
Abstract
Extracellular DNA trap formation is a cellular function of neutrophils, eosinophils, and basophils that facilitates the immobilization and killing of invading microorganisms in the extracellular milieu. To form extracellular traps, granulocytes release a scaffold consisting of mitochondrial DNA in association with granule proteins. As we understand more about the molecular mechanism for the formation of extracellular DNA traps, the in vivo function of this phenomenon under pathological conditions remains an enigma. In this article, we critically review the literature to summarize the evidence for extracellular DNA trap formation under in vivo conditions. Extracellular DNA traps have not only been detected in infectious diseases but also in chronic inflammatory diseases, as well as in cancer. While on the one hand, extracellular DNA traps clearly exhibit an important function in host defense, it appears that they can also contribute to the maintenance of inflammation and metastasis, suggesting that they may represent an interesting drug target for such pathological conditions.
Similar content being viewed by others

Facts
-
The demonstration of extracellular DNA traps in vivo requires sections of affected tissues, which are to be investigated with special staining techniques. These structures are seen in multiple inflammatory and cancer diseases.
-
Measurements of cell-free (cf) DNA either alone or as complexes with granules or other cationic proteins, do not prove the in vivo presence of extracellular traps.
-
Although neutrophil extracellular traps (NETs) contribute to pathogen clearance, excessive NET formation promotes inflammation and tissue damage.
-
There is experimental evidence that NETs can contribute to metastasis.
-
Eosinophil extracellular traps (EETs) can bind and kill bacteria. They are often seen in areas of epithelial barrier defects.
Open questions
-
Under which pathological conditions are extracellular DNA traps suitable drug targets?
-
Is there a simple biomarker that reflects extracellular DNA trap formation?
-
What is the contribution of extracellular microbe killing compared to intracellular killing following phagocytosis?
-
The mechanism of DNA trap formation is unknown.
Introduction
Since their discovery, neutrophil extracellular traps (NETs) have been implicated as playing a role in host defense since they can disarm and kill bacteria extracellularly1. Studies aiming at unraveling the underlying mechanisms of NET formation demonstrated a requirement for the production of reactive oxygen species (ROS)2,3,4,5. ROS induces actin and tubulin glutathionylation, which is tightly regulated by glutaredoxin 1 (Grx1), an enzyme required for deglutathionylation of actin and microtubulin. Thus, an intact cytoskeleton is required for the formation of NETs6. Moreover, optic atrophy 1 (OPA1), one of five GTPase dynamin family members, known to play a role in mitochondrial (mt) fusion, has recently been shown to be required for ATP production through glycolysis in neutrophils. If increases in ATP production are blocked, the assembly of the microtubule network and thus the formation of NETs do not occur7. ATP and ATP channel pannexin 1 (Panx1) contribute to NET formation and may represent therapeutic targets8.
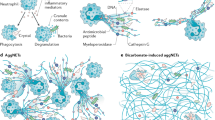
In addition to neutrophils, other granulocyte types can also form extracellular DNA traps, such as eosinophils (eosinophil extracellular traps, EETs)9,10 and basophils (basophil extracellular traps, BETs)11. Over the last decade, a number of stimuli, microbial and noninfectious stimuli, able to induce extracellular trap formation have been identified12,13,14,15,16. It should be noted that there is an ongoing scientific dispute whether NET formation requires cell death or not17,18,19,20. In this article, we focus on the current evidence of DNA traps under in vivo conditions and discuss their possible role(s) in the disease pathogenesis. These studies suggest that extracellular DNA traps exert effects beyond host defense, including allergic diseases, cancer, vascular diseases, and coagulation. This assumption is best supported by the observation that besides granulocytes, activated T cells, B cells, NK cells, and monocytes are also able to release mtDNA forming extracellular web-like structures, but the latter are devoid of bactericidal proteins. However, these structures containing mtDNA are able to provoke a rapid type I interferon (IFN-I) production in peripheral blood mononuclear cells, suggesting that lymphocytes and monocytes use mtDNA as a rapid signaling molecule to communicate danger21. Interestingly, mtDNA being enriched in unmethylated cytosine-phosphate-guanine (CpG) dinucleotide motifs similar to bacterial and viral DNA could provoke IL-10 secretion21,22, and transforming growth factor beta (TGF-β) release23. IL-10 is known to be an anti-inflammatory cytokine24, and TGF-β mediates the suppression of macrophage-directed inflammation, reducing TNF-α release25. Combination of anti-inflammatory and regulatory cytokines could dampen the excess inflammation (Fig. 1a).

a NET formation: In response to physiological stimuli or bacterial infection, viable neutrophils generate NETs in a NADPH oxidase-dependent manner. NETs consist of a mitochondrial (mt)DNA scaffold, which binds neutrophil granule proteins such as neutrophil elastase (NE) and myeloperoxidase (MPO)4,6,7,144,152. If the source of infection/stimulation persists, the released mtDNA, having similarity to viral and bacterial DNA (enriched in unmethylated CpG motifs), acts as a danger signal and triggers cytokine production for a protective and regulated immune response7,55. b Cytolysis: Under pathological conditions, such as the persistent presence of foreign antigens104, “too large to be trapped antigens”, such as fungal hyphae60,61, strong adhesion receptor activation153,154,155, presence of monosodium urate (MSU)88, or phorbol-myristate-acetate (PMA) stimulation2,12, results in an excess of reactive oxygen species (ROS), leading to neutrophil cytolysis. Similarly, excessive increases in intracellular calcium [Ca2+]i by ionomycin results in non-apoptotic neutrophil death12,81. Certain bacteria are also capable of causing neutrophil cytolysis by releasing pore-forming toxins that can directly cause plasma cell and nuclear membrane permeabilization156,157,158,159. The externalized cell exudates containing cytoplasmic and nuclear proteins together with damaged nuclear DNA can act as so-called self-antigens (Self-Ag) that are recognized and processed by antigen-presenting cells (APCs). Activated APCs produce pro-inflammatory cytokines and stimulate autoreactive T and B cells, leading to autoantibody production. The circulating autoantibodies such as anti-damaged-DNA/RNA ribonucleoprotein antibody immune complexes (RNP-ICs-Ab) can further activate neutrophils, including NET formation (not shown)13,73,74, leading to vicious cycle of chronic inflammation in genetically susceptible individuals68,74, causing autoimmune diseases such as systemic lupus erythematous (SLE).
The verification of DNA traps in vivo is challenging, as it requires special technical skills26,27. Measuring DNA concentrations in the absence or presence of granule proteins is clearly insufficient for concluding the presence of NETs or EETs, but in combination with (immuno)-histological investigations (Fig. 2), these techniques might be used for possible quantification. For example, increased DNA concentrations can occur as a consequence of a lytic granulocyte death (Fig. 1b), but also as a result of tissue damage that is a frequent phenomenon with inflammatory responses. Moreover, in contrast to in vitro conditions, the exact type of stimulus, its dosage and exposure time cannot be assessed under in vivo condition. With these limitations in mind, the reader may understand that the interpretation of the available studies is difficult and it is thus impossible, owing to space limitations, to critically evaluate all cited published findings. We sometimes just reflect the cited work, as it was reported by the original authors. Nevertheless, we often also mention a note of caution.

a Eosinophil extracellular traps (EETs) in mouse and human tissues. EETs consisting of DNA (red) and eosinophil peroxidase (EPX) (green) are depicted in 12-week-infected Citrobacter rodentium-infected mouse colon tissues55 (upper panel) and cutaneous Larva migrans-infested human skin tissue26 (lower panel). b Neutrophil extracellular traps in mouse and human tissues. NETs consisting of DNA (red) and neutrophil elastase (green) are shown in overnight infected Pseudomonas aeruginosa-mouse lung tissue7 (upper panel), and in human lung tissue from patients with bronchial asthma97 (lower panel). Extracellular traps are indicated by white arrows. Bars, 10 μm.
The formation of extracellular DNA traps in infectious diseases
The formation of extracellular DNA traps by neutrophils, eosinophils, and basophils, but also lymphocytes, has been observed in various infections of humans, mice, and additional species. Viruses, bacteria, fungi, and parasites have been shown to induce the generation of extracellular DNA traps, which can entrap and even kill the microbes through the action of associated toxic proteins.
