
Abstract
Since the discovery and definition of neutrophil extracellular traps (NETs) 14 years ago, numerous characteristics and physiological functions of NETs have been uncovered. Nowadays, the field continues to expand and novel mechanisms that orchestrate formation of NETs, their previously unknown properties, and novel implications in disease continue to emerge. The abundance of available data has also led to some confusion in the NET research community due to contradictory results and divergent scientific concepts, such as pro- and anti-inflammatory roles in pathologic conditions, demarcation from other forms of cell death, or the origin of the DNA that forms the NET scaffold. Here, we present prevailing concepts and state of the science in NET-related research and elaborate on open questions and areas of dispute.
Similar content being viewed by others

Facts
-
Neutrophil extracellular traps (NETs) are formed as a defense mechanism to immobilize invading microorganisms but also in response to sterile triggers.
-
NETs consist of a DNA scaffold decorated with granule-derived proteins, such as enzymatically active proteases and anti-microbial peptides.
-
Apart from their function in immune defense, NETs play important detrimental or beneficial roles in inflammation, autoimmunity and other pathophysiological conditions
-
NET release can be instigated by many triggers and via a multitude of distinct pathways with often unknown interdependence.
Open questions
-
Are NETs primarily formed from nuclear or mitochondrial DNA, or both? Does the source of the DNA depend on the activating stimulus and/or the specific conditions that trigger NET formation? Do NETs composed of nuclear or mitochondrial DNA reflect different pathways that are adapted to distinct physiological needs?
-
How can we unambiguously distinguish NETs from the remnants of other forms of cell death?
-
Is there a connection between NET formation, neutrophil aggregation and/or neutrophil swarming?
-
Is there a link between autophagy, necroptosis, pyroptosis and NET formation?
Introduction
Histones and other nuclear proteins organize DNA in the nucleus of eukaryotic cells into nucleosomes and higher-order chromatin by neutralizing the negative charges on DNA. Thus, protein–DNA interactions constrain the potential energy of DNA to extend into a fibrous polymer and allow it to participate in the complex choreography that defines cellular functions [1]. The uncoiling of DNA represents the release of that potential energy, as can be appreciated during the rupture of a cell, which vastly expands the volume of nuclear DNA.
In 2004, Brinkmann et al. observed that the release of nuclear chromatin can be a regulated process that results in the appearance of what they called neutrophil extracellular traps (NETs) [2]. This insight raised the possibility that the release of nuclear chromatin may have physiologically beneficial consequences by significantly contributing to host defense.
NETs consist of DNA fibers decorated with proteins normally confined to granules, including antimicrobial molecules [2,3,4]. Extracellular DNA traps have been shown to be able to contribute to the immobilization and neutralization of certain kinds of bacteria [2, 5], fungi [6,7,8], and even some viruses [9, 10].
NETs form by the release of potential energy contained in the nucleus overall [2], some parts of it [11], or mitochondrial nucleoid DNA [12]. The compact structure of nuclear chromatin may be loosened by several alternative mechanisms: one is the global transcriptional activation that unwinds the inactive chromatin at the majority of loci [13]. A second is the proteolytic degradation of histone termini that assist in folding nuclear DNA [8] and a third is the post-translational modification of positively charged residues in core and linker histones [14, 15]. These three processes may synergize with each other or take precedence under specific conditions.
In the course of the last 14 years, the number of publications involving NETs have virtually exploded (Fig. 1). A search on Pubmed (www.ncbi.nlm.nih.gov/pubmed/) for “neutrophil extracellular traps” yielded 1940 results through the end of October 2018. NETs have been implicated not only in anti-microbial defense but also in a variety of sterile inflammatory and autoimmune conditions [2, 4, 16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37].

Number of publications including the term “neutrophil extra-cellular trap” per year (according to PubMed)
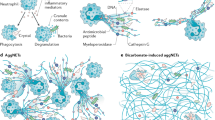
A lively discussion is currently ongoing about key aspects of NETs, their contents and morphology [38, 39], how their formation should be precisely named to reflect the different pathways of their generation [40, 41], the fate of the NETing neutrophil [42], by which triggers NET formation can be induced [3, 43], and the implications of NET formation for the host [5, 39, 44, 45]. These reports are partly overlapping, conflicting, or in direct contrast to each other. Specifically, the requirement of certain molecular pathways, the connection between NET formation and cell death, and the source of DNA in NETs are a matter of debate [38, 46, 47] (Fig. 2). The use of different methods of detection and quantification of NETs in vitro, in serum and in tissue [48,49,50,51,52,53,54,55,56] also impedes interpretation and/or comparison.

Current areas of consensus and controversy about neutrophil extracellular traps (NETs)
We have made an effort to put together a broad panel of opinion leaders and experts in the field to formulate concepts and raise further questions regarding various aspects of NET formation. The seeds for this effort germinated during a NET consensus meeting held in Erlangen, Germany in September 2016.
This paper revolves around a list of statements that summarize levels of agreements on various NET-related questions (Table 1), accompanied by a commentary that focuses on open questions and areas of scientific dispute. In particular, we list aspects of terminology and mechanisms of NET formation, and of components, triggers, physiological functions and pathological implications of NETs. Furthermore, we elaborate on minimal requirements for proper experimental designs and methodological accuracy of NET-related studies and for quantification and definition of NETs. The article also contains paragraphs penned by individual authors that describe the state-of-the art and ongoing efforts in various areas of NET research in the light of their own research (Supplementary Text). This dual structure of the paper was intended to provide a glimpse into the kaleidoscope of current NET research.
Current consensus and diverging opinions in NET research
To shed light on opinions on NET-related topics, a questionnaire with 140 statements (submitted by the authors of this paper) was sent out. Every author was to rate the level of agreement with each statement (1, agree; 2, do not agree; 0, undecided). From this list, 85 non-redundant statements with high response rates were chosen (Table 1). This listing illustrates current areas of consensus and dispute.
General statements and terminology
A large majority of the authors of this paper agree that the current literature creates confusion for lack of proper definitions of NETs (statement 1, st. 1). In many publications, extracellular DNA derived from different sources and/or after different forms of cell death pathways is collectively and erroneously equated with NETs. This confusion might partly arise due to the use of unspecific bioassays (such as measurement of extracellular DNA, measurement of extracellular elastase activity) that are used as a surrogate for NETs (st. 2). Different isolation procedures and neutrophil sources (isolation via density gradient centrifugation or magnetic cell sorting from bone marrow or from peripheral blood) or species differences between mice and humans may further complicate this issue. Also, the term “NETosis” suggests that cell death is an inevitable consequence of extrusion of DNA. Yet it is challenging to determine the exact sequence of events and the fate of the cell in retrospect when analyzing tissue sections. Furthermore, not even all pathways of NET generation elicited under controlled experimental conditions in vitro result in cell death. Therefore, in alignment with the Nomenclature Committee of Cell Death [46] the authors of this paper suggest to avoid the term”NETosis” or use it only in contexts where the demise of the neutrophil is obvious (st. 3, 4). In all other cases, we recommend to use the term “NET formation” instead.
