
Abstract
Autophagy and mitophagy act in cancer as bimodal processes, whose differential functions strictly depend on cancer ontogenesis, progression, and type. For instance, they can act to promote cancer progression by helping cancer cells survive stress or, instead, when mutated or abnormal, to induce carcinogenesis by influencing cell signaling or promoting intracellular toxicity. For this reason, the study of autophagy in cancer is the main focus of many researchers and several clinical trials are already ongoing to manipulate autophagy and by this way determine the outcome of disease therapy. Since the establishment of the cancer stem cell (CSC) theory and the discovery of CSCs in individual cancer types, autophagy and mitophagy have been proposed as key mechanisms in their homeostasis, dismissal or spread, even though we still miss a comprehensive view of how and by which regulatory molecules these two processes drive cell fate. In this review, we will dive into the deep water of autophagy, mitophagy, and CSCs and offer novel viewpoints on possible therapeutic strategies, based on the modulation of these degradative systems.
Similar content being viewed by others

Facts
-
Autophagy plays a dual role in cancer as a tumor suppressor and promoter.
-
CSCs are usually characterized by a dysregulation of autophagy/mitophagy.
-
The modulation of autophagy/mitophagy impacts on CSC generation, differentiation, plasticity, migration/invasion and pharmacological, viral and immune-resistance.
-
Targeting autophagy/mitophagy could pave the way for new therapeutic strategies to fight CSC aggressiveness.
Open questions
-
What are the key signaling pathways impacted by autophagy in CSCs?
-
Which is the role of mitophagy in the profound metabolic reprogramming of CSCs?
-
How could we manipulate autophagy to drive CSC fate?
Autophagy in cancer: an overview
Autophagy is a self-digestion mechanism, in which cytoplasmic materials, proteins (in a pathway commonly defined as macroautophagy), damaged organelles, such as mitochondria (termed mitophagy), and lipids are sequestered into vesicles, called autophagosomes, for degradation and recycling. In basal conditions, autophagy is crucial for the preservation of cell homeostasis, acting as a protein/organelle quality control mechanism; during stressful conditions, such as starvation, hypoxia, and chemo/radiotherapy, it is instead fundamental for a cancer cell survival and adaptability to the perturbations of tumor microenvironment.
Autophagy has been initially attributed both tumor-suppressive and tumor-promoting functions. Opposite functions were interpreted as autophagy being a double-edged sword in cancer, and challenged researchers to further explore its impact on oncogenesis and tumor progression. Next, research in the field clearly demonstrated that the autophagy role in cancer exhibits a significant degree of context dependency, making us aware of the need to strictly relate each finding to its own and proper experimental system: e.g., the type/stage of tumor, the local (microenvironment)/systemic extracellular milieu of the tumor, the treatment with a specific cancer therapy or the genetic context.
Indeed, the accelerated oncogenesis observed in murine models defective for autophagy strongly supports the notion that autophagy prevents malignant transformation [1,2,3]. This tumor-suppressive function mostly occurs through the maintenance of the physiological tissue homeostasis, and empowers the pre- malignant cells to escape genotoxic stress and inflammation [4, 5], which both promote tumorigenesis. Such a cytoprotective role turns into a weapon serving cancer cells, and allowing them to cope with stress (metabolic, genotoxic, and inflammatory), which occurs after the malignant transformation is induced by anticancer therapy [5, 6]. Besides safeguarding cellular homeostasis, autophagy also affects cellular processes, such as epithelial-to-mesenchymal transition and migration, with both processes driving tumor progression and metastasization [7,8,9]. Altogether, autophagy can both promote and suppress cancer progression and metastasis at several stages. Notably, while autophagy induction is often a side effect of chemotherapy [10,11,12], it also has a beneficial role in cancer therapies involving induction of immunogenic cell death [13]. Hence, in order to exploit autophagy activation/inhibition for cancer treatment, it would be crucial to carefully assess the dependence/sensitivity of each specific type of cancer to autophagy, as well as the impact of autophagy modulation on selected cancer therapies.
The cancer stem cell models
Cancer stem cells (CSCs, also known as tumor-initiating cells or tumor-propagating cells) are a small subpopulation of cancer cells that are responsible for tumor heterogeneity, displaying high metastatic potential and resistance to conventional anticancer therapy [14]. CSCs have been first identified in acute myeloid leukemia [15, 16] and then in many solid cancers, such as breast, pancreatic [17, 18], colon [19, 20], melanoma [21, 22], ovarian [23] and lung [24], and brain cancers [25, 26]. They are immortal tumor-cells that possess extraordinary self-renewal and differentiation capabilities that give rise to different phenotypes. CSCs are defined by the expression of specific cell surface markers that can be used to distinguish them from other tumor or normal cells. This opened the way to establish many in vitro and in vivo strategies to isolate and manipulate CSCs. Another important feature defining CSCs is the ability to recapitulate the original malignancy when transplanted in immune-deficient mice [14]. Breast cancer was the first human solid tumor proven to consist of heterogeneous populations of cells: non-CSCs and CSCs; specifically the CSCs subpopulation (CD44+ CD24−/low) is capable of initiating tumor growth in immune-deficient mice [27]. Besides the capability of these cells to self-renew, accumulated evidence has established that a stronger resistance than non-CSC populations to anticancer therapies characterizes them. The failure of conventional treatments is strictly related to the plasticity of CSCs that, owing to their (1) deregulated self-regeneration and differentiation proprieties, (2) proliferative potential, (3) capability to be a quiescent cell pool, are most likely responsible for tumor initiation, progression, recurrence, and invasion. Overall, the identification of molecular mechanisms implicated in CSC survival remains crucial for augmenting the efficacy of presently available treatment regimens.
At least two main different models have been proposed to account for tumor origin and heterogeneity: the stochastic model and the hierarchical model. According to the first one, all cancer cells have the capability to give rise to new tumors by converting non-CSCs to a CSC phenotype in a dynamic way and in response to specific stimuli. By contrast, the hierarchical model is based on the concept that a unique population of CSCs produces the tumor and gives rise to heterogeneity by generating both differentiated and quiescent cancer cells. Although these models seem to exclude each other, what does happen is probably a combination of both things.
One of the pivotal processes that have been strongly associated to CSCs maintenance and aggressiveness is autophagy. In this review, we describe the role of both autophagy and mitophagy in CSC biology and discuss how their targeting could interfere with CSC survival. We will thus dig into how autophagy/mitophagy act and contribute to each step of CSC physiology: generation, differentiation, plasticity, migration/invasion and pharmacological, viral and immune-resistance (Fig. 1; for a summary see Table 1).