NETs and EETs in bacterial infections
In the bronchial aspirates from patients with acute respiratory infections, bacteria and NETs were visualized using Gram stain and immunostaining. Neutrophils abundantly released NETs, and the NET length that was highest during acute infection and shortened with the recovery, correlated with clinical and laboratory signs of infection, as well as inflammatory cytokine levels in serum28. In an animal model of acute respiratory distress syndrome, in which neutrophil infiltration promotes tissue injury and sustained inflammation, the content of cf DNA in bronchoalveolar fluid was significantly higher in mice injected with LPS from Escherichia coli as compared with controls, and was decreased by glutamine treatment29. It should be noted, however, that in these two studies, NET formation and DNA concentrations were analyzed ex vivo using BAL fluids. To compensate for this shortcoming, lung tissue biopsies should be stained for NET detection in order to confirm the ex vivo data. In addition, cf DNA as well as granule and histone proteins are often quantified as “in vivo NET formation”30. Clearly, it is impossible to distinguish whether the released DNA/protein complexes are owing to NET formation or neutrophil death31,32, the latter of which was reported to occur under in vivo conditions more than 50 years ago33,34.
Genetically modified mice have been used to determine the role of specific proteins for NET formation under in vivo conditions. For instance, the role of peptidylarginine deiminase 4 (PAD4), an enzyme that catalyzes citrullination of histones, has been intensively studied in association with NET formation. Several reports have argued that PAD4 activity is essential for NET formation14,35,36,37,38,39,40,41, and contrary others disputed that PAD4 is not crucial for NET formation or the antimicrobial defense mechanism in vivo42,43,44,45. Specifically, NET formation in Klebsiella pneumonia-induced pneumonia was investigated showing NET-like structures surrounding Klebsiella bacteria at sites of immune infiltration in both Pad4−/− and Pad4+/+ mice. Moreover, both groups showed similar bacterial growth, lung inflammation, and organ injury. In conclusion, these data argue against a major role for PAD4 in NET formation, host defense, or organ injury during pneumonia-derived sepsis44.
Cystic fibrosis (CF) is characterized by a chronic inflammation of the airways associated with bacterial colonization. It has been reported that CF neutrophils have a pro-survival phenotype that allows increased NET production, which can in turn induce inflammation46. However, it should be noted that this conclusion is based mainly on ex vivo data. Both mucoid and non-mucoid Pseudomonas aeruginosa strains were demonstrated to activate neutrophils to generate NETs, a process that is promoted by macrophage migration inhibitory factor (MIF)47. Moreover, MIF protein levels in the blood of CF patients were significantly elevated compared with MIF levels in pooled human serum from healthy controls and negatively correlated with lung function47. On the other hand, the development of mucoidy (i.e., increased alginate production) is an acquired Pseudomonas aeruginosa virulence factor that is closely associated with increased severity of CF. The conversion to a mucoid phenotype coincided with a decline in susceptibility to NETs, raising the possibility that increased alginate production decreases interactions with NETs, or otherwise interferes with killing by NET-associated granule proteins48.
Staphylococcus aureus may cause serious infections, in particular when complicated by bacteremia and sepsis, and present a common health problem worldwide. In order to unravel the mechanism of organ damage, a mouse model was applied. Intravenous infection with multi-resistant Staphylococcus aureus led to a rapid sequestration of the bacteria to the liver, neutrophil recruitment and NET formation within the liver sinusoids, and subsequent liver damage14. As neutrophil elastase (NE), a component of NETs, was demonstrated to be enzymatically active and NE staining observed in areas adjacent to focal necrosis, the authors concluded that NET formation largely contributes to liver damage14. However, the authors also observed that destroying NETs by DNase treatment only partly reduced tissue injury, leaving some doubt about whether NETs are solely responsible for the immunopathology in this experimental model.
In addition, DNases are expressed by many Gram-positive bacterial pathogens, but their role in virulence is not clear. Expression of a surface endonuclease encoded by EndA is a common feature of many pneumococcal strains. EndA nuclease allows pneumococci to degrade the DNA scaffold of NETs and escape. Escaping NETs promotes spreading of pneumococci from the upper airways to the lungs and from the lungs into the blood stream during pneumonia49. Bacterial release of DNase and phosphatases contribute to defense against NET-mediated killing of Pseudomonas aeruginosa, highlighting the role of manipulating enzymes in combating NETs by microorganisms50. In addition, the pneumococcal polysaccharide capsule protects from NET trapping, but is not required for resistance to NET-mediated killing (Fig. 3)51.

a Extracellular traps consisting of a DNA scaffold and granule proteins entrap and kill microorganisms6,7,14,152. Certain pathogens have developed properties for escaping the physical entrapment by (b) encapsulation of bacteria to shield against traps51 or (c) release of endonucleases to degrade the extracellular DNA scaffold leading to less efficient killing of bacteria49,50.
In piglets infected with Streptococcus suis causing meningitis, NETs that consisted of DNA and associated NE have been detected in the cerebrospinal fluid (CSF)52. During pneumococcal meningitis, NETs in the central nervous system have been reported to hinder bacterial clearance. NETs were present in the CSF of patients with pneumococcal meningitis, but absent in other forms of meningitis with neutrophil influx in the CSF53. Pneumococci-induced NET formation in the CSF of infected rats could be cleared upon intravenous application of DNase I resulting in a disruption of NETs in the CSF followed by bacterial clearance, suggesting that NETs may contribute to pneumococcal meningitis pathogenesis in vivo53.
The formation of NETs has also been observed at cutaneous tick bite sites. Here, NETs have the potential to entrap and kill Borrelia burgdorferi, spirochetes causing Lyme disease. NETs have been observed in the upper and deep dermis after 3 and 5 days, respectively, that was not affected by either tick saliva or nucleases derived from these bacteria54.
Intestinal eosinophil infiltration and deposition of EETs, which were shown to be able to kill bacteria, were protective against microbial sepsis after cecal ligation in IL-5-transgenic, but not wild-type mice9. Moreover, infection with Citrobacter rodentium resulted in increased frequencies and activation of eosinophils and low bacterial load in the colon of wild-type mice, while eosinophil-deficient mice exhibited a high bacterial load despite the infiltration of neutrophils, Th1, and Th17 cells55. In this in vivo model showing that Citrobacter rodentium induced formation of EETs with bactericidal effects, and that EETs are present in the colon of infected mice provide further evidence for a role of EETs in controlling bacterial infection55.
NETs in viral, fungal, and parasitic infections
Influenza virus infection creates a highly pro-inflammatory lung environment and neutrophils, which are recruited to the lungs, encounter inflammatory mediators that can trigger NET formation42. Upon challenge of mice with lethal doses of influenza virus, NETs were found in infected lungs in the alveoli, and DNA fibers associated with matrix metallopeptidase (MMP)-9 and histones H2B were directed toward the alveolar epithelium and small blood vessels in areas with hemorrhagic lesions, indicating that there exists a contribution by NETs to alveolar–capillary damage56. Respiratory syncytial virus (RSV) may cause a severe lower respiratory tract disease (LRTD) in young children that is characterized by an extensive neutrophil accumulation in the lungs and occlusion of small airways by DNA-rich mucus plugs. In vitro, NETs were shown to capture RSV, and thus prevent its binding to target cells57. NET formation was also observed in the airways and lungs of children with severe RSV-LTRD. However, their effects in vivo seem double edged as demonstrated in a bovine model of RSV-LTRD revealing NET formation either with or without captured viral antigen in the dense plugs that finally occluded the airways57. Furthermore, double-stranded DNA (dsDNA) released by NETs recapitulate and promote rhinovirus-induced type-2 allergic immune responses and asthma exacerbation. Thus, NETs and their associated extracellular dsDNA contribute to the pathogenesis and may represent potential therapeutic targets of rhinovirus-induced asthma exacerbations58.
Candida albicans, a eukaryotic pathogen that is a common cause of fungal infections in humans, particularly in immunocompromised individuals, induces neutrophils to form NETs that capture and kill both hyphal and yeast form cells, for which NET-associated granule proteins are essential59. Further, it has been postulated that neutrophil responses, either phagocytosis or NET formation, are regulated by the microbial size independent of fungal surface molecule expression or enzymatic activity in vitro60,61. Correspondingly, NET release in the lungs of mice occurred upon exposure to wild-type Candida albicans that form both yeast and hyphae, whereas the yeast-locked hgc1Δ mutant, that cannot form hyphae, failed to induce NETs, and myeloperoxidase (MPO)-deficient mice able to kill via phagocytosis cleared the hgc1Δ yeast-locked strain, but not wild-type Candida albicans61. These in vivo observations indicate that NETs were not required for the clearance of yeasts, but for controlling hyphae.
