
Abstract
Cytosolic nucleic acid sensors have a critical role in detecting endogenous nucleic acids to initiate innate immune responses during microbial infections and/or cell death. Several seminal studies over the past decade have delineated the conserved mechanism of cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS) and the downstream signaling adapter stimulator of interferon genes (STING) in mediating innate immune signaling pathways as a host defense mechanism. Besides the predominant role in microbial infections and inflammatory diseases, there is an increased attention on alternative functional responses of cGAS–STING-mediated signaling. Here we review the complexity of interactions between the cGAS–STING signaling and cell death pathways. A better understanding of molecular mechanisms of this interplay is important with regard to the development of new therapeutics targeting cGAS–STING signaling in cancer, infectious, and chronic inflammatory diseases.
Similar content being viewed by others
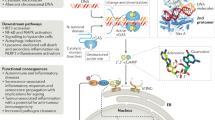

Facts
-
Cytosolic DNA stimulates various signaling pathways that often lead to different modes of cell death.
-
The cGAS–STING pathway recognizes cytosolic DNA to initiate type I interferon production.
-
Autophagy has a dual role in cGAS–STING signaling, by initiating inflammatory responses and by targeting STING for degradation.
Open questions
-
What dictates the redundancy in cell death pathways triggered by the cGAS–STING?
-
What are the mechanisms by which the cGAS–STING induces autophagy-dependent cell death?
-
Can STING-mediated cell death be targeted for treating cancer and other diseases?
Introduction
Cell death is indispensable for the development of tissues and organs, and for eliminating infected, damaged, or transformed cells to maintain organismal homeostasis. Not that long-ago cell death was broadly categorized into just two types, apoptosis (immunologically silent) and necrosis (accidental or inflammatory cell death) [1, 2]. Subsequent studies uncovered various other cell death modalities including (but not limited to) programmed necrosis, pyroptosis, ferroptosis, and autophagy-dependent cell death (ADCD) [3]. In the last two decades, the ability of DNA to initiate different types of cell death as a host protective response have emerged as a key feature of innate immunity in mammalian cells. Under normal circumstances DNA is strictly confined to the nucleus or the mitochondrion of a eukaryotic cell. Therefore, the presence of self-DNA in unusual sites such as cytoplasm or endosomes and/or the presence of exogeneous DNA from microbial challenge is considered as a threat. To counteract these danger signals, cells are equipped with various DNA sensors, such as toll-like receptor 9 (TLR9), absent in melanoma 2 (AIM2), cyclic GMP-AMP synthase (cGAS), RNA polymerase III, DNA-dependent activators of IRFs, interferon-inducible protein 16, DDX41, LSm14A [4]. Of these, only TLR9, AIM2, and cGAS are well characterized, whereas the rest of them are yet to be validated by genetic approaches [5].
TLR9 was the first DNA-sensing receptor identified in immune cells such as dendritic cells (DCs), B cells and macrophages [6]. Under basal conditions, TLR9 is retained in the endoplasmic reticulum (ER) [7, 8]. However, detection of unmethylated cytosine-guanine (CpG)-rich motifs that are relatively abundant in bacteria and DNA viruses, occurs in early endosomes that activate NF-κB via MyD88 and type I interferon (IFN) production to initiate a cascade of innate and adaptive immune responses [6, 9, 10]. Yet, mitochondrial DNA (mtDNA) can also activate TLR9 marked by p38 mitogen-activated protein kinase activation in human PMNs and immortalized podocytes [11, 12]. This is unsurprising as mitochondria evolved from bacteria that are endosymbionts in mammalian cells and therefore, mtDNA contains non-methylated CpG motifs [13, 14].
Another sensor for cytosolic DNA is AIM2 that can bind to DNA directly in a sequence-independent manner via C-terminal HIN-200 domain, where an N-terminal pyrin domain interacts with apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC) to initiate inflammasome formation [15]. AIM2 requires at least 80 bp of dsDNA to initiate inflammasome activation [16] resulting in IL-1β and IL-18 release accompanied by inflammatory cell death termed pyroptosis (discussed below) to offer protection against cancer and infections caused by DNA viruses, bacteria, and fungal pathogens [15].
In recent years, cytosolic sensing of mislocalized DNA by cGAS has emerged as a key event in cellular responses to pathogen invasion, DNA damage, and mitochondrial stress. cGAS recognizes DNA of various sizes in a sequence-independent manner, with human cGAS able to sense DNA sequences as short as 45 bp [17], however, larger DNA fragments result in better immune response [18]. cGAS-mediated stimulator of interferon genes (STING) activation drives IFN responses and also activates NF-κB (discussed in detail below). Historically, the nucleus has been regarded as ‘immune-privileged’, however, this paradigm was overturned by nuclear localization of cGAS and its association with centromeric DNA in DCs [19], and also the identification of heterogeneous nuclear ribonucleoprotein A2B1 that can bind viral DNA in the nucleus during herpes simplex virus-1 infections to drive IFN responses [20]. Notably, nuclear localized cGAS is 200-fold less efficient to nuclear DNA than extranuclear cGAS to cytosolic DNA to induce IFN responses [19], which could be due to suppression of cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) production by nucleosomes [21]. In addition, positioning of cGAS to plasma membrane could not be discounted [22]. This raises the question if localization of the cGAS affects intensity of the IFN responses. However, another persistent question is why does any cell require multiple DNA sensors? One possibility is that these sensors act as a safety checkpoints to ensure any danger signal in the form of cytoplasmic DNA is not missed in different cellular contexts. The other possibility is that if one DNA-sensing pathway becomes nonfunctional and/or inhibited by microbial or danger signals, the other DNA sensors could act as a backup. Although, mammalian cells are equipped with an array of DNA sensors, cGAS–STING-mediated IFN response is gaining increasing attention due its critical role in inflammatory and infectious diseases, and cancer [23, 24]. This is possibly due to widespread STING expression in various cell-types and the ability of cGAS–STING to regulate different programmed cell death pathways. In this review we discuss our current understanding of the interplay between the cGAS–STING signaling and different modes of cell death such as apoptosis, necroptosis, and pyroptosis. Finally, we summarize the new work linking the cGAS–STING pathway to autophagy and ADCD.
