
Abstract
Sorafenib was originally identified as an inhibitor of multiple oncogenic kinases and remains the first-line systemic therapy for advanced hepatocellular carcinoma (HCC). MicroRNAs (miRNAs) have been reported to play critical roles in the initiation, progression, and drug resistance of HCC. In this study, we aimed to identify sorafenib-induced miRNAs and demonstrate their regulatory roles. First, we identified that the expression of the tumor-suppressive miRNA miR-375 was significantly induced in hepatoma cells treated with sorafenib, and miR-375 could exert its antiangiogenic effect partially via platelet-derived growth factor C (PDGFC) inhibition. Then, we demonstrated that sorafenib inhibited PDGFC expression by inducing the expression of miR-375 and a transcription factor, achaete-scute homolog-1 (ASH1), mediated the induction of miR-375 by sorafeinb administration in hepatoma cells. Finally, we verified that the expression of miR-375 was reduced in sorafenib-resistant cells and that the restoration of miR-375 could resensitize sorafenib-resistant cells to sorafenib partially by the degradation of astrocyte elevated gene-1 (AEG-1). In conclusion, our data demonstrate that miR-375 is a critical determinant of HCC angiogenesis and sorafenib tolerance, revealing a novel miRNA-mediated mechanism underlying sorafenib treatment.
Similar content being viewed by others

Introduction
Hepatocellular carcinoma (HCC) is the most common type of liver cancer and the second leading cause of cancer-related deaths worldwide [1]. The heavy burden of HCC has provoked extensive studies to investigate the molecular mechanisms underlying this disease.
Abnormal angiogenesis is an important hallmark of HCC. The capacity to undergo angiogenesis addresses the nutrient and oxygen needs of the tumor [2]. Additionally, angiogenesis can help tumor cells spread through the bloodstream to distant organs [3, 4]. Therefore, angiogenesis plays a crucial role in tumor development and metastasis, and novel anticancer strategies focusing on molecules that possess antiangiogenic activity may provide promising breakthroughs for HCC treatment. Sorafenib is a multikinase inhibitor targeting vascular endothelial growth factor (VEGF)-mediated angiogenesis and is the first molecular targeted drug found to prolong survival in patients with advanced HCC [5, 6]. However, acquired resistance to sorafenib has been found in HCC patients, resulting in a poor prognosis. Hence, the identification of new potential targets, detailed mechanisms or therapeutic strategies to overcome this acquired resistance is necessary and valuable.
MicroRNAs (miRNAs) constitute a class of endogenous small noncoding regulatory RNAs that mainly recognize complementary sequences in the 3′-untranslated regions (UTRs) of their target genes and lead to mRNA degradation or translation inhibition [7]. MiRNA deregulation has been demonstrated to play a role in the pathologic processes of tumorigenesis. Furthermore, miRNAs serve as potential diagnostic markers, prognostic factors, and therapeutic targets. Recently, it has been reported that miRNAs contribute to HCC angiogenesis and play critical roles in the therapeutic mechanisms underlying sorafenib resistance. For example, the downregulation of miRNA-638 and miR-497 promotes angiogenesis and the growth of HCC by targeting VEGF [8, 9]. Liver-specific miR-122 expression was found to be significantly reduced in sorafenib-resistant cells, and the overexpression of miR-122 rendered drug-tolerant cells sensitive to sorafenib [10]. Similarly, miR-181a induces sorafenib resistance in HCC cells through the downregulation of RASSF1 expression [11]. Collectively, miRNAs likely play crucial roles in angiogenesis and sorafenib resistance. Therefore, therapies targeting these miRNAs are expected to overcome the loss of the efficacy of sorafenib in the treatment of HCC.
In this study, we identified miR-375 as a sorafenib-inducible miRNA and demonstrated that miR-375 suppresses pro-angiogenic activity in HCC by targeting PDGFC. Furthermore, ASH1 was further identified as an important upstream regulator of miR-375 in sorafenib-treated hepatoma cells. Finally, we found that miR-375 could reverse the acquired resistance to sorafenib by suppressing AEG-1. Collectively, these results demonstrate that miR-375 represses tumor angiogenesis and renders sorafenib-resistant HCC cells sensitive to sorafenib.
Materials and methods
Cell line and human tissue specimens
The human liver cancer cell lines Hep3B, HepG2, Huh1, and Huh7, the normal human liver cell line LO2, and the human umbilical vein endothelial cell line HUVEC were obtain from Chinese Academy of Sciences, Shanghai Institutes for Biological Sciences (Shanghai, China) and verified by STR profiling. Cells were cultured and maintained in Dulbecco’s modified Eagle’s medium (DMEM, HyClone, USA) supplemented with 10% fetal bovine serum (FBS, Gibco BRL, USA) and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin) in an atmosphere with 5% CO2 at 37 °C. Twenty paired HCC and adjacent nontumorous liver tissue samples were collected from patients undergoing resection of HCC at the General Hospital of Western Theater Command from August 2012 to June 2013. No patient received local or systemic therapy before surgery, and both tumorous and matched adjacent nontumorous tissue samples were histologically confirmed by three independent pathologists. The study was approved by the Ethics Committee of the General Hospital of Western Theater Command, and informed consent was obtained from all patients.
Establishment of sorafenib-resistant hepatoma cells
Sorafenib-resistant hepatoma cell lines were established according to previous reference [12]. The IC50 of HCC cells to sorafenib was initially determined by incubating cells with different concentrations of sorafenib, and the cell viability was measured 3 days later as described below. The cells were cultured in 96-well plates at 1 × 104 cells/well and incubated with sorafenib at a concentration slightly below their respective IC50 values. The concentration of sorafenib was slowly increased by 0.25 mmol/l per week. After 6 months, two sorafenib-resistant cell lines designated Hep3B-SR and HepG2-SR were obtained and continuously maintained by culturing in the presence of sorafenib.
miRNA-expressing plasmids, miRNA inhibitors, siRNAs, and cell transfection
The miR-375 overexpression vectors (miR-375), miR-375 inhibitors (anti-miR-375), AEG-1 overexpression vectors (AEG-1), PDGFC and ASH1 siRNAs (si-PDGFC and si-ASH1) and their matched negative controls (miR-NC, anti-miR-NC, NC, and si-NC) were purchased from Thermo Scientific. The sequences of si-PDGFC and si-ASH1 are shown in Supplementary Table 1. Cell transfection was performed using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s instructions.
miRNA microarray and quantitative real-time PCR
A differential expression profiling analysis of the miRNAs expressed in the hepatoma cells treated with sorafenib at a concentration of 10 μM or a vehicle control (DMSO < 0.1%) was conducted using a Human miRNA Microarray (Agilent Technologies, USA). The fluorescence intensity was calculated by Feature Extraction software (Agilent Technologies). The differentially expressed miRNAs were identified by arbitrarily setting the threshold at a fold change of 2.0 or above combined with P < 0.05.