Composition and morphology of NETs
There is a strong consensus that NETs contain enzymatically active neutrophil proteases (st. 7) and other anti-bacterial molecules. The source of DNA in NETs is less unambiguous. Simon, Yousefi et al. described extrusion of NETs consisting of mitochondrial DNA together with granule proteins rather than nuclear DNA [57, 58]. This mechanism, which was also shown to occur in eosinophils [12], involves an active reorganization of the cytoskeleton [59] and is ROS-dependent, but not accompanied by cell death. While it is now acknowledged by a majority of authors that NETs can be formed from both nuclear and mitochondrial origin (st. 5, 6, 54, 55), a potential mechanistic and/or physiologic demarcation between these processes is still unclear. Simon and Yousefi also claimed that NETs composed of mitochondrial DNA manifest as fibers, while the cloud-like appearance of nuclear DNA often seen after prolonged incubation of neutrophils in vitro with canonical NET instigators such as phorbol 12-myristate 13-acetate (PMA) or bacteria [5] is a result of necrotic cell death [38]. However, according to the opinion of a majority of authors, NETs created in vitro can also have a cloud-like appearance (st. 8) and morphological differences might be due to mechanical agitation of culture slides (or the lack thereof).
NET formation in vivo
There is unequivocal consensus that NET formation in response to microbial and sterile agents is a real phenomenon that occurs in vivo (st. 9–14). However, it is important to highlight that only a limited number of studies have addressed the direct effect of specific stimuli in the induction of NETs in vivo. Despite these potential limitations, stimuli considered to be inducers of NET formation in vivo are bacteria [2], fungal hyphae [7], biochemical stimuli [2, 34, 41, 60,61,62,63], some inflammatory cytokines and chemokines [2, 64, 65], immune complexes [66], and contact with activated platelets [67].
Physiological functions of NETs
An important function of NETs is the defense against bacteria, viruses and fungi (st. 15). NETs not only immobilize the opponent [68], but also are equipped with anti-microbial compounds (such as anti-microbial peptides, histones and proteases) (st. 16). They are therefore considered able to kill pathogens directly. Extracellular DNA-containing structures have also been described in zebrafish [69], in cats [70], invertebrates [71, 72], and plants [73]. Therefore, the formation of extracellular DNA traps may be considered an ancient, evolutionary conserved defense mechanism [71, 74] (st. 17). In alignment with this perspective, bacteria have evolved strategies to avoid killing through NETs, per example via expression of nucleases that degrade NETs, or even to use NETs to their advantage (per example in biofilms) [5, 68, 75] (st. 18).
Apart from their multiple enhancing functions in immune defense and autoimmunity, evidence for anti-inflammatory action of NETs is also accumulating [44, 45] (st. 19). An important part of the regulatory effect of NETs on inflammation is due to the modulation of cytokine and chemokine activity by NET-related proteases (st. 20, 24) [26, 34, 76,77,78,79].
NETs have been shown to build larger conglomerates when present in higher densities [34, 77, 80], with both detrimental and beneficial outcome for the host [44] (st. 21, 23, 24, 25). A remaining open question is the connection between neutrophil aggregation and NET formation (st. 22). The technological progress in two-photon intravital microscopy has enabled the discovery of neutrophil swarming, a phenomenon characterized by highly coordinated series of neutrophil movement, followed by cell accumulation mediated by chemoattractant signals and adhesion molecules [81]. Swarming is observed during infection and sterile inflammation in both mouse and human neutrophils [82]. Interestingly, cell death, both in the inflamed surrounding tissue and within the neutrophil cluster itself, strongly amplifies swarming and fuels immune activation [83]. It is tempting to hypothesize that neutrophil swarming and the formation of NETs might be interdependent processes, but as of now, the connection between these cellular functions remains elusive (st. 22).
NET formation has been reported in blood vessels, ductal structures, and surfaces [15, 80, 84], but also in the tissue [34, 85]. NETs constitute an anti-microbial defense mechanism and are therefore likely to be found at places with high microbial burden. Thus, the conclusion that the location of internal and external body surfaces is responsible for the (perceived) enrichment for NETs is controversial (st. 26), as is the view that NETs provide a protective coating to mucosal surfaces (st. 27).
In a recent publication, a lining of NETs was found adjacent to large necrotic areas [86]. The authors suggested that aggregated NETs wall-off lumps of material with immunostimulatory activity, such as necrotic tissue or monosodium urate crystals, thereby limiting immune reactivity and inflammation to sterile agents (st. 28). However, this isolating effect needs to be balanced against the tissue-damaging properties of NETs that have been confirmed in several studies [31, 32, 44, 87].
Triggers of NET formation
While there is a large consensus that microbial agents, biochemical stimuli, calcium influx, immune complexes, and contact with platelets (thrombocytes) and/or damage-associated molecular patterns can trigger NET formation (st. 29–35) a direct connection between lysosomal membrane instability and NET formation is still under discussion (st. 36). Munoz et al. have reported lysosomal instability and concurrent disintegration of the nuclear morphology in neutrophils upon exposure to nonpolar nanoparticles followed by NADPH oxidase-dependent chromatin externalization [63, 88]. The authors therefore introduced a model where lysosomal leakage triggers a cascade of events involving ROS production and ending in formation of NETs [63]. A direct connection between these phenomena remains, however, yet to be proven.
Pathways of NET formation
In the initially described pathway of NET formation induced by PMA or bacteria and later termed “NETosis” [89], neutrophils release nuclear DNA decorated with proteins into the extracellular milieu via an NADPH oxidase 2 (NOX2)-dependent mechanism involving the death of the neutrophil [2, 90]. Fourteen years later, it has become clear that NET release can occur via multiple distinct pathways with often unknown interdependence (st. 37, 38) [3, 43]. Since different stimulators of NET formation induce differential signaling, generalized statements about certain protein requirements should be avoided. A common denominator is however that NET release is mostly seen as an active process driven by cell-intrinsic pathways that are activated by external stimuli (st. 39).
Apart from NOX2, the requirement of neutrophil-specific serine proteases neutrophil elastase (NE) and myeloperoxidase (MPO) has also been described for the later stages of NET formation in association with cell death, more particularly for chromatin decondensation [91, 92]. In particular, NOX2-derived ROS were reported essential for the release of NE and MPO from azurophilic granules [92, 93]. However, it is now clear that some forms of NET formation occur independently of NOX2 and MPO [3] (st. 40, 41).