In mice infected with larvae of Strongyloides stercoralis, an increase of extracellular DNA in the peritoneal exudates was observed 3 h after infection. The authors argued that this observation would be suggestive evidence for NET formation, although direct proof under in vivo conditions was not provided. However, the authors demonstrated in vitro experiments, suggesting that larvae of Strongyloides stercoralis induce the release of extracellular DNA forming clot-like structures ensnaring live larvae62. In an experimental setting using human neutrophils and macrophages in the presence of complement, both cells collaborated and killed the larvae in a manner requiring NET formation. Interestingly, when mouse cells were used, killing of larvae occurred independent of NET formation62.
The protozoan Toxoplasma gondii induced NET formation by mouse and human neutrophils with parasite entrapment and killing in vitro. NET formation was also shown in vivo in a mouse intranasal infection model63. On the other hand, although infection with Leishmania mexicana resulted in NETs entrapping parasites in the tissue of infected mice, in vitro experiments revealed that the induced NETs were unable to kill this protozoan64.
NET formation in onchocerciasis (river blindness), a helminth infection, is induced by the release of the bacterial endosymbiont, Wolbachia, and not directly by the filarial nematode Onchocerca volvulus. The onchocercomata (subcutaneous nodules) of patients contain worms positive for Wolbachia and exhibited high numbers of NETs. NETs were found in zones adjacent to the nematode cuticle, whereas nodules derived from patients treated with the anti-Wolbachia drugs, doxycycline + ivermectin were depleted for Wolbachia and lacked NETs65. More recently it was reported that NETs would drive inflammation in malaria by releasing soluble NET components to facilitate parasite sequestration and tissue destruction, and inhibition of NETs as a treatment strategy in vascular infections was recommended16.
The formation of extracellular DNA traps in autoimmune and autoinflammatory diseases
The formation of NETs has been demonstrated in several autoimmune and autoinflammatory diseases. In most studies, NETs appear to be part of the pathological process. Some of the proposed mechanisms are discussed below.
NETs in systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is considered to be a prototypic systemic autoimmune disease and is characterized by loss of tolerance to self-antigens, abnormal T- and B-cell responses, and autoantibody production66,67,68,69,70. Its pathogenesis involves defective clearance of immune complexes and debris containing nucleic acids, excessive innate immune activation involving Toll-like receptors (TLR) and type I interferons, as well as aberrant lymphocyte activation71. Upon stimulation with antimicrobial72 or anti-ribonucleoprotein (RNP) antibodies13,73,74, neutrophils from SLE patients have been shown to release self-DNA associated with antimicrobial peptides able to trigger innate plasmocytoid dendritic cell (pDC) activation via TLR9 to produce IFN-Ι (Fig. 1). The immune complex-mediated glomerulonephritis in SLE has been associated with a non-lytic extrusion of NETs concomitant with clustering of neutrophils within minutes that have immunogenic properties, including enrichment for high mobility group box protein 1 (HMGB1), oxidized mtDNA, and immune complex (ICx) formation13,73,75. By applying immunofluorescence staining, NETs composed of mtDNA and MPO have been detected in affected glomeruli66. Moreover, the proportion of glomeruli infiltrated by netting neutrophils correlates with lupus nephritis activity. The fact that NETs containing MPO as well as intact neutrophils are present in the dermis and dermal blood vessels of cutaneous lupus lesions suggests that enhanced NET formation occurs in vivo in affected organs of SLE patients76. SLE patients were found to develop autoantibodies to both the self-DNA and antimicrobial peptides present in NETs, indicating that these complexes serve as autoantigens to trigger B-cell activation. NETs containing cathelicidin LL-37–DNA complexes can directly trigger human memory B cells and induce the production of anti-neutrophil and anti-LL-37 antibodies in B cells of SLE patients (Fig. 1)77.
The timely removal of NETs seems crucial to avoid presentation of self-antigens. For instance, sera of a subset of SLE patients failed to properly clear NETs due to the presence of DNase I inhibitors or and anti-NET antibodies preventing DNase I access to NETs. The resulting impaired NET degradation correlated with renal disease78. Moreover, NETs derived from SLE neutrophils exhibit lower ubiquitin concentrations and a different ubiquitinated protein pattern compared with healthy controls79. The presence of ubiquitinated MPO in NETs and anti-ubiquitinated MPO antibodies in sera of SLE patients correlated with disease severity, suggesting a role for the ubiquitination status of NETs in the pathogenesis of SLE79.
A mitochondrial ROS scavenger was administered to MRL/lpr (lupus-prone mice) resulting in reduced mtDNA release and suppressed lupus-like disease13. Metformin, which in vitro decreased the number of mtDNA copies in NETs, significantly reduced the disease activity in SLE patients when given as an add-on therapy compared with conventional therapy alone66. Moreover, inhibition of PAD4 was reported to reduce the severity of SLE in an experimental mouse model by reducing histone hypercitrullination and self-nucleosome antibodies38,80. Therefore, PAD4 has been suggested as a promising drug target, and novel PAD4 inhibitors have been developed37,38,39. However, a pharmacological approach to inhibit PAD4 in a human serum transfer model of SLE failed to ameliorate end-organ damage and concluded that PAD4 does not appear to be crucial for SLE pathogenesis45,81.
NETs in vasculitis, rheumatoid arthritis, and psoriasis
NETs have also been implicated in the pathogenesis of small vessel vasculitis (SVV) leading to inflammation and destruction of small-sized blood vessels and capillaries. Upon stimulation with anti-neutrophil autoantibodies (ANCAs), neutrophils have been shown to release NETs that contain autoantigens, such as proteinase-3 (PR3) and MPO82. On the other hand, NETs with their components mtDNA, PR3, and MPO have been shown to be taken up by myeloid dendritic cells (mDC), significantly inducing the production of anti-neutrophil cytoplasmic autoantibodies (ANCA) and anti-dsDNA autoantibodies in mice immunized with NET-loaded mDC83. Therefore, both NET-induced autoimmunity as well as autoantibody-mediated NET formation seem to represent a vicious circle in disease pathogenesis. Similarly, drug-induced formation of NETs enriched in NE serving as auto-antigen, together with the release of B-cell activating factor (BAFF) resulting in B-cell activation, and the production of ANCA directed against NE that further enhance NET formation, have been assumed to be pathogenic mechanisms in cocaine and levamisole-associated autoimmunity84. A role for NETs in ANCA-associated vasculitis is also suggested by the observation that excessive NET formation correlated with disease exacerbation85.
Increased NET formation has also been associated with rheumatoid arthritis (RA). In a mouse model of collagen-induced arthritis (CIA), injecting chloramidine (Cl-amidine), which inhibits PAD4-mediated hypercitrullination, significantly reduced NET release and attenuated clinical disease activity. NETs apparently induced the expansion of Th1 pathogenic cells through maturation of dendritic cells and production of IFN-γ86. Therefore, analogous to SLE, NETs from RA patients may exhibit antigenic properties, but may also be recognized by autoantibodies, namely anti-citrullinated antibodies (ACPA)87. On the other hand, NETs may promote the resolution of neutrophilic inflammation by degrading cytokines and chemokines and disturbing neutrophil recruitment and activation88.
NET-like DNA formations have been detected associated with MPO and IL-17 in the epidermis, particularly in Monro’s microabscesses, and associated with IL-17 and LL-37 in the dermis89. However, the observations that the morphology of the NETs varied and most of the neutrophils in Monro’s microabscesses had altered nuclear morphology, suggested that the DNA release was at least partially the result of cell death17,89.
EETs and NETs in bullous pemphigoid
Bullous pemphigoid (BP) is an autoimmune blistering skin disease characterized by an activation of autoreactive B and T cells, the production of pathogenetically relevant autoantibodies directed against the hemidesmosomal proteins BP180 and BP230, and a prominent eosinophil infiltration in the skin90. It has been shown that in prebullous lesions, a small subgroup of eosinophils have formed EETs consisting of mtDNA associated with eosinophil granular proteins26. Some of these EET were directed against the dermal–epidermal junction, the site where blister formation occurs26. Thymic stromal lymphopoietin (TSLP) has been identified as a cytokine-stimulating eosinophils to form EETs, an observation that might be relevant for BP since TSLP is expressed in the epidermis and eosinophils infiltrating BP lesions express the TSLP receptor10. In an ex vivo skin model, eosinophils stimulated with IL-5 in the presence of BP serum caused a dermal–epidermal splitting resembling BP. This splitting was significantly decreased upon adding DNase I, suggesting that EET formation plays at least a partial role90. These observations make eosinophils interesting targets for therapy91. A recent study reported the presence of NETs at sites of blister formation in BP that decreased with time following treatment in patients undergoing remission92.