The cGAS–STING signaling pathway
cGAS recognizes viral, bacterial, protozoal, mitochondrial, and self-DNA from tumor or dead cells in the cytosol to activate a cascade of signaling pathways leading to inflammation [25,26,27,28] (Fig.1). Binding of DNA to the cGAS induces conformational changes to cGAS, which in turn catalyzes the formation of 2′,3′-cGAMP, a cyclic di-nucleotide (CDN) with a unique phosphodiester linkage that uses adenosine triphosphate (ATP) and guanosine triphosphate (GTP) [29]. DNA binding to cGAS induces liquidlike droplets that act as a microreactor, enriching enzyme, and reactants to enhance cGAMP production, as demonstrated by cGAS-DNA puncta in both human fibroblast cell line and mouse embryonic fibroblasts (MEF) [30]. The generated cGAMP acts as a secondary messenger and activates STING (also known as transmembrane protein 173 (TMEM173), N-terminal methionine–proline–tyrosine–serine plasma membrane transpanner (MPYS), mediator of interferon regulatory factor 3 (IRF3) activation, and endoplasmic reticulum IFN stimulator) [31]. Notably, cGAMP can transactivate STING in neighboring cells [32] with the aid of gap junction proteins such as connexins to amplify inflammatory responses [33]. In addition, bacterial CDNs such as cGAMP, c-di-AMP and c-di-GMP can also activate STING directly [34,35,36].

a cGAS is activated by mislocalized DNA in the cytosol by various sources such as self-DNA from damaged mitochondrion or micronucleus, or exogeneous DNA derived from virus, dead cell, and bacterium. Activated cGAS uses ATP and GTP as substrates to catalyze the formation of cGAMP, which acts as a second messenger to activate STING. b Cyclic dinucleotides secreted from bacteria can also activate STING directly. c Activated STING dimerizes and translocates from the ER to the perinuclear region via Golgi to recruit and promote TBK1-IRF3-IκB assembly, which subsequently drives activation of IRF3 and NF-κB to produce IFN and inflammatory cytokines (TNF, IL-6), respectively.
In resting state, STING is contained in the ER as a transmembrane homodimer by a Ca2+ sensing transmembrane protein, stromal interaction molecule 1 (STIM1) [37]. Upon activation, STING undergoes a conformational change and translocates to the perinuclear region via an unknown mechanism, where it oligomerizes and recruit tank-binding kinase 1 (TBK1) [38]. Oligomerized STING offers a platform for trans-TBK1 autophosphorylation, and in turn gets phosphorylated by TBK1 as delineated by structural studies using human TBK1 complex with cGAMP and full-length chicken STING [38]. STING can interact with both TBK1 and the transcription factor IRF3, therefore specifying IRF3 for phosphorylation by TBK1 [39, 40]. Phosphorylated IRF3 dimerizes and translocates into the nucleus to induce the expression of inflammatory genes including IFN and IFN-stimulated genes (ISG). IFNs are indispensable for antitumor immune responses in DCs and CD8+ T cells, and mediate adaptive immunity [41, 42]. In addition, STING–TBK1 association also phosphorylates IκB kinase, leading to noncanonical activation of NF-κB that regulates the transcription of inflammatory cytokines, including IL-6 and tumor necrosis factor (TNF) [25, 26]. However, TBK1 and IκB kinase are dispensable for STING-induced NF-κB activation in mouse myeloid cells both in vivo and in vitro [43]. Notably, STING can be degraded by autophagy (discussed later in this review) to tightly regulate the cGAS–STING pathway, as aberrant activation of this pathway causes chronic inflammatory diseases such as systemic lupus erythematosus (SLE) [28]. However, IFN-independent functions of STING in antiviral responses and T cell death to control immune evasion cannot be discounted [44].
Phylogenetic analysis has revealed that both cGAS and STING homologs are present in mammals and the origin of this pathway has been traced back to a unicellular eukaryote Monosiga brevicollis, with the core CDN-binding function of STING showing evolutionary conservation over 600 million years [45]. Interestingly, STING or cGAS alone is unable to drive IFN-β expression effectively as shown following Ectromelia Virus infections in HEK293T cells [46]. This is further supported by the fact that these two proteins have coevolved, such that different species will either contain homologs of both cGAS and STING or have lost both, highlighting their crucial requirement to cooperatively mediate the inflammatory responses [45].
Interactions between the cGAS–STING pathway and apoptosis
Apoptosis is one of the most conserved and best studied cell death pathways in terms of mechanism and regulation for its vital role in development and homeostasis [47, 48]. In brief, apoptosis is mediated by caspases, a family of cysteine proteases [49]. There are three major pathways that signal caspase activation and apoptosis in mammalian cells, often termed as intrinsic, extrinsic (or mitochondrial pathway), and granzyme-mediated cell death pathway [50]. The extrinsic pathway is initiated by binding of ‘death ligands’ such as TNF, FASL, TRAIL, to TNF superfamily receptors such as TNF receptor 1 (TNFR1), FAS, death receptor 4/5, respectively, to recruit cytoplasmic adapter proteins such as Fas-associated death domain or TNF-associated death domain. These adapters in-turn recruit and activate caspase-8. Caspase-8 is the central protease in death receptor induced apoptosis, and once activated, it mediates caspase-3 activation, either directly or through (BH3 Interacting Domain Death Agonist) BID cleavage and engagement of the mitochondrial pathway [51]. The intrinsic pathway is centered on B-cell lymphoma-2 (BCL-2) family proteins that comprises of both antiapoptotic and proapoptotic members [47, 52]. Cellular stress or danger signals can either activate proapoptotic proteins such as BCL-2 associated X protein (BAX) and BCL-2 homologous antagonist/killer (BAK) or inhibit antiapoptotic proteins (BCL-2, BCL-x, BCL-w, and MCL-1) through the activation of several BH3 only domain family members (BIM, BID). This results in mitochondrial outer membrane permeabilization, resulting in cytochrome c release, that binds to an adapter apoptotic protease activating factor-1 (APAF1) to initiate the formation of the apoptosome, a complex required for recruitment and activation of caspase-9. Activated caspase-9 cleaves and activates caspase-3 and -7, which targets a number of cellular substrates to initiate apoptotic cell death [53]. Last, in the granzyme pathway cytotoxic T lymphocytes kill target cells via secreted granular proteases and perforins [54, 55]. Despite the specific requirements of each of these pathways, intrinsic, extrinsic, and granzyme B converge on execution pathway (caspase-3 and -7 activation) of apoptosis [50]. It is however worth noting that cytotoxic T lymphocytes can also induce non-apoptotic types of death in target cells [56].
