
Abstract
Here, we show for the first time that developmental cell death plays a critical role in the morphogenesis of multicellular proprioceptors in Drosophila. The most prominent multicellular proprioceptive organ in the fly larva, the pentascolopidial (LCh5) organ, consists of a cluster of five stretch-responsive sensory organs that are anchored to the cuticle via specialized attachment cells. Stable attachment of the organ to the cuticle is critical for its ability to perceive mechanical stimuli arising from muscle contractions and the resulting displacement of its attachment sites. We now show that five attachment cells are born within the LCh5 lineage, but three of them are rapidly eliminated, normally, by apoptosis. Strong genetic evidence attests to the existence of an autophagic gene-dependent safeguard mechanism that guarantees elimination of the unwanted cells upon perturbation of the apoptotic pathway prior to caspase liberation. The removal of the three superfluous cells guarantees the right ratio between the number of sensory organs and the number of attachment cells that anchor them to the cuticle. This accurate matching seems imperative for the attachment of cell growth and functionality and is thus vital for normal morphogenesis and functionality of the sensory organ.
Similar content being viewed by others


Introduction
The larval chordotonal organs (ChO) of Drosophila are sub-epidermal stretch receptors that function as proprioceptors [1]. The most prominent larval ChO is the lateral pentascolopidial (LCh5) organ. It contains five sensory neurons, each ensheathed by a scolopale cell that is stretched between two accessory cells termed the cap and ligament cells (Fig. 1a). The five cap cells and the five ligament cells anchor to the cuticle via two cap-attachment (CA) and a single ligament-attachment (LA) cell, respectively (Fig. 1a). During larval locomotion, muscle contractions induce cuticle deformations that lead to relative displacement of the CA and LA cells. The resulting mechanical strain propagates to the neuron where it is transduced into electrical neuronal activity via stretch-responsive ion channels [2, 3].

The lateral pentascolopidial (LCh5) organ and its lineage. a Schematic illustration of the embryonic LCh5 organ (on the left) and two abdominal segments of a stage 16 embryo stained with anti-αTub85E (green) and anti-Sr (magenta). Anti-αTub85E labels the two cap-attachment (CA), cap (C), ligament (L), and the single ligament-attachment (LA) cells. Anti-Sr labels the nuclei of the ligament, CA and LA cells as well as tendon cells (all the cells that are not circled). The scolopale cells (Sc) and the neurons (N) are not labeled and are represented in the scheme by empty ellipses. The cap cell of the lateral single ChO (LCh1) is also indicated (arrowhead). The scale bar denotes 20 μm. b Schematic representation of the LCh5 lineage. A single chordotonal organ precursor cell (COP) divides asymmetrically to generate two secondary precursors (pIIa and pIIb). pIIa divides asymmetrically to generate the cap (c) and CA cells. pIIb divides asymmetrically to generate the ligament (L) cell and a tertiary precursor (pIIIb) that then divides to generate the neuron (N) and scolopale (Sc) cell. c Schematic representation of the expected versus the observed number of CA cells in the mature LCh5 organ
Each of the five units (scolopidia) that together constitute the LCh5 organ originates from a single sensory organ precursor [4], which goes through four rounds of asymmetric cell divisions to give rise to a CA cell, a cap cell, a scolopale cell, a neuron, and a ligament cell [5] (Fig. 1b). The LCh5 organ is thus expected to contain five cells of each of these cell types. However, while this cluster contains quintets of neurons, scolopale cells, cap cells, and ligament cells, it contains only two CA cells (Fig. 1a–c). The reason for this discrepancy in cell numbers remained elusive until now. Two explanations could account for this discrepancy: either the LCh5 organ consists of two subtypes of lineages, only two of which form a CA cell. Alternatively, the five lineages are similar, and they all form a CA cell, but the number of CA cells is later adjusted.
Developmentally regulated elimination of superfluous cells is most commonly achieved by apoptosis [6, 7], the hallmark of which is the activation of caspases. Almost all Drosophila cells express caspases in their inactive zymogen form [8, 9], but spontaneous caspase activation is prevented by the simultaneous expression of inhibitor of apoptosis proteins (IAP), such as Diap-1 [10, 11]. Cells that are destined to die commence the transcription of the proapoptotic genes, reaper (rpr), grim and/or head involution defective (hid), which antagonize the protective function of Diap-1 by binding to it and liberating the bound caspases [10, 11]. The main apoptotic initiator caspase, Dronc, is then recruited to, and subsequently activated by, the death-associated APAF1-related killer (dark) apoptosome. Activated Dronc induces a proteolytic cascade by processing the main apoptotic effector caspases, DrIce and Dcp-1, that in turn process multiple substrates within the doomed cell leading to its destruction [12, 13].