NETs in autoinflammatory diseases
Autoinflammatory diseases manifest as recurrent fevers, various forms of systemic inflammation or sterile skin, bone, and joint inflammation without prominent fever, but involving myeloid cells that lack the stigmata of classical autoimmune diseases, such as high-titer autoantibodies or antigen-specific T cells93. Familial Mediterranean fever (FMF) is characterized by neutrophilia and neutrophil infiltration in affected tissues during inflammatory attacks induced by physical or psychological stress. In vitro, the amounts of NETs consisting of DNA associated with NE and interleukin (IL)-1β, released by PMNs isolated from FMF patients during attack were significantly higher compared with those in remission, when PMNs were resistant to stimuli usually inducing NET formation94. Abundant infiltrates of neutrophils forming NETs have been reported in Schnitzler syndrome, defined by recurrent urticarial rash, monoclonal gammopathy, and systemic inflammation95. Flares of sterile arthritis with neutrophil infiltrate and the overproduction of IL-1β are the main features of pyogenic arthritis, pyoderma gangrenosum and acne (PAPA) syndrome. NETs have been identified in the skin lesions of a patient with active PAPA syndrome in a milieu characterized by IL-1β, IL-8, and IL-17A expression, but not in skin samples from a patient with no active skin lesions96. In vitro, serum of PAPA patients induced NET formation by neutrophils from healthy donors that could be blocked by the IL-1 receptor antagonist anakinra, suggesting that IL-1β contributes to the enhanced NET formation in PAPA96.
The formation of extracellular DNA traps in eosinophilic diseases
The formation of EETs has been demonstrated in several eosinophilic diseases. In most studies, the extent of DNA trap formation correlated with disease severity. On the other hand, bacterial killing by DNA traps appears to be an important innate immune mechanism in case of the presence of a disease-promoting and/or inflammation-induced epithelial barrier defect.
EETs in asthma and rhinosinusitis
Bronchial asthma is a heterogeneous inflammatory airway disorder that involves eosinophilic and non-eosinophilic, including neutrophilic, phenotypes. In bronchial biopsies, EETs consisting of a mtDNA scaffold co-localizing with MBP were seen in all asthma patients, and their number correlated with the number of eosinophils97. A subgroup of patients expressed a high level of neutrophils and NETs, in which the extracellular mtDNA was associated with NE97. In an animal model of asthma, the treatment with deoxyribonuclease, which cleaves extracellular DNA, resulted in an improvement of airway resistance and abolished extracellular DNA content in BALF as well as goblet cell hyperplasia98. Peripheral blood eosinophils from patients with severe eosinophilic asthma (SEA) may be more activated to produce EETs than those from patients with non-severe asthma (NSA), which further induces inflammation in asthmatic airways99. Surfactant protein-D (SP-D), an epithelial cell product of the airways, is a critical immune regulatory molecule with a multimeric structure susceptible to oxidative modifications. We have demonstrated that SP-D directly binds to the eosinophil membrane, inhibits EET formation, and reduces asthma exacerbations100. NETs have also been visualized in induced sputum from patients with asthma and COPD, which compared with control sputum, exhibited higher levels of extracellular DNA and other NET components, such as cathelicidin LL-37, alpha-defensin 1–3, NE, IL-1β, and CXCL8 correlating with decreased lung function101. In stable COPD patients, extensive NET formation was observed in all sputum samples irrespective of purulence or smoking status102,103. Moreover, the presence of NETs is associated with disease severity and microbiota diversity in patients with COPD104.
In chronic rhinosinusitis with nasal polyps that is characterized by Th2-biased eosinophilic inflammation, about 8.8% of tissue eosinophils exhibited EETs correlating with IL-5 and periostin tissue levels and colonization with Staphylococcus aureus (S. aureus)105. In an ex vivo human mucosal disease tissue model, transepithelial migration at sites of epithelial defects and massive EET formation of eosinophils to entrap S. aureus has been demonstrated105. These observations, together with the finding that S. aureus can directly induce EET formation, suggested that, in case of epithelial barrier defects, eosinophils are part of the innate immune response for avoiding the invasion by bacteria10,105. In a subsequent study, it was demonstrated that EETs, but not NETs, were detected at various degrees in all tissue specimens of nasal polyps or ethmoid tissues obtained by endoscopic sinus surgery for chronic rhinosinusitis. The number of EETs correlated with that of tissue eosinophils, blood eosinophilia, severity and decreased olfactory function, regardless of the presence of nasal polyps, concomitant asthma or atopy106.
EETs in acute dermatitis responses and eosinophilic esophagitis
When specimens of various eosinophilic skin diseases were examined, EETs could be detected in atopy patch test-induced lesions, but not in biopsies from atopic dermatitis or in positive patch test reactions of patients with allergic contact dermatitis, suggesting that EET formation occurs as an acute response to allergens26. Induced lesions in a patient with bullous delayed-pressure urticaria showed a marked infiltration of eosinophils, of which >80% formed extracellular DNA traps, suggesting a pathogenic role of eosinophils and EETs107.
In eosinophilic esophagitis, EET formation occurred frequently, as it was detected in all tissue samples analyzed, and correlated with the number of tissue eosinophils108. Moreover, there was evidence for epithelial barrier defects, e.g., decreased filaggrin and protease inhibitor LEKTI expression, while LEKTI inversely correlated with the number of EETs108. These findings imply a role for eosinophils in protecting the host against invading pathogens in case of a disrupted epithelial barrier by generating EETs and thus establishing a second barrier.
The formation of extracellular DNA traps in cardiovascular diseases
Several non-microbial stimuli for NETs have been described including cholesterol, which can trigger and amplify sterile inflammation109. Such stimuli likely play a role in NET formation associated with cardiovascular diseases.
NETs in atherosclerosis
The presence of NETs has been reported in mice and human atherosclerotic lesions. Using a two photon microscopic intravital approach, luminally adhering neutrophils releasing DNA in apolipoprotein-deficient (Apoe−/−) mice fed a high-fat diet for 4–6 weeks were observed, whereas no neutrophil adhesion and hence no NET release were detected in mice receiving chow diet110. Together with NETs, associated antimicrobial peptide Cramp/LL-37 and plasmacytoid dendritic cells (pDC) have been identified in atherosclerotic lesions. Cramp/DNA complexes can stimulate pDC to produce IFN-α, that in turn promotes atherosclerotic plaque growth that is associated with enhanced anti-dsDNA antibody titers111. In order to study atherosclerosis in the absence of NETs, Apoe−/− mice lacking NE and PR3 (Apoe−/−Elane−/−Prtn3−/−) were employed and revealed reduced lesions size after 8 weeks of high-fat-diet feeding112.
Inhibition of PAD4 by Cl-amidine resulted in decreased atherosclerotic lesion size and delayed carotid artery thrombosis in the Apoe−/− mouse model80. The authors also showed that PAD4 inhibition mitigates arterial IFN-I responses, and reduces the number of netting neutrophils that infiltrate the media and adventitia of atheromatous lesions. Although the authors concluded a causative role for NETs in the development of murine atherosclerosis80, it should be again noted that a requirement for PAD4 for NET formation, and its correlation with diseases, is in dispute17,19,42,43,44,45. In fact, more recently, selective genetic deficiency of Pad4 in bone marrow-derived cells exhibited no differences in the formation and progression of atheromatous plaques compared with control mice113.
In mice, cholesterol accumulation in myeloid cells activates the NOD-like receptor protein (NLRP3) inflammasome which enhances neutrophil accumulation and NETs formation in atherosclerotic plaques114. Furthermore, activated platelets were determined as physiological stimulus that via P-selectin induce NET formation115.