Roles of autophagy in cancer stem cells (CSCs). Tumor cells are heterogeneous and include cancer stem cell (CSC) populations. CSCs are often characterized by high levels of autophagy that (1) maintain pluripotency; (2) cope with low nutrients and oxygen levels (hypoxia) in the tumor microenvironment; (3) regulate CSCs migration and invasion; (4) promote resistance to chemotherapy, (5) help to escape immunosurveillance; (6) support oncovirus capability to infect, replicate in and kill them. In this scenario, autophagy manipulation is found to be crucial for the effective targeting of cancer cells. Aa: Amino acids; Glc: Glucose; NK: Natural Killer; O2: Oxygen
Autophagy and cancer stem cells
Autophagy in the maintenance and survival of CSCs
In line with normal stem cells, CSCs are characterized by ability to self-renew and a limited differentiation capacity [14]. Pluripotency is a key feature of CSCs that allows them to indefinitely divide and maintain the undifferentiated state. By using fluorescence activated cell sorting (FACS) based on CD34 and CD38 (CD34+ CD38−) surface marker expression, John Dick isolated the first CSCs from acute myeloid leukemia (AML) [15, 16]. By studying breast CSCs, it was made clear that autophagic homeostasis is an intrinsic feature for the maintenance of pluripotency under various pathophysiological conditions (Fig. 2a, for detailed description see ref. [28]). In fact, autophagy is upregulated in the mammospheres [29, 30] when compared to adherent cells, and both BECLIN 1 and ATG4, two key autophagy proteins, are needed for their maintenance and expansion. More recently, autophagy has been related to a variety of CSCs, such as breast [31, 32], pancreatic, liver [33], osteosarcoma [34], ovarian [35], and gliobastoma [36] CSCs, in which its impairment negatively affects the expression of staminal markers and consequently the cell self-renewal capacity. In hematological malignancies, depending on the context, autophagy can act both as a chemoresistance or tumor-suppressive mechanism. It is now clear that depending on both the type of progenitors and the state of leukemia disease (initiation versus progression), autophagy could have opposite roles. For example, in CML some autophagy-related genes (such as ATG4, ATG5, or BECLIN 1 [37,38,39]) are upregulated and the silencing of both ATG7 or ATG4B affects cell survival; so, the levels of autophagy in CML seems to be closer to solid tumor CSCs. On the contrary, functional autophagy is essential for protecting the evolution of myelodysplastic syndrome (MDS) to AML [40] and many autophagy-related genes are mutated or downregulated in some AML patients [41].

Role of autophagy in breast cancer stem cells (CSCs). a In human breast cancer, two different populations of cancer cells co-exist, the breast and non-breast CSCs. While the first population is characterized by high autophagy levels, increased resistance to chemotherapy and mesenchymal phenotype, the latter shows decreased autophagy, higher sensitivity to drugs and an epithelial, rather than mesenchymal, phenotype. Also, breast and non-breast CSCs differentially express cell surface markers (CD24, CD44, and ALDH). Importantly, autophagy inhibition results into enhanced migration and stemness and alteration in IL-6 secretion. b. In a mouse model of breast tumor, two different populations of breast CSCs have been isolated: a luminal one (in pink) and a mesenchymal one (in light blue). Intriguingly, all stemness markers (ALDH, CD29, and CD61) are downregulated in both populations upon FIP200 depletion, and this event correlates with Stat3 or TGFβ2/3 signaling downregulation, respectively. Also in this case, the two different populations of CSCs are distinguished by differential expression of cell surface markers (ALDH, CD29, and CD61)
In recent years, researchers put a great deal of effort in understanding the molecular mechanisms of autophagy-dependent CSCs maintenance. Different signaling pathways have been identified: in MMTV-PyMT transgenic mice (a mouse model of breast CSCs), Yeo et al. [42] demonstrated that autophagy acts through EGFR/Stat3 and Tgfβ/Smad signaling in two distinct breast cancer stem-like cells (ALDH+ and CD29hiCD61+ , respectively, see Fig. 2b). Upon FIP200 depletion, they found a decrease in the phosphorylation of EGFR, with this resulting in decreased STAT3 activation and consequently in an impairment of ALDH+ breast cancer stem cells (BCSCs) tumorigenicity. On the other hand, autophagy inhibition leads to decreased TFGβ2 and TGFβ3 expression, inducing a defect in Smad signaling, which is indispensable for the CD29hiCD61+ CSCs phenotype. More recently, in the triple-negative type of autophagy-dependent BCSCs, it has been found that autophagy inhibition decreases the secretion of IL-6, probably through the STAT3/JAK2 pathway [43]. IL-6 secretion is crucial for CSC maintenance [44] and sufficient to induce the CD44 + /CD24 low phenotype in breast cancer cell lines and tumors, this supporting the idea that the IL-6-JAK2-STAT3 signal transduction pathway could play an important role in the conversion of non-CSCs into CSCs.
Other studies suggest a role for FOXOs in regulating the fate of CSCs [45]. Although it is well described that FOXO-dependent regulation of transcription is fundamental to preserve homeostasis of stem cell in both embryos and adults [46], it still needs to be defined how FOXO activity could affect CSCs functions. Knockdown of FOXO3 results in increased CSC self-renewal capacity in prostate, glioblastoma, ovarian, breast, liver, and colorectal cancer [47,48,49,50,51]; in contrast leukemia-initiating cells need FOXO3 for stem cell maintenance [52, 53]. In the autophagy context, FOXOs have been reported to mediate the transcription of some autophagy-related genes (ATG5, ATG8, ATG12, ATG14, BECLIN 1, ULK1, LC3, GABARAPL1, and BNIP3, reviewed in ref. [54]) and as such cytosolic FOXOs participate in autophagy regulation. Very recently, the pro-autophagic protein AMBRA1 was found to be crucial for regulatory T-cell differentiation and homeostasis, acting on FOXO3-FOXOP3 axis. This opens new scenarios that may involve autophagy in FOXO3-mediated regulation of a lot of cellular processes [55].
However, further investigation is necessary to understand how FOXO-dependent regulation of stemness and autophagy are interconnected in tumorigenesis, considering that this is not simply a linear readout.
Intriguingly, recent findings suggest a crosstalk among autophagy, NAD + biosynthesis pathway and staminal markers; Sharif et al. [56] found that any perturbation in basal autophagy (generated by using both autophagy inhibitors and activators) decreases the pluripotency of teratocarcinoma CSCs, leading to differentiation and/or senescence.
Altogether, this evidence highlights the complexity of the autophagy-dependent regulation of CSCs.
Very recently, a novel link between autophagy and stemness arises from studies in ovarian cancer stem cells (OCSCs) [35]. Peng et al. found that Forkhead Box A2 (FOXA2) is overexpressed in ovarian CSCs and regulated by autophagy activity; inhibition of autophagy by both genetic and pharmacological approaches induces FOXA2 downregulation and, consequently, impairment of self-renewal ability.
Finally, other studies suggest a role for autophagy in the regulation of chromosome stability by coordinating the ATR checkpoint and double-strand-break processing [57]. This opens the possibility that CSCs may exploit autophagy to prevent further DNA damage after an initial insult and maintain their survival.