Autophagy, primarily used as an intracellular lysosome-mediated degradation pathway, can be used as an alternative cell death mechanism that does not utilize components of the apoptotic machinery [14]. Instead, multiple autophagy-related genes (Atg genes) bring about the formation of double-membrane vesicles (autophagosomes) that deliver cytosolic components to the lysosome via fusion (reviewed in ref. [15]). The autophagosomal cargo is then digested by lysosomal hydrolases [16]. In Drosophila, certain developmental cell death events were shown to utilize both the apoptotic and the autophagic machineries [17,18,19,20,21]. The mode of interaction between the two cell-death pathways depends on the cellular context. Apoptosis and autophagy may work in parallel, or mutually inhibit each other [19, 20, 22]. Alternatively, autophagy and caspases can function in a common regulatory hierarchy in which either autophagy regulates the activity of caspases, or caspases regulate autophagy [17, 18, 20, 21, 23].
In this study, we investigated how and why is the number of CA cells adjusted during LCh5 development. We demonstrate that, initially, five CA cells are formed within the LCh5 cluster, but soon after their formation three of them are eliminated, normally, by apoptosis. Blocking apoptosis at the effector-caspase level effectively prevented the death of the unwanted cells. However, interfering with apoptosis at the level of IAP antagonists, allowed the removal of the doomed cells by an alternative autophagy gene-dependent cell-death mechanism. Removal of the superfluous CA cells in embryos lacking the IAP antagonists still required a functional dark apoptosome, as well as the presence of maternal products of the IAP antagonists’ genes. Finally, we show that accurate elimination of the superfluous CA cells is critical for the construction of a functional sensory organ.
Materials and methods
Fly strains
The following fly strains were used in this work: white1118 was used as a control strain. 5’F:2.6 ato-lacZ [24], FRT2A H99/TM6Z, darkP46/CyO, darkN28/CyO [25], UAS-P35 (#5072, Bloomington Drosophila Stock Center), UAS-Ep-Diap1/TM6Z, H99/TM6Z [26], Atg7d14/CyO, Atg7d77/CyO [27], Atg1∆3D/TM6 [28], Atg13∆81/TM6 [29], vps34∆m22/ CyO [30], UAS-Atg1KQ13A [31]. FRT79D-F ovoD1/TM3,Sb (kindly provided by K. McCall), hs-FLP122;Gl/TM3,Sb, UAS-LacZ (#1776, Bloomington Drosophila Stock Center), en-gal4/CyO [32], deiChO-GFP, deiattachment-RFP,en-gal4 [33]. The following strains were generated using standard genetic crosses: darkP46/CyO, wg-lacZ; H99/TM6B, abdA-lacZ, darkN28/CyO, wg-lacZ; H99/TM6B, AbdA-lacZ, Atg7d14/CyO, wg-lacZ; H99/TM6, abdA-lacZ, Atg7d77/CyO, wg-lacZ; H99/ TM6B, AbdA-lacZ, vps34∆m22/CyO, wg-lacZ; H99/TM6B, AbdA-lacZ. The Atg1∆3D, H99/TM6B, abdA-lacZ and H99, Atg13Δ81/TM6B, abdA-lacZ strains were generated by meiotic recombination. For the generation of embryos devoid of a maternal contribution of the H99 genes, FRT79D-F H99/TM6B virgin females were crossed to hs-FLP122/Y; FRT79D-F ovoD1/TM3, Sb males and were allowed to lay eggs for one day before being moved into a fresh vial. A heat-shock (1 h at 37 °C) was applied after the first wandering larva had emerged. In the next generation, non-TM6 non-TM3 virgin females were collected and crossed to FRT79D-F H99/TM6Z males. The progeny of this cross was collected and fixed for immunostaining. Some of the vials were not subjected to heat-shock and served as a control for female sterility.
Phenotypic analysis in 2nd instar larvae
deiChO–GFP, deiattachment–RFP, en-gal4 virgin females were crossed to either UAS-LacZ or UAS-p35, or UAS-Atg1KQ13A, or UAS-p35, UAS-Atg1KQ13A males and were allowed to lay eggs for 24 h at 29°. The parents were discarded and larvae were collected 24 h later. Larvae were fixed in 4% formaldehyde (in PBS) at 4 °C overnight without dissection. Following fixation, the larvae were washed twice in PBT and once in PBS and stored in mounting medium (DAKO, DAKO Cytomation, Denmark) at 4 °C until their mounting and examination (within a few days).
Immunohistochemistry
The primary antibodies used in this study and their working dilutions were: guinea pig anti-SrA/B [34] (1:300), rabbit anti-αTub85E [35] (1:20), mouse anti-αTub85E [36] (1:200), rabbit anti-cleaved Dcp-1 (Asp 216) and rabbit anti-cleaved Caspase-3, (Asp175) (1:50; Cell signaling Technology), mouse anti-β-galactosidase (Promega) (1:1000). The secondary antibodies were Cy3-, Cy2-, or Alexa Fluor-647-conjugated anti-mouse/rabbit/guinea pig (Jackson Laboratories, Bar-Harbor, ME, USA). Embryos were collected and immunestained using standard techniques. For each tested genotype, anti-β-galactosidase staining allowed the identification of homozygous embryos that do not carry the lac-Z-labeled balancer chromosome/s.
