeneity overlapped largely in tumor F where the cells with 8q gain fell in cluster 6, and in tumor C, where cells having a loss in chromosomes 14, 15 and 16 segregated to cluster 7. However, in tumor C, cells with a 6p gain were distributed between cluster 8 and a portion of cluster 5. Importantly, minority pre-existing subclones or rare poor prognosis cells may be missed by classical bulk sequencing because their abundance falls below the lower limit of sensitivity, while they represent the functional cell subsets that will outgrowth and drive the metastatic dissemination.
Characterization of major cell subpopulations in primary uveal melanomas
Next, to get insights into the stable transcriptional cell states, we used the Single-cell regulatory network inference and clustering (SCENIC) method [23]. SCENIC exploits transcription factors and cis-regulatory sequences, to map the activity of the regulatory networks (regulons) underlying the different gene expression signatures. This analysis disclosed 122 regulons (out of 1046) that displayed significant activity in uveal melanomas (Fig. 3a and Supplementary Fig. 8). After non-linear dimensionality reduction (t-SNE) of these data, we can observe a degree of cellular overlapping between cells from different tumors, indicating that cells with similar transcriptional program can be found in different tumors (Fig. 3B). Together, these findings further confirm the transcriptional intratumor heterogeneity.

A Heatmap of cells and regulon binary scores with hierarchical clustering inferred by SCENIC. The additional track, above, show supervised clustering by patients and the PC1 score based on the top ten genes with the highest values in PC1. The 72 regulons with the best clustering out of the 122 identified in the six primary uveal melanomas are represented. B t-SNE shows cells colored by patient (SCENIC approach). C HES6 regulon activity comprising 72 genes quantified using AUCell is represented. Regulons of predicted transcription factors in the six patients were determined to be active if they exceeded the threshold (Blue), otherwise, regulons were considered to be inactive (gray). D Kaplan–Meier survival plot of the HES6 regulon (TCGA dataset). E Kaplan–Meier survival plot of HES6 (TCGA dataset).
SCENIC heatmap also revealed clustered regulons (RELB, HES6, HSF1 and MYC) that correlated with a high PC1 score. This transcriptional state can be inferred as an invasive state as MYC and RELB have been involved in metastasis of uveal melanoma cells [24,25,26,27,28]. However, we focused our attention on HES6 (enhancer of split family basic helix-loop-helix transcription factor 6). HES6 was detected among the top ten genes with the highest PC1 values, it stimulates the invasive ability of various tumor cells [25,26,27], and its role in uveal melanomas remains to be elucidated. Cells with high HES6 regulon activity were found mainly in tumors A, C and E, but few HES6-positive cells can be found in other tumors (Fig. 3C).
Importantly, Kaplan–Meier analysis showed that the HES6 regulon (Supplementary Table 4) as well HES6 itself, which is carried by chromosome 2, were negatively correlated with overall survival (Fig. 3D, E). In keeping with this, both in tumors analyzed hereby and the TCGA dataset, HES6 expression is associated with chromosome 3 loss (Supplementary Fig. 9), which in uveal melanomas highly correlates with the metastatic risk. Notably, HES6 expression overlapped with class 1b and class 2 tumors [8], which bear worse prognosis than class 1a (Supplementary Fig. 10). Taken together these observations highlight the role of HES6 as a key marker of uveal melanoma cell metastatic potential and patient survival.
HES6 enhances growth and motile ability in vitro and in vivo of primary uveal melanoma
To validate these analyses and given the lack of high-quality HES6 antibody for immunochemistry, its expression in human patient biopsies was evaluated by RNAscope® fluorescence in situ hybridization assay. The staining confirmed that primary uveal melanomas comprised both HES6-high and HES6-low cells that were segregated or intermixed reflecting regional heterogeneity and different cell states (Fig. 4a). Negative control staining is shown (Supplementary Fig. 11). In line with the single-cell analysis, HES6 expression was higher in tumors A, C and E compared to tumors B, D and F.