The enzyme PAD4 that is highly expressed in neutrophils mediates conversion of arginine into citrulline, which results in a massive loss of positive charges on arginine residues in histones. This conversion loosens the forces between DNA and histones and thus contributes to chromatin decondensation [94]. The role of PAD4 in NET formation is, however, one of the most controversial aspects in the study of NETs. Inhibition of PAD4 was reported to decrease NET formation in response to certain stimuli and PAD4-deficient mice sometimes display impaired NET formation [15, 26, 84, 95,96,97,98]. However, it should be noted that other reports observed normal NET formation in the absence of functional PAD4 [3, 99]. Therefore, this has led to the idea that not all NET release is PAD4-dependent (st. 42–44). Some of the discrepancies on the role of PAD4 in NET formation may be explained by our limited understanding of the different functions that PAD4 may have in neutrophil biology. Per example, a recent study linked PAD4 to assembly and activation of NOX2 [100]. Although this function of PAD4 is citrullination-independent, it can be blocked by PAD4 inhibitors. In contrast, conditions in which PAD4 is catalytically active prevented activation of NOX2. These novel findings shed some light on the paradox of why PAD4 is sometimes found to be essential for NET formation under conditions in which citrullination is not detected (e.g., in PMA-induced NETs) [3, 98].
Of note, the presence of citrullinated histones in cell culture or tissue is often regarded as a strong indicator of NET formation having occurred. However, these findings could potentially also be explained by extracellular citrullination of NET-bound histones by the PAD2 enzyme that is released from the cytoplasm upon stimulation with PMA [101], although PAD2 is not directly involved in NET formation triggered by LPS or TNFα [102]. For all the above reasons, caution in extracting conclusions should be exerted when studying PAD4 and citrullination as drivers of NET formation.
Neeli and Radic reported that several pathways of NET formation converge at the level of protein kinase C [41] (st. 45). They report that PAD4 activity is dependent on PKCζ activation and that PKCα is a dominant negative repressor of histone citrullination. Still, activation of both isoforms by combinatory treatment with PMA and ionomycin leads to increased NET release without detectable citrullination. It remains unclear, what caused these synergistic effects in the absence of citrullination as a driver of chromatin decondensation and this finding contrasts reports that PAD4 activity is solely dependent on calcium [15, 84].
Over the last few years, evidence has indicated that autophagy might be required for NET formation, although the molecular mechanisms are not clearly defined yet [103]. Remijsen et al. were the first to show that a combination of autophagy and ROS production is necessary for efficient PMA-induced NET formation in human neutrophils [104]. Next, Mitroulis et al. demonstrated that neutrophils from patients with acute gouty arthritis exhibit autophagy-mediated spontaneous NET release [61]. Furthermore, pharmacological inhibition of the mTOR pathway enhanced autophagosome formation along accelerated NET release following neutrophil stimulation with the bacteria-derived peptide fMLP [105]. The first genetic evidence illustrated that silencing of Atg5 in a neutrophil-like human cell line infected with adherent–invasive Escherichia coli blocked NET formation [103]. Recently, diminished Atg5 expression due to aging was also shown to reduce the capacity of neutrophils to form NETs [106,107,108]. In apparent contrast, Atg5-knockout mouse neutrophils had reduced autophagic activity but normal capacity to release extracellular DNA [109]. Of note, pharmacological inhibition of autophagy with PI3 kinase inhibitors such as wortmannin has to be interpreted with caution, because some, but not all, studies have indicated wortmannin to also inhibit ROS production which, consequently, could also block NET formation [104, 110,111,112]. Treatment with the so-called late-autophagy inhibitors, such as bafilomycin A1 and chloroquine, had no effect on NET formation [109]. Due to these conflicting data, there is yet no consensus on the role of autophagy in NET formation (st. 46).
Non-suicidal pathways of NET formation were described, where the cell remains intact and normal cellular functions of neutrophils, such as chemotaxis and phagocytosis, are still carried out [11, 42, 57, 113] (st. 47). These processes seem to occur much more quickly than the canonical NET pathway induced by PMA [90] or other forms of NET release that result in disruption of the plasma membrane [7, 34, 62, 114], per example crystal-induced NET formation (st. 49) and thus seem to be mechanistically distinct. Suicidal and live NET formation therefore need to be looked at separately (st. 48).
The frequent use of unspecific bioassays, such as the detection of extracellular DNA as a surrogate for the presence of NETs has created confusion, since it is not able to distinguish between NET formation and other forms of cell death with a necrotic morphotype. A distinction that is followed by the majority of authors of this paper is that NET formation requires an active and regulated process (st. 39), while necrosis can also occur in a passive, unregulated way (st. 50).
Desai et al. have found that NET formation upon 2 h stimulation with PMA or crystals involves the RIPK1/RIPK3/MLKL-dependent pathway of necroptosis [115, 116] (st. 51). They therefore argue that NET formation that involves cell death is a passive process secondary to plasma membrane rupture induced by necroptosis or other forms of necrosis [117] (st. 53) This view is opposed by others who have seen RIP3/MLKL-independent extrusion of DNA choosing different experimental conditions [118] (st. 52) or who argue that the definition of NET formation includes a highly regulated and coordinated process that is different from both necroptosis and necrosis (st. 50, 53). It also needs to be mentioned that two novel studies [119, 120] have demonstrated that the rupture of the plasma membrane during ROS-dependent NET formation is mediated by gasdermin D, thus connecting NETs with pyroptosis [121, 122].
Several recent reports have demonstrated that a considerable fraction of the nucleic acids contained in NETs is of mitochondrial origin [29, 57, 123, 124]. Special caution is, however, required to distinguish NET-derived mitochondrial DNA from mitochondrial nucleic acids expulsed during incomplete neutrophil mitophagy [125]. Due to its pro-inflammatory and interferogenic properties, oxidized mitochondrial DNA has been allotted an important role in the pathogenesis of SLE [29, 124, 125]. Although the mutual interdependence of extrusion of mitochondrial and nuclear DNA in NETs has yet to be confirmed, there seem to exist pathways of NET formation that are dependent and independent of mitochondria (st. 54, 55). For the majority of the authors of this paper, however, the breakdown of the nuclear membrane is still a hallmark of NET formation (st. 56).
Extracellular traps in different cell types
Extracellular trap release from cells other than neutrophils is understudied, and the mechanisms of chromatin decondensation release remain elusive. While extracellular trap formation in neutrophils and eosinophils [12] is more or less unequivocally accepted (st. 57, 58), further research, best performed in genetic models, is needed to understand both prevalence and relevance of extracellular trap release in cell types other than granulocytes (st. 59, 60). Extracellular trap release has also been reported for mast cells [126, 127]. Although a necessity for ROS production was observed, definite cellular pathways and further details are still warranted. The caspase-1-dependent release of monocyte extracellular traps following high multiplicities of infection has been reported by Webster et al. [128] and has very recently been described to contribute to the pathogenesis of rhabdomyolysis [129]. Similar to the cell death reported by Webster et al., also pyroptosis relies on caspase-1 activity and also leads to the release of intracellular components [46]. It is therefore unclear, whether extracellular trap formation in monocytes is distinct from pyroptosis.