TUNEL assay
TUNEL assays were performed using the In Situ Cell Death Detection Kit, TMR red (Roche) following the immunostaining procedure. Stained embryos were fixed for 20 min at RT, washed five times in PBT and then incubated in TUNEL solution at 37 °C for 2 h. The embryos were then washed twice in PBT and once in PBS and stored in mounting medium until their mounting and examination. Stained embryos were examined using confocal microscope LSM-5 Exciter.
Results
Three out of five CA cells are removed by apoptosis
To test whether cell death occurs within the ChO lineage and whether the three CA cells are eliminated by apoptosis, we examined embryos homozygous for the H99 chromosomal deficiency. This deficiency removes the proapoptotic genes rpr, hid, and grim, thus blocking almost all apoptotic events that occur normally during development [26]. As shown in Fig. 2, the LCh5 of H99 embryos contained on average 2.14 CA cells (2.14 ± 0.87, n = 83; Fig. 2a, g). This slight elevation in the average number of CA cells does not support the idea that apoptosis accounts for the loss of three CA cells. Surprisingly, however, when we inhibited apoptosis by expressing the caspase inhibitor P35 [37] within the LCh5 lineage, the phenotypic outcome was very different and the number of CA cells increased significantly to 4.68 ± 0.60 cells per LCh5 organ (n = 31; Fig. 2b, g). This latter observation implies that, initially, five CA cells are born within the LCh5 cluster and suggests that, later, three of them are removed by a caspase-dependent cell death mechanism. It also points to the fact that blocking early steps in the apoptotic pathway (H99 embryos) yields a different LCh5 phenotype than blocking late stages of the apoptotic pathway (p35-expressing embryos).

Three out of five CA cells are removed by apoptosis. a-b, e-f Lateral view of stage 16 embryos stained with anti-αTub85E (magenta) and anti-Sr (green). The anti-β-gal staining used for distinguishing between homozygous and heterozygous embryos in a and f and for identifying the UAS-diap1 expressing embryos in e is not shown (the UAS-diap-1 construct and the mutant alleles were balanced over TM6B, lacZ). All images are oriented such that anterior is to the left and dorsal is up. The scale bars represent 10 µm. The anti-Sr staining is shown separately in a’, b’, e’, f ’. a Two CA cells are evident in 85.6% of the LCh5 organs of H99 homozygous embryos (circled). In 13.2% of the segments three cells and in 1.2% of the segments four CA cells were evident (n = 83). b A P35-expressing embryo displaying the typical increase in the number of CA cells (circled). c A stage 12 ato-lacZ embryo labeled with anti-βGal (blue), anti-Sr (green) and TUNEL (red). The TUNEL staining is shown separately in c’. d Three segments of a stage 12 ato-lacZ embryo stained with anti-Sr (green), anti-cleaved Dcp-1 (blue) and TUNEL (red). The Cleaved Dcp-1 staining is shown separately in d’ and TUNEL staining in d”. Activated Dcp-1 and fragmented DNA (arrows and arrowheads in c-d) are observed in the immediate surroundings of the surviving CA cells (white circles in c-d). The arrowheads in d point to cells in which the TUNEL and Dcp-1 staining overlap (approximately one third of the cells, n = 60). e An embryo over-expressing the caspase inhibitor diap-1 within the en domain. An increased number of CA cells is evident (circled). f A darkP46/N28 mutant embryo displaying a gain of CA cells (circles). g Quantification of the CA cell-number in different genetic backgrounds represented in a box-and-whiskers plot. The number of examined segments (n) is indicated for each genotype
To further test whether the doomed CA cells die by apoptosis, we looked for caspase activation and DNA fragmentation within the developing LCh5 cluster. For that purpose, we stained ato-lacZ embryos, in which the LCh5 lineage is marked with βGal, with antibodies recognizing the activated form of the effector caspase Dcp-1 and performed TUNEL analysis to detect DNA fragmentation. The two surviving CA cells were recognized by anti-Stripe immunostaining [38]. These experiments revealed TUNEL staining, and/or staining for activated caspase, in cells surrounding the two surviving CA cells in all segments of stage 12/13 embryos (Fig. 2c, d). This observation supports our conclusion that the three CA cells are eliminated by apoptosis soon after their initial formation.
Experiments in which the activity of negative and positive regulators of apoptosis was altered provided further support for this conclusion. Overexpression within the LCh5 lineage of the caspase inhibitor diap-1 led to a small but significant increase in the number of surviving CA cells (2.59 ± 0.69, n = 37; Fig. 2e, g). Similarly, an increase in the number of CA cells was observed in dark mutant embryos (2.81 ± 0.89, n = 83; Fig. 2f, g), in which caspase activation is reduced due to diminished apoptosome activity [25]. These data corroborate our conclusion that, normally, five CA cells form within the LCh5 cluster, but three of them are eliminated by apoptosis soon after their formation.
