
Abstract
Over the past decade, the Nomenclature Committee on Cell Death (NCCD) has formulated guidelines for the definition and interpretation of cell death from morphological, biochemical, and functional perspectives. Since the field continues to expand and novel mechanisms that orchestrate multiple cell death pathways are unveiled, we propose an updated classification of cell death subroutines focusing on mechanistic and essential (as opposed to correlative and dispensable) aspects of the process. As we provide molecularly oriented definitions of terms including intrinsic apoptosis, extrinsic apoptosis, mitochondrial permeability transition (MPT)-driven necrosis, necroptosis, ferroptosis, pyroptosis, parthanatos, entotic cell death, NETotic cell death, lysosome-dependent cell death, autophagy-dependent cell death, immunogenic cell death, cellular senescence, and mitotic catastrophe, we discuss the utility of neologisms that refer to highly specialized instances of these processes. The mission of the NCCD is to provide a widely accepted nomenclature on cell death in support of the continued development of the field.
Similar content being viewed by others


Introduction
For a long time, cell death has been dismissed by biologists as an inevitable and, hence, spurious consequence of cellular life. A large body of experimental evidence accumulating over the past decades, however, has unveiled and characterized in ever greater detail a set of genetically encoded mechanisms for targeted elimination of superfluous, irreversibly damaged, and/or potentially harmful cells [1,2,3,4]. Intriguingly, regulated cell death (RCD) is not unique to multicellular life forms, a setting in which RCD has an obvious advantage for organismal homeostasis in both physiological and pathological settings [5,6,7,8,9], but is also found (in simplified variants) among unicellular eukaryotes living (at least for part of their life cycle) in colonies (such as several yeast species and Dictyostelium discoideum) [10,11,12,13,14,15], and at least in some prokaryotes (e.g., Escherichia coli) [16]. In striking contrast with accidental cell death (ACD)—the instantaneous and catastrophic demise of cells exposed to severe insults of physical (e.g., high pressures, temperatures, or osmotic forces), chemical (e.g., extreme pH variations), or mechanical (e.g., shear forces) nature—RCD relies on a dedicated molecular machinery, implying that it can be modulated (i.e., delayed or accelerated) by pharmacological or genetic interventions [5, 17].
Although the underlying molecular mechanisms exhibit considerable overlap (see below), RCD is involved in two diametrically opposed scenarios. On the one hand, RCD can occur in the absence of any exogenous environmental perturbation, hence operating as a built-in effector of physiological programs for development or tissue turnover [6, 18]. These completely physiological forms of RCD are generally referred to as programmed cell death (PCD). On the other hand, RCD can originate from perturbations of the intracellular or extracellular microenvironment, when such perturbations are too intense or prolonged for adaptative responses to cope with stress and restore cellular homeostasis [5]. Importantly, stress-driven RCD also constitutes a strategy for the preservation of a biological equilibrium, hence resembling adaptative stress responses. However, while adaptative stress responses operate at the cellular level (which—by extension—promotes the maintenance of homeostasis at the level of organism or colony), RCD directly operates at the level of the organism or colony in spite of cellular homeostasis [5]. Such a homeostatic function not only reflects the elimination of useless or potentially dangerous cells, but also the ability of dying cells to expose or release molecules that alert the organism or colony about a potential threat. Such danger signals are commonly referred to as damage-associated molecular patterns (DAMPs) or alarmins [19,20,21,22].
Cell death manifests with macroscopic morphological alterations. Together with the mechanisms whereby dead cells and their fragments are disposed of, such morphotypes have historically been employed to classify cell death into three different forms: (1) type I cell death or apoptosis, exhibiting cytoplasmic shrinkage, chromatin condensation (pyknosis), nuclear fragmentation (karyorrhexis), and plasma membrane blebbing, culminating with the formation of apparently intact small vesicles (commonly known as apoptotic bodies) that are efficiently taken up by neighboring cells with phagocytic activity and degraded within lysosomes; (2) type II cell death or autophagy, manifesting with extensive cytoplasmic vacuolization and similarly culminating with phagocytic uptake and consequent lysosomal degradation; and (3) type III cell death or necrosis, displaying no distinctive features of type I or II cell death and terminating with the disposal of cell corpses in the absence of obvious phagocytic and lysosomal involvement [23, 24]. Of note, this morphological classification is still extensively employed, irrespective of multiple limitations, and caveats. Starting from 2005, the Nomenclature Committee on Cell Death (NCDD) gathered on a regular basis (1) to address the issues related to the use of a nomenclature of cell death based on morphological grounds; (2) to precisely define major cell death modalities on a genetic, biochemical, pharmacological, and functional (rather than morphological) basis; (3) to distinguish essential (causal) from accessory (correlative) aspects of the death process; and (4) to identify consensus criteria for the identification of dead cells with irreversible plasma membrane permeabilization or complete cellular fragmentation [17, 25,26,27,28].
As the field continues to progress and novel signaling pathways that orchestrate RCD are still being characterized, we propose here an updated classification of cell death modalities centered on molecular and essential aspects of the process (Fig. 1 and Box 1). A major focus will be placed on the signal transduction modules involved in the initiation, execution, and propagation of cell death, as well as on the pathophysiological relevance of each of the main types of RCD.

Major cell death subroutines. Mammalian cells exposed to unrecoverable perturbations of the intracellular or extracellular microenvironment can activate one of many signal transduction cascades ultimately leading to their demise. Each of such regulated cell death (RCD) modes is initiated and propagated by molecular mechanisms that exhibit a considerable degree of interconnectivity. Moreover, each type of RCD can manifest with an entire spectrum of morphological features ranging from fully necrotic to fully apoptotic, and an immunomodulatory profile ranging from anti-inflammatory and tolerogenic to pro-inflammatory and immunogenic. ADCD: autophagy-dependent cell death, ICD: immunogenic cell death, LDCD: lysosome-dependent cell death, MPT: mitochondrial permeability transition.
Box 1 Operational definitions
Accidental cell death (ACD). Virtually instantaneous and uncontrollable form of cell death corresponding to the physical disassembly of the plasma membrane caused by extreme physical, chemical, or mechanical cues.
Anoikis. Specific variant of intrinsic apoptosis initiated by the loss of integrin-dependent anchorage.
Autophagy-dependent cell death. A form of RCD that mechanistically depends on the autophagic machinery (or components thereof).
Autosis. A specific instance of autophagy-dependent cell death that critically relies on the plasma membrane Na+/K+-ATPase.
Cell death. Irreversible degeneration of vital cellular functions (notably ATP production and preservation of redox homeostasis) culminating in the loss of cellular integrity (permanent plasma membrane permeabilization or cellular fragmentation).
Cellular senescence. Irreversible loss of proliferative potential associated with specific morphological and biochemical features, including the senescence-associated secretory phenotype (SASP). Cellular senescence does not constitute a form of RCD.
Efferocytosis. Mechanism whereby dead cells and fragments thereof are taken up by phagocytes and disposed.
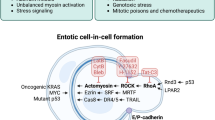
Entotic cell death. A type of RCD that originates from actomyosin-dependent cell-in-cell internalization (entosis) and is executed by lysosomes.
Extrinsic apoptosis. Specific variant of RCD initiated by perturbations of the extracellular microenvironment detected by plasma membrane receptors, propagated by CASP8 and precipitated by executioner caspases, mainly CASP3.
Ferroptosis. A form of RCD initiated by oxidative perturbations of the intracellular microenvironment that is under constitutive control by GPX4 and can be inhibited by iron chelators and lipophilic antioxidants.
Immunogenic cell death. A form of RCD that is sufficient to activate an adaptive immune response in immunocompetent hosts.
Intrinsic apoptosis. Type of RCD initiated by perturbations of the extracellular or intracellular microenvironment, demarcated by MOMP, and precipitated by executioner caspases, mainly CASP3.
Lysosome-dependent cell death. A type of RCD demarcated by primary LMP and precipitated by cathepsins, with optional involvement of MOMP and caspases.
Mitochondrial permeability transition (MPT)-driven necrosis. Specific form of RCD triggered by perturbations of the intracellular microenvironment and relying on CYPD.
Mitotic catastrophe. Oncosuppressive mechanism for the control of mitosis-incompetent cells by RCD or cellular senescence. Per se, mitotic catastrophe does not constitute a form or RCD.
Mitotic death. Specific variant of RCD (most often, intrinsic apoptosis) driven by mitotic catastrophe.
Necroptosis. A modality of RCD triggered by perturbations of extracellular or intracellular homeostasis that critically depends on MLKL, RIPK3, and (at least in some settings) on the kinase activity of RIPK1.
NETotic cell death. A ROS-dependent modality of RCD restricted to cells of hematopoietic derivation and associated with NET extrusion.
Parthanatos. A modality of RCD initiated by PARP1 hyperactivation and precipitated by the consequent bioenergetic catastrophe coupled to AIF-dependent and MIF-dependent DNA degradation.
Programmed cell death (PCD). Particular form of RCD that occurs in strictly physiological scenarios, i.e., it does not relate to perturbations of homeostasis and hence does not occur in the context of failing adaptation to stress.
Pyroptosis. A type of RCD that critically depends on the formation of plasma membrane pores by members of the gasdermin protein family, often (but not always) as a consequence of inflammatory caspase activation.
Regulated cell death (RCD). Form of cell death that results from the activation of one or more signal transduction modules, and hence can be pharmacologically or genetically modulated (at least kinetically and to some extent).
Intrinsic apoptosis
Intrinsic apoptosis is a form of RCD initiated by a variety of microenvironmental perturbations including (but not limited to) growth factor withdrawal, DNA damage, endoplasmic reticulum (ER) stress, reactive oxygen species (ROS) overload, replication stress, microtubular alterations or mitotic defects [29,30,31,32,33,34]. Apoptotic cells retain plasma membrane integrity and metabolic activity (to some degree) as the process proceeds to completion, which—in vivo—allows for the rapid clearance by macrophages or other cells with phagocytic activity (a process commonly known as efferocytosis) [35]. Importantly, intrinsic (and extrinsic, see below) apoptosis and consequent efferocytosis are not always immunologically silent, as previously thought (see below) [36, 37]. In vitro, end-stage apoptosis is generally followed by complete breakdown of the plasma membrane and the acquisition of a necrotic morphotype (secondary necrosis), unless cultured cells display phagocytic activity [38], a process that has recently been linked to the pore-forming activity of gasdermin E (GSDME; best known as DFNA5) [39].
The critical step for intrinsic apoptosis is irreversible and widespread mitochondrial outer membrane permeabilization (MOMP) [40, 41], which is controlled by pro-apoptotic and anti-apoptotic members of the BCL2, apoptosis regulator (BCL2) protein family, a group of proteins sharing one to four BCL2 homology (BH) domains (i.e., BH1, BH2, BH3, and BH4) [29, 42, 43]. In response to apoptotic stimuli, MOMP is mediated by BCL2 associated X, apoptosis regulator (BAX), and/or BCL2 antagonist/killer 1 (BAK1; best known as BAK), both of which contain four BH domains and a conserved transmembrane domain [44,45,46]. Together with BOK, BCL2 family apoptosis regulator (BOK) [47], BAX and BAK are the only BCL2 family members characterized so far in mammalian cells for their ability to form pores across the outer mitochondrial membrane (OMM) and possibly other intracellular membranes [29, 42, 43]. In physiological conditions, BAX continuously cycles between the OMM and the cytosol, where it exhibits a quiescent monomeric or inactive dimeric conformation [48,49,50]. In contrast, BAK constitutively resides at the OMM, where it inserts within the lipid bilayer via its hydrophobic C-terminal α9 helix upon interaction with voltage dependent anion channel 2 (VDAC2) [51,52,53,54]. Of note, some degree of BAK retrotranslocation from the OMM to the cytosol has been documented [55]. Upon induction of apoptosis, BAX retrotranslocation ceases as the mitochondrial pools of BAX and BAK undergo direct or indirect activation (see below) by pro-apoptotic BH3-only proteins [48, 56,57,58,59].
These pro-apoptotic members of the BCL2 protein family (which contain a single BH3 domain) are activated transcriptionally or post-translationally as specific organelles or cellular compartments experience perturbations of homeostasis, de facto operating as cellular transducers of stress signaling [60,61,62,63]. Some BH3-only proteins—such as BCL2 binding component 3 (BBC3; best known as p53-upregulated modulator of apoptosis, PUMA), BCL2 like 11 (BCL2L11; best known as BCL2-interacting mediator of cell death, BIM), and phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1; best known as NOXA)—are mainly activated by transcriptional upregulation, while others—such as BH3 interacting domain death agonist (BID)—mostly undergo post-translational activation [64,65,66,67,68,69,70]. BID, BIM, PUMA, and NOXA share the ability to physically (but transiently) interact with the mitochondrial pool of BAX and/or BAK (hence being known as “activators”) to promote a series of conformational changes [59, 64, 67, 71,72,73,74] culminating with the dissociation of the core and latch domains of BCL2 effectors [75,76,77]. The current view is that activated BAX and BAK form homodimers (also heterodimers in specific settings), resulting in the release of BH3-only proteins and further dimer-by-dimer oligomerization [76,77,78,79,80,81,82,83]. Oligomerization ultimately leads to the assembly of a toroidal lipidic pore that alters mitochondrial permeability and causes profound rearrangements of the mitochondrial ultrastructure [78, 84,85,86]. In line with this model, it has recently been shown that (1) BAX can form rings or linear/arc-shaped oligomers that perforate the OMM [84, 85], and (2) MOMP proceeds upon the formation of pores (impinging on OMM curvature stress), which can vary in size depending on the number of BAX dimers recruited [87].
MOMP is antagonized by anti-apoptotic members of the BCL2 family, including BCL2 itself, BCL2 like 1 (BCL2L1; best known as BCL-XL), MCL1, BCL2 family apoptosis regulator (MCL1), BCL2 like 2 (BCL2L2; best known as BCL-W), and BCL2 related protein A1 (BCL2A1; best known—in human—as BFL-1) [29, 42, 43]. These pro-survival proteins contain all four BH domains, are generally inserted into the OMM or the ER membrane through their α9 helix, and mainly exert anti-apoptotic functions by directly binding pro-apoptotic members of the BCL2 family, an activity that generally—but not always—depends on a hydrophobic binding groove formed by BH1, BH2, and BH3 domains [88,89,90,91,92,93,94]. In addition, some anti-apoptotic BCL2 family members have been proposed to promote cellular survival by: (1) regulating Ca2+ homeostasis at the ER [95,96,97,98,99]; (2) promoting bioenergetic metabolism upon interaction with the F1FO ATP synthase [100,101,102,103,104]; and (3) contributing to the regulation of redox homeostasis [105,106,107,108,109]. However, the importance of these functions has been challenged by the generation of cell lines that lack all major anti-apoptotic and pro-apoptotic BCL2 family members [93]. Thus, most pro-survival BCL2 family members inhibit BAX and BAK by preventing their oligomerization and pore-forming activity either directly, upon physical sequestration at the OMM, or indirectly, following the sequestration of BH3-only activators [29, 64, 79, 110]. Of note, in physiological conditions, some anti-apoptotic BCL2 proteins, such as BCL-XL, exert a protective role by promoting the retrotranslocation of BAX and (to a lesser degree) BAK from the mitochondria to the cytoplasm, thus limiting their mitochondrial pool [48, 55, 111]. Evidence from T cells and platelets suggests that such retrotranslocation occurs in vivo, resulting in the physiological inhibition of BAK by BCL-XL [112]. Importantly, some BH3-only proteins including BCL2 associated agonist of cell death (BAD), Bcl2 modifying factor (BMF), or harakiri, BCL2 interacting protein (HRK) promote MOMP in the absence of a physical interaction with BAX or BAK. These BH3-only proteins, which are sometimes referred to as “sensitizers” or “inactivators” bind to anti-apoptotic BCL2 family members and hence limit their availability to sequester BAX, BAK, or BH3-only activators [58, 93].
Different BH3-only proteins have been suggested to preferentially bind specific anti-apoptotic BCL2 family members (e.g., BID, BIM, and PUMA potently bind all anti-apoptotic BCL2 family members; BAD preferentially interacts with BCL2, BCL-XL, and BCL-W; NOXA preferentially inhibits MCL1; and HRK preferentially inhibits BCL-XL) [57, 113, 114]. In vitro results suggest that the distinction between sensitizers and activators may be much less rigid than previously thought [79, 114,115,116,117]. However, overexpression of BH3-only sensitizers induces minimal apoptosis in cells lacking BID, BIM, PUMA, and NOXA [64], suggesting that BH3-only activators function downstream of BH3-only sensitizers. Of note, the interaction between anti-apoptotic and pro-apoptotic BCL2 family members has major therapeutic implications, with BCL2 representing the pharmacological target of the FDA-approved BH3 mimetic venetoclax (also known as ABT-199) and other molecules with a similar mechanism of action that are currently under development (e.g., the MCL1 inhibitor S63845) [118, 119]. Indeed, venetoclax kills chronic lymphocytic leukemia (CLL) cells by mimicking the activity of BH3-only proteins [120]. Recently, a mechanism of resistance to BH3 mimetics has been ascribed to the tight association between BCL-XL and BH3-only activators at subcellular membranes [121, 122]. The relevance of this mechanism for CLL patients under venetoclax treatment, however, remains to be elucidated.
Confirming the essential role of BCL2 family members for MOMP and the high degree of overlap between the machineries responsible for stress-driven RCD and PCD, the co-deletion of Bax and Bak1 not only renders a large panel of cell types profoundly resistant to diverse lethal stimuli [74], a phenotype that in some settings can be exacerbated by the co-deletion of Bok [123], but also causes perinatal lethality in mice as a consequence of severe developmental defects [124]. Along similar lines, Bcl2l11−/−Bmf−/− as well as Bid−/−Bcl2l11−/−Bbc3−/− mice die prematurely or display severe developmental defects, respectively [68, 125]. However, transformed cells lacking all major BH3 activators (i.e., BID, BIM, PUMA, and NOXA) can still undergo apoptosis in response to DNA-damaging agents or downregulation of BCL2, BCL-XL, and MCL1 [64]. This observation is in line with the notion that BAX and BAK can self-activate in the absence of anti-apoptotic BCL2 family members and pro-apoptotic BH3 proteins (according to a relatively slow kinetics) [64, 93]. Perhaps, BAX and BAK can even be activated independently of BH3-only proteins by the concerted action of the prolyl isomerase peptidylprolyl cis/trans isomerase, NIMA-interacting 1 (PIN1) and either tumor protein p53 (TP53; best known as p53) [126,127,128] or ATR serine/threonine kinase (ATR) [129, 130], several proteins containing BH-like motifs [131], as well as by detergents, heat, pH changes, or specific monoclonal antibodies [132]. That said, the actual pathophysiological relevance of non-canonical BAX and BAK activation remains to be formally established. Both anti-apoptotic and pro-apoptotic BCL2 proteins are also subjected to tight transcriptional and post-translational regulation, involving (but not limited to) proteasomal degradation, phosphorylation, and subcellular (re)localization [48, 108, 133,134,135,136,137,138]. Finally, it is becoming increasingly evident that mitochondrial size and shape [139,140,141] as well as lipid composition [142, 143] can influence the likelihood of mitochondria to undergo irreversible MOMP. These observations exemplify the number of factors involved in MOMP at the level of single mitochondria. Of note, active BAX and BAK have also been proposed to (1) permeabilize ER membranes, especially in response to reticular stress, leading to release of luminal ER chaperones into the cytosol [30, 144]; and (2) favor the activation of type 1 inositol trisphosphate receptors at the ER, resulting in the cytosolic leak of Ca2+ ions and consequent mitochondrial Ca2+ uptake [96, 145]. However, the actual relevance of ER permeabilization for intrinsic apoptosis remains to be elucidated. That said, the contact sites between mitochondria and the ER (which are commonly known as mitochondria-associated ER membranes) appear to regulate a plethora of cellular processes that influence RCD or its immunological consequences, including (but not limited to) ER stress signaling, the transfer of Ca2+ ions from the ER to mitochondria, and inflammatory reactions [146,147,148].
As for BOK, it has been proposed that this BCL2 protein contributes to the regulation of ER homeostasis, as demonstrated by its prominent localization at the ER membrane [149] and the defective apoptotic response of Bok−/− cells to some ER stressors [150]. Moreover, BOK has recently been shown to induce MOMP in the absence of BAX and BAK and independently of other BCL2 family members [47, 151, 152]. In particular, BOK appears to be constitutively active and to be antagonized by an ER-associated degradation pathway rather than by anti-apoptotic BCL2 proteins [47]. BOK is also regulated by a mechanism involving the binding to inositol 1,4,5-trisphosphate (IP3) receptors, which reportedly limits its proteasomal degradation [153]. Of note, Bok−/−, Bax−/−Bok−/− as well as Bak1−/−Bok−/− mice display no obvious abnormalities (except for persistence of primordial follicle oocytes in aged Bax−/−Bok−/− females) [154, 155], implying that physiological functions of BOK can be compensated for by BAK and/or BAX.
MOMP directly promotes the cytosolic release of apoptogenic factors that normally reside in the mitochondrial intermembrane space [40, 44, 156]. These mitochondrial proteins include (but are not limited to) cytochrome c, somatic (CYCS), which usually operates as an electron shuttle in the mitochondrial respiratory chain [157,158,159,160], and diablo IAP-binding mitochondrial protein (DIABLO; also known as second mitochondrial activator of caspases, SMAC) [161,162,163]. The release of CYCS and SMAC to the cytosol is favored by mitochondrial cristae remodeling [164], which relies on the oligomerization and activation of OPA1, mitochondrial dynamin like GTPase (OPA1) [165], possibly preceded by the BAX-dependent and BAK-dependent activation of OMA1 zinc metallopeptidase (OMA1) [166, 167], and/or dynamin 1 like (DNM1L; best known as DRP1) [168]. Accordingly, nitric oxide (NO) has been shown to precipitate the release of apoptogenic factors from mitochondria upon direct nitrosylation of DRP1 (at least in some settings) [169,170,171]. The cytosolic pool of CYCS binds to apoptotic peptidase activating factor 1 (APAF1) and pro-caspase 9 (CASP9) in a deoxyATP-dependent manner to form the supramolecular complex known as apoptosome, which is responsible for CASP9 activation [160]. Recently, the structure of the apoptosome from multiple organisms including humans has been characterized at atomic resolution [172,173,174]. These studies revealed that the autocatalytic maturation of CASP9 within the apoptosome occurs through generation of CASP9 homodimers and CASP9-APAF1 heterodimers/multimers upon association of their respective caspase recruitment domains (CARDs) [175,176,177,178].
Activated CASP9 can catalyze the proteolytic activation of CASP3 and CASP7, which are widely perceived as the enzymes responsible for cell demolition during intrinsic (and extrinsic, see below) apoptosis in mammalian cells (and hence are commonly known as executioner caspases) [179, 180]. Cytosolic SMAC precipitates apoptosis by associating with members of the inhibitor of apoptosis (IAP) protein family, including X-linked inhibitor of apoptosis (XIAP) [162, 163, 181]. To acquire apoptogenic activity, SMAC must undergo a proteolytic maturation step that unleashes its latent IAP-binding domain, which is catalyzed by the inner membrane peptidase (IMP) complex [182] and perhaps by the inner mitochondrial membrane (IMM) protease presenilin associated rhomboid like (PARL) [183]. XIAP is the only IAP protein family member that counteracts the apoptotic cascade by stably binding to and hence physically blocking caspases [184, 185]. Conversely, baculoviral IAP repeat containing 2 (BIRC2; best known as c-IAP1) and BIRC3 (best known as c-IAP2) mostly do so as they (1) drive the upregulation of potent anti-apoptotic factors such as CASP8 and FADD like apoptosis regulator (CFLAR; best known as c-FLIP) [186]; (2) promote caspase inactivation by virtue of their E3 ubiquitin ligase activity [187,188,189,190,191,192,193,194,195]; (3) ubiquitinate receptor interacting serine/threonine kinase 1 (RIPK1) and hence trigger pro-survival NF-κB signaling [196,197,198]; and (4) perhaps promote SMAC degradation at mitochondria through a mechanism that depends on BCL2 proteins [199]. Of note, MOMP eventually leads to the dissipation of the mitochondrial transmembrane potential (Δψm)—mostly as a consequence of the respiratory impairment imposed by the loss of CYCS—and hence to the cessation of Δψm-dependent mitochondrial functions (including ATP synthesis and some forms of protein import) [200,201,202,203]. Intriguingly, BAK and BAX may not always be required for pro-apoptotic stimuli to promote CYTC release and consequent caspase activation, even in conditions in which mitochondrial permeability transition (MPT; see below) is disabled [204, 205]. This may suggest the existence of another—presently unidentified—mechanism for MOMP, possibly involving specific lipids like ceramide [206, 207]. The actual pathophysiological relevance of this potential mechanism remains obscure.
The catalytic activity of executioner caspases precipitates cellular demise and is responsible for many of the morphological and biochemical correlates of apoptosis, including DNA fragmentation [208], phosphatidylserine (PS) exposure [209, 210], and the formation of apoptotic bodies [211, 212]. CASP3 favors DNA fragmentation by catalyzing the proteolytic inactivation of DNA fragmentation factor subunit alpha (DFFA; best known as ICAD), hence unleashing the catalytic activity of DFFB (best known as CAD) [213,214,215]. Recent experimental evidence demonstrates that CASP3 promotes PS exposure by activating proteins involved in PS externalization, such as the phospholipid scramblases [216,217,218], or inactivating factors that mediate PS internalization, such as phospholipid flippases [219,220,221]. Thus, in response to apoptotic stimuli, active CASP3 reportedly cleaves (1) XK related protein 8 (XKR8), which interacts with basigin (BSG) or neuroplastin (NPTN) to form a phospholipid-scrambling complex responsible for PS exposure [216, 217], and (2) ATPase phospholipid transporting 11A (ATP11A) and ATP11C, resulting in inhibition of their flippase activity and PS exposure, as demonstrated by absent or reduced PS translocation on the cell surface of cells expressing a non-cleavable ATP11C or developing erythrocytes from Atp11a−/− mice [219,220,221]. That said, PS exposure may not universally accompany intrinsic (and extrinsic) apoptosis [222,223,224].
Of note, a large body of evidence suggests that executioner caspases precipitate intrinsic apoptosis, once a hitherto poorly defined point-of-no-return has been trespassed, but are not essential for it [17]. Accordingly, blocking post-mitochondrial caspase activation by genetic means or with specific pharmacological inhibitors, such as to N-benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethylketone (Z-VAD-fmk) and (3S)-5-(2,6-difluorophenoxy)-3-[[(2S)-3-methyl-1-oxo-2-[(2-quinolinylcarbonyl)amino]butyl]amino]-4-oxo-pentanoic acid hydrate (Q-VD-OPh), generally delays (but does not prevent) intrinsic apoptosis in vitro and in vivo (at least in the mammalian system), as it promotes a switch to other types of RCD [17, 225]. In addition, when MOMP affects a limited number of mitochondria, the consequent sublethal activation of caspases does not precipitate RCD but promotes genomic instability [226]. Finally, at least some cells exposed to transient apoptotic stimuli appear to survive MOMP affecting a limited number of mitochondria and the partial activation of executioner caspases by a hitherto poorly characterized process called anastasis (most likely constituting a robust adaptative response upstream of the boundary between cellular life and death) [226,227,228]. Altogether, these observations suggest that CASP3 and CASP7 mediate a facilitating, rather than indispensable, role in RCD (for an extensive discussion on this topic, please refer to ref [17]). This said, executioner caspases can positively or negatively regulate the emission of multiple DAMPs from dying cells, including immunostimulatory [229] as well as immunosuppressive [230] factors. Thus, pharmacological agents targeting executioner caspases may be unable to mediate bona fide cytoprotection, but may efficiently switch RCD modality. Interestingly, although CASP6 has long been considered as an executioner caspase based on its homology with CASP3 and CASP7, recent data on substrate specificity suggest that CASP6 may actually be involved in RCD initiation [179, 231, 232]. Additional investigation is required to elucidate the function of CASP6 in mammalian cells.
A specific variant of intrinsic apoptosis elicited by the loss of integrin-dependent attachment to the extracellular matrix is commonly known as anoikis [233, 234]. As such, anoikis is demarcated by MOMP and precipitated by the activation of executioner caspases, notably CASP3 [233]. At least in some settings, detachment from the extracellular matrix triggers MOMP upon activation of the BH3-only proteins BIM and BMF [137, 235]. Since anoikis prevents anchorage-independent proliferation and attachment to an improper matrix, it is generally considered as an oncosuppressive process [234, 236]. Accordingly, cancer cells need to acquire at least some degree of resistance to anoikis to initiate and progress though the so-called “metastatic cascade” [237,238,239]. Neoplastic cells can evade anoikis upon activation of mitogen-activated protein kinase 1 (MAPK1; best known as ERK2) caused by cellular aggregation and consequent epidermal growth factor receptor (EGFR) stabilization mediated by erb-b2 receptor tyrosine kinase 2 (ERBB2) [237, 240], or degradation of the negative ERK2 regulator BRCA1-associated protein (BRAP), which is favored by coiled-coil domain containing 178 (CCDC178) [241]. Once activated, ERK2 reportedly supports anoikis resistance by promoting the cytosolic sequestration of BIM in complex with dynein light chain LC8-type 1 (DYNLL1; best known as LC8) and beclin 1 (BECN1) [138, 238], or the transactivation of integrin subunit alpha 6 (ITGA6) via a mechanism dependent on KRAS [242].