A Sections from tumors # A–F were labeled with RNAscope probe for HES6 (red), and images were captured by confocal microscopy. Cell nuclei (blue green). Shown are the areas of high and low heterogeneity. Scale bars represent 60 μm. For each tumor, magnification of the boxed area is shown (Right). Scale bars represent 25 μm. B Western blot and RT-QPCR analysis of HES6 in primary MP46 melanoma cells transduced with a control or HES6 adenovirus expressing HES6 (HES6 OE) for 72 h. C Human primary MP46 melanoma cells were transduced with empty (control) or HES6 expressing adenovirus (HES6 OE) for 48 h before being seeded in the upper part of the Boyden chamber. Migration was examined after 24 h. Values represent means +SD of three independent experiments. **p < 0.01. D Representative images are shown. Bar = 100 μm. E Western blot and RT-QPCR analysis of HES6 in primary Mel270 melanoma cells transfected with a control siRNA (siCtl) or two different pools of multiple siRNA targeting HES6 (siHES6#1 and siHES6#2). F Migration of Mel270 cells transfected with the indicated siRNA. ***p < 0.001. G Representative images are shown. Bar = 100 μm.
Next, we aimed to portray the biological role of HES6. We first assessed the ability of HES6 to control the motile capacity of primary uveal melanoma cells. Ectopic HES6 expression enhanced migration of two different primary cell lines (Fig. 4B–D and Supplementary Fig. 12A, C). Conversely, reduced migration of primary cells was obtained with HES6 inhibition by both siRNA and shRNA (Figs. 4E–G and 5A–C). Although we searched for metastatic drivers in the primary tumor, we also asked whether HES6 could have an effect in the metastatic settings. Our results showed that HES6 gain enhanced (Supplementary Fig. 12D–F), whereas HES6 loss reduced (Supplementary Fig. 13A, B) motility of metastatic cells.

A QPCR analysis of HES6 in primary 92.1 melanoma cells expressing doxycycline-inducible control or HES6 shRNA in presence of 1 μg/ml doxycycline for 96 h in presence or absence of DLL4 1 μg/ml. B Migration of 92.1 melanoma cells expressing doxycycline-inducible control or HES6 shRNA in presence of 1 μg/ml doxycycline for 48 h in presence or absence of DLL4 1 μg/ml. ***p < 0.001. C Representative images are shown. Bar = 100 μm. D Description of the chicken embryo CAM assay. E 92.1 uveal melanoma cells expressing doxycycline-inducible control or HES6 shRNA were grafted on the CAM of 9-day-old (E9) chick embryos. The tumors were collected and weighted on day 18 (E18). Values represent means + SEM. F Genomic DNA is extracted from the lower CAM to evaluate the number of metastatic cells on day 18 and analyzed by qPCR with specific primers for human Alu sequences. Values represent means ± SEM.
HES6 inhibition by siRNA or by using an inducible shRNA strategy also prevented the ability to form colony in primary uveal melanoma cells (Supplementary Fig. 14A–C). The same held true in metastatic cells (Supplementary Fig. 14D–F). Thus, our findings indicated that HES6 might represent a valid target to limit uveal melanoma cell proliferation and migration.
HES6 is an atypical HES gene whose role as downstream effector of NOTCH signaling is unclear. Among NOTCH natural ligand, in uveal melanomas, Delta-like ligand 4 (DLL4) is the NOTCH ligand the most associated with the metastatic risk and its expression is the most inversely correlated with patient survival (Supplementary Fig. 15A). Although a role for NOTCH signaling pathway has been reported in uveal melanoma [28, 29], the effect of DLL4 has never been investigated. Our data showed that DLL4 increased NOTCH reporter activity, an effect that was inhibited by the γ-secretase inhibitor BMS-906024 (Supplementary Fig. 15B). In addition, blocking the NOTCH pathway with two NOTCH inhibitors BMS-906024 and DAPT reduced spheroid formation (Supplementary Fig. 15C). Finally, we observed that DLL4 enhanced uveal melanoma cell migration (Supplementary Fig. 15D). Thus, DLL4 activates the NOTCH signaling pathway in uveal melanoma cells and controls their growth and migration.
To delineate the role of HES6 downstream of NOTCH, we assessed the impact of HES6 knockdown upon treatment with DLL4. Compared to control, DLL4 could no longer increase the migration of HES6 knockdown 92.1 and Mel270 cell lines (Fig. 5A–C and Supplementary Fig. 16A, B). These data provide evidence that HES6 has critical tumorigenic properties downstream the NOTCH signaling pathway and mediates its effect on the motile ability of primary uveal melanoma cells.
Next, we demonstrated that HES6 knockdown in 92.1 and Mel270 cells also reduced the formation of 3D spheroids, that more faithfully model the tumor microenvironment than 2D cell cultures (Supplementary Fig. 17A–F). Further, a matrigel invasion assay showed that Mel270 cells originating from the control spheroids efficiently invaded the matrigel compared to spheroids formed with the HES6-knockdown cells (Supplementary Fig. 17G). This experiment could not be performed with 92.1 cells given that HES6 knockdown dramatically reduced sphere formation in these cells, thereby preventing spheroids for being harvested and embedded in matrigel. Thus, HES6 also controls the invasive ability of primary uveal melanoma cells.