Pathology and treatment
Owing to the multiple reports of the detrimental effects of NETs, especially in autoimmune diseases such as SLE, RA and vasculitis [4, 130,131,132,133,134], treatment with DNAse has become a promising therapeutic option (st. 67). NETs are degraded by endonucleases and DNase I-like proteins in the circulation. Apart from DNase I, also DNase I-like 3 is involved in vivo in the disintegration of NETs [135]. Removal of extracellular DNA by inhalation of recombinant human DNAse I is already a widespread and safe therapeutic option for cystic fibrosis [136]. In lupus, impairment of DNAse I function is associated with nephritis [22] and DNase I activity negatively correlates with disease activity [137]. Missense mutations in nucleases cause lupus-like disease in humans and mice [22, 138,139,140,141]. Furthermore, NET-binding proteins, such as antibodies or complement factor C1q, protect them from degradation possibly by inhibiting DNase I [22, 142]. Taken together, this argues for a beneficial role of DNAse in lupus. Similar mechanisms might be at work in other autoimmune diseases with occurrence of autoimmune reactivity to components of the nucleus. However, DNAse removes DNA from any source and its effect is not NET-specific. Furthermore, intravital imaging has revealed that when injected into circulation, DNase I is effective in the removal of DNA and decomposition of the NET-like structure but not necessarily in detachment of other components on NETs, which additionally attach to (glyco)proteins lining endothelium [143] and have potential tissue-damaging properties. Last but not least, other studies have challenged the detrimental effect of NETs on lupus-like autoimmunity and tissue damage [26, 96, 144]. Thus, caution needs to be exercised to identify the precise clinical conditions and developmental stages of diseases that warrant the in vivo use of DNAse or other therapeutic agents that aim at inhibition of NET formation.
Materials and Methods in NET-related research
PMA was initially used as one of the triggers to induce and define NETs [2, 90]. It is therefore often used as a surrogate for other NETs. PMA-induced NET formation is ROS-dependent and results in cell death (formerly called NETosis). Since then, many other pathways have been detected [3, 43], so that nowadays, the sole use of PMA is often considered limiting and the use of additional other more physiologically relevant stimuli is encouraged (st. 68, 69).
Immunocyto- and immunohistochemistry are the most widely used methods for the detection of NETs. NETs are identified as structures containing extracellular DNA co-localizing with granule-derived proteins, such as neutrophil elastase, and histones [2] (st. 70–72). NET formation can be monitored in real time via intravital microscopy [42, 143], live cell imaging [43, 51, 145,146,147] and with techniques based on DNA-intercalating dyes [11, 148] (st. 73, 74). Given the correct sample preparation and the use of proper controls, NETs can be visualized ex vivo in tissue sections and in fluid secretions [50, 149] (st. 75), although demarcation from necrosis can be challenging and caution should be taken to avoid overinterpretation of findings. Confirmation of the presence of granule proteins is encouraged also in these settings and even in in vivo settings to identify NETs. NETs can either appear cloud-like or filamentous [5] (st. 8).
Regarding neutrophil isolation for NET assays, blood might either be anti-coagulated with heparin or by chelating divalent ions, although it should be noted that a study found inhibition of NET formation by heparin [150] (st. 76, 77). Besides, isolation of neutrophils is generally performed in the absence of calcium and magnesium to prevent clumping and adhesion [151, 152] although NET generation and aggregation was reported to be inhibited by agents chelating divalent cations [34, 62] (st. 78, 79). The presence of calcium, magnesium and chelators (EGTA and EDTA) should therefore be described in the paper as they may have an impact on NET release.
Also, no real consensus yet exists about which medium to use for the storage of neutrophils prior to or during assessing NET formation in vitro, although the composition of the culture medium strongly influences the propensity to form NETs [153, 154] (st. 80). Also, NET assays must be performed under controlled CO2/HCO3-/pH balance [155,156,157] (st. 81). For the future, the introduction of standardized buffers to assess NET formation is desirable (st. 82).
As of now, the minimal requirements are that experiments on NET formation should exactly specify the culture conditions (st. 83). This constitutes the base medium, the use of serum [90, 158] or protein, the absence or presence of platelets [60] and the surface constitution of the cell culture plate [159] (st. 84). In addition, independently of the stimulus used, the source or preparation of the inducer should be stated in detail (st. 85).
Concluding remarks
Prompted by the excitement that followed the seminal paper by the Zychlinsky group that introduced NETs to the scientific community [2], a large body of data emerged that allotted major roles in defense from pathogenic microorganisms, in inflammation, and multiple pathophysiological conditions to NET formation. This wave of excitement was, and is, accompanied by doubts and criticisms. This paper illustrates current areas of consensus and dispute in the NET field (Fig. 2). The main areas of discussion are 1) the source of the DNA in NETs, 2) that demarcation from other forms of cell death is incomplete because factors that unambiguously distinguish NETs from the remnants of other forms of cell death are still missing, and 3) that NET formation can be mediated by multiple pathways. Therefore, it is unlikely that targeting a single pathway inhibits all NET formation without having a considerable impact on other aspects of cell biology and/or pathophysiology. Finally, 4) certain aspects of experimental procedures are not yet standardized. By highlighting these open questions, this paper aims to instigate further research and contribute to the harmonization of these issues.
Interestingly, defects in the signaling cascades that precipitate NET formation (such as the oxidative burst in chronic granulomatous disease or neutrophil serine proteases in Papillon–Lefèvre syndrome) are associated with pathologies characterized by chronic autoimmunity and inflammation, both of sterile and infectious origin [160,161,162,163,164]. On the other hand, treatment with PAD4 inhibitors that impedes NET formation (along with other cellular pathways) has had promising results for the treatment of autoimmune diseases [27, 165,166,167]. Thus, NET formation can be considered a major therapeutic target for the management of multiple human disorders. Understanding of the molecular mechanisms and the spatiotemporal dynamics that regulate NET formation and clearance and delineate it from other forms of cell death, will enable to fine-tune therapeutic approaches and minimize the risk of detrimental side effects and adverse outcome.
References
Kavenoff R, Zimm BH. Chromosome-sized DNA molecules from Drosophila. Chromosoma. 1973;41:1–27.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5.
Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. 2017;6.
Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra19.
Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol. 2012;198:773–83.
Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114:2619–22.
Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. 2014;15:1017–25.
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5:e1000639.
Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe. 2012;12:109–16.