CA cell removal in H99 embryos depends on maternal contribution of the H99 genes and zygotic activity of the apoptosome
The intriguing difference between the LCh5 phenotype of H99-deficient embryos and the phenotype of p35-expressing embryos could be explained in several, not mutually exclusive, ways. One possible explanation is that maternally contributed products of the rpr, hid, and grim (RHG) genes are sufficient for CA cell removal in H99 zygotic mutants. Another option is that, in the absence of the H99 genes, a different regulatory pathway is activated leading eventually to caspase activation and CA cell elimination. It is also possible that upon blocking the apoptotic pathway at its initial stages, a different cell-death mechanism takes over and eliminates the unwanted cells.
To test whether maternally deposited transcripts of the H99 genes were involved, we generated females bearing H99-deficient germline clones and crossed them to H99 heterozygous males. In this setting, all embryos were devoid of maternal contribution, and half of the embryos were additionally devoid of the zygotic contribution, of the H99 genes. The LCh5 organs of embryos that lacked maternally contributed H99 genes, but carried a zygotic copy, manifested a normal LCh5 phenotype of two CA cells (2.04 ± 0.42, n = 57; Fig. 3a, d). In contrast, the absence of both maternal and zygotic contribution of the H99 genes resulted in a significant increase in the number of surviving CA cells (3.72 ± 0.72, n = 72; Fig. 3b, d). These results support the idea that maternal transcripts of one or more of these proapoptotic genes play a role in the removal of CA cells in zygotic H99 mutant embryos.

In H99 zygotic background, the removal of superfluous CA cells depends on the maternal contribution of the H99 genes and the zygotic activity of the apoptosome. a-c Stage 16 embryos stained with anti-αTub85E (magenta) and anti-Sr (green). The anti-Sr staining is shown separately in a’-c’. The anti-β-gal staining used for distinguishing between homozygous and heterozygous embryos in a-c is not shown. The scale bar represents 10 µm. a An embryo that lacks maternal contribution, but carries a zygotic copy of the H99 genes, exhibits a normal number of CA cells (circled). b An embryo that lacks both the maternal and zygotic contribution of the H99 genes manifests an abnormally high number of CA cells (circled). c A darkP46/N28; H99 zygotic mutant embryo shows a nearly complete prevention of CA-cell elimination. d Quantification of the CA-cell number in different genetic backgrounds represented in a box-and-whiskers plot. The number of examined segments (n) is indicated for each genotype. e-h Stage 12–13 heterozygous H99/TM6, abdA-lacZ embryos e, g or homozygous H99 embryos f, h stained with anti-Sr (green), anti-βGal (blue), and either TUNEL e, f or anti-cleaved caspase-3 g, h. Homozygous embryos were identified by the lack of anti-βGal staining. Although the three CA cells are eliminated in the zygotic H99 mutant embryos, and only two CA cells are evident, the hallmarks of apoptotic cells are not evident in these embryos f, h
To determine whether other components of the apoptotic pathway are required for the removal of CA cells in zygotic H99 mutant embryos, we tested the effects of perturbing the function of the apoptosome in this background. darkP46/darkN28; H99 (maternal+ /zygotic−) embryos displayed a significant increase in the number of surviving CA cells (4.18 ± 0.62, n = 44; Fig. 3c, d), implicating the dark apoptosome in the elimination of the superfluous CA cells in zygotic H99 mutant background.
The involvement of both the maternal products of the H99 genes and the zygotic dark apoptosome in CA cell elimination in zygotic H99 mutant embryos may suggest that CA cell removal still follows the typical apoptotic pathway in this background. However, in agreement with previous publications [26], neither activated caspases nor DNA fragmentation was evident in stage 12/13 zygotic H99 embryos (Fig. 3e–h), indicating that the CA cells are not removed by classic apoptosis in this background. The complete absence of TUNEL staining suggests that although some components of the apoptotic machinery (maternal H99 products and the apoptosome) are indispensable for the elimination of CA cells in zygotic H99 embryos, other cell-death pathway/s, distinct from apoptosis, join to the aforementioned components of the apoptotic machinery to guarantee successful removal of the unwanted CA cells.