Do these findings obtained in murine models correlate with human disease? In patients with coronary artery disease (CAD) proven by CT-angiography markers of cell death, NET formation in plasma was measured, revealing significantly elevated levels of dsDNA, nucleosomes, and MPO–DNA complexes116. Moreover, a high plasma nucleosome level was identified as an independent risk factor for severe coronary stenosis, and the level of MPO–DNA complexes predicted the number of atherosclerotic vessels116. In patients with stable coronary artery disease who had high dsDNA plasma levels, the risk of experiencing a clinical endpoint defined as unstable angina, non-hemorrhagic stroke, myocardial infarction, or death was significantly increased after 2 years117. However, it still remains to be established whether circulating extracellular DNA in patients indeed reflects the extent of NET formation. Nevertheless, host DNases prevented vascular occlusion by NETs118.
NETs in atherothrombosis
Progression of atherosclerotic lesions can eventually lead to the destabilization of plaque with subsequent intraluminal atherothrombosis resulting in acute coronary syndrome or ischemic stroke, depending on the plaque location. For initiating arterial thrombosis in injured vessels, the interaction of neutrophils with endothelial cells is a critical step preceding platelet accumulation119. Activated lesional smooth muscle cells (SMCs) may attract neutrophils that undergo neutrophil death (not NET formation), releasing nuclear proteins, including histone H4, which induce lytic cell death of smooth muscle cells causing the destabilization of plaques. Neutralization of histone H4 led to a plaque stabilization, whereas blocking histone H2A or disrupting DNA structures by DNase had no effect on smooth muscle cells120. A significant decrease in infarction size and reduction of citrullinated histone 3 in infarcted tissue were demonstrated upon blocking of CCL5 and CXCL4 in mice, what was believed to be due to a reduced neutrophil recruitment121. In a model of myocardial infarction, Pad4−/− mice were significantly protected from, whereas mice without PAD4 deficiency had myocardial injury with the presence of citrullinated histone H3 at the site of injury and increased plasma levels for nucleosomes122. Our interpretation of these findings is that a lytic neutrophil death might be prevented in the absence of PAD4. Therefore, a lytic neutrophil death and not of NET formation seems to be important for atherothrombosis.
On the other hand, studies investigating atherothrombosis in humans revealed evidence for the presence of NETs in thrombi after endarterectomy. NETs were identified by immunohistochemical staining, e.g., with anti-MPO and anti-NE antibodies, in fresh and lytic, but not in organized thrombi. The presence of NETs was evident in the thrombi and hemorrhages as well as at the thrombus-plaque interface and in perivascular tissue adjacent to complicated, but not intact plaques123. In coronary thrombi, the NET burden was found to correlate with the infarct size and was negatively related to plasma DNase activity124. In addition to neutrophils, also macrophages, eosinophils, and mast cells have been observed to form DNA traps in coronary thrombi125.
NETs in venous thrombosis and thromboembolism
Deep vein thrombosis (DVT) has been linked to neutrophil activation and release of NETs based on studies investigating the pathogenic role of NETs in the pathogenesis of venous thromboembolism (VT) using genetically modified mice, various large animal models and human material assessing plasma markers or thrombi species126.
In a mouse DVT model applying intravital microscopy, neutrophils and monocytes, crawling along and adhering to the venous endothelium, have been shown to provide the initiating stimulus for DVT127. A cooperative signaling of P-selectin glycoprotein ligand (PSGL)-1 and CXCR2 in neutrophils increases their adhesion and enhances murine venous thrombosis through NET formation in flow-restricted veins128. Furthermore, platelet-derived HMGB1 as well as NK cell-dependent IFN-γ production were also reported to stimulate NET formation and thus contribute to the development of thrombi in deep vein thrombosis129,130. Thrombus-resident neutrophils bind factor XII and activate it through the release of NETs, whereas disintegration of NETs protected against DVT amplification127. In mice with DVT induced by flow restriction in the inferior vena cava, extracellular DNA was increased in plasma, and citrullinated histone H3 associated with neutrophils in venous thrombi131. Both the DNA scaffold and histones were shown to contribute to DVT131.
The analysis of 16 thrombi from patients with VTE demonstrated that NETs, defined as extracellular diffuse citrullinated histone 3 areas associated with MPO and DNA, were predominantly detected during the phase of thrombus organization, while NETs were rare in unorganized and organized thrombi122. As biomarkers of DVT, extracellular DNA and MPO have been tested. Plasma DNA levels are elevated in patients with DVT correlating with C-reactive protein, D-dimer, von Willebrand factor, and MPO132. Moreover, in elderly patients, levels of circulating extracellular DNA correlated with the extent of venous thromboembolism, inflammation as assessed by C-reactive protein and leukocytosis, and predicted mortality133. Recently, flow cytometric detection of MPO/citrullinated histone 3-positive neutrophils and serum dsDNA have been proposed for biomarker assessment30. However, as mentioned earlier, these biomarkers are not suitable for the definition of NET formation and can also not be used for mechanistic studies.
To date, clinical trials investigating whether targeting NETs prevents or treats venous thrombosis in humans are lacking. However, some established therapies of venous thromboembolism may affect extracellular DNA traps126. For instance, heparin was shown to dismantle the DNA scaffold and remove platelet aggregates from NETs134. Acetylsalicylic acid prevents NET formation in vitro by reducing the phosphorylation of the NF-κB p65 subunit135. In mice, acetylsalicylic acid inhibited tissue factor activity and NET formation followed by a marked reduction of thrombus size136. Clopidogrel, by decreasing P-selectin expression in platelets, blocks the interaction with neutrophils and thus NET formation137.
The formation of extracellular DNA traps in cancer
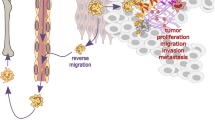
Hypothetically, NETs could have anti-tumorigenic effects, for example by actual killing of tumor cells or activating the immune system. However, there is more experimental evidence for a pro-tumorigenic activity of NETs (Fig. 4). First evidence of NET formation by tumor-associated neutrophils came from a histopathological analysis of diagnostic biopsies from Ewing sarcoma. Out of eight tissue samples, tumor-associated neutrophils were detected in six and NETs in two patients. In this study, NET formation was associated with relapsing and metastatic disease despite chemotherapy138.

Interactions between tumor cells and neutrophils determines the outcome of tumor growth, progression, and metastasis. Soluble factors and inflammatory mediators such as IL-8 and granulocyte colony-stimulating factor (G-CSF), as well as microparticles released from tumor cells prime and activate neutrophils to form NETs. NETs-associated granule proteins can contribute to tumor metastasis by releasing proteases such as matrix metalloproteases (MMPs) and neutrophil elastase (NE) that allows tumor cells to move out of the primary niche and to migrate to other organs143,151,160.
In several mouse models, NET formation has been demonstrated to be associated with tumor growth and/or metastasis (Fig. 4), e.g., NETs have been reported to enhance the growth of melanoma139, gastric cancer140, and hepatocellular cancer141. Moreover, cancer cells themselves are able to stimulate neutrophils to form NETs that facilitate cancer cell migration and invasion as shown in vitro and in vivo, indicating that they make use of a physiological host defense mechanism process to promote metastasis (Fig. 4)142. Mechanisms by which cancer cells may stimulate neutrophils to form NETs are the production of IL-8 and the release of exosomes requiring additional priming with granulocyte colony stimulating factor (G-CSF) (Fig. 4)142,143. More recently, it was reported that anaplastic thyroid cancer (ATC) cells induce the release of mitochondrial extracellular DNA traps by viable neutrophils. ATC conditioned medium (CM)–primed neutrophils promoted ATC cell proliferation in a NET-dependent manner144. Furthermore, tumor cells have been demonstrated to produce IL-8, attracting myeloid-derived suppressor cells (MDSC) and activating granulocytic MDSC to extrude DNA nets145. In addition, by applying intravital microscopy, a significant increase in the in vivo hepatic adhesion of intrasplenically injected lung or colon cancer cells was observed in the presence of NETs when compared with animals, in which NET formation had been prevented146. Although tumor-infiltrating neutrophils were rare in tissue specimens of epithelial ovarian cancer, both intact neutrophils and NETs were observed in tumors from 4 of 5 patients147. Moreover, in ascites supernatants of patients with advanced tumors, high mtDNA, and NE levels were found that correlated with reduced progression-free survival148.