Additional strategies that limit the sensitivity of malignant cells to anoikis encompass (but are not limited to): (1) activation of anti-apoptotic BCL2 proteins, including MCL1 stabilization as induced by fibroblast-derived insulin like growth factor-binding proteins (IGFBPs) [243] and increase in BCL2 expression levels as imposed by hepatitis B virus X protein [244]; (2) epigenetic silencing of adhesion-related genes, such as SHC adaptor protein 1 (SHC1) upon overexpression of the hematopoietic transcription factor IKAROS family zinc finger 3 (IKZF3; also known as AIOLOS) [245]; (3) perturbation of ITG-protein tyrosine kinase 2 (PTK2; best known as FAK) signaling, which usually suppresses anoikis [246,247,248,249]; (4) activation of the so-called “epithelial-to-mesenchymal transition” (EMT), which is associated with multiple signal transduction and metabolic modules for RCD resistance [242, 250, 251]; (5) targeting of Yes associated protein 1 (YAP1) by miR-200a or via a platelet-dependent mechanism [252, 253]; (6) increased antioxidant responses driven by the activating transcription factor 4 (ATF4)-mediated upregulation of heme oxygenase 1 (HMOX1) [254]; (7) autophagy activation [254, 255]; (8) upregulation of the molecular chaperone crystallin alpha B (CRYAB; also known as HSPB5) [256]; (9) signaling via AMPK and proliferation and apoptosis adaptor protein 15 (PEA15), which favors anchorage-independent cell growth [257]; (10) upregulation of matrix metallopeptidases (MMPs) by a mechanism involving the epidermal growth factor (EGF)-driven autocrine production of angiopoietin like 4 (ANGPTL4) [258]; (11) expression and phosphorylation of signal transducer and activator of transcription 3 (STAT3) [259]; and (12) rewiring of central carbon metabolism toward NAPDH synthesis, resulting in improved redox homeostasis [260, 261]. That said, it has become evident that the adaptation of cancer cells to the loss of attachment involves multiple processes beyond (but presumably highly interconnected to) anoikis resistance [234 262,263,264], suggesting that multiple barriers need to be overcome for the metastatic cascade to be initiated.
The NCCD proposes to define intrinsic apoptosis as a form of RCD initiated by perturbations of the intracellular or extracellular microenvironment, demarcated by MOMP and precipitated by executioner caspases, mainly CASP3 (Box 1).
Extrinsic apoptosis
Extrinsic apoptosis is an RCD modality initiated by perturbations of the extracellular microenvironment [265,266,267,268]. Extrinsic apoptosis is mostly driven by either of two types of plasma membrane receptors: (1) death receptors, whose activation depends on the binding of the cognate ligand(s), and (2) dependence receptors, whose activation occurs when the levels of their specific ligand drop below a specific threshold [267 269,270,271].
Death receptors include (but are not limited to): Fas cell surface death receptor (FAS; also known as CD95 or APO-1), and TNF receptor superfamily member 1A (TNFRSF1A; best known as TNFR1), 10a (TNFRSF10A; best known as TRAILR1 or DR4), and 10b (TNFRSF10B; best known as TRAILR2 or DR5) [269, 270, 272, 273]. As a general rule, death receptor ligation allows for the assembly of a dynamic multiprotein complex at the intracellular tail of the receptor, such as so-called “death-inducing signaling complex” (DISC), “complex I”, and “complex II”, which operate as molecular platforms to regulate the activation and functions of CASP8 (or CASP10, in a limited number of settings) [274,275,276]. In the case of FAS and TRAILRs, the cognate ligands—namely, FAS ligand (FASLG; also known as CD95L or APO-1L) and TNF superfamily member 10 (TNFSF10; best known as TRAIL), respectively—stabilize preformed receptor homotrimers to induce a conformational change at their intracellular tails that enables the death domain (DD)-dependent association of the adapter Fas associated via death domain (FADD) [277,278,279,280,281,282]. In turn, FADD drives DISC assembly by promoting the death effector domain (DED)-dependent recruitment of CASP8 (or CASP10) and multiple isoforms of c-FLIP. In contrast, TNFR1 signaling involves the association of TNFRSF1A associated via DD (TRADD), which acts as an adaptor for the assembly of complex I, generally consisting of TNF receptor associated factor 2 (TRAF2), TRAF5, c-IAP1, c-IAP2, RIPK1, and the linear ubiquitin chain assembly complex (LUBAC), a supramolecular entity consisting of SHANK associated RH domain interactor (SHARPIN), RANBP2-type, and C3HC4-type zinc finger containing 1 (RBCK1; best known as HOIL-1), and ring finger protein 31 (RNF31; best known as HOIP) [283,284,285,286,287]. Of note, the glycosylation state of some death receptors (e.g., FAS) has been shown to impact on the sensitivity of T lymphocytes to extrinsic apoptosis, hence influencing the termination of inflammatory responses [288,289,290]. The relevance of death receptor glycosylation for extrinsic apoptosis in other cell types has not been investigated in detail.
The molecular mechanisms regulating CASP8 activity upon death receptor stimulation have been extensively investigated. In particular, CASP8 maturation involves a cascade of events initiated by the binding of CASP8 to FADD at the DISC. This interaction enables the assembly of a linear filament of CASP8 molecules (depending on their DEDs) that facilitates homodimerization and consequent activation by autoproteolytic cleavage [291,292,293,294,295]. A key role in this setting is mediated by c-FLIP, which is a catalytically inactive close relative of CASP8 [296, 297]. Compelling evidence indicates that the short variant of c-FLIP (c-FLIPS) and its long counterpart (c-FLIPL) inhibit and activate CASP8, respectively, by modulating CASP8 oligomerization [298,299,300,301]. Active CASP8 reportedly cleaves c-FLIPL [302] and heterodimeric complexes of CASP8 with c-FLIPL (but not c-FLIPS) are endowed with limited enzymatic activity that favors CASP8 oligomerization and consequent activation [301]. c-FLIP isoforms and CASP8 seem to be recruited at the DISC to comparable levels [303], supporting the notion that elevated expression levels of c-FLIPL inhibit, rather than activate, extrinsic apoptosis possibly by disrupting CASP8 maturation [301, 304]. Of note, CFLAR (the gene encoding c-FLIP) is under direct transcriptional control by NF-κB, which largely contributes to pro-survival TNFR1 signaling in specific circumstances (see below) [287, 296, 305]. The enzymatic activity of CASP8 appears to be controlled by additional post-translational mechanisms, including (but not limited to): (1) phosphorylation at Y380, which inhibits the autoproteolytic activity of CASP8 upon FAS activation [306], (2) phosphorylation at T273, which is catalyzed by polo like kinase 3 (PLK3) at the DISC and promotes CASP8 apoptotic functions [307], and (3) deubiquitination, which decreases CASP8 activity and interrupts extrinsic apoptosis [302].
The execution of extrinsic apoptosis driven by death receptors follows two distinct pathways. In so-called “type I cells” (e.g., thymocytes and mature lymphocytes) the CASP8-dependent proteolytic maturation of executioner CASP3 and CASP7 suffices to drive RCD, which cannot be inhibited by the transgene-driven overexpression of anti-apoptotic BCL2 proteins, the co-deletion of Bax and Bak1, or the loss of BID [308, 309]. Conversely, in “type II cells” (e.g., hepatocytes, pancreatic β cells, and a majority of cancer cells), in which CASP3 and CASP7 activation is restrained by XIAP [310], extrinsic apoptosis requires the proteolytic cleavage of BID by CASP8 [70, 311, 312]. This leads to the generation of a truncated form of BID (tBID), which translocates to the OMM [313, 314] via a mechanism that, at least upon FAS stimulation, reportedly depends on the binding of modulator of apoptosis 1 (MOAP1) to the alleged BID receptor mitochondrial carrier 2 (MTCH2) [315, 316]. At the OMM, tBID operates as a BH3-only activator to engage BAX/BAK-dependent MOMP-driven and consequent CASP9-driven RCD. Although human CASP10 shares some degree of substrate specificity with CASP8 [317] and possibly contributes to extrinsic apoptosis in primary T cells [318], rodents including mice and rats lack a functional Casp10 gene, and the precise role of this caspase in death receptor-driven apoptosis in humans and other CASP10-proficient species remains a matter of controversy [319,320,321]. A recent study shows that—following FAS activation—CASP10 causes the dissociation of CASP8 from the DISC, thereby promoting cell survival [319]. FAT atypical cadherin 1 (FAT1) appears to mediate similar anti-apoptotic function by limiting the association of CASP8 with the DISC [322].
A large body of evidence demonstrates that death receptor ligation does not necessarily culminate in RCD. In particular, TNFR1 activation can have diverse outcomes depending on multiple variables, such as the post-translational modification status of RIPK1, which has a direct impact on the assembly of pro-survival vs. pro-death signaling complexes [323,324,325]. Thus, following tumor necrosis factor (TNF) stimulation, RIPK1 is recruited at complex I in a TRADD-independent manner, followed by RIPK1 polyubiquitination by c-IAP1, c-IAP2, and LUBAC [196, 324 326,327,328,329]. Polyubiquitinated RIPK1 promotes cell survival and inflammation by acting as a scaffold for the sequential recruitment of TGF-beta activated kinase 1/MAP3K7-binding protein 2 (TAB2), TAB3, and mitogen-activated protein kinase kinase kinase 7 (MAP3K7; best known as TAK1), which can drive mitogen-activated protein kinase (MAPK) signaling or IκB kinase (IKK)-dependent NF-κB activation [283, 287 330,331,332,333]. Moreover, the phosphorylation of RIPK1 by TAK1, the IKK complex or mitogen-activated protein kinase-activated protein kinase 2 (MAPKAPK2; best known as MK2) appears to alter its ability to interact with FADD and CASP8, hence preventing the variants of TNF-induced RCD that depends on RIPK1 kinase activity and favoring RIPK1-independent TRADD-, FADD-, and CASP8-driven apoptosis [285 334,335,336]. Conversely, in the presence of so-called “SMAC mimetics” (which de facto operate as IAP inhibitors) [337], RIPK1 is deubiquitinated by CYLD lysine 63 deubiquitinase (CYLD), favoring its release from complex I and its association with FADD and CASP8 in the cytosol to form complex II, which drives extrinsic apoptosis [338]. Complex II formation also requires TRAF2 ubiquitination by HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase 1 (HACE1) [339]. To add a further layer of complexity, the proteasomal degradation of TRAF2 appears to be prevented (at least in hepatocytes) by RIPK1, independently of its kinase activity [340, 341]. Of note, TNFR1 can also activate alternative RCD modalities, such as necroptosis (see below).
Death receptor signaling can also lead to NF-κB activation, generally resulting in cell survival associated with a robust inflammatory response [272, 342]. The ability of some death receptors including TNFR1 to promote NF-κB activation over CASP8 activation appears to depend on the degree of receptor oligomerization (i.e., trimerization vs. higher-order multimerization) [343], the scaffolding (i.e., non-enzymatic) functions of CASP8, and the consequent assembly of TNFR1-like complexes containing RIPK1 and LUBAC [272, 286, 344]. Upon TRAILR activation, LUBAC reportedly ubiquitinates both CASP8 and RIPK1 while promoting the recruitment of IKK to complex I [286], which also explains the requirement of LUBAC for the inhibition of TNF-induced cell death [345]. In line with this notion, TNF alpha-induced protein 3 (TNFAIP3; best known as A20) inhibits CASP8 activation downstream of TRAILRs in glioblastoma cells, owing to its ability to polyubiquitinate RIPK1 [346, 347]. A recent study suggests that the ability of TRAILR2 to dispatch pro-survival rather than pro-apoptotic signals may depend on its preferential localization outside of lipid rafts [348]. It remains to be demonstrated whether the same also applies to other death receptors.
The family of dependence receptors consists of approximately 20 members, including: (1) the netrin 1 (NTN1) receptors DCC netrin 1 receptor (DCC), unc-5 netrin receptor A (UNC5A), UNC5B, UNC5C, and UNC5D; (2) the neurotrophin receptor neurotrophic receptor tyrosine kinase 3 (NTRK3); and (3) the sonic hedgehog (SHH) receptor patched 1 (PTCH1) [267, 349, 350]. Intriguingly, dependence receptors promote cell survival, proliferation and differentiation in physiological conditions (when their cognate ligands are normally available), but activate distinct (and not completely elucidated) lethal signaling cascades (generally impinging on caspase activation) once ligand availability falls below a specific threshold level [350]. Thus, in the absence of their respective ligands: (1) DCC is cleaved by CASP3 and this promotes its association with adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1 (APPL1) and CASP9, resulting in the activation of the CASP9-CASP3 cascade [350, 351]; (2) PTCH1 interacts with the cytosolic adaptor four and a half LIM domains 2 (FHL2; best known as DRAL), hence favoring the assembly of a CASP9-activating complex consisting of caspase recruitment domain family member 8 (CARD8; also known as TUCAN) and neural precursor cell expressed, developmentally down-regulated 4, E3 ubiquitin protein ligase (NEDD4) [352,353,354]; (3) UNC5B enables the protein phosphatase 2 (PP2A)-mediated activating dephosphorylation of death associated protein kinase 1 (DAPK1), which is known to promote p53-dependent RCD [355,356,357]; and (4) UNC5D and NTRK3 are subjected to CASP3 cleavage generating intracellular fragments that translocate either into the nucleus to trigger the E2F transcription factor 1 (E2F1)-driven expression of pro-apoptotic genes (as in the case of UNC5D) or at mitochondria to activate CASP9 upon MOMP (as in the case of NTRK3) [358, 359].
Dependence receptor-driven RCD has been involved in multiple pathophysiological settings, and exerts robust oncosuppressive functions [350]. Accordingly, neoplastic cells often escape from dependence receptor-mediated RCD by (1) upregulating the expression of their cognate ligands such as NTN1 [360,361,362]; (2) inactivating, downregulating, or losing gene(s) encoding specific dependence receptors, including DCC, UNC5C, and NTRK3 [350 363,364,365,366,367,368,369]; or (3) silencing signal transducers operating downstream of dependence receptors—such as DAPK1—via epigenetic mechanisms [370]. That said, whether the actual pathophysiological relevance of dependence receptor signaling stems from the initiation of extrinsic apoptosis remains to be formally established. Of note, in specific cell types, some members of the toll-like receptor (TLR) protein family including toll like receptor 3 (TLR3) have also been suggested to trigger RCD by a mechanism that involves toll like receptor adaptor molecule 1 (TICAM1; best known as TRIF), and ultimately impinges on CASP8 activation [371, 372]. However, it remains unclear whether TLR3 and other TLRs actually initiate a private RCD program that directly engages CASP8, or whether they promote RCD upon the activation of an NF-κB-dependent autocrine/paracrine signaling pathway involving TNF.
We propose to define extrinsic apoptosis as a type of RCD initiated by perturbations of the extracellular microenvironment that are detected by plasma membrane receptors, propagated by CASP8 (with the optional involvement of MOMP), and precipitated by executioner caspases, mainly CASP3 (Box 1).
MPT-driven necrosis
MPT-driven necrosis is a form of RCD initiated by specific perturbations of the intracellular microenvironment such as severe oxidative stress and cytosolic Ca2+ overload, which generally manifests with a necrotic morphotype [373, 374]. The term MPT refers to an abrupt loss of the impermeability of the IMM to small solutes, resulting in rapid Δψm dissipation, osmotic breakdown of both mitochondrial membranes, and RCD [373, 374].
At the biochemical level, MPT-driven necrosis has been proposed to follow the opening of the so-called “permeability transition pore complex” (PTPC), a supramolecular complex assembled at the junctions between the IMM and OMM [103, 374]. The composition, regulation, and precise mechanism of action of the PTPC are still under intense investigation and matter of a vivid debate [373, 375]. To date, peptidylprolyl isomerase F (PPIF; best known as cyclophilin D, CYPD) is the only protein whose in vivo requirement for MPT induction has been formally validated with robust genetic tools (although there is consensus around the notion that CYPD does not constitute the pore-forming unit of the PTPC) [376,377,378,379]. Accordingly, pharmacological inhibitors of CYPD including cyclosporin A (CsA) [376 379,380,381], sanglifehrin A (SfA) [382, 383], and JW47 [384] limit MPT-driven necrosis and confer protection in multiple rodent models of disease in which oxidative stress and cytosolic Ca2+ overload constitute major etiological determinants (e.g., neuronal, cardiac, and renal ischemia/reperfusion). Along similar lines, CYPD degradation through a mechanism initiated by the overexpression of HCLS1 associated protein X-1 (HAX1) abolishes MPT-driven necrosis and limits the demise of cardiomyocytes experiencing ischemia/reperfusion in vivo [385]. Nonetheless, a large randomized clinical study completed in 2015 (the CIRCUS trial) failed to confirm previous findings from 2008 [386] on the cardioprotective effects of cyclosporine administered before percutaneous coronary intervention to patients with acute myocardial infarction [387]. Although multiple caveats linked to the methods employed to measure infarct size and the use of a specific pharmacological CsA formulation can be invoked to explain the negative results of the CIRCUS trial [388], the elevated interconnectivity of RCD subroutines (notably, intrinsic apoptosis and MPT-driven necrosis) may have played a key role in this setting.
At odds with CYPD, several other proteins that had previously been hypothesized to mediate a non-redundant role within the PTPC turned out to be dispensable for MPT in vivo, based on relatively robust genetic models [373]. Thus, an inducible cardiomyocyte-specific deletion of solute carrier family 25 member 3 (Slc25a3, which codes for the inorganic phosphate carrier) in mice does not affect the ability of mitochondria to undergo MPT in vitro, as it establishes partial PTPC desensitization in cellula and slightly mitigates cardiac injury upon ischemia/reperfusion in vivo (~10% reduction in ischemic area over area at risk) [389]. Similar findings have been obtained for distinct isoforms of the IMM integral protein adenine nucleotide translocator (ANT) and the OMM protein VDAC. In particular, the concurrent knockout or knockdown of Slc25a4 and Slc25a5, which encode ANT1 and ANT2, respectively [390], or that of Vdac1, Vdac2, and Vdac3 [391, 392] fails to prevent the induction of MPT by oxidative stress or Ca2+ overload. However, mitochondria isolated from Slc25a4−/−Slc25a5−/− mouse livers are desensitized to Ca2+-driven MPT to a similar extent than mitochondria exposed to CsA [390]. Moreover, Slc25a31 encodes another ANT isoform (i.e., ANT4), that (at least in some mouse tissues) may compensate for the absence of ANT1 and ANT2 [393, 394]. These results reflect a consistent degree of genetic and functional redundancy among the components of the molecular machinery for MPT [373].
Several lines of evidence suggest that the mitochondrial F1FO ATPase mediates a non-redundant role within the PTPC. Initially, the c-ring of the F1FO ATPase [395,396,397,398] as well as F1FO ATPase dimers [399] have been proposed to constitute the long-sought PTPC pore-forming unit. A specific interaction between CYPD and the lateral stalk of the F1FO ATPase, as well as the ability of Ca2+ ions (which are potent MPT inducers) to bind to ATP synthase, H+ transporting, mitochondrial F1 complex, beta polypeptide (ATP5B) [100], lend further support to this interpretation [395, 400, 401]. However, very recent findings seem to exclude the possibility that the F1FO ATPase constitutes the pore-forming component of the PTPC [402,403,404,405]. First, it seems unlikely for c-rings (which exist as pores across the IMM) to lose their lipid plugs in relatively physiological conditions [402]. Second, mitochondria from human cells lacking all the genes coding for the c subunit of the F1FO ATP synthase, i.e., ATP synthase, H+ transporting, mitochondrial Fo complex subunit C1 (subunit 9; ATP5G1), subunit C2 (subunit 9; ATP5G2), and subunit C3 (subunit 9; ATP5G3), reportedly retain the ability to undergo MPT in response to Ca2+ overload [403]. Finally, cells lacking ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit (ATP5O; best known as OSCP), or the membrane domain of the b subunit of the F1FO ATP synthase (encoded by ATP5F1) appear to preserve normal PTPC activity [405]. That said, the implication of the F1FO ATPase or components thereof in MPT-driven necrosis remains a matter of intensive investigation. An RNA interference (RNAi)-based screening identified SPG7, paraplegin matrix AAA peptidase subunit (SPG7) as an essential component of the PTPC acting as part of VDAC-containing and CYPD-containing hetero-oligomers [406]. Despite the availability of Spg7−/− mice, the actual involvement of SPG7 in MPT-derived necrosis in vivo remains to be validated.
Several physical or functional PTPC interactors have been shown to regulate MPT-driven necrosis. These include: (1) pro- and anti-apoptotic BCL2 family members such as BAX, BAK, and BID [407,408,409,410], as well as BCL2 and BCL-XL [411,412,413,414]; (2) DRP1, which appears to promote PTPC opening in response to chronic β adrenergic receptor stimulation, via a mechanism that relies on DRP1 phosphorylation by calcium/calmodulin dependent protein kinase II (CAMK2G; best known as CaMKII) [415]; and (3) p53, which participates in MPT-driven necrosis upon physical interaction with CYPD [416]. The latter interaction has been shown to participate in the pathogenesis of ischemic stroke in mice [416]. Its pathophysiological relevance in humans, however, remains to be elucidated. Recent findings lend additional support to the relevance of tight Ca2+ homeostasis at the mitochondrial level for cellular and organismal fitness. Thus, perturbing the activity of the IMM Ca2+ uniporter, consisting of mitochondrial calcium uniporter (MCU), single-pass membrane protein with aspartate-rich tail 1 (SMDT1; also known as EMRE), mitochondrial calcium uptake 1 (MICU1) and MICU2, reportedly affects mouse survival and liver regeneration after partial hepatectomy by promoting mitochondrial Ca2+ overload and MPT-driven necrosis [417]. Along similar lines, the loss of mitochondrial m-AAA proteases of the IMM, which regulate the assembly of the IMM Ca2+ uniporter, induces mitochondrial Ca2+ overload, PTPC opening, and neuronal cell death [418]. Adult mice subjected to the cardiomyocyte-specific deletion of Mcu are protected against cardiac ischemia/reperfusion as a consequence of MTP inhibition [419]. Moreover, the inducible cardiomyocyte-specific deletion of solute carrier family 8 member B1 (Slc8b1, which encodes a mitochondrial potassium-dependent sodium/calcium exchanger) in mice reportedly provokes sudden lethality owing to heart failure imposed by MTP-regulated necrosis upon mitochondrial Ca2+ overload [420]. Finally, rap guanine nucleotide exchange factor 3 (RAPGEF3; best known as EPAC1) appears to trigger PTPC opening by increasing mitochondrial Ca2+ levels through interaction with VDAC1, heat shock protein family A (Hsp70) member 9 (HSPA9; best known as GRP75), and inositol 1,4,5-trisphosphate receptor type 1 (ITPR1; best known as IP3R1), and the knockout of Rapgef3 protects mice against myocardial ischemia/reperfusion injury [421]. However, EPAC1 activation with bicarbonate reportedly decreases mitochondrial Ca2+ uptake, stimulates ATP production, and inhibits multiple forms of RCD including MPT-driven necrosis in rat cardiomyocytes [422]. The precise reasons underlying this apparent discrepancy remain to be elucidated.
We propose to define MPT-driven necrosis as a form of RCD triggered by perturbations of the intracellular microenvironment and relying on CYPD (Box 1).
Necroptosis
Necroptosis is a form of RCD initiated by perturbations of the extracellular or intracellular microenvironment detected by specific death receptors, including (but not limited to) FAS and TNFR1 [423,424,425,426,427], or pathogen recognition receptors (PRRs), including TLR3, TLR4, and Z-DNA binding protein 1 (ZBP1; also known as DAI) [428,429,430]. It is now clear that necroptosis (which generally manifests with a necrotic morphotype) not only mediates adaptative functions upon failing responses to stress, but also participates in developmental safeguard programs (to ensure the elimination of potentially defective organisms before parturition), as well as in the maintenance of adult T-cell homeostasis (de facto serving as a PCD subroutine, at least in specific settings) [2 431,432,433].
At the molecular level, necroptosis critically depends on the sequential activation of RIPK3 and mixed lineage kinase domain like pseudokinase (MLKL) [434, 435]. Upon necroptosis initiation by TNFR1, RIPK3 is activated by RIPK1 (provided that CASP8 is inactive, see below) through a mechanism involving the physical interaction between their respective RIP homotypic interaction motif (RHIM) domains and RIPK1 catalytic activity [436,437,438]. Accordingly, chemical inhibitors of RIPK1 including necrostatin-1 (Nec-1) and derivatives (e.g., Nec-1s) robustly inhibit TNFR1-driven necroptosis, in vitro and in vivo [425, 427]. Alternatively, RIPK3 can be activated following the RHIM-dependent interaction with (1) TRIF upon either TLR3 activation by double-stranded RNA (dsRNA) within endosomes, or TLR4 activation by lipopolysaccharide (LPS) or various DAMPs at the plasma membrane [428]; or (2) ZBP1, which operates as a sensor for cytosolic DNA-promoting type I interferon (IFN) synthesis and NF-κB activation [439,440,441]. Active RIPK3 catalyzes the phosphorylation of MLKL, resulting in the formation of MLKL oligomers (most likely trimers or tetramers) that translocate to the plasma membrane, where they bind specific phosphatidylinositol phosphate species by a roll-over mechanism and hence trigger plasma membrane permeabilization [435 442,443,444,445,446,447,448,449,450,451,452,453].
Although the essential contribution of MLKL to necroptosis has been confirmed by genetic studies [435] as well as by pharmacological (i.e., inhibition of MLKL with necrosulfonamide, NSA) interventions [442], the precise mechanism through which MLKL executes necroptosis is not completely understood. Recent studies ascribe to the heat shock protein 90 kDa alpha family class A member 1 (HSP90AA1; best known as HSP90) a specific and non-redundant role in MLKL oligomerization and translocation [454, 455]. Moreover, it has also been reported that MLKL oligomerization promotes a cascade of intracellular events involving (1) Ca2+ influx, which is presumably mediated by the MLKL target transient receptor potential cation channel subfamily M member 7 (TRPM7) [449]; and (2) PS exposure, which seems to be directly operated by MLKL [456]. This is followed by the formation of PS-exposing plasma membrane bubbles whose breakdown and release is negatively regulated—in conditions of limited MLKL activation—by the antagonistic activity of the endosomal sorting complex required for transport (ESCRT)-III machinery [456, 457]. Once localized at the plasma membrane, MLKL reportedly activates cell-surface proteases of the ADAM family, which can promote the shedding of plasma membrane-associated proteins [458], or form Mg2+ permeant channels [459]. Of note, active MLKL also appears to translocate to the nucleus, but the relevance of this phenomenon for necroptosis remains to be investigated [460]. Previous data supporting the involvement of PGAM family member 5, serine/threonine protein phosphatase, mitochondrial (PGAM5)- and DRP1-driven mitochondrial fragmentation in necroptosis [461] have been conclusively invalidated [435, 446 462,463,464,465,466], confirming that necroptotic signaling can proceed normally independent of mitochondria. Of note, the core components of necroptosis are poorly conserved across the animal kingdom, as some species lack RIPK3 and/or MLKL [467]. Moreover, a few non-canonical instances of pseudonecroptotic RCD involving MLKL (but not RIPK3) [468] or RIPK3 (but not MLKL) [469] have been described. These observations reinforce the notion that the signaling pathways leading to RCD display a hitherto incompletely understood degree of interconnectivity.
Death receptor (in particular TNFR1) engagement is the trigger for RIPK3 activation best characterized so far. As mentioned above, the biological outcome of TNFR1 signaling spans from cell survival and activation (i.e., cytokine secretion) to multiple subroutines of RCD, depending on a variety of cell-intrinsic (e.g., expression levels of the proteins involved in the process) and cell-extrinsic (e.g., intensity and duration of TNF stimulation) factors [283]. In particular, the activation of RIPK3 downstream of TNFR1 ligation relies on the formation of a RIPK1-containing and RIPK3-containing amyloid-like signaling complex commonly known as necrosome [436, 470], wherein first RIPK1 and then RIPK3 undergo a series of trans-phosphorylation or auto-phosphorylation events that are required for MLKL recruitment and necroptosis activation [437, 438, 442, 471]. Major negative regulators of the necrosome include: (1) STIP1 homology and U-box containing protein 1 (STUB1; also known as CHIP), which promotes RIPK1 and RIPK3 ubiquitination followed by lysosomal degradation [472, 473]; (2) A20, which inhibits necrosome assembly by deubiquitinating RIPK3 [473, 474]; (3) protein phosphatase, Mg2+/Mn2+ dependent 1B (PPM1B), which prevents MLKL recruitment to the necrosome by dephosphorylating RIPK3 [475]; and (4) aurora kinase A (AURKA), which mediates inhibitory function upon physical interaction with RIPK1 and RIPK3 [476]. RIPK3 activation also depends on its physical association with a HSP90-containing and cell division cycle 37 (CDC37)-containing co-chaperone complex [477]. In addition, the assembly of the necrosome upon TNFR1 stimulation impinges on two conditions: (1) pharmacological or genetic CASP8 inactivation [478, 479], and (2) RIPK1 deubiquitination-dependent phosphorylation (at least in some settings), which can be favored by exogenously provided SMAC mimetics, ensuring the release of RIPK1 from complex I (see above) [334, 335, 480, 481].