We reasoned that HES6 might be effective in driving metastatic dissemination of primary uveal melanoma cells. We thus studied tumor progression to metastasis of primary uveal melanoma cells in vivo using the CAM model (Fig. 5D). Control cells efficiently formed tumors and were overall also very efficient at forming metastasis as evidenced by cells that had disseminated to the lower CAM (Fig. 5D–F). Growth and metastatic abilities were strongly reduced by 36% and 48%, respectively, with tumors formed from HES6 knocked-down cells.
Altogether, our findings showed both in vitro and in vivo that HES6 stimulates the aggressive potential of primary uveal melanoma and their motile capacity.
Discussion
Here, we used a single-cell transcriptomic profiling strategy to address the critical questions of cell heterogeneity in primary uveal melanomas in order to identify cell subpopulations driving the metastatic process.
The data gathered hereby, while confirming the existence of an intertumor heterogeneity, also uncover a molecular and functional intratumor heterogeneity. They highlight a new signature that allows to detect tumor cells that might convey unfavorable outcome among patients classified as having a good prognosis either by using classical clinical parameters or even gene expression profile on bulk tumor. An intratumoral genomic heterogeneity has previously been suspected, since DNA extracted from several areas within the same primary uveal melanomas displayed different chromosomal abnormalities [30]. Our study discloses a transcriptomic heterogeneity that is not always supported by the genomic heterogeneity, but that reflects different transcriptional programs.
SCENIC has inferred at least three transcriptional states. One is related to cell specification due to the enrichment in SOX9, SOX10 and PAX3 regulons. This state overlaps with cells displaying intermediate activity of MITF a master regulator of melanocyte differentiation, proliferation and survival [31, 32]. SOX10 and PAX3 activity was inferred by SCENIC in cells with low PC1 score (good prognosis). In line with this, uveal melanoma patients from the TCGA cohort with high levels of both PAX3 and SOX10 have an increased overall survival (not shown).
A second transcriptional cell state, with enrichment in regulons BCL3, CEBPB and AP1 members (JUNB, JUND, FOS and FOSB), may be related to immune response and inflammation. Indeed, BCL3 and CEBPB have direct roles in the regulation of proinflammatory cytokine production by cancer cells [33,34,35,36]. Further, in cutaneous melanomas, activation of JUN leads to melanoma cell dedifferentiation via MITF downregulation that is associated with the production of proinflammatory cytokines [37, 38]. This transcriptional profile defined a primary uveal melanoma intrinsic inflammatory state that should favor immune cell infiltrate. However, none of the tumors inspected in our study showed a significant immune cell infiltration. This observation is in agreement with previous work from the TCGA network also reporting immune infiltration in a few numbers of primary uveal melanomas [39].
Finally, we focused our attention on the third transcriptional state inferred by SCENIC with invasive functionality that is driven, at least in part, by HES6. HES6, that belongs to the poor prognosis signature we discovered (top ten genes of PC1), is an atypical HES gene whose role in uveal melanomas remained totally unknown. By contrast to canonical NOTCH targets, HES6 was thought to antagonize NOTCH signaling. However, in uveal melanoma cells, HES6 knockdown impairs migration induced by DLL4, an activator of NOTCH receptors, indicating that HES6, depending on the context, may be a NOTCH effector. We demonstrate in vitro and in vivo that HES6 is a key driver of uveal melanoma proliferation and metastatic dissemination. Our data reveal that the subgroup of regulons activated in cells with a high PC1 score and therefore with poor survival prognosis displayed in addition to the HES6 regulon, the MYC regulon and was also partially enriched in JUN (JUNB and JUND) regulons. Increased MYC and JUN activities were also pinpointed in poor prognosis class 2 tumors by a previous single-cell analysis [9]. The work of Durante et al. also identified the activation of ARNT, TAF1 and TAF7 regulons in poor prognosis cells that were not spotted in our study. Conversely, HES6 and HES6 regulon, that are clearly associated with decreased survival, were not identified by Durante et al. Differences with our study can be explained by the fact that Durante et al. analyzed a mix of primary and metastatic specimens while in our study, we only focused on primary melanomas [9]. Further, they selected tumor cells using the expression of the differentiation markers DCT, MITF and MelanA [9]. Depending on the threshold, this filtering may induce biases by missing some cells in the analyses. Finally, Durante et al. analyzed a subgroup of uveal melanomas with a large immune infiltrate that could potentially affect tumoral cells transcriptomic profile [9]. Nevertheless, the work from Durante et al. shows important data about uveal melanoma ecosystem [9]. By contrast, our study, focusing on primary uveal melanomas with low immune infiltrate, which represent the vast majority of uveal melanoma (TCGA), discloses new transcriptomic signatures and pathways that are associated with prognosis and have direct impact on the biology of uveal melanoma cells.