The removal of unwanted CA cells in H99 embryos depends on autophagy genes
To test for a possible involvement of autophagy in the elimination of CA cells when apoptosis is blocked, we examined the LCh5 organs of different autophagy mutants. On their own, all of the tested mutants, each lacking the activity of one autophagy gene (Atg7, Atg1, Atg13, or vps34), displayed a normal number of CA cells (Atg7d77/d14– 2.0 ± 0.0, n = 27; vps34∆m22– 2.04 ± 0.61, n = 25; Atg1∆3D− 2.06 ± 0.23, n = 72; Atg13∆81– 2.03 ± 0.18, n = 60), indicating that under normal circumstances, autophagy is dispensable for the elimination of CA cells. However, simultaneous inhibition of both apoptosis and autophagy (Atg7d77/Atg7d14; H99, vps34∆m22; H99, H99, Atg1∆3D and H99, Atg13Δ81) prevented death of the CA cells and led to a substantial increase in the number of surviving cells (4.32 ± 0.61, n = 28; 3.87 ± 0.78, n = 30; 3.17 ± 0.87, n = 82, and 3.21 ± 0.45, n = 58, respectively; Fig. 4).

Autophagy genes are involved in CA cell elimination in H99 mutant embryos. a-h Stage 16 embryos stained with anti-αTub85E (magenta) and anti-Sr (green). The anti-Sr staining is shown separately in a’-h’. The anti-β-gal staining used for distinguishing between homozygous and heterozygous embryos is not shown. The scale bar represents 10 µm. Embryos homozygous for mutations in the autophagy-related genes Atg7 (a), Atg13 (c), Atg1 (e), and vps34 (h) manifest normal LCh5 phenotype with two CA cells (circled). Embryos carrying any of these autophagy-related mutations in an H99 background (b, d, f, h) display a significant increase in the number of surviving CA. (i) A box-and-whiskers plot showing the distribution of the CA cell number data in the different genotypes
Taken together, these findings support the idea that in zygotic H99 mutants, maternally contributed H99 gene products are essential, but insufficient, for the removal of three CA cells. Under these circumstances, the balance is tipped from apoptosis toward an autophagy-dependent removal of the unwanted cells that have already been primed for elimination by the maternal copies of the H99 genes. If apoptosis and autophagy are concomitantly blocked, CA cell death is prevented.
CA cell removal is critical for proper LCh5 morphogenesis and for maintaining organ integrity
To establish whether the elimination of three CA cells is vital for the development of functional LCh5 organs, we examined the impact of increasing the number of CA cells on LCh5 morphology. We expressed either lacZ, p35, a dominant-negative form of Atg1 (Atg1KQ13A referred to as Atg1DN), or p35 plus Atg1DN within the LCh5 organs and examined the phenotypic effects in 2nd instar larvae. The LCh5 organs were visualized in intact larvae by using ChO-specific fluorescent reporters [39]. In control, lacZ-expressing, larvae, two large CA cells were seen, each anchoring two-to-three cap cells (Fig. 5a). In agreement with our notion that autophagy is not normally involved in the process of CA cell removal, the LCh5 organs of Atg1DN-expressing larvae displayed a normal number of CA cells (Fig. 5b). In contrast, the LCh5 cluster of P35-, or P35 plus Atg1DN-expressing larvae presented an elevated number of CA cells (Fig. 5c).

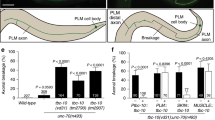
The elimination of three CA cells is critical for normal LCh5 morphogenesis and maintenance of organ integrity. a–d LCh5 organs of 2nd instar larvae harboring the deiChO-GFP, deiattachment-RFP marker chromosome, which drives GFP expression in the cap cells and RFP expression in the CA cells. Only the cap cells (labeled in green) and the CA cells (labeled in red and marked with asterisks) are shown. Two organs are shown for each genotype. a, a’ LCh5 organs of en-gal4 > lacZ larvae. b-b’ LCh5 organs of en-gal4 > Atg1DN larvae. c-c’ LCh5 organs of en-gal4 > P35 larvae. d-d’ LCh5 organs of en-gal4 > P35, Atg1DN larvae. The LCh5 organs of larvae expressing either p35 alone, or both p35 and Atg1DN, contain more than two CA cells (asterisks), the superfluous CA cells are smaller than normal, and they often detach. e A box-and-whiskers plot showing the distribution of the CA cell number data in the different genotypes in two time-points: the end of embryogenesis (green boxes, E) and the 2nd instar larval stage (red boxes, L2). The number of organs examined is shown below each box. The scale bars represent 20 µm in all panels, with the exception of (b) where it represents 50 µm
Altering the ratio between the numbers of cap and CA cells had detrimental effects on organ integrity. The supernumerary CA cells remained very small in comparison with CA cells of normal larvae (Fig. 5). Even though each of the small CA cells anchored only a single cap cell, it often failed to withstand the mechanical forces generated during larval growth and locomotion, leading to detachment from the cuticle and collapse of the attached cap cell (Fig. 5c, d). The abnormally small size of the supernumerary CA cells that anchor only a single organ could suggest that the size of a CA cell is adjusted to the number of ChOs it anchors and that this size adjustment is critical for the ability of the CA cell to remain properly fastened to the cuticle and maintain its ability to anchor the sensory organs.