The total RNA was extracted from the tissues and cells with TRIzol Reagent (Takara, Japan).
For the miRNA detection, the miRNAs were transcribed into cDNA using a miScript II RT Kit (QIAGEN, Germany) and amplified with a miScript SYBR Green PCR Kit (QIAGEN) according to the manufacturer’s recommendations.
For the mRNA detection, cDNA was synthesized using a PrimeScript RT Reagent Kit (Takara) and amplified with FastStart Universal Probe Master (Rox) (Roche, Switzerland) and Universal ProbeLibrary Set, Human (Roche) according to the manufacturer’s recommendations. The qRT-PCR primers are listed in Supplementary Tables 2–5.
The relative expression of each miRNA and mRNA was calculated using the 2−ΔΔCT method, and the relative miRNA and mRNA levels were normalized to the levels of U6 or GAPDH.
Conditioned medium preparation
To prepare the conditioned medium, we employed hepatoma cells transfected with vectors expressing miR-375 or miR-NC vectors for 24 h and cultured the cells in serum-free DMEM. After 48 h of incubation, the supernatants were collected, centrifuged to remove living cells and cellular debris, and used for subsequent functional experiments. Recombinant human PDGFC protein (R&D SYSTEMS, USA) was added at a final concentration of 400 ng/ml to conditioned medium collected from miR-375-transfected hepatoma cells for PDGFC compensation in rescue assays.
HUVEC tube network formation assay
The tube formation assay was conducted using HUVECs in a tumor cell-conditioned medium. HUVECs (3 × 104) were seeded onto a 96-well plate coated with Matrigel (BD Biosciences) in conditioned medium at 37 °C. Tube formation was found to be optimal after 3 h. Tube formation was observed, photographed and analyzed under a light microscope (Olympus, Japan).
Rat aortic ring sprouting assay
Matrigel (BD Biosciences) was thawed at 4 °C overnight. The dorsal aorta of a freshly sacrificed Male Sprague Dawley (SD) rats (200 to 250 g) (Vital River, China) was removed in a sterile manner, rinsed with ice-cold PBS and cut into 1-mm-long pieces using a surgical blade. The wells of 96-well plates were coated with 60 μl of Matrigel and incubated at 37 °C for 10 min to gel. Aortic rings were placed in the wells, sealed in place with a 20-μl Matrigel overlay, and incubated at 37 °C for 10 min to allow the gel to set. Conditioned medium (100 μl) was added. The aortic rings were incubated at 37 °C in 5% CO2 for 10 days to allow microvessel sprouting. The conditioned medium was replaced every 48 h. Images were acquired using a stereomicroscope (Olympus).
Chicken chorioallantoic membrane (CAM) assays
Fertilized white leghorn chicken eggs were incubated for 7 days at 37 °C in a humidified atmosphere. After 7 days, a small opening was generated in the shell, and a circular piece of filter paper (5 mm diameter) containing 50 μl of conditioned medium was applied to the chorioallantoic membrane. Then, the site was covered with cellophane tape before the eggs were returned to the incubator. New conditioned medium was added every 8 h. Following 3 days of incubation, the eggs were opened, and pictures were taken using a stereomicroscope (Olympus).
Bioinformatic prediction and dual-luciferase reporter gene assay
The following online miRNA target prediction algorithms were used to evaluate the potential target genes of miR-375: PicTar (http://www.pictar.org/) and TargetScan (http://www.targetscan.org/).
A dual-luciferase reporter gene assay was constructed to measure the reporter activity using a Dual-Luciferase Reporter Assay System (Promega, USA) according to the manufacturer’s protocol. HEK293 cells at 90% confluence were seeded in 96-well plates. For the PDGFC and AEG-1 3′-untranslated region (3′-UTR) luciferase reporter assay, 100 ng of wild type (PGL3-PDGFC/AEG-1 3′-UTR-WT) or mutant (PGL3-PDGFC/AEG-1 3′-UTR-Mut) reporter constructs were cotransfected into HEK293 cells in 24-well plates with 1 μg of miR-375 overexpression vectors or 1 μg of miR-NC vectors and Renilla plasmid using Lipofectamine 2000 (Invitrogen). The reporter gene assay was performed 24 h after transfection.
Western blotting and enzyme-linked immunosorbent assay
Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Millipore, USA). The membranes were blocked with 5% nonfat milk in a Tris-buffered saline solution with 0.1% TWEEN 20 (TBST) for 1 h at room temperature and incubated overnight with primary antibodies at 4 °C, followed by incubation with HRP-conjugated secondary antibodies. The following primary antibodies were used: rabbit antihuman PDGFC (catalog no. ab93899), ASH1 (catalog no. ab74065), BMP-15 (catalog no. ab108413), GDF-9 (catalog no. ab93892), P53 (catalog no. ab131442), NF-κB (catalog no. ab32536), ZEB1 (catalog no.ab203829), P-gp (catalog no. ab261736), MRP1 (catalog no. ab260038), AEG-1 (catalog no. ab227981) and GAPDH (catalog no. ab181602) (Abcam, UK).
PDGFC protein in the culture supernatant was detected by an enzyme-linked immunosorbent assay (catalog no. ab234568) (Abcam) according to the manufacturer’s instructions.
Immunohistochemistry
Serial sections (4 μm) were cut for immunohistochemistry. Rabbit antihuman PDGFC primary antibodies (catalog no. ab93899) (Abcam) and rabbit antirat CD34 primary antibodies (catalog no. ab185732) (Abcam) were applied to the tissue sections for an overnight incubation at 4 °C. The samples were further processed according to the manufacturer’s instructions, and the final sequence of procedures, consisting of incubation with peroxidase-conjugated secondary antibodies, staining, mild restaining with hematoxylin, dehydration, coverslipping, and microscopic observation, was performed.
Animal experiments
To elucidate the role of miR-375 in vivo, we constructed a recombinant lentivirus designated LV-miR-375 to generate a stable gain of function of miR-375 in cells. The recombinant lentivirus, LV-miR-375, and its negative control, LV-miR-NC, (GeneCopoeia, USA) were used to infect the cells according to the manufacturer’s instructions. Flow cytometry sorting was performed to isolate the infected cells (GFP-positive).