Apoptosis is generally regarded as silent cell death, where apoptotic bodies are rapidly phagocytosed by neighboring cells or macrophages to prevent the activation of inflammatory pathways. Therefore, not surprisingly apoptotic caspase activation has inhibitory effect on cGAS–STING inflammatory pathways. For example, both in vitro and in vivo ISG is constitutively expressed in caspase-9-deficient, and caspase-3/7 double knockout MEFs. Furthermore, STING pathway is constitutively activated in caspase-9 deficient cells [57]. Finally, mtDNA released from Bak/Bax activation is shown to be the endogenous ligand that triggers the cGAS–STING pathway activation, with caspases regulating the inflammatory responses [57]. These findings are further corroborated by an independent study that confirmed the role of mtDNA in initiating cGAS–STING-mediated IFN responses and active caspase-9 inhibiting these inflammatory responses in hematopoietic cells both in vivo and in vitro [58]. The mechanism of apoptotic caspases downregulating inflammatory responses was uncovered in a recent study, where active caspase-3 cleaved cGAS, and IRF3 in THP-1 cells during viral infections to prevent excessive IFN production [59]. Moreover, defects in clearing apoptotic cells led to the formation of apoptosis-derived membrane vesicles (ADMVs) that have been associated with autoimmune diseases such as SLE [60]. ADMVs isolated from the sera of SLE patients contained dsDNA that triggered cGAS–STING signaling pathway leading to ISG expression in THP-1 cells [61]. Based on these studies it can be argued that apoptosis prevents cGAS–STING-mediated inflammatory responses (Fig. 2). By contrast, emerging evidence suggests that ER stress associated with STING activation can trigger apoptosis. For instance, during mycobacterial infections, STING activation causes ER stress leading to BAX activation and cytochrome c release resulting in apoptosis in RAW.247 cells [62]. In addition, ER stress-mediated STING activation in mouse hepatocytes results in apoptosis in an early alcoholic liver disease model [63]. It is possible that STING regulates calcium homeostasis and ER stress, as STING gain-of-function mutant disrupts calcium homeostasis causing ER stress that primes T cell apoptosis [64]. Also, STING pathway activation by the STING agonist 10-carboxymethyl-9-acridanone (CMA) triggers apoptosis in primary and lymphoblastic leukemia murine T cells by coordinated action of IRF3 and p53 but not in murine DCs [65]. Similarly, the STING agonist cGAMP initiates ER-stress-mediated apoptosis in normal and malignant B cells, but not in MEF, B16 melanoma, Hepa 1–6 hepatoma, LL/2 Lewis lung cancer cell lines and wild-type T cells from mice [66]. Murine-specific small molecule STING agonists CMA and DMXAA also failed to trigger apoptosis in MEF, B16 melanoma, Hepa 1–6 hepatoma, LL/2 Lewis lung cancer cell lines [66]. Inability of these small molecules to activate human STING is attributed to dynamic structural differences between mouse and human STING [67]. These findings also suggest that STING-mediated apoptosis is cell- and/or context-dependent, which could be due to variation in endogenous STING expression amongst different cell types as observed in some studies [65, 66], or cell-specific kinetics of STING regulation (phosphorylation or degradation). Further mechanistic insights on how STING regulates calcium homeostasis, whether directly or indirectly by STIM1 or via other calcium signaling pathways, are required from the therapeutic viewpoint of inhibiting ER stress to prevent chronic STING activation in autoimmune diseases [64].

During apoptosis signaling the proapoptotic proteins such as BAK/BAX cause mitochondrial membrane permeabilisation and release of cytochrome c and mitochondrial DNA. Mitochondrial DNA can trigger cGAS–STING–TBK1 pathway to drive IFN-mediated immune responses. Whereas cytosolic cytochrome c drives the assembly of the Apaf1-caspaspe-9 apoptosome that subsequently activate the initiator caspases (e.g. caspase-3 and caspase-7) to mediate apoptosis. Caspase-3 negatively regulates cGAS–STING pathway resulting in cleavage of IRF3 and STING. On the other hand, STING activation causes ER-stress that ultimately results in apoptosis by via BAK/BAX activation and cytochrome c release. Red arrows indicate negative regulation.
Interplay between the cGAS–STING signaling and necroptosis
Necroptosis is a regulated form of necrosis, mediated by receptor interacting protein kinases activation and its substrate mixed lineage kinase like (MLKL) that leads to plasma membrane disruption [68,69,70]. When apoptosis is inhibited by genetic defects, viral or bacterial infections, or experimentally by caspase inhibitors, necroptosis is promoted as a mode of regulated cell death that releases DAMPS to initiate innate immune responses [71]. Importantly, IFN signaling is indispensable to trigger necroptosis and IFN-α receptor 1 (IFNAR1) deficient mice are resistant to necroptosis during Salmonella Typhimurium infections [72]. Also, in IFNAR1 deficient bone marrow-derived macrophages (BMMs) necroptosis is inhibited, despite of caspase inhibition and TLR induction by stimulation with LPS, polyinosinic-polycytidilic acid, TNFα, or IFN-β [73]. Therefore, it is logical to deduce that the cGAS–STING pathway could play a predominant role in triggering necroptosis (Fig. 3). Along the same lines, cGAS–STING-dependent TNF and IFN signaling triggers necroptosis in response to cytosolic DNA, when apoptotic caspases are inhibited in BMMs [74]. In addition, mtDNA activates the STING pathway that subsequently enhances RIPK3/MLKL expression to trigger necroptosis, when caspases are inhibited in HT29 colon cancer cell line and MEFs [75]. Furthermore, constitutive IFN levels mediated by cGAS–STING activation maintain MLKL at a threshold level to predispose BMMs to necroptosis [76]. Yet, overexpression of MLKL in BMMs from Ifnar−/− mice does not sensitize cells to necroptosis [76, 77]. This suggests the presence of additional IFN-inducible necroptotic pathway components, that are required for necroptosis, yet to identified. However, during Salmonella Typhimurium infections, IFN-mediated RIPK3 activation impairs antioxidative responses that could sensitize BMMs to necroptosis [78].

Activation of the cGAS–STING pathway by mitochondrial DNA results in production of IFN and TNF. Binding of IFN to IFNAR1 and TNF to TNFR1 can results in RIPK1–RIPK3 activation, when caspase-8 is inhibited. RIPK1–RIPK3 activates MLKL to execute necroptosis. Moreover, IFN upregulates RIPK3 and MLKL expression. It would be interesting to understand if STING agonists can initiate necroptosis to limit tumor growth when apoptotic caspases are usually inhibited in cancer cells. Green arrow indicate upregulation.
Considering that loss of apoptosis is a hallmark of cancer [79], it is rational to trigger secondary cell death pathway such as necroptosis that is mechanistically distinct. Based on some studies (as discussed above) indicating cGAS–STING signaling enhances expression of necroptotic apparatus to trigger robust cell death upon specific stimuli [75, 76], it is reasonable to consider using STING agonists to trigger necroptosis in cancer cells. However, the use of the STING agonist DMXAA combined with caspase inhibition in vivo increases TNF and IFN levels subsequently leading to necroptosis, but this resulted in increased mortality due to septic shock [74]. Although, DMXAA is a failure in clinics due to its inability to effectively activate human STING, small molecule STING agonist amidobenzimidazole (ABZI) has been reported to bind and activate both human and mouse STING [80, 81]. Therefore, such STING agonists in clinical trials will need to be assessed for their roles in triggering necroptosis without extensive secondary effects. This conclusion is reinforced by the observation that necroptotic rather than apoptotic cell death is efficient in DC-mediated cross-priming of CD8+ T cells to achieve antitumor immunity [82]. Despite the clinical interest in use of STING agonists as anticancer drug, surprisingly little is known about the role of necroptosis during STING activation, however, this is clearly an area of ongoing investigations. On the other hand, it would be of interest to examine if STING antagonists can be used to treat septic shock caused by bacterial infections. This will require further understanding of the mechanism and role of necroptosis in inducing septic shock in the context of bacterial infections.