Interestingly, the average number of superfluous CA cells observed in P35- or P35 plus Atg1DN-expressing embryos was higher than the average number of such cells observed in second instar larvae of the same genotypes (4.39 ± 0.78, n = 62; vs. 2.44 ± 0.50, n = 59; and 4.44 ± 0.67, n = 62; vs. 2.64 ± 0.55 for p35 and p35 plus Atg1DN, respectively). This observation suggests that even when CA cell removal is prevented during embryogenesis, the survival of the undead CA cells is not guaranteed and some of them are eliminated later during larval life.
Discussion
The LCh5 organs are stretched between two attachment sites in the larval body wall and are fastened to the cuticle via specialized epidermal attachment cells (CA and LA). The ability of these organs to remain stably anchored to the cuticle is critical for their function as stretch receptors. Thus, an accurate matching between the number of organs and the position, number and size of their attachment cells is vital. The current work contributes significantly to our understanding of how such coordination is achieved.
We have previously shown how the single LA cell is recruited by the ligament cells via an epidermal growth factor receptor-dependent mechanism [38]. The current work sheds light on the mechanism that guarantees the matching between the number and size of CA cells and the number of cap cells they anchor. Our data show that each of the five chordotonal lineages that collectively constitute the LCh5 organ, generate a CA cell, but three of these attachment cells are removed even before they become identifiable by CA cell-specific markers. The expression of P35 within the ChO lineages enables the survival of the three doomed cells, suggesting that, normally, they are eliminated by apoptosis. This notion is further supported by the presence of activated caspase and fragmented DNA in cells adjacent to the two surviving cells, and by the effect of positive and negative regulators of apoptosis on the elimination process. Quite surprisingly, if the apoptotic machinery is blocked in its early stages, prior to caspase liberation, the superfluous CA cells are still successfully removed. In that case, cell removal depends on the combined action of maternally deposited products of the RHG genes, a functional apoptosome and functional autophagy-related genes. Nevertheless, in spite of the requirement for maternal RHG genes and functional apoptosome, the cells are eliminated via a non-apoptotic pathway as suggested by the lack of any of the hallmarks of apoptosis.
How are the superfluous CA cells removed when apoptosis is blocked? We present strong genetic evidence for the involvement of autophagy genes in this process. However, since the three superfluous CA cells, and even the two surviving CA cells, are unidentifiable in early embryonic stages, it is impossible to characterize their phenotype at the ultrastructural level by electron microscopy. In addition, the commonly used autophagy reporters [21, 40,41,42] caused dominant-negative effects in this developmental context in the extremely sensitive background of H99 embryos. For that reason, even though we could show that the function of autophagy genes is necessary for ensuring CA cell removal when apoptosis is compromised, we were not able to determine unambiguously whether the autophagic machinery actively kills the unwanted CA cells via autolysosomal degradation or any other mechanism of autophagy-dependent cell death [43]. Alternatively, the formation of autophagosomes could allow the assembly and/or activation of alternative cell death pathway components. For instance, recent studies revealed the involvement of autophagosome membranes in switching from apoptotic to necroptotic cell death under certain experimental conditions [44, 45]. Further studies are required for revealing the actual execution machinery responsible for the removal of CA cells in the H99 background.
What is the relationship between autophagy and caspase activation in this scenario? Since no caspase activation is evident in H99 zygotic mutants, we must assume that the autophagy-related genes are not regulated by activated effector caspases. However, we cannot rule out the possibility that other proteins that affect the apoptotic pathway, e.g., maternal RHG proteins, zygotic Dark, or one of the initiator caspases, regulate (most likely indirectly) the autophagy genes in this background. A somewhat similar switch from apoptosis to autophagy-dependent cell death has been reported in etoposide-treated BAX−/−BAK−/− double-mutant mouse embryonic fibroblasts (MEF) that were resistant to apoptosis [46]. Similar to our results, the switch to autophagy did not occur in either Apaf1- or caspase-9-deficient MEFs as well as in wild-type MEFs treated with a caspase inhibitor that inhibited more advanced stages of apoptosis than BAX/BAK. Altogether, our data points to a stage-specific switch from apoptosis to autophagy gene-dependent cell death. The accurate molecular switching point is yet to be identified. However, based the available data we can already conclude that switching toward autophagy gene-dependent cell death occurs upstream to effector-caspase activation and probably upstream to initiator caspase release and activation.