BALB/c athymic nude mice (male, 4–5 weeks old and 16–20 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. and bred under pathogen-free conditions. All animal experiments were approved by the Animal Care and Use Committee of the General Hospital of Western Theater Command. The mice were randomized into five groups (n = 5 per group). Hepatoma cells (6 × 107 cells per ml) were suspended in PBS. The cells were subcutaneously injected (100 μl per mouse) into the flank of each mouse. Fifty microliters of recombinant human PDGFC protein (400 ng/ml) were injected into the xenografts every other day for PDGFC compensation in the rescue assay. On the 20th day after injection, the mice were sacrificed, and their tumors were harvested. The tumors were dissected, fixed in formalin, embedded in paraffin, and sectioned for immunohistochemistry. For the survival experiments, the mice were randomly divided into 5 groups, each containing 10 mice, and injected subcutaneously as previously described.
The experimental protocol has been described previously [13,14,15]. In the in vivo sorafenib gavage experiments, sorafenib-resistant cells were inoculated subcutaneously into the mice, which received an oral administration of 15 mg/kg sorafenib every 3 days. A lower dose of sorafenib was used to maintain the sorafenib-resistant ability of the hepatoma cells, which were maintained in culture with sorafenib. After 20 days, sorafenib was suspended in an oral vehicle containing Cremophor (Sigma-Aldrich), 95% ethanol and water in a ratio of 1:1:6 and administered orally at a dose of 30 mg/kg by gavage daily. The tumors were harvested 18 days after the start of the treatment.
Statistical analyses and workflow chart
All data are expressed as the mean ± SEM of at least three separate experiments, and each experiment was performed in triplicate unless otherwise noted. The differences between the groups were analyzed by two-tailed unpaired Student’s t tests. The Kaplan–Meier method and log-rank test were used to compare survival, which was defined as the time from injection to death. Statistical analysis was conducted using SPSS version 24.0 software. P < 0.05 was considered statistically significant. All statistical tests justified as appropriate and the data meet the assumptions of the tests. The variance is similar between the groups that are being statistically compared. No samples or mice were excluded from the analysis as outliers. Mice were randomly allocated to groups using the random number table method. Blinding and sample size estimation tests were not done for our animal studies. To clarify our experiment procedures, we provide a workflow chart to simplify these processes in supplementary Fig. 1.
Results
MiR-375 is a sorafenib-inducible miRNA in hepatoma cells
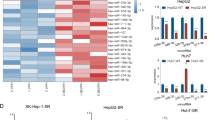
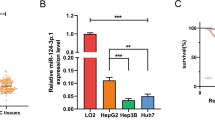
To search for sorafenib-inducible miRNAs, we performed miRNA microarray scanning of hepatoma cell lines subjected to sorafenib or DMSO (<0.1%) for 48 h. Hepatoma cells were treated with a wide range of concentrations (0–20 μM) of sorafenib. We chose a sorafenib concentration of 10 μM for the treatment of hepatoma cells in the subsequent miRNA microarray assays because the hepatoma cells were in good condition and exhibited proliferation (Supplementary Fig. 2A). Furthermore, the apoptosis detection indicated that cell growth was significantly inhibited by sorafenib without obvious cell death at this concentration (Supplementary Fig. 2B). We found that the pattern of miRNA expression in the sorafenib-treated hepatoma cells markedly differed from that in the untreated hepatoma cells (Fig. 1a). Of all differentially expressed miRNAs, 113 were upregulated and 114 were downregulated in the sorafenib-treated hepatoma cells (Fig. 1c). A heat map displaying 60 miRNAs with 5-fold or greater differential expression between the sorafenib-treated hepatoma cells and untreated hepatoma cells is shown in Fig. 1b. Among the miRNAs with significant differences (>5-fold difference) (Table 1), miR-375, miR-483-5p, and miR-638 have been fully identified as tumor-suppressive miRNAs in HCC in previous studies [8, 16, 17]. Then, to validate the microarray data, we performed qRT-PCR to quantify miR-375, miR-483-5p, and miR-638 in the sorafenib-treated hepatoma cells and untreated hepatoma cells. As demonstrated, the miR-483-5p and miR-638 expression levels were not associated with the sorafenib concentration, whereas the miR-375 expression level gradually increased in a concentration-dependent manner (Supplementary Fig. 3 and Fig. 1d). Based on these findings, we identified that miR-375 is a sorafenib-inducible miRNA in hepatoma cells. To explore the role of miR-375 in the development and progression of HCC, we examined miR-375 expression in clinical HCC specimens and hepatoma cell lines. The expression of miR-375 was significantly repressed in the HCC clinical samples compared with that in the adjacent nontumorous liver tissues (n = 20, Fig. 1e). As shown in Fig. 1f, the miR-375 levels in all four hepatoma cell lines (Hep3B, HepG2, Huh1, and Huh7) were significantly lower than those in the normal liver cell line LO2, indicating that the downregulation of miR-375 likely plays crucial roles in HCC initiation and progression.

a, b MiRNA microarray detection identified miRNAs that were differentially expressed between sorafenib-treated hepatoma cells and untreated hepatoma cells. The scale bar on top indicates the relative levels with yellow corresponding to a high expression level and blue corresponding to a low expression level. c One hundred thirteen miRNAs were upregulated, and 114 miRNAs were downregulated in both Hep3B and HepG2 cells subjected to 48 h of sorafenib treatment compared with untreated hepatoma cells. d The expression levels of miR-375 in hepatoma cells treated with a wide range of concentrations (0–20 μM) of sorafenib were detected by qRT-PCR, and the results demonstrated that sorafenib-induced miR-375 expression in a dose-dependent manner. e qRT-PCR showed that miR-375 was downregulated in human HCC tissues (HCC) compared with that in matched adjacent nontumorous liver tissues (NT). f The expression of miR-375 was downregulated in four human liver cancer cell lines (Hep3B, HepG2, Huh1, and Huh7) compared with that in a human normal hepatic cell line (LO2). HCC: hepatocellular carcinoma tissues, NT: matched adjacent nontumorous liver tissues. *P < 0.05, **P < 0.01, ***P < 0.001.