As for the first condition, compelling experimental findings demonstrate that the concerted activity of CASP8, FADD, and c-FLIPL tonically inhibits necroptosis [432, 466, 478, 479 482,483,484]. Thus, the embryonic lethality imposed on mice by the loss of Casp8 or Fadd can be rescued by concurrent ablation of Ripk3 or Mlkl, even though these double knockout animals generally display lymphoproliferative and/or systemic autoimmune disorders as adults [432, 466, 484, 485]. Of note, Cflar−/− mice require the concomitant knockout of Ripk3 and Fadd to develop into adulthood, which underscores the inhibitory role of c-FLIP in both necroptosis and extrinsic apoptosis reported above [483, 486]. Along similar lines, the concurrent deletion of Ripk3 averts perturbations of cutaneous and intestinal homeostasis imposed by the tissue-specific ablation of Fadd or Casp8 [483, 487, 488]. Moreover, the proliferative defects of Casp8−/− or Fadd−/− T cells can be rescued by the administration of the RIPK1 inhibitor Nec-1 or the concomitant ablation of Ripk3 [489]. Necroptosis is also tonically inhibited by c-IAPs, owing to their ability to ubiquitinate RIPK1 [490,491,492,493]. Accordingly, necroptosis relies on the deubiquitinating activity of CYLD [338], which is also a proteolytic target of CASP8 [494,495,496]. Finally, some components of the TNFR1 signaling cascade reportedly regulate necroptosis either in a negative manner, by catalyzing the inhibitory phosphorylation of RIPK1 (e.g., the IKK complex and MK2) [335, 336] and constitutively interacting with (and thus preventing the activation of) MLKL (e.g., TRAF2) [497], or in a positive manner, by favoring the activating phosphorylation of RIPK1 or RIPK3 upon prolonged activation (e.g., TAK1) [334, 498]. In this context, CYLD also contributes to necroptosis by deubiquitinating—and hence suppressing the anti-necroptotic activity of—TRAF2 [497].
That said, mounting evidence indicates that necroptosis driven by several stimuli—in some circumstances even TNFR1 activation—does not necessarily rely on RIPK1. Thus, in contrast to Ripk3−/− mice that are viable and fertile, the Ripk1−/− genotype causes perinatal lethality [482], which cannot be prevented by the ablation of Ripk3, Casp8, or Fadd alone, but can be rescued by the co-deletion of Ripk3 and Casp8, Fadd or Tnfrsf1a [482 499,500,501]. Moreover, Ripk1−/− cells display increased sensitivity to necroptosis and/or extrinsic apoptosis induced by a set of innate immune stimuli [499]. Conditional knockout mouse models demonstrate the key role of RIPK1 for the preservation of intestinal and cutaneous homeostasis and survival [502, 503]. In particular, mice lacking Ripk1 in intestinal epithelial cells display increased rates of spontaneous CASP8-driven apoptosis and develop severe inflammatory lesions leading to premature death, a detrimental phenotype that can be prevented by co-deleting Fadd or (to a lesser degree) Tnfrsf1a [502]. Likewise, the absence of Ripk1 from keratinocytes promotes spontaneous necroptosis and consequent cutaneous inflammation, which can be prevented by the co-deletion of Ripk3, Mlkl, or Zbp1 but not Fadd [440, 502]. Collectively, these results suggest that (at least in some settings) RIPK1 can inhibit (rather than activate) RIPK3-dependent necroptosis and/or CASP8-dependent extrinsic apoptosis [504]. At least in some settings, this reflects the major role of RIPK1 in NF-κB activation [505,506,507].
Intriguingly, the pro-survival role of RIPK1 in development seems to be independent of both its kinase activity and RIPK3 binding, as demonstrated by the fact that mice genetically engineered to express a kinase-dead variant of RIPK1 (e.g., RIPK1K45A) are viable and fertile [447, 499, 508]. Moreover, it has recently been reported that the autophagic receptor optineurin (OPTN) [509] actively regulates the proteasomal turnover of RIPK1, as the loss of OPTN induces axonal degeneration via RIPK1-dependent necroptosis [510]. Inhibitor of nuclear factor kappa B kinase subunit gamma (IKBKG; best known as NEMO) also prevents RIPK1-driven intestinal inflammation and epithelial cell death, although the underlying molecular mechanisms remain poorly understood [511] Finally, when catalytically inactive or inhibited by specific pharmacological agents such as Nec-1, RIPK1 (and, at least under certain circumstances, RIPK3) reportedly contributes to specific forms of CASP8-dependent apoptosis (see above) [335, 336, 446, 447, 481 512,513,514,515,516]. The current view ascribes the opposing roles of RIPK1 (and—at least in part—RIPK3) in promoting or inhibiting RCD to its kinase-dependent vs. kinase-independent (i.e., scaffolding) functions, respectively [4, 517].
As mentioned above, RIPK3 can be activated by proteins involved in innate immunity to invading pathogens including TRIF and ZBP1 [428, 439]. Thus, in the absence of CASP8 activity, stimulation of TLR3 or TLR4 by their respective ligands promotes necroptosis upon the interaction between TRIF and RIPK3 and the consequent activation of MLKL [428]. Accordingly, the synthetic TLR3 ligand polyinosinic-polycytidylic acid (polyI:C) or the co-administration of low-dose LPS and the caspase inhibitor Z-VAD-fmk trigger necroptosis in dendritic cells (DCs) [518] or microglial cells [519], respectively. In this context, IFN alpha and beta receptor subunit 1 (IFNAR1) and IFN gamma receptor 1 (IFNGR1) also appear to have pro-necroptotic functions [520,521,522,523]. Thus, Ifnar1−/− macrophages are resistant to RCD induced by LPS or polyI:C in the context of caspase inhibition, which would otherwise trigger a necroptotic process relying on TRIF and tonic IFN-stimulated gene factor 3 (ISGF3) signaling [523]. Genetic studies demonstrate that the lethality imposed to mice by the Ripk1−/−Tnfrsf1a−/− genotype is delayed (but not prevented) by the co-deletion of Ticam1 or Ifnar1 [482]. Moreover, Ripk1−/− cells are more sensitive to necroptosis induced by polyI:C or type I IFN [482]. However, Tnfrsf1a−/−Ripk1−/−Ripk3−/− mice develop into adulthood, suggesting the existence of additional RIPK3 activators [482].
Recently, the mechanism underlying ZBP1-mediated necroptosis and its regulation by RIPK1 has been elucidated [440, 441]. ZBP1 acts at the initial steps of necroptosis by mediating the sequential activation of RIPK3 and MLKL. Moreover, mice expressing a variant of RIPK1 mutated in the RHIM domain die perinatally, a phenotype that can be rescued by concurrent Ripk3, Mlkl, or Zbp1 (but not Ticam1) deletion, as well as by the knock-in of catalytically inactive RIPK3 or RIPK3 mutated in the RHIM domain [440, 441]. This suggests that the RHIM of RIPK1 acts as an inhibitor of ZBP1-driven necroptosis, most likely because it prevents the interaction between ZBP1 and RIPK3. Further investigation is required to clarify the mechanisms of ZBP1 activation in this context and its relevance for development and homeostatic tissue regulation. Importantly, multiple components of the molecular machinery for necroptosis—including ZBP1, RIPK3, MLKL, and TNFR1 (mainly via NF-κB)—impinge on the control of the so-called “inflammasome”, a supramolecular platform for the activation of CASP1 and consequent secretion of mature interleukin 1 beta (IL1B; best known as IL-1β) and IL18 [524,525,526,527,528,529]. Discussing in detail these links—which exemplify the complex interconnection between RCD signaling and inflammatory responses—goes beyond the scope of this review [4, 8 530,531,532].
In summary, we propose to define necroptosis as a type of RCD triggered by perturbations of extracellular or intracellular homeostasis that critically depends on MLKL, RIPK3, and (at least in some settings) on the kinase activity of RIPK1 (Box 1).
Ferroptosis
Ferroptosis is a form of RCD initiated by specific perturbations of the intracellular microenvironment, notably severe lipid peroxidation, which relies on ROS generation and iron availability [533,534,535,536]. The molecular mechanisms precipitating ferroptosis have begun to emerge [534], and (so far) ferroptotic RCD has been linked to toxic lipid peroxide accrual [537, 538]. Ferroptosis occurs independently of caspases, necrosome components and CYPD, and the molecular machinery for autophagy [539], manifests with a necrotic morphotype (with a predominance of mitochondrial alterations encompassing shrinkage, an electron-dense ultrastructure, reduced/disappeared cristae, and ruptured OMM) [374], and is potentially associated with a consistent release of immunostimulatory DAMPs [540, 541]. Interestingly, BCL2 has been suggested to limit the physiological demise of neuron progenitors failing to differentiate via a mechanism that (1) does not depend on BAX and caspases, and (2) can be suppressed by ferroptosis inhibitors [542]. The actual implication of BCL2 in the regulation of ferroptosis, however, remains to be firmly established.
Some of the molecular circuitries regulating the initial steps of ferroptosis have been recently unveiled by employing (1) specific ferroptosis-inducing agents, including erastin [543, 544], RSL3 [543, 544], and FIN56 [545]; and (2) specific ferroptosis-inhibiting agents, including ferrostatins [539, 546] and liproxstatins [547]. In particular, the reduced glutathione (GSH)-dependent enzyme glutathione peroxidase 4 (GPX4)—which is directly targeted by RSL3—has emerged as the main endogenous inhibitor of ferroptosis by virtue of its ability to limit lipid peroxidation by catalyzing the GSH-dependent reduction of lipid hydroperoxides to lipid alcohols [547,548,549,550]. In line with this notion, erastin triggers ferroptosis by (indirectly) affecting the catalytic cycle of GPX4 via a mechanism that involves the inhibition of the cystine/glutamate antiporter system xc− and consequent decrease in intracellular cysteine (which derives from cystine reduction in the cytoplasm) and GSH (which is synthesized from cysteine) [539, 548, 549, 551]. Accordingly, depleting GSH with L-buthionine sulfoximine (BSO)—an inhibitor of the glutamate–cysteine ligase complex—can induce ferroptotic RCD (at least in some cases) [547]. Moreover, the toxicity of high extracellular glutamate may depend (at least in part) on the activation of ferroptosis through cysteine imbalance [534, 538, 552]. Of relevance for cancer therapy, the pronounced addiction of triple-negative breast carcinoma to glutamine relates (at least in part) to its ability to drive cystine uptake via xc−, implying that xc− may constitute a therapeutic target in this setting [553, 554]. Moreover, the FDA-approved tyrosine kinase inhibitor sorafenib can trigger ferroptosis in distinct cellular models by depleting GSH upon system xc− inhibition [551 555,556,557], while altretamine (an FDA-approved alkylating agent) has been recently identified as a potential inhibitor of GPX4 by a regulatory network genome-wide system strategy [558]. Thus, the antineoplastic effects of sorafenib and altretamine may partially stem from the activation of ferroptosis. Notably, the demise of neurons caused by inhibition of xc− was initially referred to as oxytosis, oxidative glutamate toxicity, or excitotoxicity, and was linked to alterations in intracellular Ca2+ homeostasis [559,560,561]. It remains unclear to which extent oxytosis can be mechanistically discriminated from ferroptosis and MPT-driven necrosis in diverse cellular contexts.
Recent evidence indicates that ferroptosis involves the preferential oxidation of specific phosphatidylethanolamine-containing polyunsaturated fatty acids (PUFAs) such as arachidonic and adrenic acid [562]. In line with a critical requirement for oxidizable PUFAs, genetic and/or pharmacological inhibition of acyl-CoA synthetase long chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), both of which are involved in the incorporation of long PUFAs into cellular membranes, protects cells against ferroptosis (at least in some settings) [562,563,564,565]. Lipid hydroperoxides can be formed by autoxidation or via enzymatic reactions catalyzed by lipoxygenases (LOXs) or cyclooxygenases (COXs). In the context of ferroptosis, PUFA peroxidation seems to be mainly regulated by the mutually antagonistic activity of LOXs (which directly catalyze lipid peroxidation) and GPX4 (which indirectly inhibits it) [550, 566]. Although arachidonate 15-lipoxygenase (ALOX15) was initially thought to play a major role in this setting, the deletion of Alox15 fails to rescue the renal phenotype imposed by the Gpx4−/− genotype (see below) [547], suggesting that multiple LOXs are involved in PUFA peroxidation and consequent ferroptosis in some mouse tissues. Accordingly, oxidized PUFAs accumulate upon GPX4 inactivation and this can result in PUFA fragmentation and ferroptosis [539, 547]. This lethal cascade can be prevented by antioxidant agents such as ferrostatin-1 (Fer-1), liproxstatin-1 (Lip-1) as well as by vitamin E, coenzyme Q10 and their analogs, all of which efficiently limit lipid peroxidation by operating as ROS scavengers [539, 547, 550, 562 567,568,569]. Of note, the catalytic sites of LOXs contain di-iron centers [570]. This may explain: (1) the ferroptosis-inhibiting effect of iron depletion by either chelators [539, 543, 548] or phosphorylase kinase catalytic subunit gamma 2 (PHKG2) knockdown [566], and (2) the ferroptosis-promoting effect of increased intracellular iron availability consequent to import by the circulating iron carrier transferrin (TF) [571, 572], degradation of ferritin (a cellular iron storage complex) by a specific autophagic mechanism known as ferritinophagy [573, 574], disruption of iron homeostasis induced by nanoparticles [541], or administration of a bioavailable iron form [575]. Alternatively, the critical dependency of ferroptosis on iron can also be ascribed to the ability of this heavy metal to promote non-enzymatic lipid oxidation via lysosomal Fenton reactions [538, 572, 576, 577].
Additional ferroptosis regulators described so far include: (1) the mevalonate pathway component farnesyl-diphosphate farnesyltransferase 1 (FDFT1; best known as SQS) [545]; (2) the transsulfuration pathway enzyme cysteinyl-tRNA synthetase (CARS) [578]; (3) heat shock protein family B (small) member 1 (HSPB1; best known as HSP27) [579] and heat shock protein family A (Hsp70) member 5 (HSPA5) [580]; (4) glutaminolysis [571]; (5) components of the MAPK signaling pathway [539, 581]; (6) the nuclear factor, erythroid 2 like 2 (NFE2L2; best known NRF2) signaling pathway [582]; (7) metallothionein 1G (MT1G) [583]; (8) dipeptidyl peptidase 4 (DPP4) [584]; (9) Fanconi anemia complementation group D2 (FANCD2) [585]; and (10) CDGSH iron sulfur domain 1 (CISD1; also known as mitoNEET) [586]. Elucidating the precise role of these proteins or signaling pathways in ferroptosis requires further investigation.
Accumulating evidence demonstrates that the pro-survival functions of GPX4 contribute to development and homeostatic tissue maintenance. Gpx4−/− mice display embryonic lethality with complete penetrance [547, 550, 587, 588]. Moreover, the inducible or tissue-specific ablation of Gpx4 in mice provokes a variety of pathological conditions, including acute renal or hepatic injury [547, 563, 589], neurodegeneration [550, 590, 591], and defective immunity to infection [567], all of which can be prevented or mitigated by ferroptosis-inhibiting strategies. A similar protective effect is observed in GPX4-independent models of renal ischemic or toxic injury [540, 592], Parkinson disease [593], and other human pathologies [594]. Moreover, ferroptosis appears to operate as a bona fide oncosuppressive mechanism [548 595,596,597,598]. It has been proposed—but remains to be formally established—that part of the multipronged oncosuppressor functions of p53 may derive from the transcriptional downregulation of components of system xc−, which would impinge on specific post-translational modifications of p53 [596, 598]. Accordingly, the ability of ATF4 to upregulate system xc− and stabilize GPX4 (upon HSPA5 transactivation) is causally involved in some models of oncogenesis and chemoresistance to ferroptosis induction [580, 599]. Along similar lines, parts of the oncogenic effects of NRF2 activation driven by cancer-associated mutations in kelch like ECH associated protein 1 (KEAP1) may derive from the upregulation of system xc− [582]. Conversely, p53 appears to inhibit ferroptosis in colorectal cancer cells, at least in part by inhibiting DPP4 activity in a transcription-independent manner [584]. Of note, malignant cells with a mesenchymal phenotype (which are generally more resistant to treatment) reportedly acquire an accrued dependency on GPX4 activity, which can be exploited therapeutically [600]. Recently, a ferroptosis-like RCD subroutine has been described in plants responding to moderate heat stress, supporting some degree of evolutionary conservation and the relevance of ferroptosis for organismal homeostasis [601]. In this context, it is worth noting that the pharmacological inhibition of ferroptosis, but not necroptosis or apoptosis, protects tissues such as renal tubules from ischemia/reperfusion injury [540]. The precise role of ferroptosis in development and tissue homeostasis, however, remains to be fully elucidated.
We propose to define ferroptosis as a form of RCD initiated by oxidative perturbations of the intracellular microenvironment that is under constitutive control by GPX4 and can be inhibited by iron chelators and lipophilic antioxidants (Box 1).
Pyroptosis
Pyroptosis is a form of RCD triggered by perturbations of extracellular or intracellular homeostasis related to innate immunity (e.g., pathogen invasion) manifesting with specific morphological feature [602]. These include a peculiar form of chromatin condensation that differs from its apoptotic counterpart, as well as cellular swelling culminating with plasma membrane permeabilization [602]. The term pyroptosis was originally coined by Cookson and Brennan to define a particular type of RCD partially resembling apoptosis but dependent on inflammatory CASP1 (and hence linked to pyrexia) [603], and several names including pyronecrosis have been introduced since to define partially related processes [604, 605]. Initially, pyroptosis was thought to be relevant only for the demise of monocytes or macrophages undergoing canonical CASP1 activation [606, 607]. However, recent findings indicate that pyroptosis (1) can be also driven by several other caspases including CASP3 [608], (2) can also occur in cell types other than cells from the monocytic lineage [609], (3) has a major role in innate immunity against intracellular pathogens [602], and (4) is etiologically involved in pathological conditions such as lethal septic shock (at least as induced by LPS) [610, 611].
At a molecular level, pyroptosis generally relies on the activation of one or more caspases, including CASP1, CASP3, murine CASP11, and its human homologs CASP4 and CASP5, depending on the initiating stimulus [612, 613]. Thus, pyroptosis is often (if not always) associated with IL-1β and IL18 secretion, and hence mediates robust pro-inflammatory effects [614, 615]. A large body of evidence indicates that cytosolic LPS from invading Gram-negative bacteria is a major trigger of pyroptosis. In particular, it has been shown that CASP11 is responsible for the CASP1-independent death of macrophages responding to Gram-negative bacterial infection [616,617,618,619]. Moreover, Casp11 deletion protects mice against a challenge with cytosol-invasive bacteria [612, 620], as well as against systemic LPS administration and consequent pyroptosis-dependent endotoxic shock [616, 621]. Further experimental observations confirmed that CASP11, CASP4, and CASP5 trigger pyroptosis upon sensing cytosolic LPS [609 622,623,624], in monocytes as well as in other cell types [609, 625]. In particular, LPS-induced pyroptosis involves the physical interaction of LPS (or its lipid moiety) with the CARD domain of CASP11, CASP4, or CASP5, a highly specific binding resulting in caspase oligomerization and consequent activation [609]. Thus, CASP11, CASP4, and CASP5 act as bona fide PRRs for cytosolic LPS. Once activated beyond a specific threshold, inflammatory caspases precipitate pyroptosis by catalyzing the proteolytic cleavage of GSDMD [621, 626, 627]. However, at least in some cell types including DCs, CASP11 activation can drive IL-1β secretion in the absence of cell death [628].
In line with a critical role of GSDMD, Gsdmd−/− macrophages are resistant to LPS-induced and Gram-negative bacteria-induced pyroptosis, and Gsdmd−/− mice survive doses of LPS that induce lethal endotoxic shock in their wild-type counterparts [621, 626]. Recent findings demonstrate that GSDMD is constitutionally auto-inhibited by the binding of its C-terminal repressor domain (GSDMD-C, or RD) to its N-terminal pore-forming domain (GSDMD-N, or PFD). On pyroptosis induction, inflammatory caspases relieve this inhibition by catalyzing the proteolytic cleavage of the interdomain loop, which promotes the release of the pyroptotic inducer GSDMD-N [621, 629]. Cleaved GSDMD-N acquires the ability to translocate to the inner leaflet of the plasma membrane (or the bacterial plasma membrane), where it binds with high specificity to selected phosphoinositides (or cardiolipin) [629, 630]. Membrane targeting enables GSDMD-N oligomerization, generating a pore that is responsible for rapid plasma membrane permeabilization [629,630,631,632,633]. Of note, the GSDMD pore has recently been characterized at the ultrastructural level, consisting of 16 symmetric protomers with an inner diameter of ~10–14 nm [629].
Active CASP1 can also cleave GSDMD, suggesting that microbe-associated molecular patterns (MAMPs) other than cytosolic LPS as well as DAMPs usually stimulating canonical inflammasome signaling can initiate pyroptosis [626 634,635,636]. CASP1-driven pyroptosis limits the spreading of intracellular bacteria by (1) killing the host cell [637], and (2) generating so-called “pore-induced intracellular traps (PITs)”, which essentially are dead macrophages that can be efficiently disposed of (together with the living bacteria they trap) by efferocytosis [638]. Supporting the critical role of CASP1-driven pyroptosis for innate immune responses against invading bacteria, Nlrc4−/− mice (which are unable to normally activate CASP1) succumb to low amounts of otherwise innocuous environmental opportunists (e.g., Chromobacterium violaceum) [639]. Instances of CASP1-dependent but GSDMD-independent RCD have also been reported, including the demise of macrophages experiencing prolonged canonical inflammasome activation [621, 636]. Of note, additional substrates of inflammatory caspases have been hypothesized to participate in pyroptosis. Experiments with knockout mice suggest that the endotoxic shock caused by systemic LPS administration involves the CASP11-dependent cleavage not only of GSDMD, but also of pannexin 1 (PANX1) channels, leading to ATP release in the extracellular space and consequent activation of purinergic receptor P2X 7 (P2RX7), which further impinges on the collapse of ionic gradients and inflammasome signaling [640]. Most likely, however, these findings reflect the presence of an inactivating passenger mutation in Casp11 specifically affecting transgenic mice generated from 129/Sv-derived embryonic stem cells [641]. Moreover, CASP1 has been proposed to drive pyroptosis by causing mitochondrial damage upon cleavage of parkin RBR E3 ubiquitin protein ligase (PRKN; best known as PARKIN) [642]. However, contrasting observations have been reported [528], and the actual pathophysiological relevance of the latter mechanism remains to be established.
Accumulating evidence indicates that the (shared) N-terminal domains of other members of the gasdermin family, including GSDMA, GSDMB, GSDMC, GSDME/DFNA5, and GSDMA3 (which is encoded by the mouse—but not the human—genome), resemble those of GSDMD as they display pore-forming and pyroptotic activity [39, 613, 626, 629]. Although the mechanisms underlying the activation of GSDMA remain unknown, two recent studies demonstrate the existence of an instance of pyroptotic RCD dependent on GSDME-N/DFNA5-N that can be elicited by multiple challenges, including TNF, various DNA-damaging chemotherapeutic agents, and/or infection with the vesicular stomatitis virus [39, 613]. In this setting, CASP3 is responsible for the proteolytic cleavage of GSDME/DFNA5, which precipitates in pyroptosis rather than apoptosis [39, 613]. As GSDME/DFNA5 is often silenced in malignant cells but expressed by their normal counterparts, the activation of GSDME/DFNA5 by CASP3 contributes to the side effects of multiple chemotherapeutic agents, at least in mice [613]. Interestingly, GSDME/DFNA5 has also been involved in the acquisition of a necrotic phenotype by cells undergoing CASP3-driven apoptosis in vitro (in the absence of proficient phagocytosis) [39], further demonstrating the elevated degree of interconnectivity that exists between distinct types of RCD. The identification of multiple gasdermin family members as key factors in the late steps of pyroptosis, as well as the characterization of CASP3 as an activator of GSDME/DFNA5, expanded the relevance of this RCD form (and its definition, see below) well beyond inflammatory settings [613, 626, 629]. Of note, type I IFN and IFN gamma (IFNG) also contribute to pyroptosis by promoting: (1) the transactivation of CASP11, through an IFNAR1-dependent or IFNGR1-dependent mechanism initiated by TLR4 or IL18 signaling [620, 643, 644]; (2) TLR7, cyclic GMP-AMP synthase (CGAS), transmembrane protein 173 (TMEM173; best known as STING), DExD/H-box helicase 58 (DDX58; best known as RIG-I), or mitochondrial antiviral signaling protein (MAVS) signaling upon bacterial or viral infection [525, 645]; or (3) the expression of guanylate-binding proteins and an IFN-inducible GTPase commonly known as IRGB10 (official name Gm12250), which increase cytosolic LPS levels by mediating the lysis of vacuoles containing Gram-negative bacteria [646,647,648].
Further underscoring the complexity of the interaction between inflammation and pyroptosis [649], CASP11 can also be upregulated by a complement cascade dependent on carboxypeptidase B1 (CPB1) acting downstream of TLR4 and IFNAR1 activation [650]. Moreover, cytosolic LPS promotes the secretion of inflammatory cytokines by a mechanism involving CASP11 activation, followed by GSDMD cleavage, loss of K+ ions, and consequent activation of CASP1 by the NLR family pyrin domain containing 3 (NLRP3) inflammasome, in vitro (but perhaps not in vivo) [616, 620, 621, 626 651,652,653,654,655].
We propose to define pyroptosis as a form of RCD that critically depends on the formation of plasma membrane pores by members of the gasdermin protein family, often (but not always) as a consequence of inflammatory caspase activation. The NCCD discourages the use of alternative terms including pyronecrosis (Box 1).
Parthanatos
Parthanatos is a form of RCD driven by the hyperactivation of a specific component of the DNA damage response (DDR) machinery, namely, poly(ADP-ribose) polymerase 1 (PARP1). Notably, parthanatos appears to occur not only as a consequence of severe/prolonged alkylating DNA damage, but also in response to oxidative stress, hypoxia, hypoglycemia, or inflammatory cues [656,657,658]. In this context, reactive nitrogen species (RNS) including NO stand out as major triggers for PARP1 hyperactivation, especially in neurons [659,660,661]. PARP1 hyperactivation mediates cytotoxic effects as it causes (1) NAD+ and ATP depletion, which ultimately results in a bioenergetic and redox collapse, and (2) the accumulation of poly(ADP-ribose) polymers and poly(ADP-ribosyl)ated proteins at mitochondria, ultimately causing Δψm dissipation and MOMP [656, 657 662,663,664,665].
One of the key processes of parthanatos is the binding of poly(ADP-ribose) polymers to apoptosis inducing factor mitochondria associated 1 (AIFM1; best known as AIF). This promotes the release of AIF into the cytosol and its translocation into the nucleus, where it mediates large-scale DNA fragmentation and chromatin condensation [656, 658 664,665,666,667]. Further corroborating a key role for poly(ADP-ribosyl)ation in parthanatos, the poly(ADP-ribose)-degrading protein ADP-ribosylhydrolase like 2 (ADPRHL2; also known as ARH3) and the poly(ADP-ribose)-binding protein ring finger protein 146 (RNF146; best known as IDUNA) prevent AIF release and consequent RCD as they decrease poly(ADP-ribose) levels and availability, respectively [662, 668]. Moreover, specific pharmacological inhibitors of PARP1 efficiently delay parthanatos in multiple cell types, in vitro and in vivo [1, 669]. Parthanatotic DNA fragmentation occurs independently of apoptotic caspases and endonuclease G (ENDOG) [670], a mitochondrial nuclease that precipitates RCD by a mechanism involving its release followed by translocation to the nucleus (at least in Saccharomyces cerevisiae, Caenorhabditis elegans, and Drosophila melanogaster) [671,672,673,674]. Indeed, the actual contribution of ENDOG to RCD in mammals [675] has been questioned by the generation of Endog−/− mice, whose cells display normal sensitivity to multiple lethal triggers [676, 677]. That said, it seems that the catalytic activity of CPS-6 (the homolog of ENDOG in C. elegans) is boosted upon interaction with WAH-1 (the worm homolog of AIF) [678].
Macrophage migration inhibitory factor (MIF) has emerged as the main nuclease precipitating parthanatos in a recent screening for AIF-binding proteins [679]. Thus, cytosolic AIF reportedly promotes the translocation of MIF into the nucleus, where MIF precipitates parthanatos by catalyzing DNA cleavage. Accordingly, MIF depletion or specific mutations in its nuclease domain confer protection against parthanatos in vitro and in vivo (in an experimental model of focal cerebral ischemia) [679]. Another protein involved in parthanatos is hexokinase 1 (HK1), whose binding to poly(ADP-ribose) polymers inhibits glycolysis to cause a bioenergetic collapse that precipitates RCD [680, 681]. Recently, a non-canonical, AIF-independent instance of parthanatos, presumably centered on the impairment of energetic metabolism, has been proposed to contribute to the demise of retinal epithelial cells and consequent retinal degeneration [682]. Interestingly, some authors suggest a certain degree of interconnectivity between the parthanatotic and the necroptotic machineries. Thus, upon induction of necroptosis by TRAIL or β lapachone (an ortho naphthoquinone with antineoplastic activity) [683], activated RIPK1 and RIPK3 appear to stimulate the enzymatic activity of PARP1 and hence promote ATP depletion and/or AIF release [684, 685]. This interpretation may not hold true in all experimental settings [686].