In keeping with a role of HES6 in invasive ability, IPA analysis of the PC1 signature reveals activation of the Rho GTPase and integrin signaling pathways in cell subpopulations that convey a poor prognosis. Rho GTPases are essential in propagating integrin-mediated responses and, by tightly regulating actin cytoskeleton, offer a key signaling link through which adhesion, spreading, and migration are controlled in tumor cells [40]. Further, Rho lies downstream from GNAQ and GNA11 and stimulates YAP, which in turn controls uveal melanoma cell proliferation [41]. Of note, PAX3 can use YAP as a coactivator. Mechanistically, YAP activation can lead to the stimulation of PAX3 driven differentiation program [42], while in absence of PAX3, YAP is made available for TEAD transcription factors to drive uveal melanoma cell proliferation. This might append an additional level of heterogeneity.
Likewise, enhanced HES6 expression stimulates the invasive phenotype of prostate cancer, glioma and colorectal cancer cells [25, 26]. Conversely, HES6 knockdown has been reported to decrease migration of glioma, glioblastoma, colorectal cancer cells and of alveolar rhabdomyosarcoma [27, 43].
Whether the cell states and key transcription factors identified in primary lesions are maintained in the subsequent metastasis and play a critical role remains to be verified. However, in favor of this idea, HES6 knockdown also reduced growth and motile ability of metastatic uveal melanoma cells.
The identification of a HES6-driven transcriptional state, which is associated with high tumorigenic properties, is highly relevant for patient care, since we demonstrated that tumors classified as of good prognosis using bulk analysis, contained varying proportions of HES6-positive cells, that could negatively impact on patient outcome.
Given the lack of treatment options for metastatic uveal melanomas, HES6 or its target genes that we disclose hereby may represent actionable factors to be targeted therapeutically.
Thus, our single-cell transcriptomic profiling uncovers the existence of intratumor heterogeneity in primary uveal melanomas and leads to mechanistic insights into the regulation of the metastatic process in uveal melanomas, thereby offering unprecedentedly described biomarkers with critical implications for prognosis and therapeutic strategies.
Data availability
The experimental data from single-cell RNA sequencing, whole exome sequencing and array-CGH have been deposited in the NCBI Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) under the SuperSeries GSE138665.
References
Rietschel P, Panageas KS, Hanlon C, Patel A, Abramson DH, Chapman PB. Variates of survival in metastatic uveal melanoma. J Clin Oncol. 2005;23:8076–80.
Nabil AA, Marie S, Marc-Henri S, Nathalie C, Laurence D, Sophie PN, et al. Upcoming translational challenges for uveal melanoma. Br J Cancer. 2015;113:1249–53.
Pandiani C, Beranger GE, Leclerc J, Ballotti R, Bertolotto C. Focus on cutaneous and uveal melanoma specificities. Genes Dev. 2017;31:724–43.
Yang J, Manson DK, Marr BP, Carvajal RD. Treatment of uveal melanoma: where are we now? Ther Adv Med Oncol. 2018. https://doi.org/10.1177/1758834018757175.
Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–96.
Kemper K, Krijgsman O, Cornelissen-Steijger P, Shahrabi A, Weeber F, Song JY, et al. Intra- and inter-tumor heterogeneity in a vemurafenib-resistant melanoma patient and derived xenografts. EMBO Mol Med. 2015;7:1104–18.
Rambow F, Marine J, Goding CR. Melanoma plasticity and phenotypic diversity: therapeutic barriers and opportunities. Genes Dev. 2019;33:1295–318.
Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;64:7205–9.
Durante MA, Rodriguez DA, Kurtenbach S, Kuznetsov JN, Sanchez MI, Decatur CL, et al. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat Commun. 2020;11:496.
Karlsson J, Nilsson LM, Mitra S, Alsén S, Shelke GV, Sah VR, et al. Molecular profiling of driver events in metastatic uveal melanoma. Nat Commun. 2020;11:1894.