MiR-375 inhibits HCC angiogenesis by directly targeting PDGFC
As is well known, sorafenib is an antiangiogenic multikinase inhibitor. However, as a sorafenib-inducible miRNA, the effect of miR-375 on HCC angiogenesis has not been sufficiently demonstrated. We performed capillary tube formation assays with two hepatoma cell lines, i.e., Hep3B and HepG2, to investigate the regulatory roles of miR-375 in angiogenesis in HCC. Hepatoma cell lines were transfected with miR-375 overexpression vectors and the significant increase in miR-375 expression in these cells could be verified by qRT-PCR (Supplementary Fig. 4). Using these assays, we observed that the morphological differentiation of HUVECs was affected by the miR-375-transfected hepatoma cells (Fig. 2a). The HUVECs formed incomplete and fluffy tubular structures in the presence of conditioned medium obtained from miR-375-transfected hepatoma cells. In contrast, the treatment with the conditioned medium obtained from the negative control (miR-NC) led to the formation of elongated and robust tubular structures. We further evaluated the antiangiogenic effects of miR-375 using rat aortic ring sprouting assays and CAM assays. Our data show that the conditioned medium from the miR-375-transfected hepatoma cells markedly inhibited angiogenesis in the rat aortic rings and chicken CAMs (Fig. 2b, c). These results indicate that the overexpression of miR-375 in hepatoma cells could inhibit the pro-angiogenic activity of hepatoma cells.

a Restoration of miR-375 suppressed the hepatoma cell-promoted formation of tubes by HUVECs (Olympus, magnification ×100). b A rat aortic ring assay indicated that microvessel sprouting was reduced in aortic rings cultured with conditioned medium collected from the supernatant of miR-375-transfected hepatoma cells (Olympus, magnification ×100). c Representative images of chicken chorioallantoic membrane (CAM) blood vessels showing that CAM stimulated with conditioned medium from miR-375-transfected hepatoma cells developed fewer blood vessels than the negative control group (Olympus, magnification ×1.6). d Bioinformatics software predicted that PDGFC was a potential target gene of miR-375. e PDGFC mRNA levels were measured and compared between HCC tissues (HCC) and matched adjacent nontumorous liver tissues (NT), and a correlation between the levels of miR-375 and PDGFC mRNA was observed in HCC tissues (HCC). f, g In human HCC tissues (HCC) and matched adjacent nontumorous liver tissues (NT), the expression and distribution of PDGFC protein were detected by a Western blot analysis and immunohistochemical staining, respectively (Olympus, magnification ×100). h The mRNA and protein levels of PDGFC in four human liver cancer cell lines (Hep3B, HepG2, Huh1, and Huh7) and a human normal hepatic cell line (LO2) were examined by qRT-PCR and Western blot analyses. i–k The mRNA and protein levels of PDGFC were downregulated in hepatoma cells transfected with miR-375 overexpression vectors (miR-375) and upregulated in hepatoma cells transfected with miR-375 inhibitors (anti-miR-375) compared with their matched negative controls (miR-NC: empty plasmid vector or anti-miR-NC: synthetic RNA with scrambled sequence). The amounts of secreted PDGFC in the supernatants were analyzed by ELISA. l. The putative binding sequence between the 3′-UTR of PDGFC and miR-375. Mutations were generated in the complementary site that binds the seed region of miR-375 (top and middle of Fig. 2l). The luciferase reporter assay is shown at the bottom of Fig. 2l. The PGL3-PDGFC 3′-UTR-WT vector or the PGL3-PDGFC 3′-UTR-Mut vector was cotransfected with a miR-375 overexpression plasmid. The relative luciferase activity levels were measured 24 h after transfection and normalized by calculating the ratio of firefly luciferase to Renilla luciferase activity. Luciferase activity in the PGL3-PDGFC 3′-UTR-WT group displayed a significant decrease following the ectopic expression of miR-375, indicating that miR-375 inhibits PDGFC by targeting its 3′-UTR. m–o The human umbilical vein endothelial cell (HUVEC) tube formation, aortic ring sprouting and chicken chorioallantoic membrane (CAM) assays showed that the knockdown of PDGFC by siRNA significantly suppressed hepatoma cell-induced angiogenesis and that the administration of exogenous PDGFC (400 ng/ml) in conditioned medium collected from miR-375-transfected hepatoma cells attenuated the antiangiogenic effect of miR-375. WT: wild type, Mut: Mutant. **P < 0.01, ***P < 0.001, ****P < 0.0001.
To unravel the mechanism underlying miR-375-disrupted angiogenesis, we searched for positive regulators of angiogenesis using miRNA target prediction software (TargetScan and PicTar). PDGFC was found to be among the predicted high-confidence targets (Fig. 2d) and chosen for further validation due to its well-known role in tumor angiogenesis.
To further investigate the relationship between PDGFC and miR-375, we examined the expression of PDGFC and miR-375 in 20 paired HCC specimens. The qRT-PCR and Western blot analyses indicated that the expression of PDGFC in the HCC tissues was significantly higher than that in the matched adjacent nontumorous tissues (Fig. 2e, f). Moreover, the correlation analysis revealed that a negative correlation exists between the expression of PDGFC mRNA and miR-375 (Fig. 2e Left). Similarly, the analysis of the immunohistochemical staining showed that the PDGFC protein was found in the cytoplasm or the nucleus and that its expression in most tumor tissues was upregulated compared with that in the adjacent nontumorous tissues (Fig. 2g). Similarly, the mRNA and protein levels of PDGFC in four human liver cancer cell lines (Hep3B, HepG2, Huh1, and Huh7) were upregulated compared with those in the human normal hepatic cell line LO2, which consists of hepatoma cells and normal hepatocytes (Fig. 2h).
To confirm the regulatory effect of miR-375 on PDGFC expression in hepatoma cells, we performed gain- and loss-of-function assays in vitro. As shown in Fig. 2i, j, the transfection of cells with miR-375, which significantly increased the level of miR-375, resulted in the suppression of PDGFC at both the mRNA and protein levels. In contrast, the transfection of cells with anti-miR-375 (antisense oligonucleotide of miR-375), which decreased the level of endogenous miR-375 (Supplementary Fig. 5), led to a significant increase in the expression of PDGFC at both the mRNA and protein levels (Fig. 2i, j). This regulatory effect of miR-375 on PDGFC was also observed in PDGFC secreted from hepatoma cells to supernatants as detected by an ELISA assay (Fig. 2k). A luciferase assay was further employed to determine whether miR-375 directly suppresses PDGFC expression, and the results revealed that the cotransfection with miR-375 significantly inhibited the activity of the luciferase reporter with the wild-type 3′-UTR of PDGFC, whereas this effect was abrogated when the predicted 3’-UTR binding site was mutated (Fig. 2l). Taken together, the above data suggest that miR-375 directly suppresses PDGFC expression at the posttranscriptional level in HCC.