In order to investigate the association of severe postsurgical infection and adverse oncologic outcome, a murine model of infection using cecal ligation and puncture was applied, demonstrating microvascular NET deposition and trapping of circulating lung carcinoma cells that was associated with increased formation of hepatic metastases following tumor cell injection149. Similarly, surgical stress employing liver ischemia reperfusion resulted in an increase in NET formation with subsequent development and progression of metastatic disease, while pretreatment of mice with topical DNase application or a PAD4 inhibitor abrogated these effects150. Consistent with the observation in mice, increased postoperative NET formation inversely correlated with disease-free survival times in a cohort of patients undergoing attempted curative liver resection for metastatic colorectal cancer150. It should be noted, however, these authors measured serum levels of MPO–DNA complexes and did not directly analyze NET formation. Therefore, it could very well be that metastasis was associated with neutrophil cell death and not with NET formation.
Cancer cells remain dormant for a long time. In a mouse model of lung inflammation induced by either tobacco smoke or lipopolysaccharide, NET formation was shown to be essential for awakening dormant cancer cells. NETs facilitated the contact of associated proteases NE and MMP9 with their substrate laminin (Fig. 4), resulting in the cleavage of laminin and revealing an epitope that activated integrin-mediated signaling and thus proliferation of dormant cancer cells151.
Conclusion
A beneficial role of NETs and EETs is undisputable, as several studies have demonstrated its antimicrobial activities. On the other hand, they might be able to trigger additional immune responses. Based on recently published work, extracellular microbial killing and phagocytosis act in synergy to effectively remove intracellular and extracellular pathogens regardless of their sizes. A simple biomarker that reflects NET and/or EET formation is currently not available. However, extracellular DNA traps can be detected in inflamed tissues using fluorescence/immunofluorescence techniques. The mechanism of extracellular DNA trap formation remains highly controversial that includes questions regarding the requirement of cell death, the source of DNA, as well as intracellular signaling pathways.
References
Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535 (2004).
Fuchs, T. A. et al. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 176, 231–241 (2007).
Parker, H., Dragunow, M., Hampton, M. B., Kettle, A. J. & Winterbourn, C. C. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 92, 841–849 (2012).
Yousefi, S., Mihalache, C., Kozlowski, E., Schmid, I. & Simon, H. U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 16, 1438–1444 (2009).
Yang, H. et al. New insights into neutrophil extracellular traps: mechanisms of formation and role in inflammation. Front. Immunol. 7, 302 (2016).
Stojkov, D. et al. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J. Cell Biol. 216, 4073–4090 (2017).
Amini, P. et al. Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat. Commun. 9, 2958 (2018).
Sofoluwe, A., Bacchetta, M., Badaoui, M., Kwak, B. R. & Chanson, M. ATP amplifies NADPH-dependent and -independent neutrophil extracellular trap formation. Sci. Rep. 9, 16556 (2019).
Yousefi, S. et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 14, 949–953 (2008).
Morshed, M., Yousefi, S., Stockle, C., Simon, H. U. & Simon, D. Thymic stromal lymphopoietin stimulates the formation of eosinophil extracellular traps. Allergy 67, 1127–1137 (2012).
Morshed, M. et al. NADPH oxidase-independent formation of extracellular DNA traps by basophils. J. Immunol. 192, 5314–5323 (2014).
Kenny, E. F. et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife 6, e24437 (2017).
Lood, C. et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 22, 146–153 (2016).
Kolaczkowska, E. et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 6, 6673 (2015).
Maugeri, N. et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 12, 2074–2088 (2014).
Knackstedt, S. L. et al. Neutrophil extracellular traps drive inflammatory pathogenesis in malaria. Sci. Immunol. 4, eaaw0336 (2019).
Yousefi, S. et al. Untangling “NETosis” from NETs. Eur. J. Immunol. 49, 221–227 (2019).
Boeltz, S. et al. To NET or not to NET: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 26, 395–408 (2019).
Yousefi, S. & Simon, H. U. NETosis—does it really represent nature’s “suicide bomber”? Front. Immunol. 7, 328 (2016).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541 (2018).
Ingelsson, B. et al. Lymphocytes eject interferogenic mitochondrial DNA webs in response to CpG and non-CpG oligodeoxynucleotides of class C. Proc. Natl Acad. Sci. USA 115, E478–E487 (2018).
Melgaco, J. G. et al. Changes in cellular proliferation and plasma products are associated with liver failure. World J. Hepatol. 8, 1370–1383 (2016).
Bueno, M. et al. PINK1 attenuates mtDNA release in alveolar epithelial cells and TLR9 mediated profibrotic responses. PLoS ONE 14, e0218003 (2019).
Ip, W. K. E., Hoshi, N., Shouval, D. S., Snapper, S. & Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513–519 (2017).
Savill, J. & Fadok, V. Corpse clearance defines the meaning of cell death. Nature 407, 784–788 (2000).
Simon, D. et al. Eosinophil extracellular DNA traps in skin diseases. J. Allergy Clin. Immunol. 127, 194–199 (2011).
Nakazawa, D., Kumar, S., Desai, J. & Anders, H. J. Neutrophil extracellular traps in tissue pathology. Histol. Histopathol. 32, 203–213 (2017).
Hamaguchi, S. et al. Neutrophil extracellular traps in bronchial aspirates: a quantitative analysis. Eur. Respir. J. 43, 1709–1718 (2014).
de Oliveira, G. P. et al. Glutamine therapy reduces inflammation and extracellular trap release in experimental acute respiratory distress syndrome of pulmonary origin. Nutrients 11, E831 (2019).
Lee, K. H. et al. Quantification of NETs-associated markers by flow cytometry and serum assays in patients with thrombosis and sepsis. Int J. Lab Hematol. 40, 392–399 (2018).
Masuda, S. et al. NETosis markers: quest for specific, objective, and quantitative markers. Clin. Chim. Acta 459, 89–93 (2016).
Gould, T. J., Lysov, Z. & Liaw, P. C. Extracellular DNA and histones: double-edged swords in immunothrombosis. J. Thromb. Haemost. 13(Suppl 1), S82–S91 (2015).
Bunting, H. Interstitial desoxyribonucleic acid following cell death. Yale J. Biol. Med. 22, 521–525 (1950).
Vainio, T., Koskimies, O., Perlmann, P., Perlmann, H. & Klein, G. In vitro cytotoxic effect of lymphoid cells from mice immunized with allogeneic tissue. Nature 204, 453–455 (1964).
Wang, Y. et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 184, 205–213 (2009).
Rohrbach, A. S., Slade, D. J., Thompson, P. R. & Mowen, K. A. Activation of PAD4 in NET formation. Front Immunol. 3, 360 (2012).
Lewis, H. D. et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 11, 189–191 (2015).
Knight, J. S. et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J. Clin. Invest. 123, 2981–2993 (2013).
Knight, J. S. et al. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis. 74, 2199–2206 (2015).
Liu, Y. et al. Myeloid-specific deletion of peptidylarginine deiminase 4 mitigates atherosclerosis. Front. Immunol. 9, 1680 (2018).
Liu, Y. et al. Peptidylarginine deiminases 2 and 4 modulate innate and adaptive immune responses in TLR-7-dependent lupus. JCI Insight 3, 124729 (2018).
Hemmers, S., Teijaro, J. R., Arandjelovic, S. & Mowen, K. A. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS ONE 6, e22043 (2011).
Martinod, K. et al. PAD4-deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood 125, 1948–1956 (2015).
Claushuis, T. A. M. et al. Role of peptidylarginine deiminase 4 in neutrophil extracellular trap formation and host defense during Klebsiella pneumoniae-induced pneumonia-derived sepsis. J. Immunol. 201, 1241–1252 (2018).
Gordon, R. A. et al. Lupus and proliferative nephritis are PAD4 independent in murine models. JCI Insight 2, 92926 (2017).
Gray, R. D. et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax 73, 134–144 (2018).
Dwyer, M. et al. Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J. Innate Immun. 6, 765–779 (2014).
Young, R. L. et al. Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PLoS ONE 6, e23637 (2011).
Beiter, K. et al. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr. Biol. 16, 401–407 (2006).
Wilton, M., Halverson, T. W. R., Charron-Mazenod, L., Parkins, M. D. & Lewenza, S. Secreted phosphatase and deoxyribonuclease are required by Pseudomonas aeruginosa to defend against neutrophil extracellular traps. Infect. Immun. 86, e00403-18 (2018).
Wartha, F. et al. Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol. 9, 1162–1171 (2007).
de Buhr, N. et al. Neutrophil extracellular trap formation in the Streptococcus suis-infected cerebrospinal fluid compartment. Cell Microbiol. 19, e12649 (2017).