Funchal GA, Jaeger N, Czepielewski RS, Machado MS, Muraro SP, Stein RT, et al. Respiratory syncytial virus fusion protein promotes TLR-4-dependent neutrophil extracellular trap formation by human neutrophils. PLoS ONE. 2015;10:e0124082.
Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010;185:7413–25.
Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14:949–53.
Khan MA, Palaniyar N. Transcriptional firing helps to drive NETosis. Sci Rep. 2017;7:41749.
Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol. 2008;180:1895–902.
Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205–13.
Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10:136–44.
Cedervall J, Zhang Y, Huang H, Zhang L, Femel J, Dimberg A, et al. Neutrophil extracellular traps accumulate in peripheral blood vessels and compromise organ function in tumor-bearing animals. Cancer Res. 2015;75:2653–62.
Dwivedi N, Radic M. Citrullination of autoantigens implicates NETosis in the induction of autoimmunity. Ann Rheum Dis. 2014;73:483–91.
Dworski R, Simon HU, Hoskins A, Yousefi S. Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J Allergy Clin Immunol. 2011;127:1260–6.
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107:15880–5.
Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra20.
Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. 2010;107:9813–8.
Jenne CN, Wong CH, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, et al. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe. 2013;13:169–80.
Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15:623–5.
Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013;5:178ra140.
Kienhofer D, Hahn J, Stoof J, Csepregi JZ, Reinwald C, Urbonaviciute V, et al. Experimental lupus is aggravated in mouse strains with impaired induction of neutrophil extracellular traps. JCI Insight. 2017;2.
Knight JS, Zhao W, Luo W, Subramanian V, O’Dell AA, Yalavarthi S, et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest. 2013;123:2981–93.
Leffler J, Stojanovich L, Shoenfeld Y, Bogdanovic G, Hesselstrand R, Blom AM. Degradation of neutrophil extracellular traps is decreased in patients with antiphospholipid syndrome. Clin Exp Rheumatol. 2014;32:66–70.
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22:146–53.
Meng H, Yalavarthi S, Kanthi Y, Mazza LF, Elfline MA, Luke CE, et al. In vivo role of neutrophil extracellular traps in antiphospholipid antibody-mediated venous thrombosis. Arthritis Rheumatol. 2017;69:655–67.
Pieterse E, Rother N, Garsen M, Hofstra JM, Satchell SC, Hoffmann M, et al. Neutrophil extracellular traps drive endothelial-to-mesenchymal transition. Arterioscler Thromb Vasc Biol. 2017;37:1371–9.
Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE. 2012;7:e32366.
Sangaletti S, Tripodo C, Chiodoni C, Guarnotta C, Cappetti B, Casalini P, et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood. 2012;120:3007–18.
Schauer C, Janko C, Munoz LE, Zhao Y, Kienhofer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. 2014;20:511–7.
Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21:815–9.
Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–21.
Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. 2015;21:880–6.
Yousefi S, Simon HU. NETosis - does it really represent nature’s “Suicide Bomber. Front Immunol. 2016;7:328.
Nauseef WM, Kubes P. Pondering neutrophil extracellular traps with healthy skepticism. Cell Microbiol. 2016;18:1349–57.
Konig MF, Andrade F. A critical reappraisal of neutrophil extracellular traps and NETosis mimics based on differential requirements for protein citrullination. Front Immunol. 2016;7:461.
Neeli I, Radic M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front Immunol. 2013;4:38.
Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18:1386–93.
Hoppenbrouwers T, Autar ASA, Sultan AR, Abraham TE, van Cappellen WA, Houtsmuller AB, et al. In vitro induction of NETosis: comprehensive live imaging comparison and systematic review. PLoS ONE. 2017;12:e0176472.
Munoz LE, Leppkes M, Fuchs TA, Hoffmann M, Herrmann M. Missing in action-the meaning of cell death in tissue damage and inflammation. Immunol Rev. 2017;280:26–40.
Grayson PC, Schauer C, Herrmann M, Kaplan MJ. Review: neutrophils as invigorated targets in rheumatic diseases. Arthritis Rheumatol. 2016;68:2071–82.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541.
Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122:2784–94.
Carmona-Rivera C, Kaplan MJ. Induction and quantification of NETosis. Curr Protoc Immunol. 2016;115:14 41 11–14 41 14.
Coelho LP, Pato C, Friaes A, Neumann A, von Kockritz-Blickwede M, Ramirez M, et al. Automatic determination of NET (neutrophil extracellular traps) coverage in fluorescent microscopy images. Bioinformatics. 2015;31:2364–70.
Gavillet M, Martinod K, Renella R, Harris C, Shapiro NI, Wagner DD, et al. Flow cytometric assay for direct quantification of neutrophil extracellular traps in blood samples. Am J Hematol. 2015;90:1155–8.
Ginley BG, Emmons T, Lutnick B, Urban CF, Segal BH, Sarder P. Computational detection and quantification of human and mouse neutrophil extracellular traps in flow cytometry and confocal microscopy. Sci Rep. 2017;7:17755.
Gonzalez AS, Bardoel BW, Harbort CJ, Zychlinsky A. Induction and quantification of neutrophil extracellular traps. Methods Mol Biol. 2014;1124:307–18.
Mohanty T, Sorensen OE, Nordenfelt P. NETQUANT: automated quantification of neutrophil extracellular traps. Front Immunol. 2017;8:1999.
Vong L, Sherman PM, Glogauer M. Quantification and visualization of neutrophil extracellular traps (NETs) from murine bone marrow-derived neutrophils. Methods Mol Biol. 2013;1031:41–50.
White PC, Chicca IJ, Ling MR, Wright HJ, Cooper PR, Milward MR, et al. Characterization, quantification, and visualization of neutrophil extracellular traps. Methods Mol Biol. 2017;1537:481–97.
Thalin C, Daleskog M, Goransson SP, Schatzberg D, Lasselin J, Laska AC, et al. Validation of an enzyme-linked immunosorbent assay for the quantification of citrullinated histone H3 as a marker for neutrophil extracellular traps in human plasma. Immunol Res. 2017;65:706–12.
Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16:1438–44.
Amini P, Stojkov D, Felser A, Jackson CB, Courage C, Schaller A, et al. Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat Commun. 2018;9:2958.
Stojkov D, Amini P, Oberson K, Sokollik C, Duppenthaler A, Simon HU, et al. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J Cell Biol. 2017;216:4073–90.
Pieterse E, Rother N, Yanginlar C, Hilbrands LB, van der Vlag J. Neutrophils discriminate between lipopolysaccharides of different bacterial sources and selectively release neutrophil extracellular traps. Front Immunol. 2016;7:484.
Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, et al. Neutrophil extracellular trap formation is associated with IL-1beta and autophagy-related signaling in gout. PLoS ONE. 2011;6:e29318.
Chatfield SM, Grebe K, Whitehead LW, Rogers KL, Nebl T, Murphy JM, et al. monosodium urate crystals generate nuclease-resistant neutrophil extracellular traps via a distinct molecular pathway. J Immunol. 2018;200:1802–16.