Another question that remains open is the ultimate fate of the superfluous CA cells that survive in en > P35 + /− Atg1DN-expressing embryos. Our data show that these undead CA cells do not always survive throughout development and many of them are eliminated during larval life. One possible, simple, explanation could be a decline in the expression levels of the P35 and Atg1DN transgenes induced by the en-gal4 driver. Based on modENCODE temporal expression data, en expression declines rapidly toward the end of embryogenesis and reaches its lowest levels in the first instar larva ([47] http://flybase.org/reports/FBgn0000577). A reduce level of P35 + /− Atg1DN expression in the larva may not be sufficient for effective protection from cell death, tipping the balance toward a cell-death permissive cellular environment in which only some of the cells survive. Alternatively, global changes that make the 1st and 2nd instar larvae less susceptible than the embryo to reaper-induced apoptosis [48] may divert the undead, but primed-to-die cells, to a different cell death pathway that is not blocked by the p35 inhibitor and is not dependent on autophagosome formation.
The elimination of three CA cells is critical for normal morphogenesis and function of LCh5 organs. Unlike the normal situation in which two large CA cells anchor five cap cells, when cell death was prevented, the supernumerary CA cells failed to increase their size and to remain fastened to the cuticle. The correlation found between the number of cap cells and the size of the CA cell to which they are anchored, suggests that the process of CA cell growth is regulated by a cross talk between the cap and CA cells. Such a cross talk could be based on chemical and/or mechanical signals emanating from the cap cells. The idea is that a CA cell that anchors a larger number of cap cells is exposed to a stronger signal and as a result grows more than a CA cell that anchors a smaller number of cap cells. The ChO provides an excellent model system for studying the molecular mechanism that underlie the ability of the CA cells to sense the number of cap cells anchored to them and how this sensing is translated into proportional cell growth. Such a mechanism likely represents a common strategy that could, for example, dictate the growth of tendon cells to match the size and number of muscle fibers they anchor.
References
Caldwell JC, Miller MM, Wing S, Soll DR, Eberl DF. Dynamic analysis of larval locomotion in Drosophila chordotonal organ mutants. Proc Natl Acad Sci USA. 2003;100:16053–8.
Gong Z, Son W, Chung YD, Kim J, Shin DW, McClung CA, et al. Two interdependent TRPV channel subunits, inactive and Nanchung, mediate hearing in Drosophila. J Neurosci. 2004;24:9059–66.
Kim J, Chung YD, Park D-Y, Choi S, Shin DW, Soh H, et al. A TRPV family ion channel required for hearing in Drosophila. Nature. 2003;424:81–4.
Jarman AP, Grau Y, Jan LY, Jan YN. Atonal is a proneural gene that directs chordotonal organ formation in the Drosophila peripheral nervous system. Cell. 1993;73:1307–21.
Brewster R, Bodmer R. Origin and specification of type II sensory neurons in Drosophila. Development. 1995;121:2923–36.
Hidalgo A, Ffrench-Constant C. The control of cell number during central nervous system development in flies and mice. Mech Dev. 2003;120:1311–25.
Suzanne M, Steller H. Shaping organisms with apoptosis. Cell Death Differ. 2013;20:669–75.
Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14:32–43.
Kumar S, Doumanis J. The fly caspases. Cell Death Differ. 2000;7:1039–44.
Zachariou A, Tenev T, Goyal L, Agapite J, Steller H, Meier P. IAP-antagonists exhibit non-redundant modes of action through differential DIAP1 binding. EMBO J. 2003;22:6642–52.
Chai J, Yan N, Huh JR, Wu J-W, Li W, Hay BA, et al. Molecular mechanism of Reaper-Grim-Hid-mediated suppression of DIAP1-dependent Dronc ubiquitination. Nat Struct Biol. 2003; 10 : 892–8.
Dorstyn L, Kumar S. A biochemical analysis of the activation of the Drosophila caspase DRONC. Cell Death Differ. 2008;15:461–70.
Pang Y, Bai X, Yan C, Hao Q, Chen Z, Wang J, et al. Structure of the apoptosome: mechanistic insights into activation of an initiator caspase from Drosophila. Genes Dev. 2015;29:277–87.
Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy. 2011;7:457–65.
Eskelinen E-L, Reggiori F, Baba M, Kovács AL, Seglen PO. Seeing is believing: the impact of electron microscopy on autophagy research. Autophagy. 2011;7:935–56.
Mulakkal NC, Nagy P, Takats S, Tusco R, Juhász G, Nezis IP. Autophagy in drosophila: From historical studies to current knowledge. Biomed Res Int. 2014; 2014. https://doi.org/10.1155/2014/273473.
Akdemir F, Farkas R, Chen P, Juhasz G, Medved’ová L, Sass M, et al. Autophagy occurs upstream or parallel to the apoptosome during histolytic cell death. Development. 2006;133:1457–65.
Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19:1741–6.
Liu H, Wang J, Li S. E93 predominantly transduces 20-hydroxyecdysone signaling to induce autophagy and caspase activity in Drosophila fat body. Insect Biochem Mol Biol. 2014;45:30–39.
Liu H, Jia Q, Tettamanti G, Li S. Balancing crosstalk between 20-hydroxyecdysone-induced autophagy and caspase activity in the fat body during Drosophila larval-prepupal transition. Insect Biochem Mol Biol. 2013;43:1068–78.