Mohanty, T. et al. Neutrophil extracellular traps in the central nervous system hinder bacterial clearance during pneumococcal meningitis. Nat. Commun. 10, 1667 (2019).
Menten-Dedoyart, C. et al. Neutrophil extracellular traps entrap and kill Borrelia burgdorferi sensu stricto spirochetes and are not affected by Ixodes ricinus tick saliva. J. Immunol. 189, 5393–5401 (2012).
Arnold, I. C. et al. Eosinophils suppress Th1 responses and restrict bacterially induced gastrointestinal inflammation. J. Exp. Med. 215, 2055–2072 (2018).
Narasaraju, T. et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 179, 199–210 (2011).
Cortjens, B. et al. Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease. J. Pathol. 238, 401–411 (2016).
Toussaint, M. et al. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat. Med. 23, 681–691 (2017).
Urban, C. F., Reichard, U., Brinkmann, V. & Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 8, 668–676 (2006).
Branzk, N. & Papayannopoulos, V. Molecular mechanisms regulating NETosis in infection and disease. Semin Immunopathol. 35, 513–530 (2013).
Branzk, N. et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 15, 1017–1025 (2014).
Bonne-Annee, S. et al. Extracellular traps are associated with human and mouse neutrophil and macrophage mediated killing of larval Strongyloides stercoralis. Microbes Infect. 16, 502–511 (2014).
Abi Abdallah, D. S. et al. Toxoplasma gondii triggers release of human and mouse neutrophil extracellular traps. Infect. Immun. 80, 768–777 (2012).
Hurrell, B. P. et al. Rapid sequestration of Leishmania mexicana by neutrophils contributes to the development of chronic lesion. PLoS Pathog. 11, e1004929 (2015).
Tamarozzi, F. et al. Wolbachia endosymbionts induce neutrophil extracellular trap formation in human onchocerciasis. Sci. Rep. 6, 35559 (2016).
Wang, H., Li, T., Chen, S., Gu, Y. & Ye, S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of Metformin. Arthritis Rheumatol. 67, 3190–3200 (2015).
Arneth, B. Systemic lupus erythematosus and DNA degradation and elimination defects. Front. Immunol. 10, 1697 (2019).
Smith, C. K. & Kaplan, M. J. The role of neutrophils in the pathogenesis of systemic lupus erythematosus. Curr. Opin. Rheumatol. 27, 448–453 (2015).
Wirestam, L., Arve, S., Linge, P. & Bengtsson, A. A. Neutrophils-important communicators in systemic lupus erythematosus and antiphospholipid syndrome. Front. Immunol. 10, 2734 (2019).
O’Neil, L. J., Kaplan, M. J. & Carmona-Rivera, C. The role of neutrophils and neutrophil extracellular traps in vascular damage in systemic lupus erythematosus. J. Clin. Med. 8, 1325 (2019).
Frangou, E., Vassilopoulos, D., Boletis, J. & Boumpas, D. T. An emerging role of neutrophils and NETosis in chronic inflammation and fibrosis in systemic lupus erythematosus (SLE) and ANCA-associated vasculitides (AAV): implications for the pathogenesis and treatment. Autoimmun. Rev. 18, 751–760 (2019).
Lande, R. et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 3, 73ra19 (2011).
Caielli, S. et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 213, 697–713 (2016).
Garcia-Romo, G. S. et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3, 73ra20 (2011).
van Dam, L. S. et al. Intrinsically distinct role of neutrophil extracellular trap formation in antineutrophil cytoplasmic antibody-associated vasculitis compared to systemic lupus erythematosus. Arthritis Rheumatol. 71, 2047–2058 (2019).
Villanueva, E. et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 187, 538–552 (2011).
Gestermann, N. et al. Netting neutrophils activate autoreactive B cells in lupus. J. Immunol. 200, 3364–3371 (2018).
Hakkim, A. et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl Acad. Sci. USA 107, 9813–9818 (2010).
Barrera-Vargas, A. et al. Differential ubiquitination in NETs regulates macrophage responses in systemic lupus erythematosus. Ann. Rheum. Dis. 77, 944–950 (2018).
Knight, J. S. et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 114, 947–956 (2014).
Konig, M. F. & Andrade, F. A critical reappraisal of neutrophil extracellular traps and NETosis mimics based on differential requirements for protein citrullination. Front. Immunol. 7, 461 (2016).
Kessenbrock, K. et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 15, 623–625 (2009).
Sangaletti, S. et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood 120, 3007–3018 (2012).
Lood, C. & Hughes, G. C. Neutrophil extracellular traps as a potential source of autoantigen in cocaine-associated autoimmunity. Rheumatology 56, 638–643 (2017).
Kraaij, T. et al. Excessive neutrophil extracellular trap formation in ANCA-associated vasculitis is independent of ANCA. Kidney Int. 94, 139–149 (2018).
Papadaki, G. et al. Neutrophil extracellular traps exacerbate Th1-mediated autoimmune responses in rheumatoid arthritis by promoting DC maturation. Eur. J. Immunol. 46, 2542–2554 (2016).
Ribon, M. et al. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J. Autoimmun. 98, 122–131 (2019).
Schauer, C. et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 20, 511–517 (2014).
Lin, A. M. et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immunol. 187, 490–500 (2011).
de Graauw, E. et al. Monocytes enhance neutrophil-induced blister formation in an ex vivo model of bullous pemphigoid. Allergy 73, 1119–1130 (2018).
Simon, D. & Simon, H. U. Therapeutic strategies for eosinophilic dermatoses. Curr. Opin. Pharm. 46, 29–33 (2019).
Giusti, D. et al. NET formation in bullous pemphigoid patients with relapse is modulated by IL-17 and IL-23 interplay. Front. Immunol. 10, 701 (2019).
Manthiram, K., Zhou, Q., Aksentijevich, I. & Kastner, D. L. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat. Immunol. 18, 832–842 (2017).
Apostolidou, E. et al. Neutrophil extracellular traps regulate IL-1beta-mediated inflammation in familial Mediterranean fever. Ann. Rheum. Dis. 75, 269–277 (2016).
Bonnekoh, H. et al. Skin and systemic inflammation in schnitzler’s syndrome are associated with neutrophil extracellular trap formation. Front Immunol. 10, 546 (2019).
Mistry, P. et al. Dysregulated neutrophil responses and neutrophil extracellular trap formation and degradation in PAPA syndrome. Ann. Rheum. Dis. 77, 1825–1833 (2018).
Dworski, R., Simon, H. U., Hoskins, A. & Yousefi, S. Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J. Allergy Clin. Immunol. 127, 1260–1266 (2011).
da Cunha, A. A. et al. Recombinant human deoxyribonuclease therapy improves airway resistance and reduces DNA extracellular traps in a murine acute asthma model. Exp. Lung Res. 42, 66–74 (2016).
Choi, Y. et al. Biological function of eosinophil extracellular traps in patients with severe eosinophilic asthma. Exp. Mol. Med. 50, 104 (2018).
Yousefi, S. et al. Oxidative damage of SP-D abolishes control of eosinophil extracellular DNA trap formation. J. Leukoc. Biol. 104, 205–214 (2018).
Wright, T. K. et al. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology 21, 467–475 (2016).
Pedersen, F. et al. Neutrophil extracellular trap formation and extracellular DNA in sputum of stable COPD patients. Respir. Med. 109, 1360–1362 (2015).
Uddin, M., Watz, H., Malmgren, A. & Pedersen, F. NETopathic inflammation in chronic obstructive pulmonary disease and severe asthma. Front. Immunol. 10, 47 (2019).
Dicker, A. J. et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 141, 117–127 (2018).
Gevaert, E. et al. Extracellular eosinophilic traps in association with Staphylococcus aureus at the site of epithelial barrier defects in patients with severe airway inflammation. J. Allergy Clin. Immunol. 139, 1849–1860 e1846 (2017).
Hwang, C. S. et al. Eosinophil extracellular trap formation is closely associated with disease severity in chronic rhinosinusitis regardless of nasal polyp status. Sci. Rep. 9, 8061 (2019).
Kerstan, A., Simon, H. U., Yousefi, S. & Leverkus, M. Extensive accumulation of eosinophil extracellular traps in bullous delayed-pressure urticaria: a pathophysiological link? Br. J. Dermatol. 166, 1151–1152 (2012).