Crosstalk between the cGAS–STING pathway and pyroptosis
Pyroptosis is an inflammatory form of cell death initiated by inflammasomes, eventually leading to gasdermin-D (or E) cleavage to generate the N-terminal domain, which forms a pore in the cell membrane [83,84,85]. In brief, inflammasomes are large multiprotein complexes comprised of germline-encoded pattern recognition receptors of the Nod-like receptor (NLR) family (NLR family pyrin domain-containing 3, NLRP3; NLR family CARD domain-containing protein 4, NLRC4; and AIM2), the adapter ASC and procaspase-1. In response to pathogenic or physiological perturbation in the cytosol, procaspase-1 undergoes inflammasome-assisted proximity-induced autoproteolysis resulting in active caspase-1 [86]. Active caspase-1 converts pro-IL-1β and pro-IL-18 into its mature form and also cleaves gasdermin-D to initiate pyroptosis [87]. A considerable body of literature highlights the critical role of the AIM2 inflammasome in the host defense mechanism, and AIM2 deficient mice are highly susceptible to infections with Francisella tularensis, Mycobacterium tuberculosis, Staphylococcus aureus, and display higher mortality and bacterial burden in comparison to wild-type mice [88,89,90]. Nonetheless, the role of the cGAS–STING in these scenarios are yet to be examined.
A key question is whether both cGAS and AIM2 function concurrently or act sequentially to initiate immune responses? To address this, a study demonstrated that cGAS–STING-IFN1 pathway activation amplified AIM2 protein levels to induce robust innate immune responses during murine cytomegaloviral infections in BMMs [91]. Similarly, cGAS-mediated IFN responses increase caspase-1 and caspase-11 expression, which in turn increased IL-1β release and pyroptotic cell death during Chlamydia trachomatis infections [92]. Therefore, robust activation of both canonical and noncanonical inflammasomes are dependent on early IFN priming of inflammasome components. With these observations it can be proposed that AIM2 activation follows STING signaling, and the activation of these signaling pathways amplify the host defense response (Fig. 4). However, cGAS–STING-NLRP3 activation replaces the AIM2 function in human myeloid cells in response to DNA, as STING activation induces lysosomal cell death leading to K+ efflux, which in turn activates the NLRP3 inflammasome [93]. This finding raises the question as to whether STING is more sensitive to mislocalized DNA than AIM2? It is possible that cGAS–STING can detect lower levels of DNA in the cytosol than AIM2. Another possible explanation is that there is still an unknown factor that could determine which DNA sensor to be activated depending on the levels of DNA in the cytosol, that would correspond to the degree of the danger signal or localization of DNA.

A model that proposes DNA concentration could determine type of DNA sensor and level of innate immune responses required to maintain homeostasis. During low concentration of DNA in the cytosol, cGAS–STING could be activated to drive IFN-mediated immune responses. Whereas, when high concentrations of DNA accumulate in the cytosol, the AIM2 inflammasome could be activated to intensify the innate immune responses by IL-1β release, and pyroptosis mediated by active caspase-1 and gasdermin cleavage. Therefore, IFN release from cGAS–STING activation primes AIM2 inflammasome components as marked by increased expression of AIM2, procaspase-1/11/4/5. Conversely, to prevent hyperactivation of the immune responses, K+ efflux mediated by gasdermin-d cleavage inhibits cGAS, and active caspase-1/11/4/5 can cleave cGAS directly. Red arrows indicate negative regulation, and green arrows indicate positive regulation.
Conversely, to prevent overactivation of inflammatory responses, the innate immune system maintains a balance between different DNA sensors expression and/or activation. For instance, gasdermin-D activated by the AIM2 inflammasome inhibits IFN-β responses to cytoplasmic DNA via K+ efflux in BMM [94]. In addition, AIM2 deficient BMMs and bone marrow-derived DCs display elevated IFN-β levels in response to cytoplasmic DNA-mediated cGAS–STING activation [95]. Moreover, to prevent deleterious effects of inflammatory responses not only the DNA-sensing AIM2 inflammasomes, but also other components of inflammasomes can negatively regulate the cGAS–STING pathway. For example, the NLRC3 interacts directly with STING to prevent its translocation into the perinuclear region and binding to TBK1, that ultimately hinders IFN responses and NF-κB activation [96]. Also, caspase-1 limits cytosolic DNA-mediated cGAS–STING activation by directly cleaving cGAS during canonical inflammasome activation [97]. Similarly, during LPS-induced noncanonical inflammasome activation caspase-4, -5 (human), and -11 (mouse) can cleave human and mouse cGAS, respectively [97]. Furthermore, NLRP4 can recruit the ubiquitin ligase DTX4 to TBK1 for ubiquitination, which leads to TBK1 degradation and thereby regulation of DNA-mediated IFN responses [98]. By contrast, IFN inhibited NLRP3 inflammasome activation in STAT1-dependent manner, thereby preventing caspase-1 dependent IL-1β release [99]. Collectively, these studies highlight the sophisticated interplay between cGAS–STING and inflammasome components to prevent hyper-inflammation.