Figueiredo CR, Kalirai H, Sacco JJ, Azevedo RA, Duckworth A, Slupsky JR, et al. Loss of BAP1 expression is associated with an immunosuppressive microenvironment in uveal melanoma, with implications for immunotherapy development. J Pathol. 2020;250:420–39.
Triozzi PL, Schoenfield L, Plesec T, Saunthararajah Y, Tubbs RR, Singh AD. Molecular profiling of primary uveal melanomas with tumor-infiltrating lymphocytes. Oncoimmunology. 2014;8:e947169.
Chen PW, Murray TG, Uno T, Salgaller ML, Reddy RKB. Expression of MAGE genes in ocular melanoma during progression from primary to metastatic disease. Clin Exp Metastasis. 1997;15:509–18.
De Waard‐Siebinga I, Blom DR, Griffioen M, Schrier PI, Hoogendoorn E, Beverstock G, et al. Establishment and characterization of an uveal‐melanoma cell line. Int J Cancer. 1995;62:155–61.
Luyten GP, Naus NC, Mooy CM, Hagemeijer A, Kan-Mitchell J, Van Drunen E, et al. Establishment and characterization of primary and metastatic uveal melanoma cell lines. Int J Cancer. 1996;66:380–7.
Griewank KG, Yu X, Khalili J, Sozen MM, Stempke-Hale K, Bernatchez C, et al. Genetic and molecular characterization of uveal melanoma cell lines. Pigment Cell Melanoma Res. 2012;25:182–7.
Shain AH, Bagger MM, Yu R, Chang D, Liu S, Vemula S, et al. The genetic evolution of metastatic uveal melanoma. Nat Genet. 2019;51:1123–30.
Larribère L, Utikal J. Update on gna alterations in cancer: Implications for uveal melanoma treatment. Cancers. 2020;12:1–18.
Johansson P, Aoude LG, Wadt K, Glasson WJ, Warrier SK, Hewitt AW, et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget. 2016;7:4624–31.
Moore AR, Ceraudo E, Sher JJ, Guan Y, Shoushtari AN, Chang MT, et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat Genet. 2016;48:675–80.
Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013;32:4814–24.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401.
Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14:1083–6.
Meir T, Dror R, Yu X, Qian J, Simon I, Pe’er J, et al. Molecular characteristics of liver metastases from uveal melanoma. Invest Ophthalmol Vis Sci. 2007;48:4890–6.
Xu Y, Liu X, Zhang H, Zhu Z, Wu X, Wu X, et al. Overexpression of HES6 has prognostic value and promotes metastasis via the Wnt/beta-catenin signaling pathway in colorectal cancer. Oncol Rep. 2018;40:1261–74.
Haapa-Paananen S, Kiviluoto S, Waltari M, Puputti M, Mpindi JP, Kohonen P, et al. HES6 gene is selectively overexpressed in glioma and represents an important transcriptional regulator of glioma proliferation. Oncogene. 2012;31:1299–310.
Wickramasinghe CM, Domaschenz R, Amagase Y, Williamson D, Missiaglia E, Shipley J, et al. HES6 enhances the motility of alveolar rhabdomyosarcoma cells. Exp Cell Res. 2013;319:103–12.
Asnaghi L, Ebrahimi KB, Schreck KC, Bar EE, Coonfield ML, Bell WR, et al. Notch signaling promotes growth and invasion in uveal melanoma. Clin Cancer Res. 2012;18:654–65.
Asnaghi L, Handa JT, Merbs SL, Harbour JW, Eberhart CG. A role for Jag2 in promoting uveal melanoma dissemination and growth. Investig Ophthalmol Vis Sci. 2013;54:295–306.
Dopierala J, Damato BE, Lake SL, Taktak AF, Coupland SE. Genetic heterogeneity in uveal melanoma assessed by multiplex ligation-dependent probe amplification. Invest Ophthalmol Vis Sci. 2010;51:4898–905.
Goding CR, Arnheiter H. MITF—the first 25 years. Genes Dev. 2019;33:983–1007.
Cheli Y, Ohanna M, Ballotti R, Bertolotto C. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. 2010;23:27–40.
Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, et al. Senescent cells develop a parp-1 and nuclear factor-κB-associated secretome (PNAS). Genes Dev. 2011;25:1245–61.
Ohanna M, Cheli Y, Bonet C, Bonazzi VF, Allegra M, Giuliano S, et al. Secretome from senescent melanoma engages the STAT3 pathway to favor reprogramming of naive melanoma towards a tumor-initiating cell phenotype. Oncotarget. 2013;4:2212–24.