To confirm whether the antiangiogenic effects of miR-375 in HCC were mediated by PDGFC, we used PDGFC siRNAs to silence PDGFC (Supplementary Fig. 6). As shown in Fig. 2m–o, the suppression of PDGFC in hepatoma cells resulted in a significantly reduced capacity to promote HUVEC capillary tube formation, the sprouting of rat aortic rings and angiogenesis in chicken CAMs, which phenocopied the effects of miR-375 overexpression. Furthermore, for the PDGFC compensation in the rescue assays, recombinant human PDGFC protein was added to conditioned medium. As a result, the PDGFC administration to the miR-375 transfectants obviously abrogated the inhibitory effects of miR-375 on HUVEC capillary tube formation, the sprouting of rat aortic rings and angiogenesis in chicken CAMs. These results suggest that miR-375 represses tumor angiogenesis partially by inhibiting PDGFC in hepatoma cells.
MiR-375 inhibits tumor angiogenesis in hepatoma xenografts by targeting PDGFC in nude mice
To use HCC xenograft models to further confirm the above findings in vivo, we successfully constructed a recombinant lentiviral vector called LV-miR-375 (and LV-miR-NC as a control) to transduce hepatoma cells and increase the expression of miR-375 (Supplementary Fig. 7). We found that PDGFC accelerated the growth of tumors derived from hepatoma cells in nude mice, but the treatment with miR-375 could markedly block such tumor growth. Consistent with the results of the in vitro experiments, the PDGFC administration significantly reversed the inhibitory effects of miR-375 on tumor growth in vivo (Fig. 3a).

a Cells were injected subcutaneously into nude mice. Fifty microliters of recombinant human PDGFC protein (400 ng/ml) were injected into the xenografts every other day. Twenty days after the cell injection, the mice were sacrificed and photographed. b Neovascularization was labeled by CD34 immunohistochemical staining in serial sections of subcutaneous xenograft tumors (Olympus, magnification ×100). c The survival of the nude mice was estimated by the Kaplan–Meier method, and statistical significance was calculated using the log-rank test. n = 10 mice per group. Mock: Mice were subcutaneously injected with hepatoma cells without any treatment. PDGFC: Recombinant human PDGFC protein was injected into the xenografts in the mice in the mock group. LV-miR-NC: Mice were subcutaneously injected with hepatoma cells infected with a negative control lentivirus (LV-miR-NC: empty lentivirus vectors). LV-miR-375: Mice were subcutaneously injected with hepatoma cells stably expressing miR-375. LV-miR-375+PDGFC: Recombinant human PDGFC protein was injected into the xenografts of the mice in the LV-miR-375 group. ***P < 0.001.
To clarify the cellular mechanisms underlying the miR-375-mediated tumor suppression, we conducted IHC staining to detect CD34 in resected tissues from these subcutaneous xenograft tumors to analyze angiogenesis in HCC. The IHC analysis of the excised tumors showed that the miR-375-transduced mice developed a marked decrease in neovascularization compared with the miR-NC-transduced mice. However, the PDGFC administration significantly increased neovascularization in the miR-375-transduced hepatoma xenografts, indicating that PDGFC reversed the angiogenesis inhibition induced by miR-375 in vivo (Fig. 3b). Taken together, these results indicate that miR-375 suppressed angiogenesis in HCC, at least partially, by targeting PDGFC in vivo. Furthermore, we confirmed the regulation of PDGFC expression by miR-375 in vivo. As shown in Supplementary Fig. 8, miR-375 was successfully overexpressed in the excised tumors from the miR-375-transduced mice. As a result, the IHC analysis showed that PDGFC was significantly downregulated in the tumor xenografts in the nude mice seeded with miR-375-transduced hepatoma cells, indicating that miR-375 can also repress the expression of PDGFC in vivo. Moreover, the cumulative survival analysis revealed that the mice receiving the LV-miR-375-transfected hepatoma cells achieved significantly longer life spans than the mice receiving the negative control hepatoma cells. However, the PDGFC administration significantly decreased the survival benefits conferred on the mice by the miR-375 overexpression (Fig. 3c).
A sorafenib-ASH1-miR-375-PDGFC signaling axis exists in HCC
To further determine the relationship between miR-375 and PDGFC under sorafenib treatment, we first explored the effects of sorafenib on PDGFC. The qRT-PCR and Western blot analyses showed that the depletion of PDGFC was associated with a concomitant increase in the expression of miR-375 due to the sorafenib treatment in a dose-dependent manner (Fig. 4a–c). Given that PDGFC was confirmed as a target of miR-375 in our previous results, we hypothesized that miR-375 might play a role in regulating the reduction in PDGFC by sorafenib. To test this hypothesis, we transfected sorafenib-treated hepatoma cells with miR-375 inhibitors. We found that the inhibition of miR-375 hindered the suppressive effects of sorafenib on PDGFC expression (Fig. 4d, e), indicating that sorafenib inhibited PDGFC expression by inducing the expression of miR-375.

a–c qRT-PCR and Western blot analyses were used to analyze the miR-375 and PDGFC expression levels in HCC cells treated with a wide range of concentrations (0–25 μM) of sorafenib. d, e qRT-PCR and Western blot analyses showed that the downregulation of miR-375 by miR-375 inhibitors (anti-miR-375) in the sorafenib-treated hepatoma cells reversed the inhibitory effects of sorafenib on PDGFC expression at both the mRNA and protein levels. f ASH1 was obviously upregulated in the sorafenib-treated hepatoma cells. g The expression of miR-375 after the knockdown of ASH1 in sorafenib-treated hepatoma cells. Mock: Hepatoma cells without any treatment. Sorafenib: Hepatoma cells treated with sorafenib at a concentration of 10 μM. Sorafenib+anti-miR-NC: Hepatoma cells transfected with negative control miRNA inhibitors (anti-miR-NC: synthetic RNA with scrambled sequence) and treated with sorafenib at a concentration of 10 μM. Sorafenib+anti-miR-375: Hepatoma cells transfected with miR-375 inhibitors (anti-miR-375) and treated with sorafenib at the concentration of 10 μM. Sorafenib+si-NC: Hepatoma cells transfected with negative control siRNA (si-NC: synthetic RNA with a scrambled sequence) and treated with sorafenib at a concentration of 10 μM. Sorafenib+si-ASH1: Hepatoma cells transfected with ASH1 siRNAs (si-ASH1) and treated with sorafenib at the concentration of 10 μM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To further explore the mechanisms underlying the sorafenib-mediated induction of miR-375 in hepatoma cells, we detected the expression of potential upstream regulators, including ASH1, bone morphogenetic protein 15 (BMP-15), growth differentiation factor 9 (GDF-9), P53, nuclear factor-kB (NF-kB) and zinc finger E-box binding homeobox 1 (ZEB1), which are known to be upstream regulatory mechanisms of miR-375. As shown in Fig. 4f, there were no expression differences in BMP-15, GDF-9, P53, NF-kB, and ZEB1 between the sorafenib-treated and untreated hepatoma cells. However, ASH1, which is a basic helix-loop-helix transcription factor, was significantly upregulated in the sorafenib-treated hepatoma cells. To verify that ASH1 regulates miR-375, we further analyzed the expression changes in miR-375 when ASH1 expression was knocked down; as expected, the trend of miR-375 expression was consistent with the expression of ASH1 under the sorafenib treatment (Fig. 4g).