Parthanatos reportedly contributes to various pathological conditions, including some cardiovascular and renal disorders, diabetes, cerebral ischemia, and neurodegeneration [534, 656 687,688,689,690]. Accordingly, PARP1 inhibition by pharmacological or genetic interventions mediates robust cytoprotective effects in multiple animal models of disease [1, 669]. However, further experiments are required to clarify the actual role of parthanatos in the etiology of these (and possible other) pathologies and the true therapeutic benefits of parthanatos-inhibiting agents.
The NCCD proposes to define parthanatos as a form of RCD initiated by PARP1 hyperactivation and precipitated by the consequent bioenergetic catastrophe coupled to AIF-dependent and MIF-dependent DNA degradation (Box 1).
Entotic cell death
Entosis is a form of cell cannibalism that occurs in healthy and malignant mammalian tissues, involving the engulfment of viable cells by non-phagocytic cells of the same (homotypic) or a different (heterotypic) type [691, 692]. Often (but not always), internalization is followed by the demise of internalized cells (which are commonly referred to as “entotic cells”) [691,692,693].
Entosis is mainly triggered by the detachment of epithelial cells from the extracellular matrix and consequent loss of integrin signaling [263, 694], although alternative mechanisms have been reported. These include: (1) the deregulated expression of myosins during the formation of cell-to-cell contacts [695]; and (2) differences in the mechanical properties [696] or responses to metabolic stress [697] of cancer cells competing for proliferation. Moreover, a recent study suggests the existence of a specific form of entosis occurring in cancer cells during mitosis (entotic mitosis), which is driven by aberrant mitotic rounding (and thus reduced adhesion) in conditions of cell division cycle 42 (CDC42) depletion or upon exposure to antimitotic agents [698].
The current model proposes that the internalization of entotic cells occurs through cell invasion rather than by phagocytosis [691]. Accordingly, the uptake of entotic cells is an integrin-independent process promoted by the formation of junctions between engulfing and entotic cells that involve the adhesion proteins cadherin 1 (CDH1; also known as E-cadherin) and catenin alpha 1 (CTNNA1) [694, 699]. Actomyosin chains accumulate at the cortex of internalizing cells (at the pole opposite to the cell-to-cell contact site), via a mechanism that depends on the localized activity of ras homolog family member A (RHOA), Rho associated coiled-coil containing protein kinase 1 (ROCK1), ROCK2, and diaphanous related formin 1 (DIAPH1), and results in a contraction that promotes engulfment [695 699,700,701]. Actin drives entosis by promoting pro-invasive (non-apoptotic) cortical plasma membrane blebbing upon activation of a signaling pathway that involves myocardin-related transcription factor (MRTF) and serum response factor (SRF) culminating with ezrin (EZR) upregulation [702]. The regulation of microtubule dynamics by AURKA has also been attributed a role in cell invasion [703], but the relevance of AURKA signaling for entosis awaits experimental confirmation. In line with the actomyosin-dependent cell-in-cell invasion model, the administration of exogenous CDH1 promotes entosis among CDH1-deficient breast cancer cells, whereas forced overexpression of RHOA or ROCK1 plus ROCK2 enables the internalization of entotic cells by epithelial CDH1-expressing cells [701]. In addition, hyperactivation of contractile myosin induces entotic cell-in-cell invasion via a mechanism involving the activation of RHOA, ROCK1, and ROCK2 [695]. Intriguingly, competition in the tumor system can occur via an entotic process whose outcome is dictated by the activation of KRAS proto-oncogene, GTPase (KRAS), and Rac family small GTPase 1 (RAC1) signaling, which confers an advantage to engulfing cells by favoring myosin downregulation [696]. In this context, it has recently been demonstrated that, in conditions of glucose withdrawal, cells displaying high 5′ AMP-activated protein kinase (AMPK) activity succumb to entosis, underscoring a possible function of this process for nutrient recovery by cells with comparatively lower AMPK activity (which a priori are metabolically fitter) [697].
Once engulfed, entotic cells are often eliminated by a RCD subroutine that occurs independently of BCL2 proteins and caspases [694, 704], but relies (at least in part) on a specific autophagy-related process commonly known as LC3-associated phagocytosis (LAP) [509, 705, 706]. In this context, some (but not all) components of the macroautophagy apparatus, including microtubule associated protein 1 light chain 3 beta (MAP1LC3B; best known as LC3), autophagy related 5 (ATG5), ATG7, and phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3; best known as VPS34) are recruited to the cytosolic side of entotic cell-containing vesicles and promote their fusion with lysosomes (in the absence of bona fide autophagosome formation) [704]. Eventually, the lysosomal degradation of internalized entotic cells generates nutrients that are recovered by engulfing cells, via a mechanism that reportedly involves phosphoinositide kinase, FYVE-type zinc finger containing (PIKFYVE) [704, 707, 708].
Entotic cell death has been documented in several human neoplasms, presumably operating as an oncosuppressor mechanism [694, 695, 701, 709, 710]. Thus, abrogation of entosis by a chemical ROCK inhibitor reportedly favors the anchorage-independent growth of malignant cells [694]. However, entotic invasion has also been suggested to favor aneuploidization and polyploidization (which promote tumor progression) [711,712,713] through a mechanism involving cytokinesis failure of engulfing cells [714, 715]. A potential role for entosis in development and tissue homeostasis has recently been proposed. Thus, in the course of mammalian embryo implantation, trophoblast cells reportedly eliminate uterine luminal epithelial cells upon entosis [716]. Moreover, the spermatozoa of hibernating Chinese soft-shelled turtle appear to be degraded within Sertoli cell by entotic cell death [717]. Further experiments are required to elucidate the actual role of entosis in the pathophysiology of mammalian organisms. Importantly, entosis does not always lead to the death of invading cells within the lysosome. Thus, at least in some circumstances, entotic cells remain viable and even proliferate inside host cells or upon escape [716].
On the basis of this consideration, we propose to define entotic cell death as a form of RCD that originates from actomyosin-dependent cell-in-cell internalization and is executed by lysosomes (Box 1). In the absence of precise experimental determination of terminal cell fate, we recommend to use the term entosis to refer to the internalization process only.
NETotic cell death
The term “NETotic cell death” refers to a rather controversial type of RCD initially characterized in neutrophils for being associated with the extrusion of a meshwork of chromatin-containing and histone-containing fibers bound to granular and cytoplasmic proteins known as neutrophil extracellular traps (NETs), a process commonly referred to as NETosis [718, 719]. NETs, which are produced in response to various microbial and sterile activators or upon stimulation of specific receptors including (but not limited to) TLRs, de facto constitute a stable extracellular platform for trapping and degrading microbes [718 720,721,722,723]. Several reports demonstrate that a considerable fraction of the nucleic acids contained in NETs is of mitochondrial, rather than nuclear, origin [724,725,726,727,728]. Besides having antimicrobial effects, NETs reportedly contribute to the etiology of some human pathologies, including diabetes and cancer [729,730,731]. Of note, NET-like structures can be released by cells other than neutrophils, including mast cells [732], eosinophils [733], and basophils [734]. Importantly, NET extrusion per se does not necessarily result in cellular lysis [722, 724, 735].
Although the precise molecular mechanisms underlying NET generation are not fully elucidated, both NETotic cell death and NET extrusion in the absence of RCD appears to rely on the activity of NADPH oxidases [724, 736, 737]. NETotic cell death has been suggested to result from a signaling pathway that involves Raf-1 proto-oncogene, serine/threonine kinase (RAF1), mitogen-activated protein kinase kinases (MAP2Ks), and ERK2, culminating with NADPH oxidase activation and consequent ROS generation [736, 738, 739]. According to this model, intracellular ROS would drive NETotic cell death (1) by triggering the release of elastase, neutrophil expressed (ELANE), and myeloperoxidase (MPO) from neutrophil granules to the cytosol, followed by their translocation to the nucleus, and (2) by promoting the MPO-dependent proteolytic activity of ELANE [740]. Once activated, the cytosolic pool of ELANE would catalyze the proteolysis of F-actin, followed by an impairment of cytoskeleton dynamics [741]. Alongside, the nuclear pool of ELANE would promote the degradation of histones (and possibly of the nuclear envelope) and, in conjunction with MPO, chromatin decondensation [737 740,741,742]. This would culminate with the extrusion of chromatin fibers intermixed with cytoplasmic and nuclear components, ultimately leading to plasma membrane rupture and RCD [736]. That said, recent findings indicate that ROS drive NET extrusion by a mechanism that requires an intact cytoskeleton [743]. Moreover, ELANE is dispensable for NET formation in the course of deep vein thrombosis (in mice) [744]. Peptidyl arginine deiminase 4 (PADI4; also known as PAD4) has also been proposed to participate in chromatin dispersion [745], but its actual involvement remains a matter of debate and appears to depend on the initiating stimulus [746, 747]. Finally, NETotic cell death has been proposed to depend (at least in part) on components of the necroptotic apparatus, based on the fact that the administration of chemical RIPK1 or MLKL inhibitors (i.e., Nec-1 or NSA, respectively) as well as the Ripk3−/− genotype appeared to inhibit NET extrusion and neutrophil lysis [748]. However, Ripk3−/− neutrophils as well as neutrophils exposed to NSA were fully proficient in NET formation in another study [749]. These apparently contradicting findings call for additional studies to address the precise contribution of necroptosis to NET extrusion and NETotic cell death.
We propose to define NETotic cell death as a ROS-dependent modality of RCD restricted to cells of hematopoietic derivation and associated with NET extrusion (Box 1). That said, it is clear that NET can be formed and extruded by fully viable neutrophils, eosinophils, and basophils. Thus, the NCCD recommends to avoid the use of the term NETosis when no experimental evidence in support of cell death (vs, NET extrusion only) is available. Moreover, we discourage the use of alternative terms proposed to describe this process, including ETosis.
Lysosome-dependent cell death
Lysosome-dependent cell death is a subroutine of RCD initiated by perturbations of intracellular homeostasis and demarcated by the permeabilization of lysosomal membranes. Lysosome-dependent cell death is relevant for several pathophysiological conditions, including inflammation, tissue remodeling (e.g., mammary gland involution after lactation), aging, neurodegeneration, cardiovascular disorders, and intracellular pathogen response [750,751,752]. Moreover, a type of RCD that is highly reminiscent of lysosome-dependent cell death, which has been dubbed “germ cell death”, appears to play a critical role in the physiological elimination of a fraction of emerging male germ cells (at least in D. melanogaster) [753,754,755,756].
At a biochemical level, lysosome-dependent cell death proceeds upon lysosomal membrane permeabilization (LMP), resulting in the release of lysosomal contents, including proteolytic enzymes of the cathepsin family, to the cytoplasm [750]. The molecular mechanisms operating upstream of LMP are not fully elucidated. In some circumstances, LMP appears to occur downstream of MOMP as a result of apoptotic signaling, de facto constituting an epiphenomenon of intrinsic apoptosis [757,758,759]. In other experimental settings, however, lysosomes are permeabilized before mitochondria [752, 760, 761], via a mechanism that optionally involves BAX recruitment to the lysosomal membrane followed by the activation of its pore-forming activity [762,763,764,765]. More commonly, ROS play a prominent causal role in LMP, not only as the H2O2-driven luminal production of hydroxyl radicals by Fenton reactions destabilizes the lysosomal membrane upon lipid peroxidation [766, 767], but also as ROS reportedly favor the activation of lysosomal Ca2+ channels [768]. Primary LMP has been documented in vitro in cells responding to specific pro-apoptotic stimuli, including the administration of lysosomotropic agents such as L-leucyl-L-leucine methyl ester, ciprofloxacin, and hydroxychloroquine, TRAIL signaling, and viral infection [760, 761, 765 769,770,771,772], as well in an animal model of Parkinson's disease [764]. The p53 effector DNA damage regulated autophagy modulator 1 (DRAM1) [773] provides a major contribution to lysosome-dependent cell death in HIV1-infected T cells by linking LMP to MOMP [770]. Additional LMP triggers include lysosomotropic agents (e.g., sphingosine), calpains, and ROS [751]. Moreover, STAT3 reportedly promotes lysosome-dependent cell death during the involution of mammary gland post-lactation as it upregulates the expression of cathepsin B (CTSB) and CTSL, while downregulating their endogenous inhibitor serine (or cysteine) peptidase inhibitor, clade A, member 3G (SERPINA3G; best known as SPI2A) [774, 775].
Cytosolic cathepsins usually precipitate RCD by catalyzing the proteolytic activation or inactivation of several substrates, including BID, BAX, anti-apoptotic BCL2 family members, and XIAP [776,777,778], hence engaging a feed-forward amplification circuitry of the lethal cascade involving MOMP and caspases. Moreover, primary lysosomal dysfunction may negatively affect the mitochondrial network as a consequence of impaired mitophagic responses (which normally target damaged or dysfunctional mitochondria to lysosomes for degradation) [779, 780]. In aged neutrophils, LMP also allows for the release of proteinase 3 (PRTN3) from cytotoxic granules, where it promotes RCD by catalyzing the proteolytic activation of CASP3 [781]. Of note, lysosome-dependent cell death does not necessarily involve MOMP and caspases, and does not necessarily manifest with an apoptotic morphotype [782]. Moreover, CTSL appears to play a key role in the regulation of autophagic adaptation vs. RCD in cells responding to the LMP inducer resveratrol [783, 784]. These observations suggest that LMP and lysosome-dependent cell death are intimately interconnected with adaptative responses to stress and other RCD subroutines.
Lysosome-dependent cell death can be retarded in vitro and in vivo by inhibiting LMP or blocking cathepsin activity via pharmacological or genetic means [750, 752]. Commonly employed cathepsin-targeting molecules include endogenous protease inhibitors (cystatins and serpins), as well as various pharmacological agents specific for cysteine cathepsins (e.g., E64D and Ca-074-Me) or aspartyl cathepsins (e.g., pepstatin A) [785,786,787]. Moreover, under physiological conditions, lysosomal membranes can be stabilized by altering lysosomal cholesterol content [788] or by boosting endogenous activity of heat shock protein family A (Hsp70) member 1A (HSPA1A; best known as HSP70) [789, 790]. In line with this notion, the administration of recombinant HSP70 or the HSP70-inducing agent arimiclomol reverts lysosomal abnormalities in cellula as well as in murine models of various lysosomal storage disorders [789, 791]. Of relevance for cancer therapy, cancer cells may present an increased sensitivity to lysosomotropic agents and are generally vulnerable to LMP, which supports the clinical development of LCD-inducing agents [752 792,793,794,795].
We propose to define lysosome-dependent cell death as a form of RCD demarcated by primary LMP and precipitated by cathepsins, with optional involvement of MOMP and executioner caspases (Box 1).
Autophagy-dependent cell death
Autophagy-dependent cell death is a type of RCD that relies on the autophagic machinery or components thereof [509 796,797,798]. Proficient autophagic responses (which are under tight transcriptional and post-translational regulation) [509 799,800,801,802,803,804,805] most often operate at the hub of adaptation to stress, hence mediating cytoprotective (rather than cytotoxic) effects [806,807,808,809,810,811]. Thus, blocking autophagy with pharmacological or genetic interventions generally accelerates (rather than delays) the demise of cells responding to stress, and permanent or transient endogenous defects in autophagy have been associated with embryonic lethality, developmental defects, and multiple pathological disorders, including (but not limited to) neurodegeneration, cancer, and cardiovascular disorders [812,813,814,815,816,817]. However, in a number of developmental and pathophysiological settings, the molecular machinery for autophagy etiologically contributes to cellular demise [796, 798 818,819,820]. Thus, the term autophagy-dependent cell death does not refer to settings in which the autophagic apparatus (or components thereof) is activated alongside (rather than precipitates) RCD [821] or it favors the engagement of other RCD modalities, such as (1) ferroptosis, which is promoted by the autophagic degradation of ferritin (ferritinophagy) [573, 574]; (2) FAS-driven extrinsic apoptosis, which is enhanced by the autophagic degradation of protein tyrosine phosphatase, non-receptor type 13 (PTPN13; best known as FAP1) [822], and (3) necroptosis, which is favored by a necrosome-scaffolding function of the autophagy apparatus [823,824,825], as well as by the autophagic degradation of c-IAP1 and c-IAP2 [826].
The genetics and pathophysiological significance of autophagy-dependent cell death is now well established [818,819,820]. Thus, whereas the genetic inactivation of caspases in the midgut of developing D. melanogaster has no consequences, mutations or deletions in essential autophagy-related (Atg) genes suppress midgut tissue degradation [827,828,829]. Along similar lines, the complete removal of larval salivary glands from D. melanogaster larvae undergoing metamorphosis requires the apoptotic as well as the autophagic machinery [830,831,832]. In both these developmental scenarios, autophagy-dependent cell death is preceded by growth arrest and is controlled by ecdysone, a steroid hormone that is critically required in Drosophila to undergo the larva-to-pupa transition and subsequent metamorphosis into an adult fly [830, 833]. The autophagic machinery also precipitates germ cell and ventral cord neuron RCD during C. elegans development [834], and perhaps contributes to embryonic development in mammals, as suggested by the fact that Atg5 ablation in apoptosis-deficient (i.e., Bax−/−Bak−/−) mice further delays interdigital web clearance, aggravates cerebral abnormalities (at least in the C57BL/6 background), impairs negative selection of autoreactive thymocytes, and increases the resistance of some cell types to multiple stressors [835, 836].
Of note, the molecular machinery of autophagy-dependent cell death and adaptative autophagy exhibit some differences (at least in D. melanogaster) [837, 838]. For example, the autophagy-dependent degradation of the midgut tissue proceeds independently of Atg7, Atg3, and several other Atg genes that are required for starvation-induced autophagy in the fat body, but relies on ubiquitin activating enzyme 1 (Uba1) [837, 838]. Moreover, the developmental degradation of Drosophila salivary glands by autophagy requires the activity of: (1) Utx histone demethylase (Utx), which contributes to the transcriptional regulation of apoptosis and autophagy genes [839]; (2) miR-14, which specifically activates autophagy-dependent cell death by modulating multiple IP3-driven signaling pathways upon targeting inositol 1,4,5-triphosphate kinase (IP3K2) [840]; (3) Ras-like protein A (Rala), which ignites autophagy-dependent cell death upon Notch activation [841]; (4) Draper (Drpr), which is thought to promote the engulfment of dying salivary gland cells [842]; and (5) macroglobulin complement-related (Mcr), which promotes autophagy-dependent cell death at least in part by triggering Drpr signaling [843]. Of note, the apoptotic and autophagic machineries are highly interconnected during developmental cell death [796]. Thus, during Drosophila oogenesis apoptotic proteins, including effector caspases, regulate autophagy-dependent cell death [844], while the autophagic apparatus acts upstream of DNA fragmentation by promoting IAP degradation and caspase activation [845, 846]. In this context, the autophagic apparatus also drives the developmental clearance of apoptotic cells [796].
Autophagy-dependent cell death also appears to contribute to the pathogenesis of some human disorders. The neuron-specific deletion of Atg7 confers robust neuroprotection in a mouse model of severe neonatal hypoxia-ischemia by preventing neuronal RCD [847]. Along similar lines, abolishing autophagy by pharmacological means or by genetically depleting ATG5 or BECN1, prevents the neurotoxicity of cocaine in cultured neurons [848]. Recently, a signalome-wide RNAi-based screen identified glucosylceramidase beta (GBA) as a positive regulator of autophagy-dependent cell death in human cells, presumably linked to the ability of GBA to convert glucosylceramide to ceramide (and glucose) [849]. However, additional investigation is required to elucidate the molecular mechanisms whereby GBA drives autophagy-dependent cell death. Finally, the long non-coding RNA autophagy-promoting factor (APF) has been implicated in the pathogenesis of myocardial infarction owing to its ability to indirectly promote the expression of ATG7 [850]. Autosis is a specific variant of autophagy-dependent cell death that relies on the plasma membrane Na+/K+-ATPase [851]. Corroborating the physiological relevance of this process, the administration of Na+/K+-ATPase inhibitors such as cardiac glycosides, confers neuroprotection in a rat model of neonatal hypoxia-ischemia [851].
In summary, autophagy-dependent cell death can be defined as a form of RCD that depends on the autophagic machinery (or components thereof) (Box 1). To avoid confusion, this term should be consistently avoided (1) in the absence of robust experimental evidence mechanistically linking RCD to (ideally more than one) components of the autophagy apparatus, as well as (2) when pharmacological or genetic manipulations of the molecular machinery for autophagy impact on other RCD subroutines.
Immunogenic cell death
Immunogenic cell death (ICD) is a functionally peculiar form of RCD that is sufficient to activate an adaptive immune response specific for endogenous (cellular) or exogenous (viral) antigens expressed by dying cells [852, 853]. ICD can be initiated by a relatively restricted set of stimuli, including viral infection, some FDA-approved chemotherapeutics (e.g., anthracyclines, bortezomib), specific forms of radiation therapy, and hypericin-based photodynamic therapy [854,855,856,857,858,859,860,861]. These agents are able to stimulate the timely release of a series of DAMPs, whose recognition by PRRs expressed by innate and adaptive components of the immune system warns the organism of a situation of danger, resulting in the elicitation of an immune response generally associated with the establishment of immunological memory [853 862,863,864]. So far, six DAMPs have been mechanistically linked to the perception of RCD as immunogenic: (1) calreticulin (CALR) [865, 866], (2) ATP [867,868,869], (3) high-mobility group box 1 (HMGB1) [870,871,872], (4) type I IFN [854, 873, 874], (5) cancer cell-derived nucleic acids [864, 875], and (6) annexin A1 (ANXA1) [876].
In the course of ICD, CALR translocates from the ER, where it is involved in the maintenance of Ca2+ homeostasis [877], to the outer leaflet of the plasma membrane [865, 866]. CALR translocation occurs as an early ICD-associated event, i.e., it occurs before PS exposure [878], and is mediated (at least in the case of chemotherapy-driven ICD) by three sequential signal transduction modules: (1) an ER stress module, which involves the phosphorylation of eukaryotic translation initiation factor 2 subunit alpha (EIF2S1; best known as eIF2α) by eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3; best known as PERK) coupled to a block in protein synthesis; (2) an apoptotic module, which involves the CASP8-dependent cleavage of B-cell receptor-associated protein 31 (BCAP31), BAX, and BAK; and (3) an exocytosis module, which involves the anterograde transport of CALR from the ER to the plasma membrane via the Golgi apparatus depending on vesicle-associated membrane protein 1 (VAMP1) and synaptosomal-associated protein 25 (SNAP25) [878,879,880]. Defects at any level of this cascade compromise the immunogenicity of RCD in vivo [878]. In most instances of ICD, CALR translocates to the cell surface together with protein disulfide isomerase family A member 3 (PDIA3; best known as known as ERp57) [881]. Cell surface-exposed CALR functions (1) as an “eat me” signal for phagocytosis by macrophages, neutrophils, and DCs, which is required for subsequent antigen cross-presentation to cytotoxic T cells; and (2) as a trigger for TH17 cell priming [882]. In line with a key role of CALR in the immunogenicity of RCD, the RNAi-mediated knockdown of CALR as well as natural defects in the CALR exposure pathway reportedly abolish the ability of dying cancer cells succumbing to anthracyclines to establish protective immunity in mice, whereas the exogenous provision of recombinant CALR confers immunogenic properties to otherwise non-immunogenic variants of RCD [865, 878, 881, 883, 884]. Of note, in some preclinical and clinical instances, the activity of surface-exposed CALR is antagonized by CD47, which operates as a “don’t eat me” signal as it inhibits phagocytosis by DCs and macrophages upon interaction with signal regulatory protein alpha (SIRPA) [885, 886]. Accordingly, while CALR exposure has positive prognostic value in patients with acute myeloid leukemia (AML) [887], increased CD47 levels on the surface of malignant cells correlate with dismal prognosis in subjects with AML, esophageal carcinoma, and ovarian cancer [888,889,890,891]. That said, CD47 appears to be required for the efficient phagocytic uptake of some murine cell lines undergoing RCD [892, 893]. The reasons underlying this apparent discrepancy remain to be elucidated.
Extracellular ATP not only operates as a “find-me” signal for macrophages and DC precursors upon binding to purinergic receptor P2Y G-protein coupled (P2RY2), but also mediates immunostimulatory effects by activating the canonical inflammasome upon binding to P2RX7 [867, 868 894,895,896,897]. In the context of ICD, ATP is released through a cascade of events occurring downstream of caspase activation and involving: (1) the autophagy-dependent accumulation of ATP within autolysosomes (the organelles forming by the fusion of autophagosomes or amphisomes with lysosomes), (2) the relocalization of lysosomal-associated membrane protein 1 (LAMP1) to the plasma membrane, (3) ROCK1-dependent cellular blebbing, and (4) the opening of PANX1 channels [229, 898, 899]. Accordingly, pre-mortem autophagy is required for optimal ATP release in the course of ICD, and hence for cell death induced by several (but not all) ICD inducers to be perceived as immunogenic [867, 900, 901]. Moreover, overexpression of the ATP-degrading ectoenzymes ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1; best known as CD39) and 5′ nucleotidase, ecto (NT5E; best known as CD73) efficiently lowers extracellular ATP levels in favor of adenosine accumulation, hence abolishing the immunogenicity of cell death [902]. CD39 is expressed at high levels on the surface of immune cells endowed with immunosuppressive properties, including CD4+CD25+FOXP3+ regulatory T (TREG) cells, and this promotes tumor progression and spreading [903, 904].
Type I IFN is produced by cancer cells succumbing to ICD by a mechanism involving the detection of endogenous dsRNA by TLR3 [873], or double-stranded DNA (dsDNA) by cGAS [854, 905, 906]. Besides mediating broad immunostimulatory effects on immune cells expressing IFNAR1 [520, 907], type I IFN reportedly activates an autocrine/paracrine signaling pathway in malignant cells, culminating with the expression of a spectrum of IFN-stimulated genes (ISGs) that includes the chemoattractant for T cells C-X-C motif chemokine ligand 10 (CXCL10) [873]. Accordingly, defects in dsRNA or dsDNA detection imposed by genetic interventions, including the Tlr3−/− genotype and the transgene-driven overexpression of the three prime repair exonuclease 1 (TREX1), as well as the deletion of Ifnar1 from cancer or host cells, abolish the immunogenicity of cell death triggered by various ICD inducers [854, 873]. Of note, cancer cell-derived nucleic acids do not mediate immunostimulatory functions only by autocrine/paracrine circuitries [908]. Rather, cancer cells succumbing to ICD release dsDNA and RNA molecules that can be efficiently taken up by DCs, neutrophils, and macrophages, resulting in the activation of a potent type I IFN response driven by multiple TLRs and the cGAS-STING pathway [864 909,910,911]. In line with this notion, the enzymatic degradation of extracellular nucleic acids considerably limits the immunogenicity of RCD [864, 875].
The molecular mechanisms underlying the ICD-associated release of HMGB1 and ANXA1 remain to be fully elucidated. Once secreted, the non-histone chromatin-binding protein HMGB1 mediates potent pro-inflammatory effects by binding to TLR2, TLR4, and advanced glycosylation end product-specific receptor (AGER; best known as RAGE) [912], although TLR4 seems to be the sole HMGB1 receptor relevant to perceive cell death as immunogenic [870 913,914,915]. In particular, the ligation of HGMB1 to TLR4 on DCs promotes antigen processing and cross-presentation, yet does not induce DC maturation, which is mainly stimulated by RAGE [870, 916]. That said, it should be noted that biological activities of HMGB1 may flip from pro-inflammatory to anti-inflammatory depending on multiple variables including its oxidation state [917,918,919,920,921,922,923]. Extracellular ANXA1 reportedly acts as a DAMP and supports the activation of adaptive immune responses by engaging formyl peptide receptor 1 (FPR1) on DCs [876]. In line with these observations, RCD is not perceived as immunogenic when cancer cells are depleted of HMGB1 or ANXA1, as well as when the host lacks TLR4 or FPR1 [870, 876].
Of note, the immunogenicity of RCD is robustly suppressed by some caspases, notably CASP8 and CASP3, by a variety of mechanisms [7, 924]. These include: (1) the prominent ability of CASP8 to inhibit necroptosis (see above), which is generally associated with the establishment of robust inflammatory responses linked to NF-κB activation [496 925,926,927,928]; (2) the capacity of CASP3 to drive PS exposure (see above), which generally supports the phagocytic removal of dying and dead cells while delivering anti-inflammatory signals to macrophages and DCs [218, 219]; (3) the ability of CASP3 to boost the secretion of prostaglandin E2 from dying cells, which has robust immunosuppressive effects [230, 929, 930]; and (4) the CASP3-dependent inhibition of type I IFN signaling elicited by mitochondrial DNA release upon MOMP [931, 932]. These observations suggest that specific caspase inhibitors may be harnessed to potently boost the immunogenicity of RCD.