Simon, D., Radonjic-Hosli, S., Straumann, A., Yousefi, S. & Simon, H. U. Active eosinophilic esophagitis is characterized by epithelial barrier defects and eosinophil extracellular trap formation. Allergy 70, 443–452 (2015).
Jorch, S. K. & Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 23, 279–287 (2017).
Megens, R. T. et al. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 107, 597–598 (2012).
Doring, Y. et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 125, 1673–1683 (2012).
Warnatsch, A., Ioannou, M., Wang, Q. & Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 349, 316–320 (2015).
Franck, G. et al. Roles of PAD4 and NETosis in experimental atherosclerosis and arterial injury: Implications for superficial erosion. Circ. Res. 123, 33–42 (2018).
Westerterp, M. et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation 138, 898–912 (2018).
Etulain, J. et al. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 126, 242–246 (2015).
Borissoff, J. I. et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler. Thromb. Vasc. Biol. 33, 2032–2040 (2013).
Langseth, M. S. et al. Markers of neutrophil extracellular traps are associated with adverse clinical outcome in stable coronary artery disease. Eur. J. Prev. Cardiol. 25, 762–769 (2018).
Jimenez-Alcazar, M. et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 358, 1202–1206 (2017).
Darbousset, R. et al. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood 120, 2133–2143 (2012).
Silvestre-Roig, C. et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 569, 236–240 (2019).
Vajen, T. et al. Blocking CCL5-CXCL4 heteromerization preserves heart function after myocardial infarction by attenuating leukocyte recruitment and NETosis. Sci. Rep. 8, 10647 (2018).
Savchenko, A. S. et al. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood 123, 141–148 (2014).
Pertiwi, K. R. et al. Neutrophil extracellular traps participate in all different types of thrombotic and haemorrhagic complications of coronary atherosclerosis. Thromb. Haemost. 118, 1078–1087 (2018).
Mangold, A. et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 116, 1182–1192 (2015).
Pertiwi, K. R. et al. Extracellular traps derived from macrophages, mast cells, eosinophils and neutrophils are generated in a time-dependent manner during atherothrombosis. J. Pathol. 247, 505–512 (2019).
Kimball, A. S., Obi, A. T., Diaz, J. A. & Henke, P. K. The emerging role of NETs in venous thrombosis and immunothrombosis. Front. Immunol. 7, 236 (2016).
von Bruhl, M. L. et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 209, 819–835 (2012).
Yago, T., Liu, Z., Ahamed, J. & McEver, R. P. Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes deep vein thrombosis in mice. Blood 132, 1426–1437 (2018).
Dyer, M. R. et al. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci. Rep. 8, 2068 (2018).
Bertin, F. R. et al. Natural killer cells induce neutrophil extracellular trap formation in venous thrombosis. J. Thromb. Haemost. 17, 403–414 (2019).
Brill, A. et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 10, 136–144 (2012).
Diaz, J. A. et al. Plasma DNA is elevated in patients with deep vein thrombosis. J. Vasc. Surg. Venous Lymphat. Disord. 1, 341–348 (2013).
Jimenez-Alcazar, M. et al. Circulating extracellular DNA is an independent predictor of mortality in elderly patients with venous thromboembolism. PLoS ONE 13, e0191150 (2018).
Fuchs, T. A. et al. Extracellular DNA traps promote thrombosis. Proc. Natl Acad. Sci. USA 107, 15880–15885 (2010).
Lapponi, M. J. et al. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J. Pharm. Exp. Ther. 345, 430–437 (2013).
Tarantino, E. et al. Role of thromboxane-dependent platelet activation in venous thrombosis: aspirin effects in mouse model. Pharm. Res. 107, 415–425 (2016).
Thomas, M. R. & Storey, R. F. The role of platelets in inflammation. Thromb. Haemost. 114, 449–458 (2015).
Berger-Achituv, S. et al. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front. Immunol. 4, 48 (2013).
Demers, M. et al. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology 5, e1134073 (2016).
Kanamaru, R. et al. Low density neutrophils (LDN) in postoperative abdominal cavity assist the peritoneal recurrence through the production of neutrophil extracellular traps (NETs). Sci. Rep. 8, 632 (2018).
van der Windt, D. J. et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 68, 1347–1360 (2018).
Park, S. Y. et al. Autophagy primes neutrophils for neutrophil extracellular trap formation during sepsis. Am. J. Respir. Crit. Care Med. 196, 577–589 (2017).
Leal, A. C. et al. Tumor-derived exosomes induce the formation of neutrophil extracellular traps: implications for the establishment of cancer-associated thrombosis. Sci. Rep. 7, 6438 (2017).
Cristinziano, L. et al. Anaplastic thyroid cancer cells induce the release of mitochondrial extracellular DNA traps by viable neutrophils. J. Immunol. 204, 1362–1372 (2020).
Alfaro, C. et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin. Cancer Res. 22, 3924–3936 (2016).
Rayes, R. F. et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis promoting effects. JCI Insight 5, 128006 (2019).
Singel, K. L. et al. Mature neutrophils suppress T cell immunity in ovarian cancer microenvironment. JCI Insight 4 (2019).
Singel, K. L. et al. Mitochondrial DNA in the tumour microenvironment activates neutrophils and is associated with worse outcomes in patients with advanced epithelial ovarian cancer. Br. J. Cancer 120, 207–217 (2019).
Cools-Lartigue, J. et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Invest. 123, 3446–3458 (2013).
Tohme, S. et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 76, 1367–1380 (2016).
Albrengues, J. et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 361, eaao4227 (2018).
Amini, P. et al. NET formation can occur independently of RIPK3 and MLKL signaling. Eur. J. Immunol. 46, 178–184 (2016).
Wang, X., He, Z., Liu, H., Yousefi, S. & Simon, H. U. Neutrophil necroptosis is triggered by ligation of adhesion molecules following GM-CSF priming. J. Immunol. 197, 4090–4100 (2016).
Wang, X., Yousefi, S. & Simon, H. U. Necroptosis and neutrophil-associated disorders. Cell Death Dis. 9, 111 (2018).
Radonjic-Hoesli, S. et al. Adhesion-induced eosinophil cytolysis requires the receptor-interacting protein kinase 3 (RIPK3)-mixed lineage kinase-like (MLKL) signaling pathway, which is counterregulated by autophagy. J. Allergy Clin. Immunol. 140, 1632–1642 (2017).
Pilsczek, F. H. et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 185, 7413–7425 (2010).
Aulik, N. A., Hellenbrand, K. M., Klos, H. & Czuprynski, C. J. Mannheimia haemolytica and its leukotoxin cause neutrophil extracellular trap formation by bovine neutrophils. Infect. Immun. 78, 4454–4466 (2010).
Malachowa, N., Kobayashi, S. D., Freedman, B., Dorward, D. W. & DeLeo, F. R. Staphylococcus aureus leukotoxin GH promotes formation of neutrophil extracellular traps. J. Immunol. 191, 6022–6029 (2013).
Malachowa, N., Kobayashi, S. D., Quinn, M. T. & DeLeo, F. R. NET confusion. Front Immunol. 7, 259 (2016).
Park, J. et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 8, 361ra138 (2016).
Acknowledgements
The authors apologize to investigators whose contributions may not have been cited because of space limitations. The original work of the authors presenting here was supported by the Swiss National Science Foundation (310030_184816 to H.U.S. and 31003A_173215 to S.Y.) and the European Union Horizon 2020 Research and Innovation Program (Marie Sklodowska-Curie grant No. 642295; MEL-PLEX).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Piacentini
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yousefi, S., Simon, D., Stojkov, D. et al. In vivo evidence for extracellular DNA trap formation. Cell Death Dis 11, 300 (2020). https://doi.org/10.1038/s41419-020-2497-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-2497-x
This article is cited by
-
NLRP3 exacerbates EAE severity through ROS-dependent NET formation in the mouse brain
Cell Communication and Signaling (2024)
-
Eosinophil extracellular traps in asthma: implications for pathogenesis and therapy
Respiratory Research (2023)
-
Moonlighting chromatin: when DNA escapes nuclear control
Cell Death & Differentiation (2023)
-
Mitochondrial nucleic acids in innate immunity and beyond
Experimental & Molecular Medicine (2023)
-
Macrophages in immunoregulation and therapeutics
Signal Transduction and Targeted Therapy (2023)