In conclusion, these results indicate that a sorafenib-ASH1-miR-375-PDGFC signaling axis exists under sorafenib treatment. We show a schematic diagram of this regulatory mechanism in Fig. 7a.
MiR-375 reversed the acquired resistance to sorafenib by suppressing astrocyte elevated gene-1 (AEG-1)
In addition to the inducible effect on miR-375 expression in hepatoma cells by short-term sorafenib exposure, we also investigated the expression of miR-375 in sorafenib-resistant hepatoma cells subjected to long-term exposure to sorafenib. For this purpose, we established sorafenib-resistant hepatoma cell models by six-month chronic exposure to sorafenib. To confirm the development of drug resistance to sorafenib in hepatoma cells, we mainly examined and calculated the variation in the IC50 values in hepatoma cells subjected to chronic exposure to sorafenib by CCK-8 proliferation assays. The data showed that the sorafenib-resistant cells required higher doses of the drug to exhibit partial growth inhibition than their parental cells (Supplementary Fig. 9A). Furthermore, we expected to further strengthen the evidence regarding drug resistance development by detecting the expression of MDR-related genes, such as P-glycoprotein (P-gp) and multidrug resistance-associated protein-1 (MRP1). The analysis of the MDR-related gene expression revealed that the expression levels of both P-gp and MRP1 were increased in the sorafenib-resistant cells compared with those in their parental counterparts at both the mRNA and protein levels (Supplementary Fig. 9B-C). These data indicate that we successfully established sorafenib-resistant cell models.
Next, to determine whether miR-375 dysregulation was responsible for sorafenib resistance, we analyzed the miR-375 expression in the sorafenib-resistant cells and their parental counterparts and verified that the expression of miR-375 was reduced in the sorafenib-resistant cells (Fig. 5a). To investigate whether the restoration of miR-375 could resensitize sorafenib-resistant cells to sorafenib, we overexpressed miR-375 in sorafenib-resistant cells. Strikingly, miR-375 enhanced the sensitivity to sorafenib in the sorafenib-resistant cells in CCK-8 proliferation assays and resulted in the suppression of MDR-related genes at both the mRNA and protein levels (Fig. 5b–d). These results indicate that the overexpression of miR-375 could reverse acquired resistance to sorafenib.

a Transfection of miR-375 overexpression vectors resulted in high expression levels of miR-375 in sorafenib-resistant cells. b Enhanced response to sorafenib due to miR-375 overexpression was observed in sorafenib-resistant cells. The IC50 of each treatment is presented. c, d qRT-PCR and western blot analyses revealed decreased levels of P-gp and MRP1 in miR-375-transfected sorafenib-resistant hepatoma cells. e Conserved miR-375 cognate site in the 3′-UTR of AEG-1. f qRT-PCR analysis of miR-375 and Western blot analysis of AEG-1 in sorafenib-resistant cells and their parental counterparts. g, h. The expression of AEG-1 was downregulated in sorafenib-resistant cells transfected with miR-375 overexpression vectors (miR-375) and upregulated in sorafenib-resistant cells transfected with miR-375 inhibitors (anti-miR-375) compared with their matched negative controls (miR-NC: empty plasmid vector or anti-miR-NC: synthetic RNA with a scrambled sequence). i Mutations were generated in the complementary site that binds the seed region of miR-375. The luciferase reporter assay is shown at the bottom of Fig. 5i. The PGL3-AEG-1 3′-UTR-WT vector or the PGL3-AEG-1 3′-UTR-Mut vector was cotransfected with a miR-375 overexpression plasmid. The relative luciferase activity levels were measured 24 hours after transfection and normalized by calculating the ratio of firefly luciferase to Renilla luciferase activity. j The miR-375-induced sensitizing effect was reversed by AEG-1 in the sorafenib-resistant cells. WT: wild type, Mut: Mutant. *P < 0.01, **P < 0.01, ***P < 0.001, ***P < 0.0001.
To explore the mechanisms underlying sorafenib resistance, miRNA target prediction algorithms were again used to predict the miR-375 target genes related to drug resistance. Astrocyte elevated gene-1 (AEG-1), which was recently identified as an oncogene that plays a role in drug resistance, was a putative target of miR-375 (Fig. 5e). To confirm that AEG-1 is involved in acquired resistance to sorafenib in HCC, we explored AEG-1 expression in sorafenib-resistant cells and their parental counterparts and verified that the expression of AEG-1 was induced in the sorafenib-resistant cells (Fig. 5f). We also performed gain- and loss-of-function assays to determine whether miR-375 could regulate AEG-1 expression, and the results showed that the overexpression of miR-375 in sorafenib-resistant cells greatly reduced the expression of AEG-1, whereas the inhibition of miR-375 enhanced AEG-1 expression (Supplementary Fig. 10 and Fig. 5g, h). Furthermore, the luciferase assay showed that miR-375 could directly target its predicted binding site on AEG-1 and lead to the suppression of luciferase expression on the wild-type 3′-UTR of AEG-1, whereas miR-375 had no obvious effects on its control (Fig. 5i). Taken together, these data indicate that miR-375 negatively regulates AEG-1 expression by interacting with its 3′-UTR.
To determine whether the reduction in drug resistance by miR-375 overexpression was mediated by AEG-1 inhibition, we performed an AEG-1 rescue assay in miR-375 overexpressed sorafenib-resistant hepatoma cells. For this purpose, AEG-1 overexpression vectors were cotransfected with miR-375 overexpression vectors for AEG-1 compensation in the rescue assay (Supplementary Fig. 11). The results showed that the miR-375-induced sensitizing effect was reversed by AEG-1 in the sorafenib-resistant cells (Fig. 5j). These results suggest that miRNA-375 could reverse sorafenib resistance partially by targeting AEG-1.