Autophagy as an adaptive mechanism of CSCs in the tumor microenvironment
Accumulating evidence indicate that the CSCs behavior is regulated by both extracellular signals (including hypoxic, metabolic, and oxidative stress) and intrinsic signals within CSCs that can promote self-renewal and plasticity. In particular, environmental stresses (such as lower oxygen levels, higher lactate levels, extracellular acidosis, and depletion of nutrients) are considered crucial for the maintenance of CSCs. It is suggested that stem cells lose the possibility for continued self-renewal when removed from their environment, the stem cell niche, which implies an essential role for microenvironment in directing stem cell fate [58]. It is well known that hypoxia commonly results in autophagy, mediated by the hypoxia- inducible factor 1alpha (HIF-1α). Hypoxia-induced autophagy has been demonstrated to be crucial for survival of liver CD133+ CSCs; more interestingly, in pancreatic CSCs, HIF-1α-dependent autophagy is critical for the equilibrium between non-stem pancreatic cancer cells and pancreatic CSCs [59]. Indeed, autophagy was found to be upregulated in multiple human AML cell lines and primary blasts after prolonged exposure to hypoxia; also, inhibition of the late-stage of autophagy overcomes Leukemia Stem Cells (LSCs) survival and chemoresistance [60, 61]. This highlights the controversial role of autophagy in the death/survival of leukemic cells, in which reduced autophagy appears to be an adaptive mechanism that accelerates AML development [62].
While advances have been made in understanding the role of autophagy and hypoxia in cancer, very little is known about the potential role of hypoxia-induced autophagy in maintaining the cancer stem cell niche.
Autophagy role in migration/invasion of CSCs
EMT (epithelial-to-mesenchymal transition) is a critical event during embryonic development, determining changes in cell polarity and cell–cell contact. It is now clear that EMT signaling and CSCs phenotypes are strictly connected and a key feature of CSCs is their capability to migrate, which enhances their metastatic potential [27, 63, 64]. There is now increasing evidence that autophagy signaling and EMT are linked one to another [8, 65,66,67] and autophagy is often highly expressed in tumor cells carrying a mesenchymal signature [68]. In breast CSCs, autophagy inhibition (for instance, by ATG12 downregulation and chloroquine treatment) impairs the migratory and invasive cellular state, leading to increased expression of the epithelial marker CD24 and a decrease of vimentin (a mesenchymal cell marker) [69]. Moreover, in glioblastoma CSCs, two autophagy regulators, DRAM1 and SQSTM1 were found to be upregulated and to correlate with the expression of mesenchymal factors [67], thus supporting the idea that autophagy controls migration/invasion in CSCs [10, 11]. In some solid tumors, such as glioblastoma, pancreatic, gastric and breast CSCs, autophagy negatively correlates with EMT; autophagy inhibition, indeed, is able to impair migration and invasion, while autophagy upregulation restores the mesenchymal phenotype. There are also evidences that EMT and autophagy could be considered two distinct processes, both correlated to the heterogeneous nature of CSCs (non-cycling and cycling CSCs) [68]. Accordingly, a hierarchical model defined the existence of cycling or non-cycling CSCs that come from EMT tumor cells: EMT tumor cells would be first induced to become autophagic CSCs (non-cycling) and, subsequently, cycling CSCs (featuring low autophagy). One possible hypothesis is that autophagic CSCs could induce EMT in other tumor cells upon release of EMT-inducing paracrine factors.
Autophagy-mediated chemo and immune-resistance of CSCs
Despite advancements in radiation treatments and chemotherapy, which often target highly proliferating cells, it is clear that CSCs, living in a quiescent state and acquiring resistance to conventional therapy, are responsible for tumor recurrence. Different mechanisms through which CSCs can resist to drug-therapy have been proposed:
(1) Components of the CSC niche (immune cells, endothelial cells, fibroblasts and peri-vascular cells) protect CSCs from therapeutic interventions; (2) cellular plasticity; (3) high efficiency in repairing DNA damage; (4) high levels of MDR (multi-drug resistance) gene expression, or (5) inhibition of apoptosis. A relationship between CSCs and drug resistance has been found in many human cancers such as leukemia, melanoma, brain, breast, pancreatic, and colorectal cancers [70]. One of the model proposed is based on the existence of an intrinsic chemoresistance; this results in the persistence of a population of cancer cells that then leads to relapse following treatment. Among the mechanisms that have been proposed to confer resistance to chemotherapeutics, autophagy seems to be crucial. Moreover, it is well recognized that chemotherapeutic treatments are per se able to induce autophagy in cancer cells [71]. Several experimental approaches reveal that combining cytotoxic drugs and autophagy inhibitors increase CSCs sensitivity [72]. For example, in glioblastoma stem cells, Bevacizumab, a blocker of EGFR, or Temozolomide, in combination with chloroquine (a late-stage autophagy inhibitor), enhance drug toxicity, thus affecting glioblastoma CSCs survival [73, 74]. Very recently, Li et al. [75] found that in gastric CSCs the triple combination of 5-fluorouracil, chloroquine, and Notch inhibitor decreases cell viability and treatment resistance. Along similar lines, JAK-mediated autophagy was found responsible for preservation of stemness in cisplatin-resistant bladder cancer cells [76]; in agreement with this modulation, in glioma (GSCs) or AML CSCs, knockdown of ATG7 potentiates the inhibitory effect of salinomycin on cell survival [77]. In CML, Bellodi et al. [78] showed that the combination of autophagy inhibitors such as chloroquine or bafilomycin A1 (another late-stage autophagy inhibitor) with tyrosine kinase inhibitors (TKI) affects CML cell survival.
On the other hand, there are some evidence of the role that autophagy may play in drug-mediated cytotoxicity. In particular, resveratrol acts on breast CSC survival by inhibiting the Wnt pathway that, in turn, induces autophagy [79]; By contrast, the inactivation of mTOR stimulates neuroblastoma and glioma CSCs differentiation [80, 81]. In sum, uncovering the individual contribution of autophagy to CSCs drug resistance remains crucial for the development of novel antineoplastic therapies.
Besides the CSC resistance to chemotherapy, it is now emerging that CSCs are able to escape from the immune system through a process termed immunoresistance. Immune system is able to detect and interact with tumor cells through a mechanism named immunosurveillance. However, CSCs have been shown to be less immunogenic by both evading immune recognition and manipulating the immune system to stimulate their own growth; indeed, they achieve this by (1) producing immunosuppressive factors, (2) recruiting immunosuppressive cell types, (3) losing the expression of tumor antigens [82], through activation of distinctive cellular pathways such as Notch and Wnt. Moreover, CSCs produce cytokines to inhibit immune response: breast and glioblastoma CSCs are able to produce more TGFβ than normal cancer cells, and colon CSCs secrete IL-4 to inhibit antitumor immune responses. Of note, recent findings have revealed that autophagy contributes to immunosuppressive-related chemoresistance and promotes the capability of the tumor to avoid immune detection [83, 84]. Along the same line, enhanced autophagy has been observed in advanced stages of metastatic diseases, characterized by low levels of tumor-infiltrating T lymphocytes (TILs) [85]. Although this area of research is still poorly explored, autophagy could be considered a critical process for counteracting CSCs resistance to immunotherapeutic approaches.