Nezis IP, Shravage BV, Sagona AP, Johansen T, Baehrecke EH, Stenmark H. Autophagy as a trigger for cell death: Autophagic degradation of inhibitor of apoptosis dBruce controls DNA fragmentation during late oogenesis in Drosophila. Autophagy. 2010;6:1214–5.
Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015; 16. https://doi.org/10.1038/nrm3999.
Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014. https://doi.org/10.1038/nrm3735.
Sun Y, Jan LY, Jan YN. Transcriptional regulation of atonal during development of the Drosophila peripheral nervous system. Development. 1998;125:3731–40.
Srivastava M, Scherr H, Lackey M, Xu D, Chen Z, Lu J, et al. ARK, the Apaf-1 related killer in Drosophila, requires diverse domains for its apoptotic activity. Cell Death Differ. 2007;14:92–102.
White K, Grether ME, Abrams JM, Young L, Farrell K, Steller H. Genetic control of programmed cell death in Drosophila. Science. 1994;264:677–83.
Juhász G, Érdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21:3061–6.
Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7:167–78.
Chang Y-Y, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell. 2009;20:2004–14.
Juhász G, Hill JH, Yan Y, Sass M, Baehrecke EH, Backer JM, et al. The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J Cell Biol. 2008;181:655–66.
Scott RC, Juhász G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol. 2007;17:1–11.
Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–15.
Hassan A, Timerman Y, Hamdan R, Sela N, Avetisyan A, Halachmi N, et al. An RNAi screen identifies new genes required for normal morphogenesis of larval chordotonal organs. G3. 2018;8:1871 LP–1884.
Becker S, Pasca G, Strumpf D, Min L, Volk T. Reciprocal signaling between Drosophila epidermal muscle attachment cells and their corresponding muscles. Development. 1997;124:2615–22.
Klein Y, Halachmi N, Egoz-Matia N, Toder M, Salzberg A. The proprioceptive and contractile systems in Drosophila are both patterned by the EGR family transcription factor Stripe. Dev Biol. 2010;337:458–70.
Nachman A, Halachmi N, Matia N, Manzur D, Salzberg A. Deconstructing the complexity of regulating common properties in different cell types: lessons from the delilah gene. Dev Biol. 2015;403:180–91.
Hay Ba, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1994;120:2121–9.
Inbal A, Volk T, Salzberg A. Recruitment of ectodermal attachment cells via an EGFR-dependent mechanism during the organogenesis of Drosophila proprioceptors. Dev Cell. 2004;7:241–50.
Halachmi N, Nachman A, Salzberg A. A newly identified type of attachment cell is critical for normal patterning of chordotonal neurons. Dev Biol. 2016;411:61–71.
Barth JMI, Szabad J, Hafen E, Köhler K. Autophagy in Drosophila ovaries is induced by starvation and is required for oogenesis. Cell Death Differ. 2011;18:915–24.
McPhee CK, Logan Ma, Freeman MR, Baehrecke EH. Activation of autophagy during cell death requires the engulfment receptor Draper. Nature. 2010;465:1093–6.
Yacobi-Sharon K, Namdar Y, Arama E. Alternative germ cell death pathway in drosophila involves HtrA2/Omi, lysosomes, and a caspase-9 counterpart. Dev Cell. 2013;25:29–42.
Bialik S, Dasari SK, Kimchi A. Autophagy-dependent cell death – where, how and why a cell eats itself to death. J Cell Sci. 2018;131:jcs215152.
Basit F, Cristofanon S, Fulda S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013;20:1161–73.
Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, et al. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell. 2016;37:337–49.
Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–8.
Gramates LS, Marygold SJ, Dos Santos G, Urbano JM, Antonazzo G, Matthews BB, et al. FlyBase at 25: Looking to the future. Nucleic Acids Res. 2017. https://doi.org/10.1093/nar/gkw1016.
Kang Y, Bashirullah A. A steroid-controlled global switch in sensitivity to apoptosis during drosophila development. Dev Biol. 2014. https://doi.org/10.1016/j.ydbio.2013.12.005.
Acknowledgements
We thank E. Arama, E. Kurant, T. Volk, K. McCall and the Bloomington Drosophila Stock Center (NIH P40OD018537) and the Developmental Studies Hybridoma Bank at the University of Iowa for fly strains and antibodies. We are grateful to E. Arama for his insightful advice. This research was supported by grants (nos. 499/12 and 674/17) from The Israel Science Foundation. AS is also supported by the Rappaport Institute for Research in the Medical Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by E. Baehrecke
Rights and permissions
About this article

Cite this article
Avetisyan, A., Salzberg, A. Accurate elimination of superfluous attachment cells is critical for the construction of functional multicellular proprioceptors in Drosophila. Cell Death Differ 26, 1895–1904 (2019). https://doi.org/10.1038/s41418-018-0260-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0260-y