The NCCD proposes to define ICD as a type of RCD that is sufficient to activate an adaptive immune response in immunocompetent hosts (Box 1).
Non-lethal processes
The molecular machinery for RCD is involved in several processes that have been mistakenly considered as bona fide instances of cell death over the past decades, including cellular senescence, mitotic catastrophe, and multiple cases of terminal differentiation.
Cellular senescence
The term “cellular senescence” refers to a pathophysiological process by which the cells permanently lose their proliferative capacity while remaining viable and metabolically active [933,934,935]. Senescent cells exhibit specific morphological traits including flattening, intracellular vacuolization, cellular/nuclear enlargement, and altered chromatin structure. At the biochemical level, cellular senescence is often characterized by: (1) increased lysosomal galactosidase beta 1 (GLB1) activity; (2) inhibition of multiple cyclin-dependent kinases (CDKs) and consequent dephosphorylation of various members of the retinoblastoma (RB) protein family—including RB transcriptional corepressor 1 (RB1), RB transcriptional corepressor like 1 (RB1L; best known as p107) and RB2L (best known as p130)—upon upregulation of cyclin dependent kinase inhibitor 1A (CDKN1A; best known as p21) [936] and/or the CDKN2A products p16 (a powerful inhibitor of CDK4 and CDK6) [937, 938] and ARF (an activator of p53) [939, 940]; (3) absence of proliferation markers, such as marker of proliferation Ki-67 (MKI67); (4) activation of the DDR machinery, generally as a consequence of telomere erosion; and (5) presence of so-called “senescence-associated heterochromatic foci” (SAHF) [934, 941]. Senescent cells secrete a variety of immunomodulatory and mitogenic cytokines, chemokines, growth, and MMPs [941,942,943]. Although such a senescence-associated secretory phenotype (SASP) appears to be involved in the immunological clearance of senescent cells, it also affects the biology of neighboring cells with an intact proliferative potential (and this has major implications for senescence-inducing anticancer agents) [941,942,943,944,945].
Waves of cellular senescence (followed by RCD) appear to contribute to developmental embryogenesis (although in a dispensable manner) [6, 946, 947] as well as to multiple pathophysiological processes in the adult, including tissue repair and regeneration, immunity, preservation of the stem cell compartment, and oncosuppression [948,949,950,951,952,953,954,955,956,957]. In particular, this failsafe cellular senescence reportedly occurs in response to (1) potentially carcinogenic events including oncogene activation or oncosuppressor gene inactivation; and (2) several sublethal cellular insults, including telomere shortening, DNA damage, mitochondrial dysfunction, defective/stalled DNA replication and epigenetic, lysosomal, mechanical, metabolic, mitotic, oxidative, or proteotoxic challenges [933, 958, 959]. Mounting evidence, however, indicates that senescent cells accumulate during organismal aging due to their increased generation coupled to inefficient clearance [935, 941, 960, 961]. Accordingly, chronic cellular senescence has been involved in natural aging, lifespan shortening, tissue deterioration, and the etiology of multiple age-related diseases, including atherosclerosis and osteoarthritis [935, 945, 948 962,963,964,965,966,967,968,969]. Moreover, senescent cells have been implicated in the adverse effects of some chemotherapeutic regimens as well as in the recurrence of specific neoplasms, at least in mice [969]. Thus, senescence stands out as an attractive therapeutic target for extending healthy lifespan [941, 970]. In this context, one promising senolytic regimen relies on the elevated vulnerability of senescent cells to inhibitors of pro-survival BCL2 family members (in particular BCL-XL), reflecting the elevated dependence of senescent cells on these proteins for survival [971,972,973]. The role of acute cellular senescence in multiple physiological processes, however, casts doubts on the actual feasibility of this approach [958, 968].
For these reasons, cellular senescence cannot be considered as a form of RCD (Box 1).
Mitotic catastrophe
Mitotic catastrophe is a regulated oncosuppressive mechanism that impedes the proliferation and/or survival of cells that are unable to complete mitosis owing to extensive DNA damage, problems with the mitotic machinery, and/or failure of mitotic checkpoints [974, 975]. Mitotic catastrophe is morphologically defined by unique nuclear changes, including multinucleation and macronucleation (two potential consequences of chromosomal missegregation) as well as micronucleation (perhaps resulting from the persistence of lagging or acentric chromosomes) [974, 975]. Mitotic defects can derive from: (1) exogenous sources, including a large panel of xenobiotics that alter DNA replication, cell cycle checkpoints, chromosome segregation, and/or microtubular dynamics [976]; or (2) endogenous sources, such as high levels of DNA replication stress or mitotic stress caused by an aberrant ploidy or by deregulated expression/activity of factors involved in DNA replication or chromosome segregation [977, 978]. Of note, the primary alterations that drive catastrophic mitoses can originate in other phases of the cell cycles, including the S phase (e.g., a premature entry in mitosis caused by failure of the intra-S-phase checkpoint) [979, 980]. The precise molecular mechanisms through which mitotic alterations are sensed and trigger mitotic catastrophe cascade are unclear, but presumably involve p53 (at least in many cell types) [975]. A large body of experimental evidence suggests that—at least in specific experimental settings—mitotic catastrophe is precipitated by a signal transduction cascade that relies on CASP2 activation, often (but not always) triggering a variant of intrinsic apoptosis regulated by the BCL2 protein family and demarcated by MOMP [974 981–984]. In line with a key role for CASP2 in the control of mitotic proficiency, the bone marrow of Casp2−/− mice accumulates aneuploid cells with aging [985], and Casp2−/− malignant cells exhibit increased levels of aneuploidy as compared to their wild-type counterparts [982, 986, 987]. Moreover, Casp2−/− mice are more susceptible than their wild-type littermates to oncogenesis in a multitude of experimental settings [982 985,986,987,988,989]. In the absence of p53, however, mitotic defects appear to drive a necrotic variant of RCD independent of CASP2 signaling (at least in some settings) [990,991,992].
Of note, the ultimate fate of cells undergoing mitotic catastrophe seems to be dictated (at least in part) by the time spent under mitotic arrest and their capability to slip out of aberrant mitoses [993]. Thus, while cells arrested in aberrant mitosis for prolonged periods often undergo intrinsic apoptosis, cells escaping the mitotic block and reaching interphase can undergo a similar fate, enter cellular senescence on activating specific cell cycle checkpoints mediated by p53, and/or Hippo signaling, or simply be outcompeted by their proliferating counterparts [978 994,995,996,997,998,999,1000,1001,1002]. Importantly, the abrogation of mitotic catastrophe is a key event during neoplastic transformation and progression, mainly as it allows for the generation and/or survival of polyploid and aneuploid cells [975], whereas cancer (stem) cells reportedly display increased sensitivity to mitotic defects [1003]. However, mitotic catastrophe appears to constitute a major mechanism of action of anticancer chemotherapeutics, mostly reflecting the increased resistance of neoplastic cells to the induction of intrinsic apoptosis [1004,1005,1006,1007,1008,1009]. Moreover, recent data indicate that cancer cells escaping mitotic catastrophe efficiently promote the secretion of type I IFN following the detection of cytosolic dsDNA by cGAS, potentially resulting in their elimination by immunological mechanisms [905, 906]. The latter observation lends further support to the notion that extracellular homeostasis in mammalian organisms is preserved by a plethora of mechanisms that are initiated at the cell-intrinsic level but only operate once cellular homeostasis is compromised.
Since mitotic catastrophe does not always result in RCD (but can also drive cellular senescence), it cannot be considered as a form of RCD per se. We propose the use of the term mitotic death to indicate the specific variant of RCD (most often intrinsic apoptosis) driven by mitotic catastrophe (Box 1).
Terminal differentiation and others
Multiple components of the signal transduction cascades that regulate or precipitate RCD are involved in the terminal differentiation of a variety of cell types [157, 180 1010,1011,1012,1013], including (but presumably not limited to) neurons [1014,1015,1016,1017], granulocytes [1018], megakaryocytes [1019], erythroblasts [1020], osteoclasts [1021], sperm cells [1022], skeletal myocytes [1023], lens cells [1024], and the keratinized epithelium [1025]. The latter process, which is commonly known as cornification (or keratinization) and critically relies on CASP14 [1026, 1027] and multiple isoforms of transglutaminase [1025, 1028, 1029], has long been considered as a form of PCD [1025, 1030]. However, the NCCD suggests to keep PCD and terminal differentiation conceptually well discriminated from each other. Indeed, dead cells are disposed of (and hence cease to have a function) in the course of PCD. Conversely, when terminal differentiation involves cellular demise, as in the case of cornification, dead cells become integral part of a tissue (and hence mediate a specific physiological function). Along similar lines, the NCCD discourages the use of the term eryptosis, which has been coined to indicate the demise of erythrocytes exposed to stress [1031]. Irrespectively of the unquestionable relevance of this process for human pathophysiology [1032,1033,1034], it is indeed extremely complex from a conceptual standpoint to define the death of entities that—in physiological conditions—exist in a debatable state between life and death (such as erythrocytes and viruses).
Concluding remarks
As amply discussed above, RCD plays a major role in development, tissue homeostasis, inflammation, immunity, and multiple pathophysiological conditions. On the one hand, RCD constitutes a primary etiological determinant in diseases associated with the irreversible loss of post-mitotic tissues (e.g., myocardial infarction, neurodegeneration) [687, 752, 851, 869, 1035, 1036]. On the other hand, defects in the signaling cascades that precipitate RCD are associated with pathologies characterized by uncontrolled cell expansion or accumulation (e.g., some autoimmune disorders, cancer) [44, 263, 267, 537 1037,1038,1039,1040,1041,1042]. Thus, RCD stands out as a major therapeutic target for the management of multiple human disorders [1, 2, 649, 1043].
Over the past two decades tremendous efforts have been dedicated to the development of cytoprotective strategies aimed at interrupting RCD signaling after the initiation of the process (a clinically relevant scenario for most ischemic disorders) [1044, 1045], with relatively deceiving results (despite multiple clinical trials, no drug based on this concept has ever been approved for use in humans by regulatory agencies) [387]. Conversely, the BCL2 inhibitor venetoclax is currently available for the treatment of CLL patients who fail to obtain clinical benefit from conventional therapies [1046], and several other molecules with a similar mechanism of action are currently in clinical development [118] (source https://clinicaltrials.gov/). Why does the specific activation of RCD (that should not be confounded with the alteration of normal cellular functions, although this also can lead to RCD) appear as a much simpler clinical objective than its inhibition?
Besides potential issues linked to the pharmacokinetics and pharmacodynamics of the compounds tested so far, this discrepancy likely reflects the high interconnectivity of the signaling modules involved in RCD control in mammalian organisms (which has begun to emerge only recently) [374 1047,1048,1049]. Thus, while tilting the balance toward RCD appears as a relatively easy task, blocking it—once a hitherto poorly defined point-of-no-return has been trespassed—may require the simultaneous inhibition of several signal transduction modules, and hence may be hardly achievable (Fig. 2). Moreover, the community has focused for a long time on specific enzymes that were thought to have a key causal role in RCD execution, but in a majority of scenarios only appear to accelerate (rather than causally determine) cellular demise (e.g., caspases) [17, 374]. Indeed, cell death in all its forms (including ACD) is ultimately associated with a bioenergetic and redox crisis that may constitute its actual cause [17, 374]. In this scenario, true cytoprotection may be achieved only by interventions that counteract such crisis or the causes (rather than the epiphenomena) thereof. Interestingly, one of the most rapid consequences of potentially lethal ATP depletion in D. discoideum is an abrupt nucleolar disorganization coupled to an irreversible block in ribosomal RNA and DNA synthesis [1050]. A similar process has also been observed in mammalian and plant cells succumbing to multiple forms of RCD, perhaps suggesting that nucleolar stress plays a key role in RCD execution across different species [1051]. This possibility, however, remains to be formally addressed.

Interconnectivity of cell death from a therapeutic perspective. On the basis of the assumption that each regulated cell death (RCD) subroutine would operate in a virtually isolated manner (a), considerable efforts have been dedicated to the development of pharmacological agents that would interrupt RCD by operating on a single signal transduction module (b). It is now clear that the molecular mechanisms underlying distinct RCD modalities exhibit a considerable degree of interconnectivity (c). This implies that robust cytoprotection may not be achieved by targeting a single RCD subroutine, but only upon the simultaneous inhibition of multiple signal transduction modules (assuming that these modules are the actual cause of cell death, and not simple epiphenomena of RCD signaling (d).
Only recently, it has become clear that the modality through which an individual cell succumbs to stress may have a major impact on how RCD affects the local and systemic microenvironment [36, 852, 1052]. This opened an entirely new therapeutic perspective for the field, involving two major approaches: (1) the development of approaches aimed at switching RCD modality, rather than increasing or limiting the incidence of RCD (which may be problematic in both directions) [856 1053,1054,1055,1056]; and (2) the development of agents that intercept DAMPs or regulate DAMP-dependent signaling pathways [20 1057,1058,1059]. In this context, ACD may also constitute a therapeutic target. Indeed, although ACD occurs in a limited number of human disorders (e.g., trauma, severe burns) and it cannot be pharmacologically inhibited (by definition), the molecules released by cells undergoing ACD may be blocked (at least theoretically) with specific interventions, and this may have a positive impact on long-term disease outcome [21 1060,1061,1062]. These observations exemplify the complexity of targeting primary RCD or ACD (the death of cells succumbing to primary environmental perturbations), secondary RCD (the death of cells succumbing to the microenvironmental conditions established, directly or indirectly, by neighboring cells undergoing primary RCD or ACD), and RCD-driven or ACD-driven DAMP signaling for therapeutic purposes [1063] (Fig. 3).

Causal vs. accessory aspects of cell death from a therapeutic perspective. Cells exposed to very harsh environmental conditions disassemble in a virtually instantaneous and uncontrollable manner, a process that is referred to as accidental cell death (ACD). Conversely, relatively mild perturbations of exogenous or endogenous origin promote adaptative stress responses aimed at the restoration of cellular homeostasis. If such responses fail, cells generally activate one or more of multiple, highly interconnected signal transduction modules that precipitate regulated cell death (RCD). ACD cannot be retarded by pharmacological or genetic interventions, and most (if not all) strategies conceived so far to block RCD in mammalian organisms fail to efficiently do so, at least in part owing to the elevated interconnectivity of the process. Conversely, some agents that de facto promote RCD by primarily targeting the underlying molecular machinery (rather than by targeting normal cellular functions) are already available for use in the clinic. The events that follow primary cell death—including a secondary wave of RCD in neighboring cells established (directly or indirectly) by molecules released from the cells succumbing to the primary insult, as well as danger-associated molecular pattern (DAMP) signaling—may also be targets for pharmacological interventions. Finally, although altering quantitatively the percentage of cells succumbing to primary RCD remains challenging (especially when a hitherto poorly defined point-of-no-return of the process has been trespassed), favoring the use of specific signaling modules over others may have prominent effects on long-term disease outcome.
In conclusion, targeting RCD holds great promise for the treatment of several human disorders and considerable efforts are being made to generate RCD modulators for clinical use, but additional studies are required to devise the most efficient strategies in that sense. We are confident that a correct, but flexible, use of the RCD-related terms defined herein will strongly support the progress of the field toward such an ambitious goal. To avoid confusion, it will be important to incorporate neologisms into the scientific literature only for novel RCD subroutines clearly relying on signal transduction modules and effector mechanisms that show little or no overlap with known types of RCD. Along these lines, we believe that terms mostly referring to morphological features of cellular demise and/or indicating considerable mechanistic overlap with well-established RCD forms, such as autoschizis [1064, 1065], should be dismissed. The NCCD surmises that this is the only way for new cell death-related terms to acquire genuine utility and be broadly adopted by the scientific community.
References
Conrad M, et al. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15:348–66.
Weinlich R, et al. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18:127–36.
Fuchs Y, et al. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol. 2015;16:329–44.
Pasparakis M, et al. Necroptosis and its role in inflammation. Nature. 2015;517:311–20.
Galluzzi L, et al. Regulated cell death and adaptive stress responses. Cell Mol Life Sci. 2016;73:2405–10.
Fuchs Y, et al. Programmed cell death in animal development and disease. Cell. 2011;147:742–58.
Galluzzi L, et al. Caspases connect cell-death signaling to organismal homeostasis. Immunity. 2016;44:221–31.
Jorgensen I, et al. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17:151–64.
Nagata S, et al. Programmed cell death and the immune system. Nat Rev Immunol. 2017;17:333–40.
Cornillon S, et al. Programmed cell death in Dictyostelium. J Cell Sci. 1994;107(Pt 10):2691–704.
Olie RA, et al. Apparent caspase independence of programmed cell death in Dictyostelium. Curr Biol. 1998;8:955–58.
Cornillon S, et al. An insertional mutagenesis approach to Dictyostelium cell death. Cell Death Differ. 1998;5:416–25.
Madeo F, et al. A yeast mutant showing diagnostic markers of early and late apoptosis. J Cell Biol. 1997;139:729–34.
Eisenberg T, et al. The mitochondrial pathway in yeast apoptosis. Apoptosis. 2007;12:1011–23.
Buttner S, et al. Why yeast cells can undergo apoptosis: death in times of peace, love, and war. J Cell Biol. 2006;175:521–25.
Green DR, et al. Just so stories about the evolution of apoptosis. Curr Biol. 2016;26:R620–27.
Galluzzi L, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58–73.
Conradt B. Genetic control of programmed cell death during animal development. Annu Rev Genet. 2009;43:493–523.
West AP, et al. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017;17:363–75.
Krysko DV, et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–75.
Galluzzi L, et al. Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol. 2012;13:780–88.
McDonald B, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–66.
Schweichel JU, et al. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7:253–66.
Galluzzi L, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–43.
Kroemer G, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005;12(Suppl 2):1463–67.
Kroemer G, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11.
Galluzzi L, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20.
Galluzzi L, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–107.
Czabotar PE, et al. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63.
Pihan P, et al. BCL-2 family: integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017;24:1478–87.
Roos WP, et al. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016;16:20–33.
Vitale I, et al. DNA damage in stem cells. Mol Cell. 2017;66:306–19.
Nunez G, et al. Deregulated Bcl-2 gene expression selectively prolongs survival of growth factor-deprived hemopoietic cell lines. J Immunol. 1990;144:3602–610.
Brumatti G, et al. Crossing paths: interactions between the cell death machinery and growth factor survival signals. Cell Mol Life Sci. 2010;67:1619–30.
Green DR, et al. The clearance of dying cells: table for two. Cell Death Differ. 2016;23:915–26.
Yatim N, et al. Dying cells actively regulate adaptive immune responses. Nat Rev Immunol. 2017;17:262–75.
Green DR, et al. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353–63.
Vanden Berghe T, et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–30.
Rogers C, et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128.
Tait SW, et al. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–32.
Galluzzi L, et al. Mitochondrial regulation of cell death: a phylogenetically conserved control. Microb Cell. 2016;3:101–08.
Moldoveanu T, et al. Many players in BCL-2 family affairs. Trends Biochem Sci. 2014;39:101–11.
Shamas-Din A, et al. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5:a008714.
Delbridge AR, et al. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16:99–109.
Luna-Vargas MP, et al. Physiological and pharmacological control of BAK, BAX, and beyond. Trends Cell Biol. 2016;26:906–17.
Aouacheria A, et al. Evolution of Bcl-2 homology motifs: homology versus homoplasy. Trends Cell Biol. 2013;23:103–11.
Llambi F, et al. BOK Is a Non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell. 2016;165:421–33.
Edlich F, et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell. 2011;145:104–16.
Garner TP, et al. An autoinhibited dimeric form of BAX regulates the BAX activation pathway. Mol Cell. 2016;63:485–97.
Schellenberg B, et al. Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol Cell. 2013;49:959–71.
Cheng EH, et al. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–17.
Lazarou M, et al. Inhibition of Bak activation by VDAC2 is dependent on the Bak transmembrane anchor. J Biol Chem. 2010;285:36876–883.
Naghdi S, et al. Motifs of VDAC2 required for mitochondrial Bak import and tBid-induced apoptosis. Proc Natl Acad Sci U S A. 2015;112:E5590–99.
Ma SB, et al. Bax targets mitochondria by distinct mechanisms before or during apoptotic cell death: a requirement for VDAC2 or Bak for efficient Bax apoptotic function. Cell Death Differ. 2014;21:1925–35.
Todt F, et al. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 2015;34:67–80.
Kuwana T, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–35.
Chen L, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403.
Letai A, et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–92.
Kim H, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–58.
Bouillet P, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–38.
Bouillet P, et al. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 2002;415:922–26.
Villunger A, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–38.
Galluzzi L, et al. Organelle-specific initiation of cell death. Nat Cell Biol. 2014;16:728–36.
Chen HC, et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol. 2015;17:1270–81.
Dai H, et al. Evaluation of the BH3-only protein Puma as a direct Bak activator. J Biol Chem. 2014;289:89–99.
Moldoveanu T, et al. BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol. 2013;20:589–97.
Dai H, et al. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J Cell Biol. 2011;194:39–48.
Ren D, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330:1390–93.
Li MX, et al. BAK alpha6 permits activation by BH3-only proteins and homooligomerization via the canonical hydrophobic groove. Proc Natl Acad Sci U S A. 2017;114:7629–34.
Luo X, et al. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–90.
Gavathiotis E, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–81.
Gavathiotis E, et al. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell. 2010;40:481–92.
Kim H, et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36:487–99.
Wei MC, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30.
Alsop AE, et al. Dissociation of Bak alpha1 helix from the core and latch domains is required for apoptosis. Nat Commun. 2015;6:6841.
Brouwer JM, et al. Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell. 2014;55:938–46.
Czabotar PE, et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–31.
Bleicken S, et al. Structural model of active Bax at the membrane. Mol Cell. 2014;56:496–05.
Subburaj Y, et al. Bax monomers form dimer units in the membrane that further self-assemble into multiple oligomeric species. Nat Commun. 2015;6:8042.
Zhang Z, et al. BH3-in-groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J. 2016;35:208–36.
Ma S, et al. Assembly of the Bak apoptotic pore: a critical role for the Bak protein alpha6 helix in the multimerization of homodimers during apoptosis. J Biol Chem. 2013;288:26027–38.
Dewson G, et al. Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ. 2012;19:661–70.
Oh KJ, et al. Conformational changes in BAK, a pore-forming proapoptotic Bcl-2 family member, upon membrane insertion and direct evidence for the existence of BH3-BH3 contact interface in BAK homo-oligomers. J Biol Chem. 2010;285:28924–37.
Salvador-Gallego R, et al. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016;35:389–401.
Grosse L, et al. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 2016;35:402–13.
Aluvila S, et al. Organization of the mitochondrial apoptotic BAK pore: oligomerization of the BAK homodimers. J Biol Chem. 2014;289:2537–51.
Gillies LA, et al. Visual and functional demonstration of growing Bax-induced pores in mitochondrial outer membranes. Mol Biol Cell. 2015;26:339–49.
Hardwick JM, et al. Multiple functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol 2013; 5:a008722.
Barclay LA, et al. Inhibition of pro-apoptotic BAX by a noncanonical interaction mechanism. Mol Cell. 2015;57:873–86.
Antonsson B, et al. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277:370–72.
Oltvai ZN, et al. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–19.
Yin XM, et al. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 1994;369:321–23.
O’Neill KL, et al. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016;30:973–88.
Cheng EH, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–11.
Rong Y, et al. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol. 2008;70:73–91.
Scorrano L, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–39.
White C, et al. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol. 2005;7:1021–28.
Monaco G, et al. The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J Biol Chem. 2015;290:9150–161.
Vervliet T, et al. Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene. 2016;35:5079–92.
Chen YB, et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195:263–76.
Green DR, et al. Cell biology. Metabolic control of cell death. Science. 2014;345:1250256.
Perciavalle RM, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14:575–83.
Bonora M, et al. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene. 2015;34:1475–86.
Alavian KN, et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol. 2011;13:1224–33.
Chen ZX, et al. Bcl-2 induces pro-oxidant state by engaging mitochondrial respiration in tumor cells. Cell Death Differ. 2007;14:1617–27.
Clement MV, et al. Decrease in intracellular superoxide sensitizes Bcl-2-overexpressing tumor cells to receptor and drug-induced apoptosis independent of the mitochondria. Cell Death Differ. 2003;10:1273–85.
Chen ZX, et al. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell Death Differ. 2010;17:408–20.
Low IC, et al. Ser70 phosphorylation of Bcl-2 by selective tyrosine nitration of PP2A-B56delta stabilizes its antiapoptotic activity. Blood. 2014;124:2223–34.
Velaithan R, et al. The small GTPase Rac1 is a novel binding partner of Bcl-2 and stabilizes its antiapoptotic activity. Blood. 2011;117:6214–26.
Llambi F, et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44:517–31.
Bleicken S, et al. Quantitative interactome of a membrane Bcl-2 network identifies a hierarchy of complexes for apoptosis regulation. Nat Commun. 2017;8:73.
Lee EF, et al. Physiological restraint of Bak by Bcl-xL is essential for cell survival. Genes Dev. 2016;30:1240–50.
Birkinshaw RW, et al. The BCL-2 family of proteins and mitochondrial outer membrane permeabilisation. Semin Cell Dev Biol 2017;72:152–162.
Bhola PD, et al. Mitochondria-Judges and executioners of cell death sentences. Mol Cell. 2016;61:695–704.
Hockings C, et al. Bid chimeras indicate that most BH3-only proteins can directly activate Bak and Bax, and show no preference for Bak versus Bax. Cell Death Dis. 2015;6:e1735.
Du H, et al. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chem. 2011;286:491–01.
Merino D, et al. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol. 2009;186:355–62.
Kotschy A, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–82.
Roberts AW, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:311–22.
Green DR. A BH3 mimetic for killing cancer cells. Cell. 2016;165:1560.
Aranovich A, et al. Differences in the mechanisms of proapoptotic BH3 proteins binding to Bcl-XL and Bcl-2 quantified in live MCF-7 cells. Mol Cell. 2012;45:754–63.
Pecot J, et al. Tight sequestration of BH3 proteins by BCL-xL at subcellular membranes contributes to apoptotic resistance. Cell Rep. 2016;17:3347–λ.
Ke F, et al. Impact of the combined loss of BOK, BAX and BAK on the hematopoietic system is slightly more severe than compound loss of BAX and BAK. Cell Death Dis. 2015;6:e1938.
Lindsten T, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–99.
Labi V, et al. Deregulated cell death and lymphocyte homeostasis cause premature lethality in mice lacking the BH3-only proteins Bim and Bmf. Blood. 2014;123:2652–62.
Chipuk JE, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–14.
Mihara M, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–90.
Vaseva AV, et al. The mitochondrial p53 pathway. Biochim Biophys Acta. 2009;1787:414–20.
Follis AV, et al. Pin1-induced proline isomerization in cytosolic p53 mediates BAX activation and apoptosis. Mol Cell. 2015;59:677–84.
Hilton BA, et al. ATR plays a direct antiapoptotic role at mitochondria, which is regulated by prolyl isomerase Pin1. Mol Cell. 2015;60:35–46.
Aouacheria A, et al. Redefining the BH3 death domain as a 'short linear motif'. Trends Biochem Sci. 2015;40:736–48.
Iyer S, et al. Identification of an activation site in Bak and mitochondrial Bax triggered by antibodies. Nat Commun. 2016;7:11734.
Del ReDP, et al. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Mol Cell. 2014;54:639–50.
Dumitru R, et al. Human embryonic stem cells have constitutively active Bax at the Golgi and are primed to undergo rapid apoptosis. Mol Cell. 2012;46:573–83.
Inuzuka H, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104–09.
Wertz IE, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–14.
Puthalakath H, et al. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293:1829–32.
Puthalakath H, et al. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell. 1999;3:287–96.
Pyakurel A, et al. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol Cell. 2015;58:244–54.
Renault TT, et al. Mitochondrial shape governs BAX-induced membrane permeabilization and apoptosis. Mol Cell. 2015;57:69–82.
Weaver D, et al. Distribution and apoptotic function of outer membrane proteins depend on mitochondrial fusion. Mol Cell. 2014;54:870–78.
Luo L, et al. Integration and oligomerization of Bax protein in lipid bilayers characterized by single molecule fluorescence study. J Biol Chem. 2014;289:31708–18.
Chipuk JE, et al. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell. 2012;148:988–1000.
Wang X, et al. Bcl-2 proteins regulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis. Cell Death Differ. 2011;18:38–47.
Oakes SA, et al. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2005;102:105–10.
Sassano ML, et al. Mitochondria-associated membranes as networking platforms and regulators of cancer cell fate. Front Oncol. 2017;7:174.
Bassoy EY, et al. ER-mitochondria contacts control surface glycan expression and sensitivity to killer lymphocytes in glioma stem-like cells. EMBO J. 2017;36:1493–512.
Phillips MJ, et al. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol. 2016;17:69–82.
Echeverry N, et al. Intracellular localization of the BCL-2 family member BOK and functional implications. Cell Death Differ. 2013;20:785–99.