Autophagy and oncolytic virotherapy in CSCs
A new frontier in cancer treatment is represented by oncolytic viruses (OVs, including adenoviruses, herpes simplex virus, measles virus, reovirus, and Newcastle disease virus) that bypass the above-mentioned mechanisms of chemoresistance, thus efficiently killing CSCs in some cancer types [86]. OVs have the capability to infect, replicate in and kill cancer cells that express high levels of some virus-receptors, including CAR, CD46, or CD155. In the context of autophagy, there is accumulating evidence that, indeed, a role for OVs in the perturbation of cellular autophagy does exist [87]; on the other hand, the increased autophagy of CSCs could be exploited to target these cells by OVs. It has been proposed that a range of factors can promote or inhibit autophagy during the process of oncolytic adenoviral therapy, including infection, replication, and cell lysis [87]. During the replication step, adenoviral proteins induce autophagy via the upregulation of ATG5 and LC3 [88] or by stimulating BECLIN 1- Bcl-2 dissociation [89, 90]; by contrast, the E4 protein blocks autophagy by acting directly on mTOR signaling. Remarkably, high levels of autophagy in CSCs could represent an useful feature to counteract their resistance to conventional therapy. In 2013, Tong et al. [91] found that an oncolytic adenovirus encoding BECLIN 1 is able to kill LSCs. Interestingly, some OVs have been displayed to regulate autophagy to stimulate both innate and adaptive immune responses, by contributing to antigen presentation and cytokine production during the oncolytic processes. For instance, the autophagy-mediated secretion of ATP in infected-tumor cells activates dendritic cells to produce IL-1β that stimulates IFN-γ-dependent T lymphocytes [92]. Overall, OVs represent a great promise for CSC counteraction, and they can be used for both autophagy-inducing and -inhibiting strategies, in order to modulate the CSCs fate.
Mitophagy in CSCs: a role in metabolic reprogramming
Mitochondria are the organelles responsible for energy production through OXPHOS, synthesis of biomolecules, maintenance of calcium homeostasis, production of reactive oxygen species (ROS), and apoptotic activation [93]. Metabolic reprogramming through the regulation of mitochondrial activities is a key feature of CSCs. It orchestrates their self-renewal capacity, stemness, resistance to toxic agents and also their migration abilities [94,95,96]. Indeed, it is widely known that many cancer cells rely on aerobic glycolysis, a phenomenon defined as “Warburg effect”, with a prominent decrease in oxidative phosphorylation (OXPHOS) and, therefore mitochondrial functions, even in the presence of functional mitochondria [97, 98]. At variance with this notion, CSCs show a unique metabolic adaptation that is determined by the surrounding environment, such as the hypoxic niche of solid tumors, regions with adequate levels of oxygen (normoxia), active growing regions of the tumor and metastatic sites [94, 95, 99,100,101]. In a few instances, this wide adaptation has generated controversial results: Vlashi et al. [102] showed that glioma CSCs relied mainly on OXPHOS for energy supply, while other studies described that glioma CSCs are driven by a glycolytic reprogramming, exhibiting more fragmented mitochondria than neuronal stem cells and downregulation of mitochondrial respiratory activity in GSCs [103]. Although CSCs denote an elevated degree of metabolic plasticity, growing evidence suggests that these cells rely more on OXPHOS for energy production, with this being a far more efficient process in ATP generation than glycolysis (as recently reviewed in refs. [94, 95, 101]). In fact, several studies in different tumor types, such as CD133+ cells of glioblastoma and pancreatic ductal adenocarcinoma, LSCs, lung cancer side population cells, and breast cancer, strongly support an OXPHOS phenotype [94, 95, 101].
In fact, in both AML and CML stem/progenitor cells, the inhibition of OXPHOS (obtained by means of inhibitors of complex I or by blocking amino acid metabolism/mitochondrial translation) induces cell death, while in AML it promotes cell differentiation; thus, these data highlight the existing connection between CSCs fate and mitochondrial metabolism [104,105,106,107].
Which is thus the role of mitophagy, the selective removal of damaged or superfluous mitochondria by autophagy [108], in such a profound metabolic reprogramming of CSCs (Fig. 3)?

Mitophagy in CSCs, a working model. In brain tumor-initiating cells (BTICs), CDK5-dependent DRP1 activation promotes mitochondrial fission, and (hypothetically) mitophagy, to sustain self-renewal and growth. In leukemia stem cells (LSCs), activated AMPK induces FIS1-mediated mitophagy to guarantee removal of damaged mitochondria, keeping ROS levels under tight control, and, thus, contributing to LSC proliferation. In fact, AMPK or FIS1 loss leads to accumulation of damaged mitochondria and increase of ROS production, which promote GSK3 activation that drives cell cycle arrest and differentiation. Furthermore, in hepatic CSCs, PINK1 on the one hand activates p53 through phosphorylation on the mitochondrial membrane and, on the other hand, mediates mitophagy-dependent p53 degradation, thus favouring NANOG expression and hepatic CSCs proliferation. The suppression of mitophagy efficiency entails the accumulation of activated p53 that translocates to the nucleus, where it inhibits NANOG expression, hindering hepatic CSC proliferation. Moreover, BNIP3L-mediated mitophagy contributes to doxorubicin resistance in colorectal CSCs. A hypothetical model upon hypoxia conditions: hypoxia activates HIF-1α that drives the CSCs metabolic reprogramming. We can hypothesize that HIF-1α endorses BNIP3 and BNIP3L expression and, in turn, these factors mediate mitochondrial degradation and contribute to the switch from oxidative to glycolytic metabolism
Indeed, mitochondria are highly dynamic structures, undergoing constant fission (regulated by GTPase MFN1,-2 and OPA1) and fusion (regulated by GTPase DRP1 and its accessory factors, including FIS1) to adapt their structure to the energetic and physiological needs of the cell or to ensure their transport or elimination in case of damage [109]. Mitochondrial dynamics also play an important role in regulating mitophagy [110]. It has recently been described that DRP1-driven mitochondrial fragmentation contributes to the acquisition and maintenance of stem cell pluripotency [111, 112] and that this seems to be true also in CSCs [113]. Brain tumor-initiating cells (BTICs), which can be considered a type of neuronal CSCs, boost up mitochondrial fission through CDK5-dependent DRP1 activation to prevent cell death and, thus, to sustain self-renewal and growth [114]. In fact, DRP1 activation in BTICs correlates with poor glioblastoma patient survival [114].
Furthermore, emerging evidence suggests CSC increase antioxidant defense to counterattack ROS production, derived by enhancement of OXPHOS rate; this mechanism assists in the maintenance of their stemness and tumorigenic capacities [94, 101]. To the same aim, CSCs may rely on mitophagy to guarantee the degradation of damaged mitochondria, thus keeping ROS levels under tight control and preventing the activation of programmed cell death [93]. In fact, mitophagy is a potent pro-survival pathway: several mitophagic mechanisms have been identified (reviewed in ref. [115]) and, in some cases, they show some redundancy to ensure efficient and proper elimination of selected mitochondria. Alteration of mitophagy efficiency due, for example, to genetic mutations of key genes, such as PINK1 or PRKN (PARKIN), has been associated with neurological disorders (e.g., Parkinson disease, cancer, heart failure, and aging [116]). Moreover, mitophagy plays a prime role in the maintenance of stem cell pool restoration and homeostasis, and in counteracting senescence by limiting ROS-induced genome damage [117].