MiR-375 enhances the efficacy of sorafenib in treating sorafenib-resistant HCC tumors in vivo
In the in vivo sorafenib gavage experiments, the sorafenib-resistant cells formed xenograft tumors more efficiently in BALB/c athymic nude mice than their parental counterparts (Fig. 6). To identify the antitumorigenic roles of miR-375 in sorafenib-resistant HCC in vivo, sorafenib-resistant cells overexpressing miR-375 or miR-NC were injected into nude mice, and then, the tumor growth was evaluated. The tumors were harvested 18 days after the start of the sorafenib treatment; there was a significantly greater decrease in the tumor size with the expression of miR-375 (Supplementary Fig. 12 and Fig. 6). These results suggest that miR-375 likely contributes to sensitizing sorafenib-resistant cells to sorafenib in vivo.

Transplantation assay of parental cells, sorafenib-resistant cells and miR-375-transfected sorafenib-resistant cells subcutaneously injected into mice. Subcutaneous tumors were established in mice that received treatment for 18 days as described in the Materials and Methods. ***P < 0.001.
Taken together, we demonstrated that miR-375 attenuates drug resistance to sorafenib in sorafenib-resistant cells partially by AEG-1 inhibition. We also provide a schematic diagram of this regulatory mechanism in Fig. 7b.

a Schematic diagram of the sorafenib-ASH1-miR-375-PDGFC signaling axis under sorafenib treatment. b Proposed molecular mechanisms by which miR-375 attenuates drug resistance to sorafenib in sorafenib-resistant cells partially by AEG-1 inhibition.
Discussion
The clinical approval of sorafenib marked a new era of molecular targeted therapy for advanced HCC. As a multikinase inhibitor, sorafenib can inhibit angiogenesis by targeting the hepatocyte factor receptor (c-Kit), Fms-like tyrosine kinase (FLT-3), vascular endothelial growth factor receptor (VEGFR)−2, VEGFR-3, platelet-derived growth factor receptor (PDGFR-β) and other tyrosine kinases [18, 19]. Many miRNAs have been found to be involved in the pathological processes of angiogenesis. However, the effects of sorafenib on miRNAs in HCC are still unclear.
In this study, we examined the shift in the expression profile of miRNAs in hepatoma cells after sorafenib treatment using microarrays. Among the miRNAs with significant differences, miR-375, miR-483-5p and miR-638 were the most markedly upregulated in the sorafenib-treated hepatoma cells, and their roles in HCC have been fully documented as tumor suppressive in previous studies [8, 16, 17]. In our study, miR-375 was identified to be significantly induced by sorafenib treatment. In contrast, in the sorafenib-resistant hepatoma cells, miR-375 was found to be downregulated. The restoration of miR-375 in these cells could reverse the acquired resistance to sorafenib. As a tumor-suppressive miRNA, miR-375 has been reported to be significantly downregulated in multiple types of cancer and shown to suppress core hallmarks of cancer by targeting several important oncogenes, such as AEG-1, YAP1, IGF1R, and PDK1 [17, 20,21,22]. The reduced expression of miR-375 in tissues or circulation may indicate the presence of neoplasia and a poor prognosis in many malignant cancers [23, 24]. Moreover, miR-375 represents a promising direction for developing targeted therapies due to its capacity to inhibit tumor cell growth in vitro and in vivo [25]. Our study demonstrates that miR-375 likely exhibits different expression patterns and tumor suppressive functions in different phases of sorafenib treatment. Here, we mainly focus on the expression and function of miR-375 in hepatoma cells during the initial treatment stage of sorafenib (48 hours after sorafenib administration) and sorafenib-resistant hepatoma cells after chronic drug exposure (6 months after sorafenib exposure). However, as is well known, treatment with sorafenib is a continuous process. When and how the changes in miR-375 expression occur are still largely unknown and need to be further investigated by detecting the expression of miR-375 at more time points from 48 hours to 6 months in our next study.
Previous studies have shown that miR-375 plays important roles in acquired resistance. The delivery of miR-375 and doxorubicin hydrochloride by lipid-coated hollow mesoporous silica nanoparticles could overcome multiple drug resistance in HCC. Lipid-coated cisplatin nanoparticles coloaded with miR-375 decreased cell proliferation and enhanced the antitumor effect of cisplatin in chemotherapy-resistant HCC cells [26]. Among the potential targets of miR-375, AEG-1 is a well characterized multidrug resistance-related gene. In cancer cells, AEG-1 promotes the expression of MDR1, resulting in increased efflux and decreased accumulation of chemotherapeutics, leading to drug resistance [27, 28]. Our study demonstrates that miR-375 negatively regulates AEG-1 expression by interacting with its 3′-UTR and reverses acquired resistance to sorafenib partially by suppressing AEG-1. To investigate the inhibitory effect of miR-375 on drug resistance in sorafenib-resistant hepatoma cells, we primarily examined and calculated the changes in the IC50 values by a CCK-8 proliferation assay. Furthermore, we aimed to further strengthen the evidence concerning the development of drug resistance by detecting the expression of MDR-related genes, such as P-gp and MRP1, according to the literature. We found that miR-375 overexpression resulted in the reduced expression of P-gp and MRP1. However, as is well known, drug resistance to sorafenib is determined not only by the expression of drug-resistant genes but also, more importantly, by its function. To determine whether miR-375 could attenuate drug resistance to sorafenib by downregulating MDR1 expression, it is essential to investigate the functional changes in MDR1 as an efflux transporter of sorafenib after miR-375 overexpression. However, to date, we still have no data to prove this hypothesis, and we expect to learn more about the transporter function of MDR1 affected by miR-375 in our future study.
In conclusion, our study found that miR-375 inhibits tumor angiogenesis by targeting PDGFC and that miR-375 could serve as a therapeutic target to overcome sorafenib resistance by targeting AEG-1 in the treatment of HCC. Our findings demonstrate the suppressive effects of miR-375 on tumor-induced angiogenesis in HCC and the underlying molecular mechanisms of these effects, and we further identified a miR-375-mediated pathway involved in the effects of sorafenib treatment, providing a potential therapeutic strategy for overcoming acquired resistance to sorafenib in HCC.
Data availability
Data generated for the current study are available from the corresponding author on reasonable request.
References
Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2016. Ca-Cancer J Clin. 2016;66:7–30.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Gordon MS, Mendelson DS, Kato G. Tumor angiogenesis and novel antiangiogenic strategies. Int J Cancer. 2010;126:1777–87.
Xue G, Yan HL, Zhang Y, Hao LQ, Zhu XT, Mei Q, et al. c-Myc-mediated repression of miR-15-16 in hypoxia is induced by increased HIF-2alpha and promotes tumor angiogenesis and metastasis by upregulating FGF2. Oncogene. 2015;34:1393–406.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl J Med. 2008;359:378–90.
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34.
Diederichs S, Haber DA. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell. 2007;131:1097–108.