Carpio MA, et al. BCL-2 family member BOK promotes apoptosis in response to endoplasmic reticulum stress. Proc Natl Acad Sci U S A. 2015;112:7201–206.
Fernandez-Marrero Y, et al. The membrane activity of BOK involves formation of large, stable toroidal pores and is promoted by cBID. FEBS J. 2017;284:711–24.
Einsele-Scholz S, et al. Bok is a genuine multi-BH-domain protein that triggers apoptosis in the absence of Bax and Bak. J Cell Sci. 2016;129:2213–23.
Schulman JJ, et al. The stability and expression level of Bok are governed by binding to inositol 1,4,5-trisphosphate receptors. J Biol Chem. 2016;291:11820–28.
Ke F, et al. BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ. 2012;19:915–25.
Ke F, et al. Consequences of the combined loss of BOK and BAK or BOK and BAX. Cell Death Dis. 2013;4:e650.
Tait SW, et al. Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol. 2013;5:a008706.
Galluzzi L, et al. Non-apoptotic functions of apoptosis-regulatory proteins. EMBO Rep. 2012;13:322–30.
Liu X, et al. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–57.
Li K, et al. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell. 2000;101:389–99.
Li P, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89.
Chai J, et al. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature. 2000;406:855–62.
Verhagen AM, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53.
Du C, et al. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42.
Scorrano L, et al. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67.
Frezza C, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–89.
Jiang X, et al. Activation of mitochondrial protease OMA1 by Bax and Bak promotes cytochrome c release during apoptosis. Proc Natl Acad Sci U S A. 2014;111:14782–787.
Varanita T, et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015;21:834–44.
Otera H, et al. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol. 2016;212:531–44.
Cho DH, et al. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–05.
Barsoum MJ, et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–911.
Yuan H, et al. Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ. 2007;14:462–71.
Cheng TC, et al. A near atomic structure of the active human apoptosome. Elife. 2016;5:e17755.
Zhou M, et al. Atomic structure of the apoptosome: mechanism of cytochrome c- and dATP-mediated activation of Apaf-1. Genes Dev. 2015;29:2349–61.
Pang Y, et al. Structure of the apoptosome: mechanistic insights into activation of an initiator caspase from Drosophila. Genes Dev. 2015;29:277–87.
Hu Q, et al. Molecular determinants of caspase-9 activation by the Apaf-1 apoptosome. Proc Natl Acad Sci U S A. 2014;111:16254–61.
Li Y, et al. Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc Natl Acad Sci U S A. 2017;114:1542–47.
Wu CC, et al. The Apaf-1 apoptosome induces formation of caspase-9 homo- and heterodimers with distinct activities. Nat Commun. 2016;7:13565.
Riedl SJ, et al. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–13.
Julien O, et al. Caspases and their substrates. Cell Death Differ. 2017;24:1380–89.
Shalini S, et al. Old, new and emerging functions of caspases. Cell Death Differ. 2015;22:526–39.
Salvesen GS, et al. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol. 2002;3:401–10.
Burri L, et al. Mature DIABLO/Smac is produced by the IMP protease complex on the mitochondrial inner membrane. Mol Biol Cell. 2005;16:2926–33.
Saita S, et al. PARL mediates Smac proteolytic maturation in mitochondria to promote apoptosis. Nat Cell Biol. 2017;19:318–28.
Eckelman BP, et al. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem. 2006;281:3254–60.
Eckelman BP, et al. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–94.
Silke J, et al. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb Perspect Biol. 2013;5:a008730.
Ditzel M, et al. Inactivation of effector caspases through nondegradative polyubiquitylation. Mol Cell. 2008;32:540–53.
Lee TV, et al. Drosophila IAP1-mediated ubiquitylation controls activation of the initiator caspase DRONC independent of protein degradation. PLoS Genet. 2011;7:e1002261.
Lisi S, et al. Diverse domains of THREAD/DIAP1 are required to inhibit apoptosis induced by REAPER and HID in Drosophila. Genetics. 2000;154:669–78.
Morizane Y, et al. X-linked inhibitor of apoptosis functions as ubiquitin ligase toward mature caspase-9 and cytosolic Smac/DIABLO. J Biochem. 2005;137:125–32.
Schile AJ, et al. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008;22:2256–66.
Suzuki Y, et al. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc Natl Acad Sci U S A. 2001;98:8662–67.
Wilson R, et al. The DIAP1 RING finger mediates ubiquitination of Dronc and is indispensable for regulating apoptosis. Nat Cell Biol. 2002;4:445–50.
Chai J, et al. Molecular mechanism of Reaper-Grim-Hid-mediated suppression of DIAP1-dependent Dronc ubiquitination. Nat Struct Biol. 2003;10:892–98.
Kamber Kaya HE, et al. An inhibitory mono-ubiquitylation of the Drosophila initiator caspase Dronc functions in both apoptotic and non-apoptotic pathways. PLoS Genet. 2017;13:e1006438.
Varfolomeev E, et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem. 2008;283:24295–99.
Feltham R, et al. The small molecule that packs a punch: ubiquitin-mediated regulation of RIPK1/FADD/caspase-8 complexes. Cell Death Differ. 2017;24:1196–204.
Witt A, et al. Diverse ubiquitin linkages regulate RIP kinases-mediated inflammatory and cell death signaling. Cell Death Differ. 2017;24:1160–71.
Hamacher-Brady A, et al. Bax/Bak-dependent, Drp1-independent targeting of X-linked inhibitor of apoptosis protein (XIAP) into inner mitochondrial compartments counteracts Smac/DIABLO-dependent effector caspase activation. J Biol Chem. 2015;290:22005–18.
Nunnari J, et al. Mitochondria: in sickness and in health. Cell. 2012;148:1145–59.
Zamzami N, et al. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exp Med. 1995;181:1661–72.
Zamzami N, et al. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–77.
Schmidt O, et al. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11:655–67.
Mizuta T, et al. A Bax/Bak-independent mechanism of cytochrome c release. J Biol Chem. 2007;282:16623–30.
Zamorano S, et al. A BAX/BAK and cyclophilin D-independent intrinsic apoptosis pathway. PLoS One. 2012;7:e37782.
Colombini M. Ceramide channels and mitochondrial outer membrane permeability. J Bioenerg Biomembr. 2017;49:57–64.
Siskind LJ, et al. The lipids C2- and C16-ceramide form large stable channels. Implications for apoptosis. J Biol Chem. 2000;275:38640–44.
Nagata S. DNA degradation in development and programmed cell death. Annu Rev Immunol. 2005;23:853–75.
Naito M, et al. Phosphatidylserine externalization is a downstream event of interleukin-1 beta-converting enzyme family protease activation during apoptosis. Blood. 1997;89:2060–66.
Martin SJ, et al. Phosphatidylserine externalization during CD95-induced apoptosis of cells and cytoplasts requires ICE/CED-3 protease activity. J Biol Chem. 1996;271:28753–56.
Sebbagh M, et al. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001;3:346–52.
Coleman ML, et al. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–45.
Enari M, et al. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50.
Kawane K, et al. Impaired thymic development in mouse embryos deficient in apoptotic DNA degradation. Nat Immunol. 2003;4:138–44.
Sakahira H, et al. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99.
Suzuki J, et al. Xkr8 phospholipid scrambling complex in apoptotic phosphatidylserine exposure. Proc Natl Acad Sci U S A. 2016;113:9509–14.
Suzuki J, et al. Exposure of phosphatidylserine by Xk-related protein family members during apoptosis. J Biol Chem. 2014;289:30257–67.
Suzuki J, et al. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341:403–06.
Segawa K, et al. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344:1164–68.
Segawa K, et al. Human type IV P-type ATPases that work as plasma membrane phospholipid flippases and their regulation by caspase and calcium. J Biol Chem. 2016;291:762–72.
Yabas M, et al. Mice deficient in the putative phospholipid flippase ATP11C exhibit altered erythrocyte shape, anemia, and reduced erythrocyte life span. J Biol Chem. 2014;289:19531–7.
Fadeel B, et al. Phosphatidylserine exposure during apoptosis is a cell-type-specific event and does not correlate with plasma membrane phospholipid scramblase expression. Biochem Biophys Res Commun. 1999;266:504–11.
Qu X, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–46.
Mellen MA, et al. Autophagy is not universally required for phosphatidyl-serine exposure and apoptotic cell engulfment during neural development. Autophagy. 2009;5:964–72.
Marsden VS, et al. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature. 2002;419:634–37.
Ichim G, et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell. 2015;57:860–72.
Sun G, et al. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J Cell Biol. 2017;216:3355–68.
Tang HL, et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell. 2012;23:2240–52.
Martins I, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014;21:79–91.
Huang Q, et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 2011;17:860–66.
Gray DC, et al. Activation of specific apoptotic caspases with an engineered small-molecule-activated protease. Cell. 2010;142:637–46.
Julien O, et al. Quantitative MS-based enzymology of caspases reveals distinct protein substrate specificities, hierarchies, and cellular roles. Proc Natl Acad Sci U S A. 2016;113:E2001–10.
Paoli P, et al. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta. 2013;1833:3481–98.
Buchheit CL, et al. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer. 2014;14:632–41.
Mailleux AA, et al. BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev Cell. 2007;12:221–34.
Buchheit CL, et al. The regulation of cancer cell death and metabolism by extracellular matrix attachment. Semin Cell Dev Biol. 2012;23:402–11.
Rayavarapu RR, et al. The role of multicellular aggregation in the survival of ErbB2-positive breast cancer cells during extracellular matrix detachment. J Biol Chem. 2015;290:8722–33.
Buchheit CL, et al. Anoikis evasion in inflammatory breast cancer cells is mediated by Bim-EL sequestration. Cell Death Differ. 2015;22:1275–86.
Simpson CD, et al. Inhibition of the sodium potassium adenosine triphosphatase pump sensitizes cancer cells to anoikis and prevents distant tumor formation. Cancer Res. 2009;69:2739–47.
de La Motte Rouge T, et al. A novel epidermal growth factor receptor inhibitor promotes apoptosis in non-small cell lung cancer cells resistant to erlotinib. Cancer Res. 2007;67:6253–62.
Hu X, et al. CCDC178 promotes hepatocellular carcinoma metastasis through modulation of anoikis. Oncogene. 2017;36:4047–59.
Zhang K, et al. Oncogenic K-Ras upregulates ITGA6 expression via FOSL1 to induce anoikis resistance and synergizes with alphaV-Class integrins to promote EMT. Oncogene. 2017;36:5681–5694.
Weigel KJ, et al. CAF-secreted IGFBPs regulate breast cancer cell anoikis. Mol Cancer Res. 2014;12:855–66.
Xu J, et al. Hepatitis B virus X protein confers resistance of hepatoma cells to anoikis by up-regulating and activating p21-activated kinase 1. Gastroenterology. 2012;143:199–212. e194
Li X, et al. Aiolos promotes anchorage independence by silencing p66Shc transcription in cancer cells. Cancer Cell. 2014;25:575–89.
Alanko J, et al. Integrin endosomal signalling suppresses anoikis. Nat Cell Biol. 2015;17:1412–21.
Aslan B, et al. The ZNF304-integrin axis protects against anoikis in cancer. Nat Commun. 2015;6:7351.
Vivo M, et al. p14ARF interacts with the focal adhesion kinase and protects cells from anoikis. Oncogene. 2017;36:4913–28.
Zheng Y, et al. Protein tyrosine kinase 6 protects cells from anoikis by directly phosphorylating focal adhesion kinase and activating AKT. Oncogene. 2013;32:4304–12.
Frisch SM, et al. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci. 2013;126:21–9.
Amelio I, et al. Exploiting tumour addiction with a serine and glycine-free diet. Cell Death Differ. 2017;24:1311–13.
Yu SJ, et al. MicroRNA-200a promotes anoikis resistance and metastasis by targeting YAP1 in human breast cancer. Clin Cancer Res. 2013;19:1389–99.
Haemmerle M, et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat Commun. 2017;8:310.
Dey S, et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J Clin Invest. 2015;125:2592–608.
Cai Q, et al. Anoikis resistance is a critical feature of highly aggressive ovarian cancer cells. Oncogene. 2015;34:3315–24.
Malin D, et al. ERK-regulated alphaB-crystallin induction by matrix detachment inhibits anoikis and promotes lung metastasis in vivo. Oncogene. 2015;34:5626–34.
Sundararaman A, et al. Calcium-oxidant signaling network regulates AMP-activated protein kinase (AMPK) activation upon matrix deprivation. J Biol Chem. 2016;291:14410–29.
Liao YH, et al. Epidermal growth factor-induced ANGPTL4 enhances anoikis resistance and tumour metastasis in head and neck squamous cell carcinoma. Oncogene. 2017;36:2228–42.
Fofaria NM, et al. STAT3 induces anoikis resistance, promotes cell invasion and metastatic potential in pancreatic cancer cells. Carcinogenesis. 2015;36:142–50.
Schafer ZT, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–13.
Jiang L, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature. 2016;532:255–58.
Yu LG. Cancer cell resistance to anoikis: MUC1 glycosylation comes to play. Cell Death Dis. 2017;8:e2962.
Mason JA, et al. Metabolism during ECM Detachment: Achilles Heel of Cancer Cells? Trends Cancer. 2017;3:475–81.
Piyush T, et al. MUC1 O-glycosylation contributes to anoikis resistance in epithelial cancer cells. Cell Death Discov. 2017;3:17044.
Ashkenazi A, et al. Death receptors: signaling and modulation. Science. 1998;281:1305–08.
Flusberg DA, et al. Surviving apoptosis: life-death signaling in single cells. Trends Cell Biol. 2015;25:446–58.
Gibert B, et al. Dependence receptors and cancer: addiction to trophic ligands. Cancer Res. 2015;75:5171–75.
Strasser A, et al. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30:180–92.
Aggarwal BB, et al. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 2012;119:651–65.
Wajant H. The Fas signaling pathway: more than a paradigm. Science. 2002;296:1635–36.
Mehlen P, et al. Dependence receptors: from basic research to drug development. Sci Signal. 2011;4:mr2.
von Karstedt S, et al. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat Rev Cancer. 2017;17:352–66.
Fleten KG, et al. hvTRA, a novel TRAIL receptor agonist, induces apoptosis and sustained growth retardation in melanoma. Cell Death Discov. 2016;2:16081.
Boldin MP, et al. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–15.
Dickens LS, et al. The 'complexities' of life and death: death receptor signalling platforms. Exp Cell Res. 2012;318:1269–77.
Muzio M, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death--inducing signaling complex. Cell. 1996;85:817–27.
Boldin MP, et al. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J Biol Chem. 1995;270:7795–98.
Chinnaiyan AM, et al. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–12.
Kischkel FC, et al. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. 2000;12:611–20.
Scott FL, et al. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature. 2009;457:1019–22.
Chan FK, et al. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288:2351–54.
Fu Q, et al. Structural basis and functional role of intramembrane trimerization of the Fas/CD95 death receptor. Mol Cell. 2016;61:602–13.
Brenner D, et al. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015;15:362–74.
Micheau O, et al. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90.
Ting AT, et al. More to life than NF-kappaB in TNFR1 signaling. Trends Immunol. 2016;37:535–45.
Lafont E, et al. The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. EMBO J. 2017;36:1147–66.
Liu ZG, et al. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 1996;87:565–76.
Toscano MA, et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol. 2007;8:825–34.
Lichtenstein RG, et al. Glycobiology of cell death: when glycans and lectins govern cell fate. Cell Death Differ. 2013;20:976–86.
Matarrese P, et al. Galectin-1 sensitizes resting human T lymphocytes to Fas (CD95)-mediated cell death via mitochondrial hyperpolarization, budding, and fission. J Biol Chem. 2005;280:6969–85.
Fu TM, et al. Cryo-EM Structure of caspase-8 tandem DED filament reveals assembly and regulation mechanisms of the death-inducing signaling complex. Mol Cell. 2016;64:236–50.
Dickens LS, et al. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol Cell. 2012;47:291–305.
Schleich K, et al. Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model. Mol Cell. 2012;47:306–19.
Oberst A, et al. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J Biol Chem. 2010;285:16632–42.
Kallenberger SM, et al. Intra- and interdimeric caspase-8 self-cleavage controls strength and timing of CD95-induced apoptosis. Sci Signal. 2014;7:ra23.
Yeh WC, et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12:633–42.
Scaffidi C, et al. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999;274:1541–48.
Kavuri SM, et al. Cellular FLICE-inhibitory protein (cFLIP) isoforms block CD95- and TRAIL death receptor-induced gene induction irrespective of processing of caspase-8 or cFLIP in the death-inducing signaling complex. J Biol Chem. 2011;286:16631–46.
Fricker N, et al. Model-based dissection of CD95 signaling dynamics reveals both a pro- and antiapoptotic role of c-FLIPL. J Cell Biol. 2010;190:377–89.
Micheau O, et al. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J Biol Chem. 2002;277:45162–71.
Hughes MA, et al. Co-operative and hierarchical binding of c-FLIP and caspase-8: a unified model defineshow c-FLIP isoforms differentially control cell fate. Mol Cell. 2016;61:834–49.
Koenig A, et al. The c-FLIPL cleavage product p43FLIP promotes activation of extracellular signal-regulated kinase (ERK), nuclear factor kappaB (NF-kappaB), and caspase-8 and T cell survival. J Biol Chem. 2014;289:1183–91.
Majkut J, et al. Differential affinity of FLIP and procaspase 8 for FADD's DED binding surfaces regulates DISC assembly. Nat Commun. 2014;5:3350.
Schleich K, et al. Molecular architecture of the DED chains at the DISC: regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ. 2016;23:681–94.
You Z, et al. Nuclear factor-kappa B-inducible death effector domain-containing protein suppresses tumor necrosis factor-mediated apoptosis by inhibiting caspase-8 activity. J Biol Chem. 2001;276:26398–404.
Powley IR, et al. Caspase-8 tyrosine-380 phosphorylation inhibits CD95 DISC function by preventing procaspase-8 maturation and cycling within the complex. Oncogene. 2016;35:5629–40.
Helmke C, et al. Ligand stimulation of CD95 induces activation of Plk3 followed by phosphorylation of caspase-8. Cell Res. 2016;26:914–34.
Barnhart BC, et al. The CD95 type I/type II model. Semin Immunol. 2003;15:185–93.
Strasser A, et al. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 1995;14:6136–47.
Jost PJ, et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature. 2009;460:1035–39.
Yin XM, et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–91.
Li H, et al. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501.
Gross A, et al. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274:1156–63.
Huang K, et al. Cleavage by caspase 8 and mitochondrial membrane association activate the BH3-only protein Bid during TRAIL-induced apoptosis. J Biol Chem. 2016;291:11843–851.
Tan CT, et al. MOAP-1 mediates Fas-induced apoptosis in liver by facilitating tBid recruitment to mitochondria. Cell Rep. 2016;16:174–85.
Zaltsman Y, et al. MTCH2/MIMP is a major facilitator of tBID recruitment to mitochondria. Nat Cell Biol. 2010;12:553–62.
Fischer U, et al. Unique and overlapping substrate specificities of caspase-8 and caspase-10. Oncogene. 2006;25:152–59.
Backus KM, et al. Proteome-wide covalent ligand discovery in native biological systems. Nature. 2016;534:570–74.
Horn S, et al. Caspase-10 negatively regulates caspase-8-mediated cell death, switching the response to CD95L in favor of NF-kappaB activation and cell survival. Cell Rep. 2017;19:785–97.
Tanzer MC, et al. Combination of IAP antagonist and IFNgamma activates novel caspase-10- and RIPK1-dependent cell death pathways. Cell Death Differ. 2017;24:481–91.
Sprick MR, et al. Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute caspase-8. EMBO J. 2002;21:4520–30.
Kranz D, et al. A synthetic lethal screen identifies FAT1 as an antagonist of caspase-8 in extrinsic apoptosis. EMBO J. 2014;33:181–97.
O’Donnell MA, et al. Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr Biol. 2007;17:418–24.
Li H, et al. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem. 2006;281:13636–43.
Gerlach B, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–96.
Ea CK, et al. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–57.
Bertrand MJ, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700.
Peltzer N, et al. Holding RIPK1 on the ubiquitin leash in TNFR1 signaling. Trends Cell Biol. 2016;26:445–61.
Haas TL, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36:831–44.
DiDonato JA, et al. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–54.
Zandi E, et al. Direct phosphorylation of IkappaB by IKKalpha and IKKbeta: discrimination between free and NF-kappaB-bound substrate. Science. 1998;281:1360–63.
Rothwarf DM, et al. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395:297–300.
Zandi E, et al. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–52.
Geng J, et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun. 2017;8:359.
Dondelinger Y, et al. NF-kappaB-independent role of IKKalpha/IKKbeta in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol Cell. 2015;60:63–76.
Jaco I, et al. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol Cell. 2017;66:698–710. e695
Fulda S, et al. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11:109–24.
Hitomi J, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–23.
Tortola L, et al. The tumor suppressor Hace1 is a critical regulator of TNFR1-mediated cell fate. Cell Rep. 2016;15:1481–92.
Schneider AT, et al. RIPK1 suppresses a TRAF2-dependent pathway to liver cancer. Cancer Cell. 2017;31:94–109.
Gentle IE, et al. In TNF-stimulated cells, RIPK1 promotes cell survival by stabilizing TRAF2 and cIAP1, which limits induction of non-canonical NF-kappaB and activation of caspase-8. J Biol Chem. 2011;286:13282–91.
Nguyen-Chi M, et al. TNF signaling and macrophages govern fin regeneration in zebrafish larvae. Cell Death Dis. 2017;8:e2979.
O’ Reilly LA, et al. Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature. 2009;461:659–63.
Henry CM, et al. Caspase-8 acts in a non-enzymatic role as a scaffold for assembly of a pro-inflammatory "FADDosome" complex upon TRAIL stimulation. Mol Cell. 2017;65:715–29. e715
Peltzer N, et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 2014;9:153–65.
Bellail AC, et al. A20 ubiquitin ligase-mediated polyubiquitination of RIP1 inhibits caspase-8 cleavage and TRAIL-induced apoptosis in glioblastoma. Cancer Discov. 2012;2:140–55.
Lork M, et al. CYLD, A20 and OTULIN deubiquitinases in NF-kappaB signaling and cell death: so similar, yet so different. Cell Death Differ. 2017;24:1172–83.
Shlyakhtina Y, et al. Dual role of DR5 in death and survival signaling leads to TRAIL resistance in cancer cells. Cell Death and Disease. 2017;8:e3025.
Goldschneider D, et al. Dependence receptors: a new paradigm in cell signaling and cancer therapy. Oncogene. 2010;29:1865–82.
Mehlen P, et al. Dependence receptors and colorectal cancer. Gut. 2014;63:1821–29.
Liu J, et al. Mediation of the DCC apoptotic signal by DIP13 alpha. J Biol Chem. 2002;277:26281–5.
Joubert O, et al. Functional studies of membrane-bound and purified human Hedgehog receptor Patched expressed in yeast. Biochim Biophys Acta. 2009;1788:1813–21.
Fombonne J, et al. Patched dependence receptor triggers apoptosis through ubiquitination of caspase-9. Proc Natl Acad Sci U S A. 2012;109:10510–15.
Mille F, et al. The Patched dependence receptor triggers apoptosis through a DRAL-caspase-9 complex. Nat Cell Biol. 2009;11:739–46.
Llambi F, et al. The dependence receptor UNC5H2 mediates apoptosis through DAP-kinase. EMBO J. 2005;24:1192–201.
Guenebeaud C, et al. The dependence receptor UNC5H2/B triggers apoptosis via PP2A-mediated dephosphorylation of DAP kinase. Mol Cell. 2010;40:863–76.
Raveh T, et al. DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat Cell Biol. 2001;3:1–7.
Zhu Y, et al. Dependence receptor UNC5D mediates nerve growth factor depletion-induced neuroblastoma regression. J Clin Invest. 2013;123:2935–47.
Ichim G, et al. The dependence receptor TrkC triggers mitochondria-dependent apoptosis upon Cobra-1 recruitment. Mol Cell. 2013;51:632–46.
Fitamant J, et al. Netrin-1 expression confers a selective advantage for tumor cell survival in metastatic breast cancer. Proc Natl Acad Sci U S A. 2008;105:4850–55.
Grandin M, et al. Structural decoding of the Netrin-1/UNC5 interaction and its therapeutical implications in cancers. Cancer Cell. 2016;29:173–85.
Harter PN, et al. Netrin-1 expression is an independent prognostic factor for poor patient survival in brain metastases. PLoS One. 2014;9:e92311.
Bernet A, et al. Inactivation of the UNC5C Netrin-1 receptor is associated with tumor progression in colorectal malignancies. Gastroenterology. 2007;133:1840–48.
Castets M, et al. DCC constrains tumour progression via its dependence receptor activity. Nature. 2011;482:534–37.
Krimpenfort P, et al. Deleted in colorectal carcinoma suppresses metastasis in p53-deficient mammary tumours. Nature. 2012;482:538–41.
Broutier L, et al. Targeting netrin-1/DCC interaction in diffuse large B-cell and mantle cell lymphomas. EMBO Mol Med. 2016;8:96–104.
Coissieux MM, et al. Variants in the netrin-1 receptor UNC5C prevent apoptosis and increase risk of familial colorectal cancer. Gastroenterology. 2011;141:2039–46.
Genevois AL, et al. Dependence receptor TrkC is a putative colon cancer tumor suppressor. Proc Natl Acad Sci U S A. 2013;110:3017–22.
Luo Y, et al. NTRK3 is a potential tumor suppressor gene commonly inactivated by epigenetic mechanisms in colorectal cancer. PLoS Genet. 2013;9:e1003552.
Grandin M, et al. Inhibition of DNA methylation promotes breast tumor sensitivity to netrin-1 interference. EMBO Mol Med. 2016;8:863–77.
Ruckdeschel K, et al. Signaling of apoptosis through TLRs critically involves toll/IL-1 receptor domain-containing adapter inducing IFN-beta, but not MyD88, in bacteria-infected murine macrophages. J Immunol. 2004;173:3320–28.
Kaiser WJ, et al. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol. 2005;174:4942–52.
Izzo V, et al. Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol. 2016;26:655–67.
Vanden Berghe T, et al. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–47.
Giorgio V, et al. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 2017.
Baines CP, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62.
Nakagawa T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8.
Basso E, et al. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–61.
Schinzel AC, et al. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–10.
Mukherjee R, et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut. 2016;65:1333–46.
Kwong JQ, et al. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015;21:206–14.
Clarke SJ, et al. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem. 2002;277:34793–99.
Jang S, et al. Elucidating mitochondrial electron transport chain supercomplexes in the heart during ischemia-reperfusion. Antioxid Redox Signal. 2017;27:57–69.
Warne J, et al. Selective inhibition of the mitochondrial permeability transition pore protects against neurodegeneration in experimental multiple sclerosis. J Biol Chem. 2016;291:4356–73.
Lam CK, et al. HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart. Proc Natl Acad Sci U S A. 2015;112:E6466–75.
Piot C, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–81.
Cung TT, et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N Engl J Med. 2015;373:1021–31.
Linkermann A, et al. Catch me if you can: targeting the mitochondrial permeability transition pore in myocardial infarction. Cell Death Differ. 2016;23:1–2.
Kwong JQ, et al. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014;21:1209–17.
Kokoszka JE, et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–65.
Baines CP, et al. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–55.
Galluzzi L, et al. Mitochondrial apoptosis without VDAC. Nat Cell Biol. 2007;9:487–89.
Brower JV, et al. Evolutionarily conserved mammalian adenine nucleotide translocase 4 is essential for spermatogenesis. J Biol Chem. 2007;282:29658–66.
Rodic N, et al. DNA methylation is required for silencing of ant4, an adenine nucleotide translocase selectively expressed in mouse embryonic stem cells and germ cells. Stem Cells. 2005;23:1314–23.
Alavian KN, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014;111:10580–5.
Bonora M, et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle. 2013;12:674–83.
Bonora M, et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 2017;18:1077–89.
Elustondo PA, et al. Mitochondrial permeability transition pore induction is linked to formation of the complex of ATPase C-subunit, polyhydroxybutyrate and inorganic polyphosphate. Cell Death Discov. 2016;2:16070.
Giorgio V, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110:5887–92.
Giorgio V, et al. Ca2+ binding to F-ATP synthase beta subunit triggers the mitochondrial permeability transition. EMBO Rep. 2017;18:1065–76.
Giorgio V, et al. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem. 2009;284:33982–8.
Gerle C. On the structural possibility of pore-forming mitochondrial FoF1 ATP synthase. Biochim Biophys Acta. 2016;1857:1191–6.
He J, et al. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci U S A. 2017;114:3409–14.
Zhou W, et al. Atomistic simulations indicate the c-subunit ring of the F1Fo ATP synthase is not the mitochondrial permeability transition pore. Elife. 2017;6:e23781.
He J, et al. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc Natl Acad Sci U S A. 2017;114:9086–91.
Shanmughapriya S, et al. SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Mol Cell. 2015;60:47–62.
Karch J, et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife. 2013;2:e00772.
Whelan RS, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A. 2012;109:6566–71.
Marzo I, et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–31.
Zamzami N, et al. Bid acts on the permeability transition pore complex to induce apoptosis. Oncogene. 2000;19:6342–50.