One of the most well-characterized mitophagy pathways is represented by the PINK1-PARKIN cascade [118]. PINK1 accumulates specifically on depolarized mitochondria and, through direct phosphorylation, stimulates PARKIN’s E3 ligase activity and recruitment to mitochondria [118]. Therefore, the PINK1-PARKIN system tags damaged mitochondria with ubiquitin to allow recognition by autophagy cargo receptors, such as SQSTM1/p62, optineurin (OPTN), NDP52 and AMBRA1, which, in turn, promote the engulfment of mitochondrion by autophagosomal membrane by binding with LC3/GABARAP family members [115, 118]. Interestingly, loss of PINK1- dependent mitophagy is sufficient to dramatically decrease the efficiency and speed of induced pluripotent stem cell (iPSC) reprogramming from mouse embryonic fibroblasts [119]; this indicates that mitophagy is directly responsible for determining the fate of stem cells. Likewise, in haematopoietic stem cells, mitophagy is fundamental to promote healthy mitochondria degradation and so to maintain quiescence and stemness, mainly to preserve the regenerative capacity of old haematopoietic stem cells [120].
Similarly, LSCs showed a constitutive activation of AMPK, a central regulator of energy and mitochondrial homeostasis that coordinates initiation of autophagy and mitophagy through ULK1 activation [121]. In LSCs, AMPK endorses FIS1-mediated mitophagy to maintain a healthy mitochondrial network and preserve LSC stemness [122]. Intriguingly, inhibition of the AMPK-FIS1 axis leads to a plethora of consistent phenomena, ranging from accumulation of damaged mitochondria, and presumably increased ROS levels, to GSK3 inactivation and cell cycle arrest. All these events drive inhibition of proliferation and induce differentiation of LSCs [122].
Also, Liu et al. [123] discovered that mitophagy positively regulated hepatic CSCs in an unexpected way, by promoting transcriptional activation of NANOG, a key transcription factor known to be required for self-renewal and maintenance of cell stemness [124]. In fact, mitophagy activation fosters autophagosome-mediated p53 sequestration and degradation; PINK directly phosphorylates p53, regulating its localization and consequently its capability to suppress the expression of NANOG [124]. As mentioned above, CSC metabolic adaptation is mainly a result of their surrounding cell environment. Hypoxia is a common condition for those CSCs that are located inside a tumor niche; here, CSCs are able to trigger the switch from OXPHOS to glycolytic metabolism and undergo quiescence [95, 125] in order to increase their fitness and self-renewal potential. Hypoxia leads to activation of both HIF-1α and HIF-2α that are generally inhibited by VHL upon normoxic conditions [126, 127]. Thus, HIF-1α drives CSCs metabolic reprogramming [126] and promotes the expression of several glycolytic proteins inducing in the meantime cell survival [128]. We may also hypothesize that, upon these hypoxic conditions, CSCs may bio-energetically take advantage of BNIP3, BNIP3L/NIX, or FUNDC1-dependent mitophagy, through the activation of HIF-1α, in order to guarantee the reduction of mitochondrial mass and avoid activation of apoptosis. In fact, all these three proteins, which act as mitophagy receptors by interacting directly with LC3 through their LIR motif [129], are transcriptionally upregulated by HIF-1α during hypoxia [126, 130]. A recent study reports that in the context of somatic cell reprogramming, a BNIP3L-dependent mitophagy-mediated metabolic shift toward glycolysis is essential [131], indicating that BNIP3L, but also BNIP3 and FUNDC1, require further investigation in order to elucidate their role in regulating CSCs fate.
So far, we have discussed mitophagy as a positive mechanism that sustains CSCs in adverse conditions or during their metabolic shift. However, an excessive rate of mitophagy flux can also confer chemoresistance: CD133+ /CD44+ CSCs from HCT8 human colorectal cancer cells CSCs have been shown to use BNIP3L-mediated mitophagy to escape from doxorubicin-induced cell death [132].
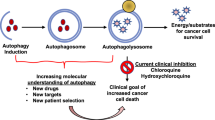
Autophagy, CSCs, and clinical implications
In general, it is well defined that autophagy and mitophagy could represent a promising target for counteracting CSCs aggressiveness. However, it is important to remind that chloroquine and its derivatives (mainly hydroxychloroquine) are used in many clinical trials, often in combination with conventional anticancer treatments [133].
Further and of the highest importance, CSC heterogeneity and patient-specificity makes the situation more complex than previously thought. We are still far away from dealing with useful tools to set up novel drug combinations allowing us to eradicate CSCs or at least to inhibit their proliferation. One of the main ambitions of the actual worldwide efforts is to integrate latest discoveries into innovative treatment strategies. Recent experimental observations suggest that autophagy inhibition and autophagy activation may both be used as encouraging approaches for sensitizing CSCs to therapy. In light of the findings described above, the efficacy of chloroquine application in anti-CSCs therapy could depend on the tumor type that is being treated and autophagy-dependency of CSCs might also play a role in this context. Nowadays much more specific and potent lysosome inhibitors than chloroquine are being established, such as Concanamycin A, a selective inhibitor of V-ATPase that prevents lysosome and endosome acidification, or E64d, an inhibitor of cathepsins B, H, and L, or pepstatin A, inhibitor of cathepsins D and E [134]; these drugs offer the opportunity to develop a wide combinations of different therapies. However, the prevention of autophagosome degradation does not affect autophagosome formation and cargo sequestration. As mentioned above, mitophagy mediates the removal of damaged mitochondria preventing oxidative stress and activation of apoptotic cell death [135]; indeed, lysosomal inhibitors may not be able to reduce the rate of mitochondrial sequestration by autophagosomes, and this could limit the drug efficacy on CSC effects that rely on mitophagy. Thus, in this context, the use of drugs against the initial phases of autophagy, such as VPS34 (i.e., SAR405 [136] or PIK-III [137]) or ULK1 (i.e., MRT68921 [138]) inhibitor, may provide better results.
Moreover, recent findings prompt us to hypothesize new and intriguing scenarios based on the regulation of autophagy in the microenvironment surrounding CSCs. For example, malignant tumor cells induce autophagy in the microenvironment and distal tissues to support their own growth by increasing the availability of recycled nutrients. Autophagy inhibition within the tumor causes a moderate effect on tumor progression, while autophagy inhibition through oral administration of chloroquine leads to a more noticeable reduction in tumor growth and invasion [139]. There is a metabolic crosstalk between CSCs, non-CSCs and cancer- associated fibroblasts (CAFs) that generate metabolic symbiosis [140]; thus, we can hypothesize that targeting non-CSCs and CAFs with autophagic inhibitors may lead to a reduction of nutrient availability and this, in turn, may negatively impact on CSC innate resistance mechanisms against chemotherapy. However, interventions that inhibit autophagy could have unintended side effects on anticancer immune surveillance; in recent years, it turned out that fasting- and caloric-restriction-dependent autophagy induction improve anticancer immune surveillance, hence promoting tumor growth arrest and improvement of the chemotherapeutic outcome [141].
Consequently, getting insights into the regulatory factors and the molecular mechanisms by which autophagy exploits its function in CSCs is fundamental for developing more effective and safe antitumor strategies. Besides the use of autophagy inhibitors/activators in combination with chemotherapeutic drugs, the investigation of autophagy in both immune and virotherapy could be crucial to identify new strategies against CSCs that escapes conventional therapy. In addition, it is central to remind that solid tumors usually grow in low oxygen environments, which create a niche to protect CSCs, and make them more aggressive and resistant to cell death. Further investigations are thus necessary to dive into the role of autophagy in the crosstalk between stromal cells, endothelial cells, and tumor-infiltrating innate and adaptive immune cells. Importantly, these cells may have divergent necessities for autophagy that could make difficult to conceive autophagy-targeting therapy.