cGAS–STING and autophagy
The evolutionarily conserved catabolic process of autophagy is essential for cellular health and organismal homeostasis [100, 101]. Autophagy involves a complex machinery that targets damaged or long-lived organelles and/or proteins packaged in double membrane vesicles termed autophagosome, to lysosomes for degradation and/or recycling. Based on the mode of cargo delivery to lysosomes, autophagy can be broadly classified into macroautophagy (cytoplasmic contents are delivered to lysosome by autophagosomes), microautophagy (direct lysosomal membrane invagination), and chaperone-mediated (direct translocation across lysosomal membrane), which is a non-conserved process only observed in mammalian cells [102]. Macroautophagy, henceforth referred to as autophagy, involves several steps: initiation, nucleation or phagophore formation, elongation of phagophore membrane to form autophagosome, fusion of autophagosome with the lysosomes, and degradation of autophagosome contents. Each of these steps involves a series of autophagy-related gene (Atg) products. Briefly, autophagy initiation is assisted by the Atg1/ULK1 serine-threonine complex comprising ULK1, ATG13, ATG101, and FAK-family interacting protein (FLIP2000) [103]. The class III phosphatidylinositol 3-kinase (PI3K) complex, consisting of ATG15, vacuolar protein sorting (VPS) 15, VPS 34, and Beclin1-related proteins mediate nucleation by generating phosphatidylinositol 3-phosphate (PI3P) [104]. ATG9 vesicles offer a portion of the autophagosomal membrane, whilst WIPI proteins and its interacting partners (ATG2a or ATG2b), and two transmembrane ER proteins, vacuole membrane protein 1 and TMEM41 B are necessary to form isolation membrane [105,106,107,108]. Two unique ubiquitin-like protein conjugation systems, ATG12-ATG5-ATG16L complex and ubiquitin-like microtubule-associated protein 1 light chain 3 (MAP1LC3B/ATG8) are conjugated to a lipid molecule phosphatidylethanolamine to aid in elongation [109]. LC3 is synthesized in its precursor form as pro-LC3, which is cleaved by ATG4 to LC3-I [110, 111]. ATG3 and ATG7 convert LC3-I to LC3-II, which aids in elongating autophagosome membrane [112,113,114]. While the autophagosome closure remains poorly understood [115], autophagosomes fusion with the lysosomes requires specific SNARE proteins, small GTPases and their effectors [116] to initiate degradation of cargo by a series of lysosomal enzymes. Although, autophagy is regarded as an unselective process, selective autophagy is mediated by ubiquitin-binding proteins (p62, autophagy receptor optineurin, and NDP52), or proteins with transmembrane domains such as Nix (NIP3-like protein X as mitophagy receptor) [117, 118].
In recent years growing evidence has indicated interactions between autophagy machinery and the cGAS–STING pathway. Over 10 years ago Akira et al. [119] identified that ATG9a and LC3-II co-localize with STING in vesicles after dsDNA stimulation in MEFs. In addition, deletion of ATG9a increases STING–TBK1 assembly upon sensing dsDNA, suggesting the possible roles of autophagy proteins in STING regulation [119]. Subsequent studies demonstrated interactions between autophagy and cGAS–STING using both genetic and biochemical approaches [120,121,122]. It is possible that STING activation can initiate autophagy and follow the key steps of autophagy pathway (discussed above), or STING–TBK1 can be translocated directly to autophagosome via an unknown mechanism independent of p62 (Fig. 5). As an example, cGAS-Beclin-1 interaction initiates autophagy by releasing the autophagy negative regulator, Rubicon to allow PI3K complex activation upon dsDNA recognition during HSV infections [123]. Furthermore, STING-induced autophagy requires WIPI2 and ATG5 proteins [122], ATG3, p62 and increase in LC3-II conversion followed by degradation of STING via conventional autophagy [121]. On the other hand, STING interacts directly with LC3 leading to degradation of STING itself and pTBK1, highlighting unconventional means of autophagy activation [120]. Undoubtedly, there is a high degree of complexity associated with re-localization of STING with different autophagy proteins. It should be noted that most of the STING-autophagy studies used V-ATPase inhibitors such as Bafilomycin or concanamycin A, or chloroquine to inhibit autophagy pathway that could affect other signaling pathways. Also, studies involving genetic manipulation of autophagy should account for the non-autophagic role of autophagy proteins in cell survival, maintenance, and death [124].

Autophagy process involves five key steps, (1) initiation, (2) nucleation, (3) elongation, (4) autophagosome formation, and (5) degradation, where each step is regulated by specific ATG proteins as highlighted, or explicit proteins such as ULK1, Beclin-1, P13P, LC3 conjugation system as mentioned. cGAS–STING activation can initiate autophagy and follow the five key steps triggering its own degradation, however the requirement of specific autophagy components at each step is ambiguous. In addition, cGAS–STING activation mediated by cGAS–STING–TBK1–IRF3 assembly complex can translocate directly to autophagosomes via an unknown mechanism. Also, cGAS can interact directly with Beclin-1 to initiate autophagy independent and/or absence of STING. Furthermore, ULK1 can phosphorylate STING and prevent its activation.
During Mycobacterium tuberculosis infection bacterial DNA-mediated STING–TBK1 activation targets a subpopulation of bacteria to autophagosomes in BMMs [125] that is likely to attenuate virulence and pathogenicity in murine lungs [126]. However, during Mycobacterium tuberculosis infections, NF-κB-induced expression of the DNA damage-regulated autophagy modulator DRAM1, which then localized with p62 and STING in zebrafish and human macrophages initiating autophagic defense responses [127]. It is possible that autophagy prevents sustained STING phosphorylation but not NF-κB activation [128]. Although, the NF-κB activation is dependent on MyD88, the role of STING in NF-κB activation cannot be overlooked [127].
Although p62 was demonstrated to be crucial for translocation of STING to lysosome-associated compartments for STING degradation, it should be noted that p62 knockout MEFs and THP-1 cells show a delay rather than abrogation of STING degradation in comparison to wild-type cells [121]. This suggests that ubiquitination by other ubiquitin ligases such as RNF5 and TRIM30 can also target STING for proteasomal degradation [129, 130]. Additional studies are required to understand the role of ubiquitin ligases in autophagy-dependent STING activation and regulation. Collectively, STING activation initiates autophagy followed by its own degradation. However, cell-type, concentration of cytoplasmic DNA and the strength of inflammatory responses may dictate how and whether autophagy components mediate STING pathway signaling. Importantly, it is possible that STING-mediated autophagy can be a potential way to clear infections effectively, and further studies are warranted to understand the role of STING agonists to treat bacterial infections.
Some studies have reported that rather than STING, it is cGAS that interacts with autophagy components, thereby increasing the complexity of interactions between the cGAS–STING pathway and autophagy. In keeping with this concept, cGAS−/− mice display increased susceptibility to Mycobacterium tuberculosis than their wild-type or STING-deficient counterparts [131]. In addition, cGAS-induced autophagy protects hepatocytes that lack STING [132] from hypoxia-induced ischemia-reperfusion injury, independent of Beclin-1 [133]. Overall, it could be envisaged that cGAS can activate STING-dependent and -independent autophagy as host protective response depending on the intensity of the threat posed, and the degree of inflammatory responses required to clear danger signals. One possibility is that STING, but not its activation is required for basal autophagy as STING-dependent autophagy occurs independent of TBK1 activation and IFN production [122]. In contrast, cGAS–STING pathway activation leading to inflammatory responses as a host defense mechanism is required to counteract a severe threat. Alternatively, there could be an unknown sensor that licenses STING-dependent versus STING-independent autophagy in a cell-type or cytosolic DNA concentration context [134].