Vander Heiden MG, et al. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J Biol Chem. 2001;276:19414–9.
Shimizu S, et al. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–7.
Tsujimoto Y, et al. Bcl-2 and Bcl-xL block apoptosis as well as necrosis: possible involvement of common mediators in apoptotic and necrotic signal transduction pathways. Leukemia. 1997;11(Suppl 3):380–2.
Vander Heiden MG, et al. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol Cell. 1999;3:159–67.
Xu S, et al. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat Commun. 2016;7:13189.
Vaseva AV, et al. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536–48.
Antony AN, et al. MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nat Commun. 2016;7:10955.
Konig T, et al. The m-AAA protease associated with neurodegeneration limits MCU activity in mitochondria. Mol Cell. 2016;64:148–62.
Luongo TS, et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 2015;12:23–34.
Luongo TS, et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature. 2017;545:93–97.
Fazal L, et al. Multifunctional mitochondrial Epac1 controls myocardial cell death. Circ Res. 2017;120:645–57.
Wang Z, et al. A cardiac mitochondrial cAMP signaling pathway regulates calcium accumulation, permeability transition and cell death. Cell Death Dis. 2016;7:e2198.
Vercammen D, et al. Tumour necrosis factor-induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine. 1997;9:801–08.
Vercammen D, et al. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188:919–30.
Degterev A, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–19.
Galluzzi L, et al. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol. 2014;35:24–32.
Degterev A, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21.
Kaiser WJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–79.
Upton JW, et al. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–97.
Upton JW, et al. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–13.
Kaczmarek A, et al. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–23.
Zhang X, et al. MLKL and FADD are critical for suppressing progressive lymphoproliferative disease and activating the NLRP3 inflammasome. Cell Rep. 2016;16:3247–59.
Dara L, et al. Questions and controversies: the role of necroptosis in liver disease. Cell Death Discov. 2016;2:16089.
Linkermann A, et al. Necroptosis. N Engl J Med. 2014;370:455–65.
Murphy JM, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–53.
Vandenabeele P, et al. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:re4.
Li J, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–50.
Cho YS, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23.
Maelfait J, et al. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 2017;36:2529–43.
Lin J, et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature. 2016;540:124–28.
Newton K, et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature. 2016;540:129–33.
Sun L, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27.
Zhao J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109:5322–7.
Rodriguez DA, et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016;23:76–88.
Wu J, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994–1006.
Remijsen Q, et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 2014;5:e1004.
Newton K, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–60.
Wang H, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–46.
Cai Z, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65.
Chen X, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–21.
Hildebrand JM, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A. 2014;111:15072–77.
Quarato G, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Mol Cell. 2016;61:589–601.
Dondelinger Y, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–81.
Zhao XM, et al. Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis. 2016;7:e2089.
Jacobsen AV, et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 2016;7:e2051.
Gong YN, et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell. 2017;169:286–300. e216
Yoon S, et al. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity. 2017;47:51–65. e57
Cai Z, et al. Activation of cell-surface proteases promotes necroptosis, inflammation and cell migration. Cell Res. 2016;26:886–900.
Xia B, et al. MLKL forms cation channels. Cell Res. 2016;26:517–28.
Yoon S, et al. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016;23:253–60.
Wang Z, et al. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–43.
Lu W, et al. Mitochondrial protein PGAM5 regulates mitophagic protection against cell necroptosis. PLoS One. 2016;11:e0147792.
Moriwaki K, et al. The mitochondrial phosphatase PGAM5 is dispensable for necroptosis but promotes inflammasome activation in macrophages. J Immunol. 2016;196:407–15.
Moujalled DM, et al. Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis. 2014;5:e1086.
Tait SW, et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013;5:878–85.
Alvarez-Diaz S, et al. The pseudokinase MLKL and the kinase RIPK3 have distinct roles in autoimmune disease caused by loss of death-receptor-induced apoptosis. Immunity. 2016;45:513–26.
Dondelinger Y, et al. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol. 2016;26:721–32.
Gunther C, et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J Clin Invest. 2016;126:4346–60.
Zhang T, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22:175–82.
Grootjans S, et al. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ. 2017;24:1184–95.
Wu XN, et al. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014;21:1709–20.
Seo J, et al. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat Cell Biol. 2016;18:291–302.
Gyrd-Hansen M. All roads lead to ubiquitin. Cell Death Differ. 2017;24:1135–36.
Onizawa M, et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol. 2015;16:618–27.
Chen W, et al. Ppm1b negatively regulates necroptosis through dephosphorylating Rip3. Nat Cell Biol. 2015;17:434–44.
Xie Y, et al. Inhibition of Aurora kinase A induces necroptosis in pancreatic carcinoma. Gastroenterology. 2017;153:1429–1443.
Li D, et al. A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci U S A. 2015;112:5017–22.
Kaiser WJ, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–72.
Oberst A, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–67.
Dondelinger Y, et al. Poly-ubiquitination in TNFR1-mediated necroptosis. Cell Mol Life Sci. 2016;73:2165–76.
Dondelinger Y, et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013;20:1381–92.
Dillon CP, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157:1189–202.
Weinlich R, et al. Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell Rep. 2013;5:340–8.
Zhao Q, et al. RIPK3 mediates necroptosis during embryonic development and postnatal inflammation in Fadd-deficient mice. Cell Rep. 2017;19:798–808.
Liu Y, et al. RIP1 kinase activity-dependent roles in embryonic development of Fadd-deficient mice. Cell Death Differ. 2017;24:1459–69.
Dillon CP, et al. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012;1:401–07.
Bonnet MC, et al. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity. 2011;35:572–82.
Welz PS, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–4.
Lu JV, et al. Complementary roles of Fas-associated death domain (FADD) and receptor interacting protein kinase-3 (RIPK3) in T-cell homeostasis and antiviral immunity. Proc Natl Acad Sci U S A. 2011;108:15312–7.
McComb S, et al. cIAP1 and cIAP2 limit macrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell Death Differ. 2012;19:1791–801.
Moulin M, et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 2012;31:1679–91.
Vanlangenakker N, et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2011;18:656–65.
Yabal M, et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 2014;7:1796–808.
Kupka S, et al. SPATA2-mediated binding of CYLD to HOIP enables CYLD recruitment to signaling complexes. Cell Rep. 2016;16:2271–80.
Moquin DM, et al. CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One. 2013;8:e76841.
O’Donnell MA, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. 2011;13:1437–42.
Petersen SL, et al. TRAF2 is a biologically important necroptosis suppressor. Cell Death Differ. 2015;22:1846–57.
Morioka S, et al. TAK1 kinase switches cell fate from apoptosis to necrosis following TNF stimulation. J Cell Biol. 2014;204:607–23.
Kaiser WJ, et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci U S A. 2014;111:7753–58.
Rickard JA, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157:1175–88.
Zhang H, et al. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471:373–76.
Dannappel M, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513:90–94.
Takahashi N, et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature. 2014;513:95–99.
Orozco S, et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014;21:1511–21.
Hsu H, et al. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4:387–96.
Ting AT, et al. RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J. 1996;15:6189–96.
Kelliher MA, et al. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303.
Berger SB, et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol. 2014;192:5476–80.
Galluzzi L, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–36.
Ito Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603–08.
Vlantis K, et al. NEMO prevents RIP kinase 1-mediated epithelial cell death and chronic intestinal inflammation by NF-kappaB-dependent and -independent functions. Immunity. 2016;44:553–67.
Mandal P, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56:481–95.
Duprez L, et al. Intermediate domain of receptor-interacting protein kinase 1 (RIPK1) determines switch between necroptosis and RIPK1 kinase-dependent apoptosis. J Biol Chem. 2012;287:14863–72.
Lawlor KE, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282.
Cook WD, et al. RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ. 2014;21:1600–12.
Newton K, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23:1565–76.
Kondylis V, et al. NEMO prevents steatohepatitis and hepatocellular carcinoma by inhibiting RIPK1 kinase activity-mediated hepatocyte apoptosis. Cancer Cell. 2015;28:582–98.
Zou J, et al. Poly IC triggers a cathepsin D- and IPS-1-dependent pathway to enhance cytokine production and mediate dendritic cell necroptosis. Immunity. 2013;38:717–28.
Fricker M, et al. Caspase inhibitors protect neurons by enabling selective necroptosis of inflamed microglia. J Biol Chem. 2013;288:9145–52.
Zitvogel L, et al. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405–14.
Robinson N, et al. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13:954–62.
Thapa RJ, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A. 2013;110:E3109–18.
McComb S, et al. Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc Natl Acad Sci U S A. 2014;111:E3206–13.
Conos SA, et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci U S A. 2017;114:E961–E9.
Kuriakose T, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1:aag2045.
Vince JE, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36:215–27.
Kang TB, et al. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40.
Zhong Z, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164:896–910.
Greten FR, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130:918–31.
Vince JE, et al. The intersection of cell death and inflammasome activation. Cell Mol Life Sci. 2016;73:2349–67.
Moriwaki K, et al. Necroptosis-independent signaling by the RIP kinases in inflammation. Cell Mol Life Sci. 2016;73:2325–34.
Chan FK, et al. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol. 2015;33:79–106.
Dixon SJ. Ferroptosis: bug or feature? Immunol Rev. 2017;277:150–7.
Yang WS, et al. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–76.
Xie Y, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79.
Stockwell BR, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Angeli JPF, et al. Ferroptosis inhibition: mechanisms and opportunities. Trends Pharmacol Sci. 2017;38:489–98.
Dixon SJ, et al. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17.
Dixon SJ, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Linkermann A, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014;111:16836–41.
Kim SE, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. 2016;11:977–85.
Gascon S, et al. Identification and successful negotiation of a metabolic checkpoint in direct neuronal reprogramming. Cell Stem Cell. 2016;18:396–409.
Yang WS, et al. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–45.
Dolma S, et al. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–96.
Shimada K, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497–503.
Hofmans S, et al. Novel ferroptosis inhibitors with improved potency and ADME properties. J Med Chem. 2016;59:2041–53.
Friedmann Angeli JP, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91.
Yang WS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Brigelius-Flohe R, et al. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830:3289–303.
Seiler A, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–48.
Dixon SJ, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523.
Latunde-Dada GO. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta. 2017;1861:1893–900.
Timmerman LA, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24:450–65.
Muir A, et al. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife. 2017;6:e27713.
Louandre C, et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356:971–77.
Lachaier E, et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014;34:6417–22.
Louandre C, et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer. 2013;133:1732–42.
Woo JH, et al. Elucidating compound mechanism of action by network perturbation analysis. Cell. 2015;162:441–51.
Tan S, et al. Oxytosis: a novel form of programmed cell death. Curr Top Med Chem. 2001;1:497–506.
Piani D, et al. Involvement of the cystine transport system xc- in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J Immunol. 1994;152:3578–85.
Park HA, et al. Inhibition of Bcl-xL prevents pro-death actions of DeltaN-Bcl-xL at the mitochondrial inner membrane during glutamate excitotoxicity. Cell Death Differ. 2017;24:1963–74.
Kagan VE, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90.
Doll S, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–98.
Dixon SJ, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604–09.
Yuan H, et al. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478:1338–43.
Yang WS, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113:E4966–75.
Matsushita M, et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212:555–68.
Yagoda N, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–68.
Zilka O, et al. On the mechanism of cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. 2017;3:232–43.
Abeysinghe RD, et al. The environment of the lipoxygenase iron binding site explored with novel hydroxypyridinone iron chelators. J Biol Chem. 1996;271:7965–72.
Gao M, et al. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308.
Torii S, et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J. 2016;473:769–77.
Hou W, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–28.
Gao M, et al. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–32.
Wang H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66:449–65.
Kurz T, et al. Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. FEBS J. 2006;273:3106–17.
Dielschneider RF, et al. Lysosomes as oxidative targets for cancer therapy. Oxid Med Cell Longev. 2017;2017:3749157.
Hayano M, et al. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23:270–78.
Sun X, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 2015;34:5617–25.
Zhu S, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017;77:2064–77.
Poursaitidis I, et al. Oncogene-selective sensitivity to synchronous cell death following modulation of the amino acid nutrient cystine. Cell Rep. 2017;18:2547–56.
Sun X, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–84.
Sun X, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64:488–500.
Xie Y, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20:1692–704.
Song X, et al. FANCD2 protects against bone marrow injury from ferroptosis. Biochem Biophys Res Commun. 2016;480:443–9.
Yuan H, et al. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun. 2016;478:838–44.
Imai H, et al. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun. 2003;305:278–86.
Yant LJ, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496–502.
Carlson BA, et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016;9:22–31.
Hambright WS, et al. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017;12:8–17.
Chen L, et al. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem. 2015;290:28097–106.
Martin-Sanchez D, et al. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrol. 2017;28:218–29.
Do Van B, et al. Ferroptosis, a newly characterized form of cell death in Parkinson's disease that is regulated by PKC. Neurobiol Dis. 2016;94:169–78.
Skouta R, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–6.
Ou Y, et al. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. 2016;113:E6806–E12.
Wang SJ, et al. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 2016;17:366–73.
Jennis M, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016;30:918–30.
Jiang L, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Chen D, et al. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. 2017;36:5593–08.
Viswanathan VS, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–7.
Distefano AM, et al. Heat stress induces ferroptosis-like cell death in plants. J Cell Biol. 2017;216:463–76.
Jorgensen I, et al. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265:130–42.
Cookson BT, et al. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–4.
Willingham SB, et al. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe. 2007;2:147–59.
Kepp O, et al. Pyroptosis - a cell death modality of its kind? Eur J Immunol. 2010;40:627–30.
Bergsbaken T, et al. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109.
Zychlinsky A, et al. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358:167–9.
Shi J, et al. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42:245–54.
Shi J, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–92.
Aziz M, et al. Revisiting caspases in sepsis. Cell Death Dis. 2014;5:e1526.
Vanden Berghe T, et al. Simultaneous targeting of IL-1 and IL-18 is required for protection against inflammatory and septic shock. Am J Respir Crit Care Med. 2014;189:282–91.
Aachoui Y, et al. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr Opin Microbiol. 2013;16:319–26.
Wang Y, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103.
Brough D, et al. Caspase-1-dependent processing of pro-interleukin-1beta is cytosolic and precedes cell death. J Cell Sci. 2007;120:772–81.
Franchi L, et al. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–7.
Kayagaki N, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21.
Yang J, et al. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol. 2015;32:78–83.
Ng TM, et al. Revisiting caspase-11 function in host defense. Cell Host Microbe. 2013;14:9–14.
Kip E, et al. Impact of caspase-1/11, -3, -7, or IL-1beta/IL-18 deficiency on rabies virus-induced macrophage cell death and onset of disease. Cell Death Discov. 2017;3:17012.
Aachoui Y, et al. Canonical inflammasomes drive IFN-gamma to prime caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe. 2015;18:320–32.
Kayagaki N, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71.
Casson CN, et al. Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc Natl Acad Sci U S A. 2015;112:6688–93.
Kayagaki N, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–49.
Hagar JA, et al. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–53.
Ding J, et al. SnapShot: the noncanonical inflammasome. Cell. 2017;168:544–44. e541
Shi J, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
Qiu S, et al. 'Hints' in the killer protein gasdermin D: unveiling the secrets of gasdermins driving cell death. Cell Death Differ. 2017;24:588–96.
Zanoni I, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science. 2016;352:1232–6.
Ding J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–6.
Liu X, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8.
Aglietti RA, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. 2016;113:7858–63.
Chen X, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016;26:1007–20.
Sborgi L, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766–78.
Lan P, et al. TNF superfamily receptor OX40 triggers invariant NKT cell pyroptosis and liver injury. J Clin Invest. 2017;127:2222–34.
Eichholz K, et al. Immune-complexed adenovirus induce AIM2-mediated pyroptosis in human dendritic cells. PLoS Pathog. 2016;12:e1005871.
He WT, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25:1285–98.
Miao EA, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–42.
Jorgensen I, et al. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med. 2016;213:2113–28.
Maltez VI, et al. Inflammasomes coordinate pyroptosis and natural killer cell cytotoxicity to clear infection by a ubiquitous environmental bacterium. Immunity. 2015;43:987–97.
Yang D, et al. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity. 2015;43:923–32.
Vanden Berghe T, et al. Passenger mutations confound interpretation of all genetically modified congenic mice. Immunity. 2015;43:200–09.
Yu J, et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci U S A. 2014;111:15514–9.
Broz P, et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. 2012;490:288–91.
Rathinam VA, et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150:606–19.
Man SM, et al. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat Immunol. 2015;16:467–75.
Meunier E, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature. 2014;509:366–70.
Pilla DM, et al. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci U S A. 2014;111:6046–51.
Man SM, et al. IRGB10 liberates bacterial ligands for sensing by the AIM2 and caspase-11-NLRP3 inflammasomes. Cell. 2016;167:382–96. e317
Wallach D, et al. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science. 2016;352:aaf2154.
Napier BA, et al. Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. J Exp Med. 2016;213:2365–82.
Case CL, et al. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc Natl Acad Sci U S A. 2013;110:1851–56.
Man SM, et al. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci Rep. 2017;7:45126.
Kepp O, et al. Mitochondrial control of the NLRP3 inflammasome. Nat Immunol. 2011;12:199–200.
Coll RC, et al. Questions and controversies in innate immune research: what is the physiological role of NLRP3? Cell Death Discov. 2016;2:16019.
Martin-Sanchez F, et al. Lytic cell death induced by melittin bypasses pyroptosis but induces NLRP3 inflammasome activation and IL-1beta release. Cell Death Dis. 2017;8:e2984.
Fatokun AA, et al. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol. 2014;171:2000–16.
Virag L, et al. Poly(ADP-ribose) signaling in cell death. Mol Aspects Med. 2013;34:1153–67.
David KK, et al. Parthanatos, a messenger of death. Front Biosci (Landmark Ed). 2009;14:1116–28.
Lipton SA, et al. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–32.
Zhang J, et al. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–9.
Dawson VL, et al. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci U S A. 1991;88:6368–71.
Andrabi SA, et al. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat Med. 2011;17:692–9.
Andrabi SA, et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–13.
Yu SW, et al. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A. 2006;103:18314–9.
Yu SW, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–63.
Wang Y, et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci Signal. 2011;4:ra20.
Wang H, et al. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci. 2004;24:10963–73.
Mashimo M, et al. ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc Natl Acad Sci U S A. 2013;110:18964–9.
Curtin NJ, et al. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol Aspects Med. 2013;34:1217–56.
Xu Z, et al. Endonuclease G does not play an obligatory role in poly(ADP-ribose) polymerase-dependent cell death after transient focal cerebral ischemia. Am J Physiol Regul Integr Comp Physiol. 2010;299:R215–21.
Buttner S, et al. Endonuclease G regulates budding yeast life and death. Mol Cell. 2007;25:233–46.
Buttner S, et al. Endonuclease G mediates alpha-synuclein cytotoxicity during Parkinson's disease. EMBO J. 2013;32:3041–54.
Wang X, et al. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science. 2002;298:1587–92.
Parrish J, et al. Mitochondrial endonuclease G is important for apoptosis in C. elegans. Nature. 2001;412:90–4.
Li LY, et al. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–9.
David KK, et al. EndoG is dispensable in embryogenesis and apoptosis. Cell Death Differ. 2006;13:1147–55.
Irvine RA, et al. Generation and characterization of endonuclease G null mice. Mol Cell Biol. 2005;25:294–302.
Lin JL, et al. Oxidative stress impairs cell death by repressing the nuclease activity of mitochondrial endonuclease G. Cell Rep. 2016;16:279–87.
Wang Y, et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science. 2016;354:aad6872.
Andrabi SA, et al. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A. 2014;111:10209–14.
Fouquerel E, et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 2014;8:1819–31.
Jang KH, et al. AIF-independent parthanatos in the pathogenesis of dry age-related macular degeneration. Cell Death Dis. 2017;8:e2526.
Pardee AB, et al. Cancer therapy with beta-lapachone. Curr Cancer Drug Targets. 2002;2:227–42.
Park EJ, et al. beta-Lapachone induces programmed necrosis through the RIP1-PARP-AIF-dependent pathway in human hepatocellular carcinoma SK-Hep1 cells. Cell Death Dis. 2014;5:e1230.
Jouan-Lanhouet S, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012;19:2003–14.
Sosna J, et al. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol Life Sci. 2014;71:331–48.
Lee Y, et al. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat Neurosci. 2013;16:1392–400.
Sahaboglu A, et al. PARP1 gene knock-out increases resistance to retinal degeneration without affecting retinal function. PLoS One. 2010;5:e15495.
Eliasson MJ, et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–95.
Kim J, et al. Loss of poly(ADP-ribose) polymerase 1 attenuates renal fibrosis and inflammation during unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2011;301:F450–59.
Krishna S, et al. Mechanisms and consequences of entosis. Cell Mol Life Sci. 2016;73:2379–86.
Florey O, et al. Entosis: cell-in-cell formation that kills through entotic cell death. Curr Mol Med. 2015;15:861–6.
Perez E, et al. Intercellular cannibalism fuels tumor growth. Cell Death Differ. 2017;24:759–60.
Overholtzer M, et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell. 2007;131:966–79.
Wan Q, et al. Regulation of myosin activation during cell-cell contact formation by Par3-Lgl antagonism: entosis without matrix detachment. Mol Biol Cell. 2012;23:2076–91.
Sun Q, et al. Competition between human cells by entosis. Cell Res. 2014;24:1299–310.
Hamann JC, et al. Entosis is induced by glucose starvation. Cell Rep. 2017;20:201–10.
Durgan J, et al. Mitosis can drive cell cannibalism through entosis. Elife 2017; 6:e27134.
Wang M, et al. Impaired formation of homotypic cell-in-cell structures in human tumor cells lacking alpha-catenin expression. Sci Rep. 2015;5:12223.
Purvanov V, et al. G-protein-coupled receptor signaling and polarized actin dynamics drive cell-in-cell invasion. Elife. 2014;3:e02786.
Sun Q, et al. Induction of entosis by epithelial cadherin expression. Cell Res. 2014;24:1288–98.
Hinojosa LS, et al. MRTF transcription and Ezrin-dependent plasma membrane blebbing are required for entotic invasion. J Cell Biol. 2017;216:3087–95.
Xia P, et al. Aurora A orchestrates entosis by regulating a dynamic MCAK-TIP150 interaction. J Mol Cell Biol. 2014;6:240–54.
Florey O, et al. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol. 2011;13:1335–43.
Kim SE, et al. Autophagy proteins regulate cell engulfment mechanisms that participate in cancer. Semin Cancer Biol. 2013;23:329–36.
Sanjuan MA, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–57.
Krajcovic M, et al. mTOR regulates phagosome and entotic vacuole fission. Mol Biol Cell. 2013;24:3736–45.
Krishna S, et al. PIKfyve regulates vacuole maturation and nutrient recovery following engulfment. Dev Cell. 2016;38:536–47.
Wang S, et al. Internalization of NK cells into tumor cells requires ezrin and leads to programmed cell-in-cell death. Cell Res. 2009;19:1350–62.
Wen S, et al. Androgen receptor enhances entosis, a non-apoptotic cell death, through modulation of Rho/ROCK pathway in prostate cancer cells. Prostate. 2013;73:1306–15.
Jamal-Hanjani M, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376:2109–21.
Vitale I, et al. Illicit survival of cancer cells during polyploidization and depolyploidization. Cell Death Differ. 2011;18:1403–13.
Vitale I, et al. Karyotypic aberrations in oncogenesis and cancer therapy. Trends Cancer. 2015;1:124–35.
Krajcovic M, et al. A non-genetic route to aneuploidy in human cancers. Nat Cell Biol. 2011;13:324–30.
Krajcovic M, et al. Mechanisms of ploidy increase in human cancers: a new role for cell cannibalism. Cancer Res. 2012;72:1596–601.
Li Y, et al. Entosis allows timely elimination of the luminal epithelial barrier for embryo implantation. Cell Rep. 2015;11:358–65.
Ahmed N, et al. Entosis acts as a novel way within Sertoli cells to eliminate spermatozoa in seminiferous tubule. Front Physiol. 2017;8:361.
Brinkmann V, et al. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol. 2012;198:773–83.
Remijsen Q, et al. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 2011;18:581–8.
Brinkmann V, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5.
Branzk N, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. 2014;15:1017–25.
Clark SR, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–9.
Csomos K, et al. Protein cross-linking by chlorinated polyamines and transglutamylation stabilizes neutrophil extracellular traps. Cell Death Dis. 2016;7:e2332.
Yousefi S, et al. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16:1438–44.
McIlroy DJ, et al. Mitochondrial DNA neutrophil extracellular traps are formed after trauma and subsequent surgery. J Crit Care. 2014;29:e1131–5. 1133
Wang H, et al. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. 2015;67:3190–200.
Caielli S, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. 2016;213:697–713.
Lood C, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22:146–53.
Cedervall J, et al. Tumor-induced NETosis as a risk factor for metastasis and organ failure. Cancer Res. 2016;76:4311–5.
Demers M, et al. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology. 2016;5:e1134073.
Wong SL, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21:815–9.
Wartha F, et al. ETosis: a novel cell death pathway. Sci Signal. 2008;1:pe25.
Yousefi S, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14:949–53.
Morshed M, et al. NADPH oxidase-independent formation of extracellular DNA traps by basophils. J Immunol. 2014;192:5314–23.
Yipp BG, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18:1386–93.
Fuchs TA, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–41.
Parker H, et al. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J Leukoc Biol. 2012;92:841–9.
Hakkim A, et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol. 2011;7:75–7.
Remijsen Q, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21:290–304.
Papayannopoulos V, et al. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–91.
Metzler KD, et al. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014;8:883–96.
Metzler KD, et al. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood. 2011;117:953–9.
Stojkov D, et al. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J Cell Biol. 2017;216:4073–4090.
Martinod K, et al. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J Thromb Haemost. 2016;14:551–8.
Li P, et al. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207:1853–62.
Desai J, et al. Matters of life and death. How neutrophils die or survive along NET release and is "NETosis" = necroptosis? Cell Mol Life Sci. 2016;73:2211–19.
Hemmers S, et al. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One. 2011;6:e22043.
Desai J, et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur J Immunol. 2016;46:223–9.
Amini P, et al. NET formation can occur independently of RIPK3 and MLKL signaling. Eur J Immunol. 2016;46:178–84.
Aits S, et al. Lysosomal cell death at a glance. J Cell Sci. 2013;126:1905–12.
Gomez-Sintes R, et al. Lysosomal cell death mechanisms in aging. Ageing Res Rev. 2016;32:150–68.
Serrano-Puebla A, et al. Lysosomal membrane permeabilization in cell death: new evidence and implications for health and disease. Ann N Y Acad Sci. 2016;1371:30–44.
Yacobi-Sharon K, et al. Alternative germ cell death pathway in Drosophila involves HtrA2/Omi, lysosomes, and a caspase-9 counterpart. Dev Cell. 2013;25:29–42.
Yang H, et al. The regulated elimination of transit-amplifying cells preserves tissue homeostasis during protein starvation in Drosophila testis. Development. 2015;142:1756–66.
Lu KL, et al. Germ cell connectivity enhances cell death in response to DNA damage in the Drosophila testis. Elife. 2017;6:e27960.
Kutscher LM, et al. Non-apoptotic cell death in animal development. Cell Death Differ. 2017;24:1326–36.
Huai J, et al. TNFalpha-induced lysosomal membrane permeability is downstream of MOMP and triggered by caspase-mediated NDUFS1 cleavage and ROS formation. J Cell Sci. 2013;126:4015–25.
Oberle C, et al. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 2010;17:1167–78.
Plotegher N, et al. Mitochondrial dysfunction and neurodegeneration in lysosomal storage disorders. Trends Mol Med. 2017;23:116–34.
Boya P, et al. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med. 2003;197:1323–34.
Boya P, et al. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene. 2003;22:3927–36.
Feldstein AE, et al. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1339–46.
Chen F, et al. The octyl ester of ginsenoside Rh2 induces lysosomal membrane permeabilization via Bax translocation. Nutrients. 2016;8:E244.
Bove J, et al. BAX channel activity mediates lysosomal disruption linked to Parkinson disease. Autophagy. 2014;10:889–900.
Guan JJ, et al. DRAM1 regulates apoptosis through increasing protein levels and lysosomal localization of BAX. Cell Death Dis. 2015;6:e1624.
Kurz T, et al. Lysosomes and oxidative stress in aging and apoptosis. Biochim Biophys Acta. 2008;1780:1291–303.
Kurz T, et al. Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell Biol. 2008;129:389–406.
Sumoza-Toledo A, et al. TRPM2: a multifunctional ion channel for calcium signalling. J Physiol. 2011;589:1515–25.
Werneburg NW, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) protein-induced lysosomal translocation of proapoptotic effectors is mediated by phosphofurin acidic cluster sorting protein-2 (PACS-2). J Biol Chem. 2012;287:24427–37.
Laforge M, et al. DRAM triggers lysosomal membrane permeabilization and cell death in CD4(+) T cells infected with HIV. PLoS Pathog. 2013;9:e1003328.
Maejima I, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013;32:2336–47.
Hornung V, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–56.
Crighton D, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34.