Cheng J, Chen Y, Zhao P, Liu X, Dong J, Li J, et al. Downregulation of miRNA-638 promotes angiogenesis and growth of hepatocellular carcinoma by targeting VEGF. Oncotarget. 2016;7:30702–11.
Yan JJ, Zhang YN, Liao JZ, Ke KP, Chang Y, Li PY, et al. MiR-497 suppresses angiogenesis and metastasis of hepatocellular carcinoma by inhibiting VEGFA and AEG-1. Oncotarget. 2015;6:29527–42.
Xu Y, Huang J, Ma L, Shan J, Shen J, Yang Z, et al. MicroRNA-122 confers sorafenib resistance to hepatocellular carcinoma cells by targeting IGF-1R to regulate RAS/RAF/ERK signaling pathways. Cancer Lett. 2016;371:171–81.
Azumi J, Tsubota T, Sakabe T, Shiota G. miR-181a induces sorafenib resistance of hepatocellular carcinoma cells through downregulation of RASSF1 expression. Cancer Sci. 2016;107:1256–62.
He C, Dong X, Zhai B, Jiang X, Dong D, Li B, et al. MiR-21 mediates sorafenib resistance of hepatocellular carcinoma cells by inhibiting autophagy via the PTEN/Akt pathway. Oncotarget. 2015;6:28867–81.
Ma L, Li G, Zhu H, Dong X, Zhao D, Jiang X, et al. 2-Methoxyestradiol synergizes with sorafenib to suppress hepatocellular carcinoma by simultaneously dysregulating hypoxia-inducible factor-1 and -2. Cancer Lett. 2014;355:96–105.
Zhao D, Zhai B, He C, Tan G, Jiang X, Pan S, et al. Upregulation of HIF-2alpha induced by sorafenib contributes to the resistance by activating the TGF-alpha/EGFR pathway in hepatocellular carcinoma cells. Cell Signal. 2014;26:1030–9.
Zhai B, Hu F, Jiang X, Xu J, Zhao D, Liu B, et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther. 2014;13:1589–98.
Li F, Ma N, Zhao R, Wu G, Zhang Y, Qiao Y, et al. Overexpression of miR-483-5p/3p cooperate to inhibit mouse liver fibrosis by suppressing the TGF-beta stimulated HSCs in transgenic mice. J Cell Mol Med. 2014;18:966–74.
He XX, Chang Y, Meng FY, Wang MY, Xie QH, Tang F, et al. MicroRNA-375 targets AEG-1 in hepatocellular carcinoma and suppresses liver cancer cell growth in vitro and in vivo. Oncogene. 2012;31:3357–69.
Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–109.
Tanaka S, Arii S. Molecular targeted therapies in hepatocellular carcinoma. Semin Oncol. 2012;39:486–92.
Zhang ZW, Men T, Feng RC, Li YC, Zhou D, Teng CB. miR-375 inhibits proliferation of mouse pancreatic progenitor cells by targeting YAP1. Cell Physiol Biochem. 2013;32:1808–17.
Luo J, Wu J, Li Z, Qin H, Wang B, Wong TS, et al. miR-375 suppresses IGF1R expression and contributes to inhibition of cell progression in laryngeal squamous cell carcinoma. Biomed Res Int. 2014;2014:374598.
Zhou J, Song S, He S, Zhu X, Zhang Y, Yi B, et al. MicroRNA-375 targets PDK1 in pancreatic carcinoma and suppresses cell growth through the Akt signaling pathway. Int J Mol Med. 2014;33:950–6.
Yi J, Jin L, Chen J, Feng B, He Z, Chen L, et al. MiR-375 suppresses invasion and metastasis by direct targeting of SHOX2 in esophageal squamous cell carcinoma. Acta Biochim Biophys Sin (Shanghai). 2017;49:159–69.
Hu C, Lv L, Peng J, Liu D, Wang X, Zhou Y, et al. MicroRNA-375 suppresses esophageal cancer cell growth and invasion by repressing metadherin expression. Oncol Lett. 2017;13:4769–75.
Shi W, Yang J, Li S, Shan X, Liu X, Hua H, et al. Potential involvement of miR-375 in the premalignant progression of oral squamous cell carcinoma mediated via transcription factor KLF5. Oncotarget. 2015;6:40172–85.
Xue H, Yu Z, Liu Y, Yuan W, Yang T, You J, et al. Delivery of miR-375 and doxorubicin hydrochloride by lipid-coated hollow mesoporous silica nanoparticles to overcome multiple drug resistance in hepatocellular carcinoma. Int J Nanomed. 2017;12:5271–87.
Yoo BK, Chen D, Su ZZ, Gredler R, Yoo J, Shah K, et al. Molecular mechanism of chemoresistance by astrocyte elevated gene-1. Cancer Res. 2010;70:3249–58.
Emdad L, Das SK, Dasgupta S, Hu B, Sarkar D, Fisher PB. AEG-1/MTDH/LYRIC: signaling pathways, downstream genes, interacting proteins, and regulation of tumor angiogenesis. Adv Cancer Res. 2013;120:75–111.
Acknowledgements
Infrastructure for this research was supported by Institute of Aging and Immunity, Chengdu Medical College, Chengdu 610083, Sichuan Province, China.
Funding information
The research was funded by the Scientific Research Foundation of the Science and Technology Department of Sichuan Province (Grants 2017SZ0066 and 2015FZ0073), the National Natural Science Foundation of China (Grant 81101634), the Scientific Research Project of the Health Department of Sichuan Province (No. 16PJ026) and the Scientific Research Project of the Administration of Traditional Chinese Medicine of Sichuan Province (No. 2014K057).
Author information
Authors and Affiliations
Contributions
D.L., T.W., F.-F.S. and T.Z.: conceived the study, conducted experiments, acquired and analysed data, and wrote the manuscript; J.-Q.F., J.-J.P., H.L., C.W., D.W., Y.L., Y.-D.B., M.-L.S.: responsible for conception and supervision of the study, and wrote the manuscript. All authors corrected draft versions and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of General Hospital of Western Theater Command, and informed consent was obtained from all patients, which is accredited by the National Council on Ethics in Human Research.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, D., Wang, T., Sun, FF. et al. MicroRNA-375 represses tumor angiogenesis and reverses resistance to sorafenib in hepatocarcinoma. Cancer Gene Ther 28, 126–140 (2021). https://doi.org/10.1038/s41417-020-0191-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-020-0191-x
This article is cited by
-
Emerging role of MicroRNA-Based theranostics in Hepatocellular Carcinoma
Molecular Biology Reports (2023)
-
MALAT1-miRNAs network regulate thymidylate synthase and affect 5FU-based chemotherapy
Molecular Medicine (2022)