ADCD following cGAS–STING activation
As autophagy removes damaged organelles and protects cells under nutrient-limiting conditions, it is primarily a cytoprotective process. However, autophagy often accompanies cell death and has been shown to be a driver of cell death in specific contexts [135]. ADCD is defined by The Nomenclature Committee of Cell Death as ‘a form of regulated cell death that mechanistically depends on the autophagy components’ [3]. To be termed as ADCD, genetic or chemical inhibition of autophagy should prevent cell death [102, 136]. Also, more than one autophagy proteins must be involved in triggering cell death [135].
To date, the most robust genetic evidence for ADCD comes from studies involving the degradation of larval midgut during larval-pupal transition in Drosophila [137, 138]. Genetic studies have demonstrated that larval midgut degradation can occur in the complete absence of the apoptosis machinery in Drosophila but has an essential requirement for autophagy induction [131, 134, 135]. Studies in mammalian and other model systems have also identified instances of ADCD in specific developmental or pathophysiological context [135]. For example, autosis is a specific form of ADCD that involves the Na+/K+-ATPase [139]. In another instance, the removal of interdigital web cells during embryonic development is further delayed in Atg5/Bax/Bak triple deficient mice, compared to Bax/Bak double knockouts, suggesting a role for autophagy in removing interdigital web in mammals [140]. Notably, invertebrates lack C-terminal tail of STING that recruits TBK1 and IRF3 and can still mediate ADCD [141]. However, recent studies link self-DNA stimulated cGAS–STING to excessive autophagy leading to ADCD [139, 140]. One study demonstrated that cell crisis due to telomeric damage results in release of DNA fragments into the cytoplasm, that trigger autophagy as observed by increased in LC3-II or decrease in p62 [142]. Inhibition of autophagy or STING pathway components prevented cell crisis resulting in unstable genome and chromosomal aberrations, suggesting that this pathway is involved in deleting damaged and potentially premalignant cells [142]. In other recent study the infection by Burkholderia pseudomallei, that causes host cell fusion, was shown to activate cGAS–STING activation, independent of bacterial ligands [140]. Infection induced cell fusion lead to aberrant mitosis, formation of micronuclei, which colocalised with cGAS, followed by the activation of cGAS–STING pathway and ADCD [140]. These two studies demonstrate that cell crisis or bacterial-mediated cell fusion results in cGAS–STING-mediated autophagy and that subsequent ADCD limits accumulation of damaged cells [142, 143]. Thus, these studies suggest that one of the functions of cGAS–STING-dependent ADCD is to prevent tumorigenesis. However, autophagy is known to have dual role in cancer [144] and therefore, further mechanistic studies are required to understand context-dependent ADCD. Although, excessive autophagy can clearly trigger cell death, further work is warranted to establish if different molecular factors are involved in developmental versus stress-induced ADCD.
Conclusion and perspectives
Activation of the cGAS–STING pathway is a major responder to viral and bacterial infections. However, this pathway can be activated by self-DNA and can act as a double-edged sword, where it can be beneficial or detrimental to the host depending on the context of its activation (self-DNA, infection, and chronic inflammation), cell-type, cell death pathways activated, and the magnitude and duration of activation. Structural studies of STING have shown that a closed conformational change in the ‘lid’ loop of STING upon binding to CDN is necessary for its activation. However, studies with the newly identified ABZI STING agonist demonstrated that the activation can occur in an open conformation. Identification of such novel small molecules also gives a perspective that structures other than CDN with distinct physiochemical and biological properties could bypass cGAS to activate STING and provides more options for the clinical development of STING agonists. Considering that multiple cell death pathways can be triggered following cGAS–STING signaling, a better understanding of the regulatory mechanism distinct to cell-types, context and/or stimulus-dependency will require further investigations. Moreover, the role of STING in the co-existence of various cell death pathways such as the newly described PANopoptosis [145] and switch between cell death pathways remain to be studied. Further knowledge will help answer questions such as what dictates the redundancy in cell death pathways triggered by the cGAS–STING. Any direct role of cGAS–STING signaling in ADCD and the mechanisms of ADCD also require further investigations. More importantly, answers to these questions will eventually open new avenues to target STING-mediated cell death pathways for treating cancer and infectious diseases.
References
Sun EW, Shi YF. Apoptosis: the quiet death silences the immune system. Pharm Ther. 2001;92:135–45.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
Unterholzner L. The interferon response to intracellular DNA: why so many receptors? Immunobiology. 2013;218:1312–21.
Vance RE. Cytosolic DNA sensing: the field narrows. Immunity. 2016;45:227–8.
Kumagai Y, Takeuchi O, Akira S. TLR9 as a key receptor for the recognition of DNA. Adv Drug Deliv Rev. 2008;60:795–804.
Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5:190–8.
Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7:49–56.
Marongiu L, Gornati L, Artuso I, Zanoni I, Granucci F. Below the surface: the inner lives of TLR4 and TLR9. J Leukoc Biol. 2019;106:147–60.
Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature 2008;456:658–62.
Bao W, Xia H, Liang Y, Ye Y, Lu Y, Xu X, et al. Toll-like receptor 9 can be activated by endogenous mitochondrial DNA to induce podocyte apoptosis. Sci Rep. 2016;6:22579.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7.
Gray MW. Mitochondrial evolution. Cold Spring Harb Perspect Biol. 2012;4:a011403.
Liu B, Du Q, Chen L, Fu G, Li S, Fu L, et al. CpG methylation patterns of human mitochondrial DNA. Sci Rep. 2016;6:23421.
Man SM, Karki R, Kanneganti TD. AIM2 inflammasome in infection, cancer, and autoimmunity: role in DNA sensing, inflammation, and innate immunity. Eur J Immunol. 2016;46:269–80.
Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012;36:561–71.
Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, Fischer ES, et al. Structure of the human cGAS-DNA complex reveals enhanced control of immune surveillance. Cell. 2018;174:300–11 e11.
Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity. 2013;39:1019–31.
Gentili M, Lahaye X, Nadalin F, Nader GPF, Puig Lombardi E, Herve S, et al. The N-terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep. 2019;26:2377–93 e13.
Wang L, Wen M, Cao X. Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science. 2019;365:6454.
Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, Funabiki H. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell. 2019;178:302–15 e23.
Barnett KC, Coronas-Serna JM, Zhou W, Ernandes MJ, Cao A, Kranzusch PJ, et al. Phosphoinositide interactions position cGAS at the plasma membrane to ensure efficient distinction between self- and viral DNA. Cell. 2019;176:1432–46 e11.
Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760–70.
Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS–STING pathway in health and disease. Nat Rev Genet. 2019;20:657–74.
Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–92.
Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–8.
Gall A, Treuting P, Elkon KB, Loo YM, Gale M Jr., Barber GN, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. 2012;36:120–31.