Kreuzaler PA, et al. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol. 2011;13:303–09.
Sargeant TJ, et al. Stat3 controls cell death during mammary gland involution by regulating uptake of milk fat globules and lysosomal membrane permeabilization. Nat Cell Biol. 2014;16:1057–68.
Droga-Mazovec G, et al. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J Biol Chem. 2008;283:19140–50.
Bidere N, et al. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem. 2003;278:31401–11.
Taniguchi M, et al. Lysosomal ceramide generated by acid sphingomyelinase triggers cytosolic cathepsin B-mediated degradation of X-linked inhibitor of apoptosis protein in natural killer/T lymphoma cell apoptosis. Cell Death Dis. 2015;6:e1717.
Green DR, et al. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109–12.
Youle RJ, et al. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14.
Loison F, et al. Proteinase 3-dependent caspase-3 cleavage modulates neutrophil death and inflammation. J Clin Invest. 2014;124:4445–58.
Brojatsch J, et al. Distinct cathepsins control necrotic cell death mediated by pyroptosis inducers and lysosome-destabilizing agents. Cell Cycle. 2015;14:964–72.
Hsu KF, et al. Cathepsin L mediates resveratrol-induced autophagy and apoptotic cell death in cervical cancer cells. Autophagy. 2009;5:451–60.
Trincheri NF, et al. Resveratrol induces cell death in colorectal cancer cells by a novel pathway involving lysosomal cathepsin D. Carcinogenesis. 2007;28:922–31.
Turk V, et al. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta. 2012;1824:68–88.
Turk V, et al. Cystatins: biochemical and structural properties, and medical relevance. Front Biosci. 2008;13:5406–20.
Gooptu B, et al. Conformational pathology of the serpins: themes, variations, and therapeutic strategies. Annu Rev Biochem. 2009;78:147–76.
Appelqvist H, et al. Sensitivity to lysosome-dependent cell death is directly regulated by lysosomal cholesterol content. PLoS One. 2012;7:e50262.
Kirkegaard T, et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463:549–53.
Nylandsted J, et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200:425–35.
Kirkegaard T, et al. Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci Transl Med. 2016;8:355ra118.
Groth-Pedersen L, et al. Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett. 2013;332:265–74.
Petersen NH, et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell. 2013;24:379–93.
Gyparaki MT, et al. Lysosome: the cell's 'suicidal bag' as a promising cancer target. Trends Mol Med. 2014;20:239–41.
Piao S, et al. Targeting the lysosome in cancer. Ann N Y Acad Sci. 2016;1371:45–54.
Zhang H, et al. Eaten alive: novel insights into autophagy from multicellular model systems. Trends Cell Biol. 2015;25:376–87.
Sharma K, et al. Cytotoxic autophagy in cancer therapy. Int J Mol Sci. 2014;15:10034–51.
Das G, et al. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol 2012; 4: a008813.
Fullgrabe J, et al. Transcriptional regulation of mammalian autophagy at a glance. J Cell Sci. 2016;129:3059–66.
Baek SH, et al. Epigenetic control of autophagy: nuclear events gain more attention. Mol Cell. 2017;65:781–5.
Pietrocola F, et al. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol. 2013;23:310–22.
Fullgrabe J, et al. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol. 2014;15:65–74.
Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222.
Galluzzi L, et al. Metabolic control of autophagy. Cell. 2014;159:1263–76.
Kaur J, et al. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–72.
Galluzzi L, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–80.
Levy JMM, et al. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528–42.
Mulcahy Levy JM, et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife. 2017;6:e19671.
Pagotto A, et al. Autophagy inhibition reduces chemoresistance and tumorigenic potential of human ovarian cancer stem cells. Cell Death Dis. 2017;8:e2943.
Sica V, et al. Organelle-specific initiation of autophagy. Mol Cell. 2015;59:522–39.
Liu P, et al. High autophagic flux guards ESC identity through coordinating autophagy machinery gene program by FOXO1. Cell Death Differ. 2017;24:1672–80.
Gatica D, et al. Molecular mechanisms of autophagy in the cardiovascular system. Circ Res. 2015;116:456–67.
Menzies FM, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93:1015–34.
Menzies FM, et al. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–57.
Galluzzi L, et al. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16:487–511.
Bravo-San Pedro JM, et al. Autophagy and mitophagy in cardiovascular disease. Circ Res. 2017;120:1812–24.
Galluzzi L, et al. Autophagy in acute brain injury. Nat Rev Neurosci. 2016;17:467–84.
Anding AL, et al. Autophagy in cell life and cell death. Curr Top Dev Biol. 2015;114:67–91.
Denton D, et al. Autophagy as a pro-death pathway. Immunol Cell Biol. 2015;93:35–42.
Denton D, et al. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95.
Saleh T, et al. Autophagy is not uniformly cytoprotective: a personalized medicine approach for autophagy inhibition as a therapeutic strategy in non-small cell lung cancer. Biochim Biophys Acta. 2016;1860:2130–6.
Gump JM, et al. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat Cell Biol. 2014;16:47–54.
Goodall ML, et al. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell. 2016;37:337–49.
Dey A, et al. Inhibition of BMI1 induces autophagy-mediated necroptosis. Autophagy. 2016;12:659–70.
Basit F, et al. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013;20:1161–73.
He W, et al. A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene. 2014;33:3004–13.
Denton D, et al. Larval midgut destruction in Drosophila: not dependent on caspases but suppressed by the loss of autophagy. Autophagy. 2010;6:163–5.
Denton D, et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19:1741–6.
Xu T, et al. Characterization of autophagic responses in Drosophila melanogaster. Methods Enzymol. 2017;588:445–5.
Berry DL, et al. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–48.
Mills K, et al. The Drosophila melanogaster Apaf-1 homologue ARK is required for most, but not all, programmed cell death. J Cell Biol. 2006;172:809–15.
Daish TJ, et al. Drosophila caspase DRONC is required for specific developmental cell death pathways and stress-induced apoptosis. Dev Cell. 2004;7:909–15.
Denton D, et al. Relationship between growth arrest and autophagy in midgut programmed cell death in Drosophila. Cell Death Differ. 2012;19:1299–307.
Wang H, et al. Autophagy activity contributes to programmed cell death in Caenorhabditis elegans. Autophagy. 2013;9:1975–82.
Arakawa S, et al. Role of Atg5-dependent cell death in the embryonic development of Bax/Bak double-knockout mice. Cell Death Differ. 2017;24:1598–608.
Shimizu S, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–8.
Xu T, et al. Distinct requirements of Autophagy-related genes in programmed cell death. Cell Death Differ. 2015;22:1792–802.
Chang TK, et al. Uba1 functions in Atg7- and Atg3-independent autophagy. Nat Cell Biol. 2013;15:1067–78.
Denton D, et al. UTX coordinates steroid hormone-mediated autophagy and cell death. Nat Commun. 2013;4:2916.
Nelson C, et al. miR-14 regulates autophagy during developmental cell death by targeting ip3-kinase 2. Mol Cell. 2014;56:376–88.
Tracy K, et al. Ral GTPase and the exocyst regulate autophagy in a tissue-specific manner. EMBO Rep. 2016;17:110–21.
McPhee CK, et al. Activation of autophagy during cell death requires the engulfment receptor Draper. Nature. 2010;465:1093–6.
Lin L, et al. Complement-related regulates autophagy in neighboring cells. Cell. 2017;170:158–71. e158
Hou YC, et al. Effector caspase Dcp-1 and IAP protein Bruce regulate starvation-induced autophagy during Drosophila melanogaster oogenesis. J Cell Biol. 2008;182:1127–39.
Nezis IP, et al. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol. 2010;190:523–31.
Nezis IP, et al. Cell death during Drosophila melanogaster early oogenesis is mediated through autophagy. Autophagy. 2009;5:298–302.
Xie C, et al. Neuroprotection by selective neuronal deletion of Atg7 in neonatal brain injury. Autophagy. 2016;12:410–23.
Guha P, et al. Cocaine elicits autophagic cytotoxicity via a nitric oxide-GAPDH signaling cascade. Proc Natl Acad Sci U S A. 2016;113:1417–22.
Dasari SK, et al. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017;24:1288–302.
Wang K, et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat Commun. 2015;6:6779.
Liu Y, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A. 2013;110:20364–71.
Galluzzi L, et al. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111.
Kepp O, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3:e955691.
Vanpouille-Box C, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618.
Buytaert E, et al. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim Biophys Acta. 2007;1776:86–107.
Galluzzi L, et al. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol. 2017;14:247–58.
Adkins I, et al. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology. 2014;3:e968434.
Kroemer G, et al. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72.
Garg AD, et al. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med. 2016;8:328ra327.
Bezu L, et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol. 2015;6:187.
Galluzzi L, et al. Immunological mechanisms underneath the efficacy of cancer therapy. Cancer Immunol Res. 2016;4:895–902.
Lotze MT, et al. Damage associated molecular pattern molecules. Clin Immunol. 2007;124:1–4.
Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–5.
Garg AD, et al. Pathogen response-like recruitment and activation of neutrophils by sterile immunogenic dying cells drives neutrophil-mediated residual cell killing. Cell Death Differ. 2017;24:832–43.
Obeid M, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61.
Gardai SJ, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–34.
Michaud M, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–7.
Elliott MR, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–6.
Ghiringhelli F, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–78.
Apetoh L, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–59.
Scaffidi P, et al. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–95.
Conte A, et al. High mobility group A1 protein modulates autophagy in cancer cells. Cell Death Differ. 2017;24:1948–62.
Sistigu A, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20:1301–09.
Hunger A, et al. Reestablishment of p53/Arf and interferon-beta pathways mediated by a novel adenoviral vector potentiates antiviral response and immunogenic cell death. Cell Death Discov. 2017;3:17017.
Chiba S, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13:832–42.
Vacchelli E, et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science. 2015;350:972–8.
Gelebart P, et al. Calreticulin, a Ca2+-binding chaperone of the endoplasmic reticulum. Int J Biochem Cell Biol. 2005;37:260–6.
Panaretakis T, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28:578–90.
Kepp O, et al. eIF2alpha phosphorylation as a biomarker of immunogenic cell death. Semin Cancer Biol. 2015;33:86–92.
Kranz P, et al. PDI is an essential redox-sensitive activator of PERK during the unfolded protein response (UPR). Cell Death Dis. 2017;8:e2986.
Panaretakis T, et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008;15:1499–509.
Pawaria S, et al. CD91-dependent programming of T-helper cell responses following heat shock protein immunization. Nat Commun. 2011;2:521.
Garg AD, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012;31:1062–79.
Garg AD, et al. Resistance to anticancer vaccination effect is controlled by a cancer cell-autonomous phenotype that disrupts immunogenic phagocytic removal. Oncotarget. 2015;6:26841–60.
Barclay AN, et al. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. 2014;32:25–50.
Sockolosky JT, et al. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc Natl Acad Sci U S A. 2016;113:E2646–54.
Fucikova J, et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood. 2016;128:3113–24.
Wang H, et al. Expression and significance of CD44, CD47 and c-met in ovarian clear cell carcinoma. Int J Mol Sci. 2015;16:3391–404.
Suzuki S, et al. CD47 expression regulated by the miR-133a tumor suppressor is a novel prognostic marker in esophageal squamous cell carcinoma. Oncol Rep. 2012;28:465–72.
Chao MP, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med. 2010;2:63ra94.
Majeti R, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138:286–99.
Tada K, et al. Tethering of apoptotic cells to phagocytes through binding of CD47 to Src homology 2 domain-bearing protein tyrosine phosphatase substrate-1. J Immunol. 2003;171:5718–26.
Nilsson A, et al. CD47 promotes both phosphatidylserine-independent and phosphatidylserine-dependent phagocytosis of apoptotic murine thymocytes by non-activated macrophages. Biochem Biophys Res Commun. 2009;387:58–63.
Ma Y, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38:729–41.
Zitvogel L, et al. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol. 2012;13:343–51.
Trautmann A. Extracellular ATP in the immune system: more than just a "danger signal". Sci Signal. 2009;2:pe6.
Ma Y, et al. Autophagy and cellular immune responses. Immunity. 2013;39:211–27.
Wang Y, et al. Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy. 2013;9:1624–25.
Chekeni FB, et al. Pannexin 1 channels mediate 'find-me' signal release and membrane permeability during apoptosis. Nature. 2010;467:863–7.
Garg AD, et al. Autophagy-dependent suppression of cancer immunogenicity and effector mechanisms of innate and adaptive immunity. Oncoimmunology. 2013;2:e26260.
Garg AD, et al. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy. 2013;9:1292–307.
Antonioli L, et al. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer. 2013;13:842–57.
Chalmin F, et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity. 2012;36:362–73.
Sun X, et al. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology. 2010;139:1030–40.
Mackenzie KJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461–5.
Harding SM, et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466–70.
McNab F, et al. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15:87–103.
Corrales L, et al. The host STING pathway at the interface of cancer and immunity. J Clin Invest. 2016;126:2404–11.
Deng L, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843–52.
Woo SR, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–42.
Fuertes MB, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–16.
Sims GP, et al. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–88.
Tittarelli A, et al. Toll-like receptor 4 gene polymorphism influences dendritic cell in vitro function and clinical outcomes in vaccinated melanoma patients. Cancer Immunol Immunother. 2012;61:2067–77.
Gast A, et al. Association of inherited variation in Toll-like receptor genes with malignant melanoma susceptibility and survival. PLoS One. 2011;6:e24370.
Bergmann C, et al. Toll-like receptor 4 single-nucleotide polymorphisms Asp299Gly and Thr399Ile in head and neck squamous cell carcinomas. J Transl Med. 2011;9:139.
Dumitriu IE, et al. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol. 2005;174:7506–15.
Tang D, et al. A Janus tale of two active high mobility group box 1 (HMGB1) redox states. Mol Med. 2012;18:1360–62.
Venereau E, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209:1519–28.
Yang H, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med. 2012;18:250–9.
Tang D, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29:5299–310.
Kazama H, et al. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32.
Rubartelli A, et al. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007;28:429–36.
Kang R, et al. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1–116.
Connolly PF, et al. Viral hijacking of host caspases: an emerging category of pathogen-host interactions. Cell Death Differ. 2017;24:1401–10.
Giampazolias E, et al. Mitochondrial permeabilization engages NF-kappaB-dependent anti-tumour activity under caspase deficiency. Nat Cell Biol. 2017;19:1116–29.
Kearney CJ, et al. An inflammatory perspective on necroptosis. Mol Cell. 2017;65:965–73.
Gunther C, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–9.
Yatim N, et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science. 2015;350:328–34.
Buque A, et al. Trial Watch-Small molecules targeting the immunological tumor microenvironment for cancer therapy. Oncoimmunology. 2016;5:e1149674.
Zelenay S, et al. Reducing prostaglandin E2 production to raise cancer immunogenicity. Oncoimmunology. 2016;5:e1123370.
Rongvaux A, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563–77.
White MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–62.
Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705.
Sharpless NE, et al. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397–408.
van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–46.
Harper JW, et al. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–16.
Serrano M, et al. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–7.
Kamb A, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436–40.
Kamijo T, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–59.
Zhang Y, et al. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–34.
He S, et al. Senescence in health and disease. Cell. 2017;169:1000–11.
Acosta JC, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–90.
Coppe JP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68.
Wiley CD, et al. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell. 2017;16:1043–50.
Baar MP, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017;169:132–47. e116
Munoz-Espin D, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–18.
Storer M, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–30.
Baker DJ, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184–89.
Demaria M, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722–33.
Garcia-Prat L, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529:37–42.
Jeon OH, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23:775–81.
Jun JI, et al. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12:676–85.
Li T, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–83.
Kang TW, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–51.
Sharpless NE, et al. The differential impact of p16(INK4a) or p19(ARF) deficiency on cell growth and tumorigenesis. Oncogene. 2004;23:379–85.
Chiche A, et al. Injury-induced senescence enables in vivo reprogramming in skeletal muscle. Cell Stem Cell. 2017;20:407–14. e404
Mosteiro L, et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. 2016;354:aaf4445.
Childs BG, et al. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017;16:718–35.
Ewald JA, et al. Therapy-induced senescence in cancer. J Natl Cancer Inst. 2010;102:1536–46.
Lopez-Otin C, et al. The hallmarks of aging. Cell. 2013;153:1194–217.
Lopez-Otin C, et al. Metabolic control of longevity. Cell. 2016;166:802–21.
Bernet JD, et al. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat Med. 2014;20:265–71.
Eggert T, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. 2016;30:533–47.
Ruhland MK, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016;7:11762.
Baker DJ, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6.
Childs BG, et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472–7.
Hoare M, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol. 2016;18:979–2.
Sturmlechner I, et al. Cellular senescence in renal ageing and disease. Nat Rev Nephrol. 2017;13:77–89.
Demaria M, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7:165–76.
Childs BG, et al. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424–35.
Chang J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78–83.
Yosef R, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. 2016;7:11190.
Zhu Y, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016;15:428–35.
Castedo M, et al. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–37.
Vitale I, et al. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–92.
Dominguez-Brauer C, et al. Targeting mitosis in cancer: emerging strategies. Mol Cell. 2015;60:524–36.
Neelsen KJ, et al. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol. 2013;200:699–708.
Vitale I, et al. Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J. 2010;29:1272–84.
Castedo M, et al. Cyclin-dependent kinase-1: linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002;9:1287–93.
Vitale I, et al. Inhibition of Chk1 kills tetraploid tumor cells through a p53-dependent pathway. PLoS One. 2007;2:e1337.
Castedo M, et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene. 2004;23:4362–70.
Dawar S, et al. Caspase-2-mediated cell death is required for deleting aneuploid cells. Oncogene. 2017;36:2704–14.
Fava LL, et al. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev. 2017;31:34–45.
Lopez-Garcia C, et al. BCL9L dysfunction impairs caspase-2 expression permitting aneuploidy tolerance in colorectal cancer. Cancer Cell. 2017;31:79–93.
Dawar S, et al. Impaired haematopoietic stem cell differentiation and enhanced skewing towards myeloid progenitors in aged caspase-2-deficient mice. Cell Death Dis. 2016;7:e2509.
Puccini J, et al. Loss of caspase-2 augments lymphomagenesis and enhances genomic instability in Atm-deficient mice. Proc Natl Acad Sci U S A. 2013;110:19920–25.
Dorstyn L, et al. Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ. 2012;19:1288–98.
Ho LH, et al. A tumor suppressor function for caspase-2. Proc Natl Acad Sci U S A. 2009;106:5336–41.
Shalini S, et al. Caspase-2 deficiency accelerates chemically induced liver cancer in mice. Cell Death Differ. 2016;23:1727–36.
Mansilla S, et al. Mitotic catastrophe results in cell death by caspase-dependent and caspase-independent mechanisms. Cell Cycle. 2006;5:53–60.
Denisenko TV, et al. Mitotic catastrophe and cancer drug resistance: A link that must to be broken. Drug Resist Updat. 2016;24:1–12.
Surova O, et al. Various modes of cell death induced by DNA damage. Oncogene. 2013;32:3789–97.
Gascoigne KE, et al. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–22.
Furth N, et al. The LATS1 and LATS2 tumor suppressors: beyond the Hippo pathway. Cell Death Differ. 2017;24:1488–501.
Castedo M, et al. Apoptosis regulation in tetraploid cancer cells. EMBO J. 2006;25:2584–95.
Crockford A, et al. Cyclin D mediates tolerance of genome-doubling in cancers with functional p53. Ann Oncol. 2017;28:149–56.
Ganem NJ, et al. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell. 2014;158:833–48.
Hinchcliffe EH, et al. Chromosome missegregation during anaphase triggers p53 cell cycle arrest through histone H3.3 Ser31 phosphorylation. Nat Cell Biol. 2016;18:668–75.
Lambrus BG, et al. A USP28-53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol. 2016;214:143–53.
Li M, et al. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:14188–93.
Meitinger F, et al. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol. 2016;214:155–66.
Thompson SL, et al. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol. 2010;188:369–81.
Manic G, et al. CHK1-targeted therapy to deplete DNA replication-stressed, p53-deficient, hyperdiploid colorectal cancer stem cells. Gut 2017;
Huun J, et al. Effects of concomitant inactivation of p53 and pRb on response to doxorubicin treatment in breast cancer cell lines. Cell Death Discov. 2017;3:17026.
Michels J, et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res. 2013;73:2271–80.
Shibue T, et al. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–29.
Galluzzi L, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–83.
Galluzzi L, et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257.
Casinelli G, et al. N-Myc overexpression increases cisplatin resistance in neuroblastoma via deregulation of mitochondrial dynamics. Cell Death Discov. 2016;2:16082.
Tsapras P, et al. Caspase involvement in autophagy. Cell Death Differ. 2017;24:1369–79.
Galluzzi L, et al. No death without life: vital functions of apoptotic effectors. Cell Death Differ. 2008;15:1113–23.
Aram L, et al. CDPs: caspase-dependent non-lethal cellular processes. Cell Death Differ. 2017;24:1307–10.
Nakajima YI, et al. Caspase-dependent non-apoptotic processes in development. Cell Death Differ. 2017;24:1422–30.
Fernando P, et al. Neural stem cell differentiation is dependent upon endogenous caspase 3 activity. FASEB J. 2005;19:1671–73.
Aranha MM, et al. Caspases and p53 modulate FOXO3A/Id1 signaling during mouse neural stem cell differentiation. J Cell Biochem. 2009;107:748–58.
Ohsawa S, et al. Maturation of the olfactory sensory neurons by Apaf-1/caspase-9-mediated caspase activity. Proc Natl Acad Sci U S A. 2010;107:13366–71.
Mukherjee A, et al. More alive than dead: non-apoptotic roles for caspases in neuronal development, plasticity and disease. Cell Death Differ. 2017;24:1411–21.
Solier S, et al. Non-apoptotic functions of caspases in myeloid cell differentiation. Cell Death Differ. 2017;24:1337–47.
De Botton S, et al. Platelet formation is the consequence of caspase activation within megakaryocytes. Blood. 2002;100:1310–7.
Zermati Y, et al. Caspase activation is required for terminal erythroid differentiation. J Exp Med. 2001;193:247–54.
Szymczyk KH, et al. Active caspase-3 is required for osteoclast differentiation. J Cell Physiol. 2006;209:836–44.
Arama E, et al. Caspase activity and a specific cytochrome C are required for sperm differentiation in Drosophila. Dev Cell. 2003;4:687–97.
Fernando P, et al. Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci U S A. 2002;99:11025–30.
Ishizaki Y, et al. A role for caspases in lens fiber differentiation. J Cell Biol. 1998;140:153–8.
Candi E, et al. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6:328–40.
Lippens S, et al. Epidermal differentiation does not involve the pro-apoptotic executioner caspases, but is associated with caspase-14 induction and processing. Cell Death Differ. 2000;7:1218–4.
Denecker G, et al. Caspase-14 protects against epidermal UVB photodamage and water loss. Nat Cell Biol. 2007;9:666–74.
Zeeuwen PL. Epidermal differentiation: the role of proteases and their inhibitors. Eur J Cell Biol. 2004;83:761–73.
Mousa A, et al. Transglutaminases factor XIII-A and TG2 regulate resorption, adipogenesis and plasma fibronectin homeostasis in bone and bone marrow. Cell Death Differ. 2017;24:844–54.
Costanzo A, et al. Programmed cell death in the skin. Int J Dev Biol. 2015;59:73–78.
Lang KS, et al. Mechanisms of suicidal erythrocyte death. Cell Physiol Biochem. 2005;15:195–202.
Lang PA, et al. Suicidal death of erythrocytes in recurrent hemolytic uremic syndrome. J Mol Med (Berl). 2006;84:378–88.
Kempe DS, et al. Suicidal erythrocyte death in sepsis. J Mol Med (Berl). 2007;85:273–81.
Kaestner L, et al. The potential of erythrocytes as cellular aging models. Cell Death Differ. 2017;24:1475–77.
Galluzzi L, et al. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009;10:481–94.
Kers J, et al. An overview of pathways of regulated necrosis in acute kidney injury. Semin Nephrol. 2016;36:139–52.
Vaux DL, et al. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–2.
Strasser A, et al. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–3.
Strasser A, et al. bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell. 1991;67:889–99.
Strasser A, et al. DNA damage can induce apoptosis in proliferating lymphoid cells via p53-independent mechanisms inhibitable by Bcl-2. Cell. 1994;79:329–39.
Strasser A, et al. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci U S A. 1991;88:8661–5.
Dillon CP, et al. Molecular cell biology of apoptosis and necroptosis in cancer. Adv Exp Med Biol. 2016;930:1–23.
Galluzzi L, et al. Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol. 2017;12:103–30.
Dorn GW 2nd. Novel pharmacotherapies to abrogate postinfarction ventricular remodeling. Nat Rev Cardiol. 2009;6:283–91.
Galluzzi L, et al. Targeting post-mitochondrial effectors of apoptosis for neuroprotection. Biochim Biophys Acta. 2009;1787:402–13.
Ashkenazi A, et al. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. 2017;16:273–84.
Lalaoui N, et al. The molecular relationships between apoptosis, autophagy and necroptosis. Semin Cell Dev Biol. 2015;39:63–9.
Green DR, et al. Cell death signaling. Cold Spring Harb Perspect Biol. 2015;7:a006080.
Ashkenazi A, et al. Regulated cell death: signaling and mechanisms. Annu Rev Cell Dev Biol. 2014;30:337–56.
Luciani MF, et al. Early nucleolar disorganization in Dictyostelium cell death. Cell Death Dis. 2017;8:e2528.
Golstein P Conserved nucleolar stress at the onset of cell death. FEBS J 2017;284:3791–3800.
Linkermann A, et al. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol. 2014;14:759–67.
Dudek AM, et al. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013;24:319–33.
Inoue H, et al. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 2014;21:39–49.
Vanden Berghe T, et al. Disruption of HSP90 function reverts tumor necrosis factor-induced necrosis to apoptosis. J Biol Chem. 2003;278:5622–29.
Vanlangenakker N, et al. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011;2:e230.
Garg AD, et al. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ. 2014;21:26–38.
Vandenabeele P, et al. Immunogenic apoptotic cell death and anticancer immunity. Adv Exp Med Biol. 2016;930:133–49.
Hernandez C, et al. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene. 2016;35:5931–41.
Zhang Q, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7.
Sun S, et al. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS One. 2013;8:e59989.
Wenceslau CF, et al. Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am J Physiol Heart Circ Physiol. 2015;308:H768–77.
Soares MP, et al. Disease tolerance and immunity in host protection against infection. Nat Rev Immunol. 2017;17:83–96.
Gilloteaux J, et al. Ultrastructural aspects of autoschizis: a new cancer cell death induced by the synergistic action of ascorbate/menadione on human bladder carcinoma cells. Ultrastruct Pathol. 2001;25:183–92.
Jamison JM, et al. Autoschizis: a novel cell death. Biochem Pharmacol. 2002;63:1773–83.
Acknowledgements
We are indebted with Dr. Avi Ashkenazi (Genentech Inc.) for his constructive suggestions. We apologize to the authors of several high-quality articles dealing with mechanistic or pathophysiological aspects of cell death that we were not able to properly discuss. L.G. is supported by an intramural startup from the Department of Radiation Oncology of Weill Cornell Medical College (New York, US), and by Sotio a.c. (Prague, Czech Republic). I.V. is supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC, IG 2017 grant number 20417), Ministero Italiano della Salute (grant number RF_GR-2011-02351355), Ministero Italiano dell’Istruzione, dell’Università e della Ricerca (MIUR, Programma per i Giovani Ricercatori “Rita Levi Montalcini” 2010). G.K. is supported by the French Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR)—Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Institut Universitaire de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LeDucq Foundation; the LabEx Immuno-Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
Author contributions
L.G. conceived the article as part of the NCCD mission, which has been actively perpetrated over the past decade by L.G., G.M., and G.K. L.G. and I.V. prepared the first version of the manuscript and figures. L.G. integrated inputs from all co-authors (listed in alphabetical order) with the help of I.V. All co-authors provided constructive feedback to the preparation of the article, and explicitly approved its final content.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing financial interests.
Additional information
Lorenzo Galluzzi and Ilio Vitale contributed equally to this work.
Edited by F. Pentimalli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. If you remix, transform, or build upon this article or a part thereof, you must distribute your contributions under the same license as the original. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
Galluzzi, L., Vitale, I., Aaronson, S. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25, 486–541 (2018). https://doi.org/10.1038/s41418-017-0012-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-017-0012-4
This article is cited by
-
Dysregulated dendritic cells in sepsis: functional impairment and regulated cell death
Cellular & Molecular Biology Letters (2024)
-
The application of nanoparticles-based ferroptosis, pyroptosis and autophagy in cancer immunotherapy
Journal of Nanobiotechnology (2024)
-
Calcium signaling from sarcoplasmic reticulum and mitochondria contact sites in acute myocardial infarction
Journal of Translational Medicine (2024)
-
Exploration of programmed cell death-associated characteristics and immune infiltration in neonatal sepsis: new insights from bioinformatics analysis and machine learning
BMC Pediatrics (2024)
-
Exploring the molecular mechanisms of ferroptosis-related genes in periodontitis: a multi-dataset analysis
BMC Oral Health (2024)