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31.
Chang TP, Vancurova I. Bcl3 regulates pro-survival and pro-inflammatory gene expression in cutaneous T-cell lymphoma. Biochim Biophys Acta - Mol Cell Res. 2014;1843:2620–30.
Riesenberg S, Groetchen A, Siddaway R, Bald T, Reinhardt J, Smorra D, et al. MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat Commun. 2015;6:8755.
Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. 2012;490:412–6.
Robertson AG, Shih J, Yau C, Gibb EA, Oba J, Mungall KL, et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell. 2017;32:204–20 e15.
Lawson CD, Burridge K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases. 2014;5:e27958.
Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell. 2014;25:831–45.
Miskolczi Z, Smith MP, Rowling EJ, Ferguson J, Barriuso J, Wellbrock C. Collagen abundance controls melanoma phenotypes through lineage-specific microenvironment sensing. Oncogene. 2018;37:3166–82.
Roma J, Masià A, Reventós J, De Toledo JS, Gallego S. Notch pathway inhibition significantly reduces rhabdomyosarcoma invasiveness and mobility in vitro. Clin Cancer Res. 2011;17:505–13.
Trolet J, Hupe P, Huon I, Lebigot I, Decraene C, Delattre O, et al. Genomic profiling and identification of high-risk uveal melanoma by array CGH analysis of primary tumors and liver metastases. Invest Ophthalmol Vis Sci. 2009;50:2572–80.
Acknowledgements
We thank the Single Cell Core Facility at IPMC and the imaging facility of C3M. The authors thank all patients with primary uveal melanoma for participating as well as Frédéric Reinier for the bioinformatics analysis at the beginning of the project. We also thank Dr. M. Jager (Leiden, The Netherlands) for providing the primary uveal melanoma cell lines.
Funding
This work was supported by the French government, INSERM, La Ligue Nationale contre le cancer, INCA PLBio to CB and PB (INCA-12824), Conseil Départemental des Alpes Maritimes (2016-294DGADSH-CV), the National Research Agency under the Investments for the Futur program UCAJEDI «ANR-15-IDEX-01», National Infrastructure France Génomique «ANR-10-INBS-09-03» and «ANR-10-INBS-09-02», Canceropole PACA, Club Francophone des spécialistes de la rétine (CFSR), La Ville de Nice, Mickaël Fulci, La Fondation ARC to CB (20171206312) and La Fondation pour la Recherche Médicale for CP and GG. ID is an ‘équipe labélisée’ of the French League against Cancer.
Author information
Authors and Affiliations
Contributions
CB and RB designed and supervised the study, prepared the figures and wrote the manuscript. VM carried out scRNA-seq experiments. KLB and NN performed the computational analyses and assisted in data analysis in discussions with PB who also critically reviewed the manuscript. CP, TS, YC and GB performed patient sample processing and functional experiments. KB, CH, MD and MI provided technical assistance. GG and ID carried out the RNAscope assays. SL and PH coordinated patient sample collection, maintained IRB approval and performed histological analysis. FP provided cytogenetic analyses. AM, SNE, CM, JPC and SB gathered patients’ consent, provided the samples and clinical data.
Corresponding author
Ethics declarations
Ethics
The study was approved by the hospital ethics committee (Nice Hospital Center and University of Nice Côte d’Azur). The study was performed in accordance with the Declaration of Helsinki.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Blagosklonny
Supplementary information
Rights and permissions
About this article

Cite this article
Pandiani, C., Strub, T., Nottet, N. et al. Single-cell RNA sequencing reveals intratumoral heterogeneity in primary uveal melanomas and identifies HES6 as a driver of the metastatic disease. Cell Death Differ 28, 1990–2000 (2021). https://doi.org/10.1038/s41418-020-00730-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-00730-7
This article is cited by
-
Advances in single-cell RNA sequencing and its applications in cancer research
Journal of Hematology & Oncology (2023)
-
Endoplasmic reticulum stress: molecular mechanism and therapeutic targets
Signal Transduction and Targeted Therapy (2023)
-
Single-cell transcriptomics of the ocular anterior segment: a comprehensive review
Eye (2023)
-
ITGB2-ICAM1 axis promotes liver metastasis in BAP1-mutated uveal melanoma with retained hypoxia and ECM signatures
Cellular Oncology (2023)
-
Integrated analysis reveals the dysfunction of signaling pathways in uveal melanoma
BMC Cancer (2022)