Zhou R, Xie X, Li X, Qin Z, Wei C, Liu J, et al. The triggers of the cGAS–STING pathway and the connection with inflammatory and autoimmune diseases. Infect Genet Evol. 2020;77:104094.
Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013;153:1094–107.
Du M, Chen ZJ. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science. 2018;361:704–9.
Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, et al. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–4.
Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493–8.
Pepin G, De Nardo D, Rootes CL, Ullah TR, Al-Asmari SS, Balka KR, et al. Connexin-dependent transfer of cGAMP to phagocytes modulates antiviral responses. mBio. 2020;11:e03187–19.
Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–8.
Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. 2010;328:1703–5.
Danilchanka O, Mekalanos JJ. Cyclic dinucleotides and the innate immune response. Cell. 2013;154:962–70.
Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, et al. The Ca(2+) sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol. 2019;20:152–62.
Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature. 2019;567:394–8.
Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012;5:ra20.
Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347:aaa2630.
Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003.
Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–16.
Balka KR, Louis C, Saunders TL, Smith AM, Calleja DJ, D’Silva DB, et al. TBK1 and IKKepsilon act redundantly to mediate STING-induced NF-kappaB responses in myeloid cells. Cell Rep. 2020;31:107492.
Wu J, Dobbs N, Yang K, Yan N. Interferon-independent activities of mammalian STING mediate antiviral response and tumor immune evasion. Immunity. 2020;53:115–26 e5.
Wu X, Wu FH, Wang X, Wang L, Siedow JN, Zhang W, et al. Molecular evolutionary and structural analysis of the cytosolic DNA sensor cGAS and STING. Nucleic Acids Res 2014;42:8243–57.
Cheng WY, He XB, Jia HJ, Chen GH, Jin QW, Long ZL, et al. The cGAS–STING signaling pathway is required for the innate immune response against ectromelia virus. Front Immunol. 2018;9:1297.
Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20:175–93.
Strasser A, O’Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–45.
Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases. Cell Death Differ. 2015;22:526–39.
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516.
Strasser A, Vaux DL. Viewing BCL2 and cell death control from an evolutionary perspective. Cell Death Differ. 2018;25:13–20.
Reed JC, Jurgensmeier JM, Matsuyama S. Bcl-2 family proteins and mitochondria. Biochim Biophys Acta. 1998;1366:127–37.
Porter AG. Flipping the safety catch of procaspase-3. Nat Chem Biol. 2006;2:509–10.
Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol. 2006;6:940–52.
Martinvalet D, Zhu P, Lieberman J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity. 2005;22:355–70.
Lieberman J. Granzyme A activates another way to die. Immunol Rev. 2010;235:93–104.
Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563–77.
White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–62.
Ning X, Wang Y, Jing M, Sha M, Lv M, Gao P, et al. Apoptotic caspases suppress type I interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol Cell. 2019;74:19–31 e7.
Fehr EM, Spoerl S, Heyder P, Herrmann M, Bekeredjian-Ding I, Blank N, et al. Apoptotic-cell-derived membrane vesicles induce an alternative maturation of human dendritic cells which is disturbed in SLE. J Autoimmun. 2013;40:86–95.
Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, et al. Apoptosis-derived membrane vesicles drive the cGAS–STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis. 2018;77:1507–15.
Cui Y, Zhao D, Sreevatsan S, Liu C, Yang W, Song Z, et al. Mycobacterium bovis induces endoplasmic reticulum stress mediated-apoptosis by activating IRF3 in a murine macrophage cell line. Front Cell Infect Microbiol. 2016;6:182.
Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci USA. 2013;110:16544–9.
Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med. 2019;216:867–83.
Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, et al. Signalling strength determines proapoptotic functions of STING. Nat Commun. 2017;8:427.
Tang CH, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, et al. Agonist-mediated activation of STING induces apoptosis in malignant B cells. Cancer Res. 2016;76:2137–52.
Shih AY, Damm-Ganamet KL, Mirzadegan T. Dynamic structural differences between human and mouse STING lead to differing sensitivity to DMXAA. Biophys J. 2018;114:32–9.
Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature 2015;517:311–20.
Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65.
Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–21.
Cho YS. The role of necroptosis in the treatment of diseases. BMB Rep. 2018;51:219–24.
Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13:954–62.
McComb S, Cessford E, Alturki NA, Joseph J, Shutinoski B, Startek JB, et al. Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc Natl Acad Sci USA. 2014;111:E3206–13.
Brault M, Olsen TM, Martinez J, Stetson DB, Oberst A. Intracellular nucleic acid sensing triggers necroptosis through synergistic type I IFN and TNF signaling. J Immunol. 2018;200:2748–56.
Chen D, Tong J, Yang L, Wei L, Stolz DB, Yu J, et al. PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc Natl Acad Sci USA. 2018;115:3930–5.
Sarhan J, Liu BC, Muendlein HI, Weindel CG, Smirnova I, Tang AY, et al. Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ. 2019;26:332–47.
Legarda D, Justus SJ, Ang RL, Rikhi N, Li W, Moran TM, et al. CYLD proteolysis protects macrophages from TNF-mediated auto-necroptosis induced by LPS and licensed by type I IFN. Cell Rep. 2016;15:2449–61.
Hos NJ, Ganesan R, Gutierrez S, Hos D, Klimek J, Abdullah Z, et al. Type I interferon enhances necroptosis of Salmonella Typhimurium-infected macrophages by impairing antioxidative stress responses. J Cell Biol. 2017;216:4107–21.
Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87.
Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018;564:439–43.
Chipurupalli S, Ganesan R, Dhanabal SP, Kumar MS, Robinson N. Pharmacological STING activation is a potential alternative to overcome drug-resistance in melanoma. Front Oncol. 2020;10:758.
Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science. 2015;350:328–34.
Voet S, Srinivasan S, Lamkanfi M, van Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med. 2019;11:e10248.
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20.
Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017;277:61–75.
Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–87.
He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25:1285–98.
Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010;11:385–93.
Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, et al. Critical role of AIM2 in Mycobacterium tuberculosis infection. Int Immunol. 2012;24:637–44.
Hanamsagar R, Aldrich A, Kielian T. Critical role for the AIM2 inflammasome during acute CNS bacterial infection. J Neurochem. 2014;129:704–11.
Swanson KV, Junkins RD, Kurkjian CJ, Holley-Guthrie E, Pendse AA, El Morabiti R, et al. A noncanonical function of cGAMP in inflammasome priming and activation. J Exp Med. 2017;214:3611–26.
Webster SJ, Brode S, Ellis L, Fitzmaurice TJ, Elder MJ, Gekara NO, et al. Detection of a microbial metabolite by STING regulates inflammasome activation in response to Chlamydia trachomatis infection. PLoS Pathog. 2017;13:e1006383.
Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, et al. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell. 2017;171:1110–24 e18.
Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, et al. Gasdermin D restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity. 2018;49:413–26 e5.