Of note, more work is also necessary for developing new and reliable methods for quantifying autophagy flux in patient samples. Certainly, the isolation of CSCs from the blood of the patients may represent a powerful way for monitoring basal autophagy. Moreover, taking advantage from the RNA sequencing approach, a revolutionary tool for transcriptome analysis, it could be possible to predict the state of autophagy activation based on the expression profile of these cells.
References
Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol. 2015; 17. https://doi.org/10.1038/ncb3072.
Mainz L, Rosenfeldt MT. Autophagy and cancer—insights from mouse models. FEBS J. 2018;285:792–808.
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20.
Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol. 2012;13:343–51.
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–75.
Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017;25:1037–43.
Qiang L, Zhao B, Ming M, Wang N, He T-C, Hwang S, et al. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci. 2014;111:9241–6.
Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR, et al. Autophagy is activated by TGF-β and potentiates TGF-β-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 2009;69:8844–52.
Sharifi MN, Mowers EE, Drake LE, Collier C, Chen H, Zamora M, et al. Autophagy promotes focal adhesion disassembly and cell motility of metastatic tumor cells through the direct interaction of paxillin with LC3. Cell Rep. 2016;15:1660–72.
Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68:1485–94.
Liu D, Yang Y, Liu Q, Wang J. Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med Oncol. 2011;28:105–11.
Shimgu T, Fujiwara K, Bogler O, Akiyama Y, Meritake K, Shinojima N, et al. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity In human malignant glioma cells. Int J Cancer. 2009;124:1060–71.
Galluzzi L, JMBS Pedro, Demaria, Formenti S, Kroemer SC, Activating G. autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol. 2017;14:247–58.
Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675–99.
Bonnet D, Dick JE. Human acute lyeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Gr. 1997;3:730–7.
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J. et al. Selectivity of MHC-encoded peptide transportes from human, mouse and rat. Nature. 1994;367:645–8.
Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–23.
Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Am Assoc Cancer Res J. 2007;67:1030–8.
O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10.
Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–5.
Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–9.
Boiko AD, Razorenova OV, Van De Rijn M, Swetter SM, Johnson DL, Ly DP, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466:133–7.
Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311–20.
Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–14.
Singh AK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T. et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401.
Chen T, Yuan D, Wei B, Jiang J, Kang J, Ling K, et al. E-cadherin-mediated cell-cell contact is critical for induced pluripotent stem cell generation. Stem Cells. 2010;28:1315–25.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. 2003;100:3983–8.
Han Y, Fan S, Qin T, Yang J, Sun Y, Lu Y, et al. Role of autophagy in breast cancer and breast cancer stem cells (Review). Int J Oncol. 2018;52:1057–70.
Gong C, Song E, Codogno P, Mehrpour M. The roles of BECN1 and autophagy in cancer are context dependent. Autophagy. 2012;8:1853–5.
Wolf J, Dewi DL, Fredebohm J, Müller-decker K, Flechtenmacher C, Hoheisel JD, et al. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013;15:R109.
Gong C, Bauvy C, Tonelli G, Yue W, Deloménie C, Nicolas V, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2013;32:2261–72.
Chaterjee M, van Golen KL. Breast cancer stem cells survive periods of farnesyl-transferase inhibitor-induced dormancy by undergoing autophagy. Bone Marrow Res. 2011;2011:1–7.
Jiao SY, Shan ZS, Ling GX, Sun K, Peng HZ, Li R, et al. Autophagy contributes to the survival of CD133+liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013;339:70–81.
Zhang D, Zhao Q, Sun H, Yin L, Wu J, Xu J, et al. Defective autophagy leads to the suppression of stem-like features of CD271+osteosarcoma cells. J Biomed Sci. 2016;23:1–12.
Peng Q, Qin J, Zhang Y, Cheng X, Wang X, Lu W, et al. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J Exp Clin Cancer Res. 2017;36:1–12.
Buccarelli M, Marconi M, Pacioni S, De Pasqualis I, D’Alessandris QG, Martini M, et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018; 9. https://doi.org/10.1038/s41419-018-0864-7.
Altman BJ, Jacobs SR, Mason EF, Michalek RD, MacIntyre AN, Coloff JL, et al. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene. 2011;30:1855–67.
Karvela M, Baquero P, Kuntz EM, Mukhopadhyay A, Mitchell R, Allan EK. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-. Autophagy. 2016;12:936–48.
Rothe K, Lin H, Lin KBL, Leung A, Wang HM, Malekesmaeili M, et al. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem / progenitor cells. Blood. 2014;123:3622–35.
Houwerzijl EJ, Pol H, Blom NR, Want JJL, Van Der, Wolf J, De, Vellenga E. Erythroid precursors from patients with low-risk myelodysplasia demonstrate ultrastructural features of enhanced autophagy of mitochondria. Leukemia. 2009;23:886–91.
Auberger P, Puissant A. Blood Spotlight Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood. 2018;129:547–53.
Yeo SK, Wen J, Chen S, Guan JL. Autophagy differentially regulates distinct breast cancer stem-like cells in murine models via EGFR/Stat3 and Tgfβ/Smad signaling. Cancer Res. 2016;76:3397–410.
Maycotte P, Jones KL, Goodall ML, Thorburn J, Thorburn A. Autophagy supports breast cancer stem cell maintenance by regulating IL6 secretion. Mol Cancer Res. 2015;13:651–8.
Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci. 2011;108:1397–402.
van Doeselaar S, Burgering BMT. FOXOs maintaining the equilibrium for better or for worse. Curr Top Dev Biol. 2018; 127. https://doi.org/10.1016/bs.ctdb.2017.10.003.
Liang R, Ghaffari S. Stem cells seen through the FOXO lens: An evolving paradigm. Curr Top Dev Biol. 2018;127:23–47.
Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira S-M, Garcia-Echeverria C, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci. 2009;106:268–73.
Ning Y, Luo C, Ren K, Quan M, Cao J. FOXO3a-mediated suppression of the self-renewal capacity of sphere-forming cells derived from the ovarian cancer SKOV3 cell line by 7-difluoromethoxyl-5, 4’-di-n-octyl genistein. Mol Med Rep. 2014;9:1982–8.
Prabhu VV, Allen JE, Dicker DT, El-Deiry WS. Small-molecule ONC201/TIC10 targets chemotherapy-resistant colorectal cancer stem-like cells in an akt/Foxo3a/TRAIL-dependent manner. Cancer Res. 2015;75:1423–32.
Smit L, Berns K, Spence K, Ryder WD, Zeps N, Madiredjo M, et al. An integrated genomic approach identifies that the PI3K/AKT/FOXO pathway is involved in breast cancer tumor initiation. Oncotarget. 2016;7:2596–610.
Sunayama J, Sato A, Matsuda KI, Tachibana K, Watanabe E, Seino S, et al. FoxO3a functions as a key integrator of cellular signals that control glioblastoma stem-like cell differentiation and tumorigenicity. Stem Cells. 2011;29:1327–37.
Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, et al. TGF-Β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463:676–80.
Pellicano F, Scott MT, Helgason GV, Hopcroft LEM, Allan EK, Aspinall-O’Dea M, et al. The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem Cells. 2014;32:2324–37.
Van Der Vos KE, Coffer PJ. FOXO-binding partners: It takes two to tango. Oncogene. 2008;27:2289–99.
Becher J, Simula L, Volpe E, Locatelli F, Battistini L, Cecconi F, et al. AMBRA1 controls regulatory T-cell differentiation and homeostasis upstream of the FOXO3-FOXP3 axis. Dev Cell. 2018;47:592–607.
Sharif T, Martell E, Dai C, Kennedy BE, Murphy P, Clements DR, et al. Autophagic homeostasis is required for the pluripotency of cancer stem cells. Autophagy. 2017;13:264–84.
Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471:74–9.
Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611.
Zhu H, Wang D, Liu Y, Su Z, Zhang L, Chen F, et al. Role of the hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133 + pancreatic cancer stem-like cells. Cancer Cell Int. 2013;13:1–8.
Evangelisti C, Evangelisti C, Chiarini F, Lonetti A, Buontempo F, Neri LM, et al. Autophagy in acute leukemias: A double-edged sword with important therapeutic implications. Biochim Biophys Acta-Mol Cell Res. 2015;1853:14–26.
Dykstra KM, Hanekamp DW, Johnson M, Guzman ML, Wang ES. Abstract 2864: Inhibition of the late stages of autophagy overcomes hypoxia-induced chemoresistance and targets leukemic stem cells in acute myeloid leukemia. Cancer Res. 2018;78:2864–4.
Wang Z, Cao L, Kang R, Yang M, Liu L, Zhao Y, et al. Autophagy regulates myeloid cell differentiation byp62/SQSTM1-mediated degradation of PML-RAR α oncoprotein. Autophagy. 2011;7:401–11.
Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE. 2008;3:1–7.
Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–73.
Amaravadi RK. Cell cycle news & views autophagy and tumor cell invasion. Cell Cycle. 2012;11:3718–9.
Macintosh RL, Timpson P, Thorburn J, Anderson KI, Thorburn A, Ryan KM. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle. 2012;11:2022–9.
Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A, et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene. 2013;32:699–712.
Marcucci F, Ghezzi P, Rumio C. The role of autophagy in the cross-talk between epithelial-mesenchymal transitioned tumor cells and cancer stem-like cells. Mol Cancer. 2017;16:1–8.
Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Autophagy positively regulates the CD44+CD24-/lowbreast cancer stem-like phenotype. Cell Cycle. 2011;10:3871–85.
Abdullah LN, Chow EK-H. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med. 2013;2:3.
Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013; 4. https://doi.org/10.1038/cddis.2013.350.
Sun R, Shen S, Zhang Y, Xu C, Cao Z, Wen L, et al. Biomaterials Nanoparticle-facilitated autophagy inhibition promotes the ef fi cacy of chemotherapeutics against breast cancer stem cells. Biomaterials. 2016;103:44–55.
Huang H, Song J, Liu Z, Pan L, Xu G. Autophagy activation promotes bevacizumab resistance in glioblastoma by suppressing Akt/mTOR signaling pathway. Oncol Lett. 2018;15:1487–94.
Golden EB, Cho H-Y, Jahanian A, Hofman FM, Louie SG, Schönthal AH, et al. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg Focus. 2014;37:E12.
Li L, Pan D, Zhang S, Xie D, Zheng X, Chen H. Autophagy regulates chemoresistance of gastric cancer stem cells via the Notch signaling pathway. Eur Rev Med Pharmacol Sci. 2018;22:3402–7.
Ojha R, Singh SK, Bhattacharyya S. JAK-mediated autophagy regulates stemness and cell survival in cisplatin resistant bladder cancer cells. Biochim Biophys Acta-Gen Subj. 2016;1860:2484–97.
Yue W, Hamai A, Tonelli G, Bauvy C, Nicolas V, Tharinger H, et al. Inhibition of the autophagic flux by salinomycin in breast cancer stem-like/progenitor cells interferes with their maintenance. Autophagy. 2013;9:714–29.
Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, et al. Targeting autophagy potentiates tyrosine kinase inhibitor – induced cell death in Philadelphia chromosome–positive cells, including primary CML stem cells. J Clin Invest. 2009;119:1109–23.
Fu Y, Chang H, Peng X, Bai Q, Yi L, Zhou Y, et al. Resveratrol inhibits breast cancer stem-like cells and induces autophagy via suppressing Wnt/β-catenin signaling pathway. PLoS ONE. 2014;9:1–8.
Zeng M, Zhou JN. Roles of autophagy and mTOR signaling in neuronal differentiation of mouse neuroblastoma cells. Cell Signal. 2008;20:659–65.
Zhao Y, Huang Q, Yang J, Lou M, Wang A, Dong J, et al. Autophagy impairment inhibits differentiation of glioma stem/progenitor cells. Brain Res. 2010;1313:250–8.
Codony-Servat J, Rosell R. Cancer stem cells and immunoresistance: clinical implications and solutions. Transl Lung Cancer Res. 2015;4:689–703.
Viry E, Paggetti J, Baginska J, Mgrditchian T, Berchem G, Moussay E, et al. Autophagy: An adaptive metabolic response to stress shaping the antitumor immunity. Biochem Pharmacol. 2014;92:31–42.
Lorin S, Hamai A, Mehrpour M, Codogno P. Autophagy regulation and its role in cancer. Semin Cancer Biol. 2013;23:361–79.
Zarogoulidis P, Petanidis S, Domvri K, Kioseoglou E, Anestakis D, Freitag L, et al. Autophagy inhibition upregulates CD4+tumor infiltrating lymphocyte expression via miR-155 regulation and TRAIL activation. Mol Oncol. 2016;10:1234.
Chaurasiya S, Chen NG, Warner SG. Oncolytic virotherapy versus cancer stem cells: A review of approaches and mechanisms. Cancers (Basel). 2018;10:1–19.
Tazawa H, Kuroda S, Hasei J, Kagawa S, Fujiwara T. Impact of autophagy in oncolytic adenoviral therapy for cancer. Int J Mol Sci. 2017;18:1–13.
Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene. 2008;27:4860–4.
Piya S, White EJ, Klein SR, Jiang H, McDonnell TJ, Gomez-Manzano C, et al. The E1B19K oncoprotein complexes with beclin 1 to regulate autophagy in adenovirus-infected cells. PLoS ONE. 2011;6:1–7.
Klein SR, Piya S, Lu Z, Xia Y, Alonso MM, White EJ, et al. C-Jun N-terminal kinases are required for oncolytic adenovirus-mediated autophagy. Oncogene. 2015;34:5295–301.
Tong Y, You L, Liu H, Li L, Meng H, Qian Q. Potent antitumor activity of oncolytic adenovirus expressing Beclin-1 via induction of autophagic cell death in leukemia ABSTRACT. Oncotarget. 2013;4:860–74.