Munoz LE, Bilyy R, Biermann MH, Kienhofer D, Maueroder C, Hahn J, et al. Nanoparticles size-dependently initiate self-limiting NETosis-driven inflammation. Proc Natl Acad Sci USA. 2016;113:E5856–65.
Alfaro C, Teijeira A, Onate C, Perez G, Sanmamed MF, Andueza MP, et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res: Off J Am Assoc Cancer Res. 2016;22:3924–36.
Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, et al. Cytokines induced neutrophil extracellular traps formation: implication for the inflammatory disease condition. PLoS ONE. 2012;7:e48111.
Behnen M, Leschczyk C, Moller S, Batel T, Klinger M, Solbach W, et al. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcgammaRIIIB and Mac-1. J Immunol. 2014;193:1954–65.
Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122:2661–71.
Menegazzi R, Decleva E, Dri P. Killing by neutrophil extracellular traps: fact or folklore? Blood. 2012;119:1214–6.
Palic D, Ostojic J, Andreasen CB, Roth JA. Fish cast NETs: neutrophil extracellular traps are released from fish neutrophils. Dev Comp Immunol. 2007;31:805–16.
Wardini AB, Guimaraes-Costa AB, Nascimento MT, Nadaes NR, Danelli MG, Mazur C, et al. Characterization of neutrophil extracellular traps in cats naturally infected with feline leukemia virus. J Gen Virol. 2010;91(Pt 1):259–64.
Robb CT, Dyrynda EA, Gray RD, Rossi AG, Smith VJ. Invertebrate extracellular phagocyte traps show that chromatin is an ancient defence weapon. Nat Commun. 2014;5:4627.
Zhang X, Zhuchenko O, Kuspa A, Soldati T. Social amoebae trap and kill bacteria by casting DNA nets. Nat Commun. 2016;7:10938.
Hawes MC, Curlango-Rivera G, Wen F, White GJ, Vanetten HD, Xiong Z. Extracellular DNA: the tip of root defenses? Plant Sci: Int J Exp Plant Biol. 2011;180:741–5.
Wartha F, Henriques-Normark B. ETosis: a novel cell death pathway. Sci Signal. 2008;1:pe25.
Azzouz L, Cherry A, Riedl M, Khan M, Pluthero FG, Kahr WHA, et al. Relative antibacterial functions of complement and NETs: NETs trap and complement effectively kills bacteria. Mol Immunol. 2018;97:71–81.
Clancy DM, Sullivan GP, Moran HBT, Henry CM, Reeves EP, McElvaney NG, et al. Extracellular neutrophil proteases are efficient regulators of IL-1, IL-33, and IL-36 cytokine activity but poor effectors of microbial killing. Cell Rep. 2018;22:2937–50.
Reinwald C, Schauer C, Csepregi JZ, Kienhofer D, Weidner D, Malissen M, et al. Reply to “Neutrophils are not required for resolution of acute gouty arthritis in mice”. Nat Med. 2016;22:1384–6.
Shpacovitch V, Feld M, Hollenberg MD, Luger TA, Steinhoff M. Role of protease-activated receptors in inflammatory responses, innate and adaptive immunity. J Leukoc Biol. 2008;83:1309–22.
Hahn J, Schauer C, Czegley C, Kling L, Petru L, Schmid B, et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J 2018; fj201800752R.
Leppkes M, Maueroder C, Hirth S, Nowecki S, Gunther C, Billmeier U, et al. Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis. Nat Commun. 2016;7:10973.
Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmuller W, Parent CA, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498:371–5.
Kienle K, Lammermann T. Neutrophil swarming: an essential process of the neutrophil tissue response. Immunol Rev. 2016;273:76–93.
Stephen J, Scales HE, Benson RA, Erben D, Garside P, Brewer JM. Neutrophil swarming and extracellular trap formation play a significant role in Alum adjuvant activity. NPJ Vaccin. 2017;2:1.
Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207:1853–62.
Marzo E, Vilaplana C, Tapia G, Diaz J, Garcia V, Cardona PJ. Damaging role of neutrophilic infiltration in a mouse model of progressive tuberculosis. Tuberc (Edinb). 2014;94:55–64.
Bilyy R, Fedorov V, Vovk V, Leppkes M, Dumych T, Chopyak V, et al. Neutrophil extracellular traps form a barrier between necrotic and viable areas in acute abdominal inflammation. Front Immunol. 2016;7:424.
Wang J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018;371:531–9.
Biermann MH, Podolska MJ, Knopf J, Reinwald C, Weidner D, Maueroder C, et al. Oxidative burst-dependent NETosis is implicated in the resolution of necrosis-associated sterile inflammation. Front Immunol. 2016;7:557.
Steinberg BE, Grinstein S. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Science’s STKE: Signal Transduct Knowl Environ. 2007;2007:pe11.
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–41.
Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, et al. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood. 2011;117:953–9.
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–91.
Akong-Moore K, Chow OA, von Kockritz-Blickwede M, Nizet V. Influences of chloride and hypochlorite on neutrophil extracellular trap formation. PLoS ONE. 2012;7:e42984.
Leshner M, Wang S, Lewis C, Zheng H, Chen XA, Santy L, et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol. 2012;3:307.
Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363–71.
Gordon RA, Herter JM, Rosetti F, Campbell AM, Nishi H, Kashgarian M, et al. Lupus and proliferative nephritis are PAD4 independent in murine models. JCI Insight. 2017;2:10.
Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. 2015;11:189–91.
Tatsiy O, McDonald PP. Physiological stimuli induce PAD4-dependent, ROS-independent netosis, with early and late events controlled by discrete signaling pathways. Front Immunol. 2018;9:2036.
Hosseinzadeh A, Thompson PR, Segal BH, Urban CF. Nicotine induces neutrophil extracellular traps. J Leukoc Biol. 2016;100:1105–12.
Zhou Y, An LL, Chaerkady R, Mittereder N, Clarke L, Cohen TS, et al. Evidence for a direct link between PAD4-mediated citrullination and the oxidative burst in human neutrophils. Sci Rep. 2018;8:15228.
Spengler J, Lugonja B, Ytterberg AJ, Zubarev RA, Creese AJ, Pearson MJ, et al. Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol. 2015;67:3135–45.
Bawadekar M, Shim D, Johnson CJ, Warner TF, Rebernick R, Damgaard D, et al. Peptidylarginine deiminase 2 is required for tumor necrosis factor alpha-induced citrullination and arthritis, but not neutrophil extracellular trap formation. J Autoimmun. 2017;80:39–47.
Skendros P, Mitroulis I, Ritis K. Autophagy in neutrophils: from granulopoiesis to neutrophil extracellular traps. Front Cell Dev Biol. 2018;6:109.
Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21:290–304.
Itakura A, McCarty OJ. Pivotal role for the mTOR pathway in the formation of neutrophil extracellular traps via regulation of autophagy. Am J Physiol Cell Physiol. 2013;305:C348–54.
Hazeldine J, Harris P, Chapple IL, Grant M, Greenwood H, Livesey A, et al. Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging Cell. 2014;13:690–8.
Vieira da Silva Pellegrina D, Severino P, Vieira Barbeiro H, Maziero Andreghetto F, Tadeu Velasco I, Possolo de Souza H, et al. Septic shock in advanced age: transcriptome analysis reveals altered molecular signatures in neutrophil granulocytes. PLoS ONE. 2015;10:e0128341.
Xu F, Zhang C, Zou Z, Fan EKY, Chen L, Li Y, et al. Aging-related Atg5 defect impairs neutrophil extracellular traps formation. Immunology. 2017;151:417–32.
Germic N, Stojkov D, Oberson K, Yousefi S, Simon HU. Neither eosinophils nor neutrophils require ATG5-dependent autophagy for extracellular DNA trap formation. Immunology. 2017;152:517–25.
Dreiem A, Myhre O, Fonnum F. Involvement of the extracellular signal regulated kinase pathway in hydrocarbon-induced reactive oxygen species formation in human neutrophil granulocytes. Toxicol Appl Pharmacol. 2003;190:102–10.
Makni-Maalej K, Chiandotto M, Hurtado-Nedelec M, Bedouhene S, Gougerot-Pocidalo MA, Dang PM, et al. Zymosan induces NADPH oxidase activation in human neutrophils by inducing the phosphorylation of p47phox and the activation of Rac2: involvement of protein tyrosine kinases, PI3Kinase, PKC, ERK1/2 and p38MAPkinase. Biochem Pharmacol. 2013;85:92–100.
Mihalache CC, Yousefi S, Conus S, Villiger PM, Schneider EM, Simon HU. Inflammation-associated autophagy-related programmed necrotic death of human neutrophils characterized by organelle fusion events. J Immunol. 2011;186:6532–42.
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–9.
Maueroder C, Schall N, Meyer F, Mahajan A, Garnier B, Hahn J, et al. Capability of neutrophils to form NETs Is Not Directly Influenced By A CMA-targeting peptide. Front Immunol. 2017;8:16.
Desai J, Foresto-Neto O, Honarpisheh M, Steiger S, Nakazawa D, Popper B, et al. Particles of different sizes and shapes induce neutrophil necroptosis followed by the release of neutrophil extracellular trap-like chromatin. Sci Rep. 2017;7:15003.
Desai J, Kumar SV, Mulay SR, Konrad L, Romoli S, Schauer C, et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur J Immunol. 2016;46:223–9.
Desai J, Mulay SR, Nakazawa D, Anders HJ. Matters of life and death. How neutrophils die or survive along NET release and is “NETosis” = necroptosis? Cell Mol life Sci: CMLS. 2016;73:2211–9.
Amini P, Stojkov D, Wang X, Wicki S, Kaufmann T, Wong WW, et al. NET formation can occur independently of RIPK3 and MLKL signaling. Eur J Immunol. 2016;46:178–84.
Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol. 2018;3.
Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3.
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
McIlroy DJ, Jarnicki AG, Au GG, Lott N, Smith DW, Hansbro PM, et al. Mitochondrial DNA neutrophil extracellular traps are formed after trauma and subsequent surgery. J Crit Care. 2014;29:1133 e1131–5.
Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. 2015;67:3190–200.
Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. 2016;213:697–713.
Naqvi N, Ahuja K, Selvapandiyan A, Dey R, Nakhasi H, Puri N. Role of mast cells in clearance of Leishmania through extracellular trap formation. Sci Rep. 2017;7:13240.
von Kockritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood. 2008;111:3070–80.
Webster SJ, Daigneault M, Bewley MA, Preston JA, Marriott HM, Walmsley SR, et al. Distinct cell death programs in monocytes regulate innate responses following challenge with common causes of invasive bacterial disease. J Immunol. 2010;185:2968–79.
Okubo K, Kurosawa M, Kamiya M, Urano Y, Suzuki A, Yamamoto K, et al. Macrophage extracellular trap formation promoted by platelet activation is a key mediator of rhabdomyolysis-induced acute kidney injury. Nat Med. 2018;24:232–8.
Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol. 2017;2.
Gupta S, Kaplan MJ. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol. 2016;12:402–13.
Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol. 2011;7:691–9.
Lightfoot YL, Kaplan MJ. Disentangling the role of neutrophil extracellular traps in rheumatic diseases. Curr Opin Rheumatol. 2017;29:65–70.
Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123:2768–76.
Jimenez-Alcazar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science. 2017;358:1202–6.
Aitken ML. Clinical trials of recombinant human DNase in cystic fibrosis patients. Monaldi Arch Chest Dis. 1993;48:653–6.
Skiljevic D, Jeremic I, Nikolic M, Andrejevic S, Sefik-Bukilica M, Stojimirovic B, et al. Serum DNase I activity in systemic lupus erythematosus: correlation with immunoserological markers, the disease activity and organ involvement. Clin Chem Lab Med. 2013;51:1083–91.
Bodano A, Amarelo J, Gonzalez A, Gomez-Reino JJ, Conde C. Novel DNASE I mutations related to systemic lupus erythematosus. Arthritis Rheum. 2004;50:4070–1.
Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. 2011;43:1186–8.
Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000;25:177–81.
Gray EE, Treuting PM, Woodward JJ, Stetson DB. Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of Aicardi-Goutieres Syndrome. J Immunol. 2015;195:1939–43.
Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. 2012;188:3522–31.
Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673.
Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med. 2012;4:157ra141.
Gupta S, Chan DW, Zaal KJ, Kaplan MJ. A high-throughput real-time imaging technique to quantify NETosis and distinguish mechanisms of cell death in human neutrophils. J Immunol. 2018;200:869–79.
Kraaij T, Tengstrom FC, Kamerling SW, Pusey CD, Scherer HU, Toes RE, et al. A novel method for high-throughput detection and quantification of neutrophil extracellular traps reveals ROS-independent NET release with immune complexes. Autoimmun Rev. 2016;15:577–84.
van der Linden M, Westerlaken GHA, van der Vlist M, van Montfrans J, Meyaard L. Differential signalling and kinetics of neutrophil extracellular trap release revealed by quantitative live imaging. Sci Rep. 2017;7:6529.
Gupta AK, Giaglis S, Hasler P, Hahn S. Efficient neutrophil extracellular trap induction requires mobilization of both intracellular and extracellular calcium pools and is modulated by cyclosporine A. PLoS ONE. 2014;9:e97088.