Corrales L, Woo SR, Williams JB, McWhirter SM, Dubensky TW Jr., Gajewski TF. Antagonism of the STING pathway via activation of the AIM2 inflammasome by intracellular DNA. J Immunol. 2016;196:3191–8.
Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, et al. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity. 2014;40:329–41.
Wang Y, Ning X, Gao P, Wu S, Sha M, Lv M, et al. Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity. 2017;46:393–404.
Cui J, Li Y, Zhu L, Liu D, Songyang Z, Wang HY, et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol. 2012;13:387–95.
Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–23.
Clarke AJ, Simon AK. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol. 2019;19:170–83.
Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–64.
Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–36.
Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017;61:585–96.
Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–50.
Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32.
Morishita H, Mizushima N. Diverse cellular roles of autophagy. Annu Rev Cell Dev Biol. 2019;35:453–75.
Molejon MI, Ropolo A, Vaccaro MI. VMP1 is a new player in the regulation of the autophagy-specific phosphatidylinositol 3-kinase complex activation. Autophagy. 2013;9:933–5.
Moretti F, Bergman P, Dodgson S, Marcellin D, Claerr I, Goodwin JM, et al. TMEM41B is a novel regulator of autophagy and lipid mobilization. EMBO Rep. 2018;19:e45889.
Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:859–64.
Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19:4651–9.
Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–12.
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8.
Tsuboyama K, Koyama-Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016;354:1036–41.
Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–92.
Noda T, Fujita N, Yoshimori T. The late stages of autophagy: how does the end begin? Cell Death Differ. 2009;16:984–90.
Nakamura S, Yoshimori T. New insights into autophagosome-lysosome fusion. J Cell Sci. 2017;130:1209–16.
Behrends C, Fulda S. Receptor proteins in selective autophagy. Int J Cell Biol. 2012;2012:673290.
Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–96.
Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA. 2009;106:20842–6.
Liu D, Wu H, Wang C, Li Y, Tian H, Siraj S, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 2019;26:1735–49.
Prabakaran T, Bodda C, Krapp C, Zhang BC, Christensen MH, Sun C, et al. Attenuation of cGAS–STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. 2018;37:e97858.
Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;567:262–6.
Liang Q, Seo GJ, Choi YJ, Kwak MJ, Ge J, Rodgers MA, et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. 2014;15:228–38.
Subramani S, Malhotra V. Non-autophagic roles of autophagy-related proteins. EMBO Rep. 2013;14:143–51.
Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 2012;150:803–15.
Dey B, Dey RJ, Cheung LS, Pokkali S, Guo H, Lee JH, et al. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat Med. 2015;21:401–6.
van der Vaart M, Korbee CJ, Lamers GE, Tengeler AC, Hosseini R, Haks MC, et al. The DNA damage-regulated autophagy modulator DRAM1 links mycobacterial recognition via TLR-MYD88 to autophagic defense [corrected]. Cell Host Microbe. 2014;15:753–67.
Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155:688–98.
Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity. 2009;30:397–407.
Wang Y, Lian Q, Yang B, Yan S, Zhou H, He L, et al. TRIM30alpha is a negative-feedback regulator of the intracellular DNA and DNA virus-triggered response by targeting STING. PLoS Pathog. 2015;11:e1005012.
Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, et al. Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host Microbe. 2015;17:820–8.
Thomsen MK, Nandakumar R, Stadler D, Malo A, Valls RM, Wang F, et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology. 2016;64:746–59.
Lei Z, Deng M, Yi Z, Sun Q, Shapiro RA, Xu H, et al. cGAS-mediated autophagy protects the liver from ischemia-reperfusion injury independently of STING. Am J Physiol Gastrointest Liver Physiol. 2018;314:G655–G67.
Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B, et al. STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell. 2017;171:809–23 e13.
Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019;26:605–16.
Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222.
Denton D, Aung-Htut MT, Kumar S. Developmentally programmed cell death in Drosophila. Biochim Biophys Acta. 2013;1833:3499–506.
Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol 2009;19:1741–6.
Liu Y, Shoji-Kawata S, Sumpter RM Jr., Wei Y, Ginet V, Zhang L, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci USA. 2013;110:20364–71.
Arakawa S, Tsujioka M, Yoshida T, Tajima-Sakurai H, Nishida Y, Matsuoka Y, et al. Role of Atg5-dependent cell death in the embryonic development of Bax/Bak double-knockout mice. Cell Death Differ. 2017;24:1598–608.
Margolis SR, Wilson SC, Vance RE. Evolutionary origins of cGAS–STING signaling. Trends Immunol. 2017;38:733–43.
Nassour J, Radford R, Correia A, Fuste JM, Schoell B, Jauch A, et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature. 2019;565:659–63.
Ku JWK, Chen Y, Lim BJW, Gasser S, Crasta KC, Gan YH. Bacterial-induced cell fusion is a danger signal triggering cGAS-STING pathway via micronuclei formation. Proc Natl Acad Sci USA. 2020;117:15923–34.
Lin L, Baehrecke EH. Autophagy, cell death, and cancer. Mol Cell Oncol. 2015;2:e985913.
Samir P, Malireddi RKS, Kanneganti TD. The PANoptosome: a deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol. 2020;10:238.
Acknowledgements
The work in our laboratories is supported by the National Health & Medical Research Council (NHMRC) project grants (1144500, 1156601), a NHMRC Senior Principal Research Fellowship (1103006) to SK, and the University of South Australia internal research support to SK and NR.
Author information
Authors and Affiliations
Contributions
AMVM drafted, edited, and revised the manuscript text and prepared figures, NR provided intellectual input and edited the manuscript, SK conceptualized, edited, and revised the manuscript, and contributed intellectual input.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Melino
Rights and permissions
About this article

Cite this article
Murthy, A.M.V., Robinson, N. & Kumar, S. Crosstalk between cGAS–STING signaling and cell death. Cell Death Differ 27, 2989–3003 (2020). https://doi.org/10.1038/s41418-020-00624-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-00624-8
This article is cited by
-
The dual function of cGAS-STING signaling axis in liver diseases
Acta Pharmacologica Sinica (2024)
-
Combination of IFN-gamma with STING agonist and PD-1 immune checkpoint blockade: a potential immunotherapy for gastric cancer
Medical Oncology (2024)
-
STING upregulation mediates ferroptosis and inflammatory response in lupus nephritis by upregulating TBK1 and activating NF-κB signal pathway
Journal of Biosciences (2024)
-
Investigation into the role of the MITA-TRIM38 interaction in regulating pyroptosis and maintaining immune tolerance at the maternal-fetal interface
Cell Death & Disease (2023)
-
Understanding nucleic acid sensing and its therapeutic applications
Experimental & Molecular Medicine (2023)