Hou W, Zhang Q, Yan Z, Chen R, Zeh HJ, Kang R, et al. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 2013;4:e966–10.
Held NM, Houtkooper RH. Mitochondrial quality control pathways as determinants of metabolic health. Bioessays. 2015;37:867–76.
Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res. 2016;18:1–10.
Peixoto J, Lima J. Metabolic traits of cancer stem cells. Dis Model Mech. 2018;11:dmm033464.
Lleonart ME, Abad E, Graifer D, Lyakhovich A. ROS-mediated autophagy defines the fate of cancer stem cells. Antioxid Redox Signal. 2017;7223:1066–1079.
Menendez JA, Joven J, Cufí S, Corominas-Faja B, Oliveras-Ferraros C, Cuyàs E, et al. The warburg effect version 2.0: Metabolic reprogramming of cancer stem cells. Cell Cycle. 2013;12:1166–79.
Pacini N, Borziani F. Cancer stem cell theory and the warburg effect, two sides of the same Coin? Int J Mol Sci. 2014;15:8893–930.
Shen YA, Wang CY, Hsieh YT, Chen YJ, Wei YH. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle. 2015;14:86–98.
Yi M, Li J, Chen S, Cai J, Ban Y, Peng Q, et al. Emerging role of lipid metabolism alterations in Cancer stem cells. J Exp Clin Cancer Res. 2018;118;1–18.
Snyder V, Reed-Newman TC, Arnold L, Thomas SM, Anant S. Cancer stem cell metabolism and potential therapeutic targets. Front Oncol. 2018;8:1–9.
Vlashi E, Pajonk F. The metabolic state of cancer stem cells-A valid target for cancer therapy? Free Radic Biol Med. 2015;79:264–8.
Zhou K, Yao Y-L, He Z-C, Chen C, Zhang X-N, Yang K-D, et al. VDAC2 interacts with PFKP to regulate glucose metabolism and phenotypic reprogramming of glioma stem cells. Cell Death Dis. 2018;9:988.
Jones CL, Stevens BM, Alessandro AD, Degregori J, Pollyea DA, Jordan CT, et al. Inhibition of amino acid metabolism selectively article inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. 2018;34:724–40.
Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, et al. Article inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20:674–88.
Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24:1036–46.
Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med. 2017;23:1234–40.
Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo‐San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–36.
Kasahara A, Scorrano L. Mitochondria: From cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014;24:761–70.
Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal. 2011;14:1939–51.
Son MY, Choi H, Han YM, Cho YS. Unveiling the critical role of REX1 in the regulation of human stem cell pluripotency. Stem Cells. 2013;31:2374–87.
Martin AV, Cufí S, Faja BC, Oliveras C, Vellon L, Menendez JA. Mitochondrial fusion by pharmacological manipulation impedes somatic cell reprogramming to pluripotency: New Insight into role mitophagy cell stemness. Aging. 2012;4:1–9.
Chen H, Chan DC. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 2017;26:39–48.
Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. 2015;18:501–10.
Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28:R170–85.
Redmann M, Dodson M, Boyer-Guittaut M, Darley-Usmar V, Zhang J. Mitophagy mechanisms and role in human diseases. Int J Biochem Cell Biol. 2014;53:127–33.
Boya P, Codogno P, Rodriguez-Muela N. Autophagy in stem cells: repair, remodelling and metabolic reprogramming. Development. 2018;145:dev146506.
Pickrell AM, Youle RJ. Review the roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–73.
Vazquez-Martin A, Van den Haute C, Cufí S, Corominas-Faja B, Cuyàs E, Lopez-Bonet E, et al. Mitophagy-driven mitochondrial rejuvenation regulates stem cell fate. Aging. 2016;8:1330–52.
Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature. 2017;543:205–10.
Herzig S, Shaw RJ. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19:121–35.
Pei S, Minhajuddin M, Adane B, Khan N, Stevens BM, Mack SC, et al. AMPK/FIS1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell. 2018;23:86–100.
Liu K, Lee J, Kim JY, Wang L, Tian Y, Chan ST, et al. Mitophagy controls the activities of tumor suppressor p53 to regulate hepatic cancer stem cells. Mol Cell. 2017;68:281–92.
Lin R, Iacovitti L. Classic and novel stem cell niches in brain homeostasis and repair. Brain Res. 2015;1628:327–42.
Carnero A, Lleonart M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. Bioessays. 2016;38:S65–74.
Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669–73.
Maxwell PH, Wlesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5.
Peng G, Liu Y. Hypoxia-inducible factors in cancer stem cells and inflammation. Trends Pharmacol Sci. 2015; 36; 374–83.
Hamacher-Brady A, Brady NR. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci. 2016;73:775–95.
Springer MZ, Macleod KF. In brief: Mitophagy: mechanisms and role in human disease. J Pathol. 2016;240:253–5.
Naik PP, Birbrair A, Bhutia SK. Mitophagy-driven metabolic switch reprograms stem cell fate. Cell Mol Life Sci. 2018. https://doi.org/10.1007/s00018-018-2922-9.
Yan C, Luo L, Guo CY, Goto S, Urata Y, Shao JH, et al. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017;388:34–42.
Verbaanderd C, Maes H, Schaaf MB, Sukhatme VP, Pantziarka P, Sukhatme V, et al. Repurposing drugs in oncology (ReDO) - Chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience. 2017;11:1–35.
Yang Y, Hu L, Zheng H, Mao C, Hu W, Xiong K, et al. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin. 2013;34:625–35.
Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy: The yin and yang of cell death control. Circ Res. 2012;111:1208–21.
Pasquier B. SAR405, a PIK3C3/VPS34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy. 2015;11:725–6.
Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16:1069–79.
Petherick KJ, Conway OJL, Mpamhanga C, Osborne SA, Kamal A, Saxty B, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015;290:11376–83.
Katheder NS, Khezri R, O’Farrell F, Schultz SW, Jain A, Schink MKO, et al. Microenvironmental autophagy promotes tumour growth. Nature. 2017;541:417–20.
Yoshida GJ. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: From pathophysiology to treatment. J Hematol Oncol. 2017;10:1–14.
Pietrocola F, Pol J, Kroemer G. Fasting improves anticancer immunosurveillance via autophagy induction in malignant cells. Cell Cycle. 2016;15:3327–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by F. Pentimalli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nazio, F., Bordi, M., Cianfanelli, V. et al. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ 26, 690–702 (2019). https://doi.org/10.1038/s41418-019-0292-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-019-0292-y
This article is cited by
-
RGS20 promotes non-small cell lung carcinoma proliferation via autophagy activation and inhibition of the PKA-Hippo signaling pathway
Cancer Cell International (2024)
-
Liposome-lentivirus for miRNA therapy with molecular mechanism study
Journal of Nanobiotechnology (2024)
-
Selective autophagy in cancer: mechanisms, therapeutic implications, and future perspectives
Molecular Cancer (2024)
-
Autophagy-mediated ID1 turnover dictates chemo-resistant fate in ovarian cancer stem cells
Journal of Experimental & Clinical Cancer Research (2024)
-
Targeting Hepatic Cancer Stem Cells (CSCs) and Related Drug Resistance by Small Interfering RNA (siRNA)
Cell Biochemistry and Biophysics (2024)