Brinkmann V, Abu Abed U, Goosmann C, Zychlinsky A. Immunodetection of NETs in paraffin-embedded Tissue. Front Immunol. 2016;7:513.
Manfredi AA, Rovere-Querini P, D’Angelo A, Maugeri N. Low molecular weight heparins prevent the induction of autophagy of activated neutrophils and the formation of neutrophil extracellular traps. Pharmacol Res. 2017;123:146–56.
Kuhns DB, Long Priel DA, Chu J, Zarember KA. Isolation and functional analysis of human neutrophils. Curr Protoc Immunol. 2015;111:7 23 21–16.
Oh H, Siano B, Diamond S. Neutrophil isolation protocol. J Vis Exp. 2008. pii: 745. https://doi.org/10.3791/745.
Hoffmann JH, Schaekel K, Gaiser MR, Enk AH, Hadaschik EN. Inter-individual variation of NETosis in healthy donors: introduction and application of a refined method for extracellular trap quantification. Experi Dermatol. 2016;25:895–900.
Nadesalingam A, Chen JHK, Farahvash A, Khan MA. Hypertonic saline suppresses NADPH oxidase-dependent neutrophil extracellular trap formation and promotes apoptosis. Front Immunol. 2018;9.
Behnen M, Möller S, Brozek A, Klinger M, Laskay T. Extracellular acidification inhibits the ROS-dependent formation of neutrophil extracellular traps. Front Immunol. 2017;8.
Maueröder C, Mahajan A, Paulus S, Gößwein S, Hahn J, Kienhöfer D, et al. Ménage-à-Trois: the ratio of bicarbonate to CO2 and the pH regulate the capacity of neutrophils to form NETs. Front Immunol. 2016;7.
Naffah de Souza C, Breda LCD, Khan MA, de Almeida SR, Camara NOS, Sweezey N, et al. Alkaline pH promotes NADPH oxidase-independent neutrophil extracellular trap formation: a matter of mitochondrial reactive oxygen species generation and citrullination and cleavage of histone. Front Immunol. 2017;8:1849.
Kamoshida G, Kikuchi-Ueda T, Nishida S, Tansho-Nagakawa S, Kikuchi H, Ubagai T, et al. Spontaneous formation of neutrophil extracellular traps in serum-free culture conditions. FEBS Open Bio. 2017;7:877–86.
Sperling C, Fischer M, Maitz MF, Werner C. Neutrophil extracellular trap formation upon exposure of hydrophobic materials to human whole blood causes thrombogenic reactions. Biomater Sci. 2017;5:1998–2008.
Sorensen OE, Clemmensen SN, Dahl SL, Ostergaard O, Heegaard NH, Glenthoj A, et al. Papillon-Lefevre syndrome patient reveals species-dependent requirements for neutrophil defenses. J Clin Invest. 2014;124:4539–48.
Magnani A, Brosselin P, Beaute J, de Vergnes N, Mouy R, Debre M, et al. Inflammatory manifestations in a single-center cohort of patients with chronic granulomatous disease. J Allergy Clin Immunol. 2014;134:655–62 e658.
Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Med (Baltim). 2000;79:170–200.
De Ravin SS, Naumann N, Cowen EW, Friend J, Hilligoss D, Marquesen M, et al. Chronic granulomatous disease as a risk factor for autoimmune disease. J Allergy Clin Immunol. 2008;122:1097–103.
Roberts H, White P, Dias I, McKaig S, Veeramachaneni R, Thakker N, et al. Characterization of neutrophil function in Papillon-Lefevre syndrome. J Leukoc Biol. 2016;100:433–44.
Chumanevich AA, Causey CP, Knuckley BA, Jones JE, Poudyal D, Chumanevich AP, et al. Suppression of colitis in mice by Cl-amidine: a novel peptidylarginine deiminase inhibitor. Am J Physiol Gastrointest Liver Physiol. 2011;300:G929–38.
Willis VC, Banda NK, Cordova KN, Chandra PE, Robinson WH, Cooper DC, et al. Protein arginine deiminase 4 inhibition is sufficient for the amelioration of collagen-induced arthritis. Clin Exp Immunol. 2017;188:263–74.
Wei L, Wasilewski E, Chakka SK, Bello AM, Moscarello MA, Kotra LP. Novel inhibitors of protein arginine deiminase with potential activity in multiple sclerosis animal model. J Med Chem. 2013;56:1715–22.
Acknowledgements
SB, JH, LM, MHH, and MH received support from the Collaborative Research Center 1181 of the German Research Foundation (project # CRC1181-C03). HJA, JD, and DN were supported by funding from the German Research Foundation (projects # DFG AN372/14-3 and 24-1). JSK was supported by funding from the National Institutes of Health (R01HL134846) and the Lupus Research Alliance. EK was supported by the National Science Centre of Poland (project # 2014/15/B/NZ6/02519). Research in the Radic lab (MR, IN, and ND) on this topic was funded by the Lupus Research Alliance of New York, NY (USA). AH is supported by Fundación Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC). The CNIC is supported by the Ministerio de Ciencia, Innovacion y Universidades (MCIU) and the Pro CNIC Foundation, and is a Severo Ochoa Center of Excellence (MEIC award SEV-2015-0505). PS, IM, and KR were supported by BMBF/GSRT German-Greek Bilateral Research and Innovation grant no. T2DGE-0101. FA was supported by the Jerome L. Greene Foundation and the National Institutes of Health (R01 AR069569).
Authors contributions
SB, CM and MHH prepared draft versions of the paper and the table. MHH and MH integrated inputs from all co-authors (listed in alphabetical order). All co-authors filled out the questionnaire, provided constructive feedback for the preparation of the article, and approved its content.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
FA is an inventor on issued patent no. 8,975,033 held by The Johns Hopkins University that covers “Human autoantibodies specific for PAD3 which are cross-reactive with PAD4 and their use in the diagnosis and treatment of rheumatoid arthritis and related diseases” and serves as a consultant for Bristol-Myers Squibb. The other authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Piacentini
Supplementary information
Rights and permissions
About this article

Cite this article
Boeltz, S., Amini, P., Anders, HJ. et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ 26, 395–408 (2019). https://doi.org/10.1038/s41418-018-0261-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0261-x
This article is cited by
-
Age-dependent inflammatory response is altered in an ex vivo model of bacterial pneumonia
Respiratory Research (2024)
-
What is the actual relationship between neutrophil extracellular traps and COVID-19 severity? A longitudinal study
Respiratory Research (2024)
-
IDH1/MDH1 deacetylation promotes acute liver failure by regulating NETosis
Cellular & Molecular Biology Letters (2024)
-
Neutrophil extracellular traps: a catalyst for atherosclerosis
Molecular and Cellular Biochemistry (2024)
-
Neutrophil extracellular traps primed intercellular communication in cancer progression as a promising therapeutic target
Biomarker Research (2023)
