
Abstract
Background
Epigenomic dysregulation has been linked to solid tumour malignancies, including ovarian cancers. Profiling of re-programmed enhancer locations associated with disease has the potential to improve stratification and thus therapeutic choices. Ovarian cancers are subdivided into histological subtypes that have significant molecular and clinical differences, with high-grade serous carcinoma representing the most common and aggressive subtype.
Methods
We interrogated the enhancer landscape(s) of normal ovary and subtype-specific ovarian cancer states using publicly available data. With an initial focus on H3K27ac histone mark, we developed a computational pipeline to predict drug compound activity based on epigenomic stratification. Lastly, we substantiated our predictions in vitro using patient-derived clinical samples and cell lines.
Results
Using our in silico approach, we highlighted recurrent and privative enhancer landscapes and identified the differential enrichment of a total of 164 transcription factors involved in 201 protein complexes across the subtypes. We pinpointed SNS-032 and EHMT2 inhibitors BIX-01294 and UNC0646 as therapeutic candidates in high-grade serous carcinoma, as well as probed the efficacy of specific inhibitors in vitro.
Conclusion
Here, we report the first attempt to exploit ovarian cancer epigenomic landscapes for drug discovery. This computational pipeline holds enormous potential for translating epigenomic profiling into therapeutic leads.
Similar content being viewed by others


Introduction
The last decades have witnessed a significant increase in functional genomic approaches, high-throughput data availability and innovative computational methodologies [1]. Thriving in this data-intensive environment, in silico strategies have been widely used for drug development, discovery and repositioning [2]. In silico approaches take advantage of vast genomic datasets to stratify patients into distinguishable molecular sub-groups, enabling the delivery of more accurate personalised care to patients [3, 4]. To date, this field has largely been driven by genomics, whereas now there is a surge in the exploitation of epigenomics, which offers a more dynamic view of disease, and therefore personalised treatment options [5, 6].
In contrast to genomics, epigenomics encompasses a set of covalent modifications that occur at chromatin level with no alteration to the underlying DNA sequence [7]. Different patterns of epigenetic alterations have fundamental effects on cancer progression, providing cancer cells with a degree of plasticity that allows them to adapt to their environment [8]. The precise temporal and spatial epigenomic regulation of gene transcription is orchestrated by cis-acting regulatory elements that include promoter regions surrounding the transcriptional start site of genes, and enhancers, generally located many kilobases away from the gene body [9]. Active enhancers are highlighted by acetylated histone 3 at lysine 27 (H3K27ac), an epigenetic mark commonly associated with open chromatin regions [10]. Enhancers play a central role in regulating transcription where disease-specific alterations are a critical aspect of cancer development, as aberrant enhancer landscapes alter the expression of oncogenes and tumour suppressor genes [11]. Re-programmed enhancer landscapes have been identified in several cancers, including ovarian cancer (OC) [12, 13].
OC malignancies are subdivided into histological subtypes that exhibit distinct gene expression profiles, clinical features, response to chemotherapy and outcome [14]. High-grade serous carcinomas (HGSC) are the most common (~75%) and most aggressive subtype, while other histological presentations are much less prevalent, for example clear-cell (CC, ~5%) and mucinous carcinomas (MC, 2–3%) [14]. Despite OC heterogeneity, the first line of standard treatment is generally comprised of debulking surgery followed by platinum-based chemotherapy [15]. Although most cases initially display a positive response to this treatment strategy, 40–50% patients develop platinum resistance, leading to an unfavourable prognosis [16]. At present, only a few targeted therapies have been approved for use in the management of OC and include PARP and VEGF inhibitors, however specific resistance mechanisms have also emerged against these treatment strategies [17, 18].
Here, we present a novel computational strategy designed to identify potential and much-needed candidate drugs for OC treatment. Interrogating publicly available epigenomic ChIP-Seq data, this pipeline mapped differential enhancer regions between diseased and healthy samples. Using these regions as input for the ‘Mining Algorithm for Genetic Controllers’ algorithm (MAGIC) [19], specific transcription factor (TF) binding enrichment was determined. Finally, candidate drug compounds were identified using the ‘Search Tool for Interactions of Chemicals’ (STITCH) [20], and experimental validation was conducted using patient-derived OC samples. Among the predicted top-ranked candidates specific for HGSC we identified CDK inhibitor SNS-032 and EHMT2 inhibitor UNC0646, both of which exhibited promising in vitro results. Importantly, our novel framework can now be exploited in multiple disease settings, offering a unique approach to drug discovery through profiling of epigenomic landscapes.
Methods
Dataset download and quality control assessment
ChIP-Seq datasets were downloaded from repositories GSE68104, GSE121103 and GSE156275 [12, 21,22,23]. The sample size was determined by public availability. Fastq files were downloaded using the prefetch and fasterq-dump functions from the SRA toolkit. Prior to analysis, the quality of raw data was assessed using fastqc [24]. Read mapping statistics were obtained directly from Bowtie2 and STAR aligners for ChIP-Seq and RNA-Seq respectively [25, 26]. For ChIP-Seq samples (H3K27ac), read quality was assessed using the phantompeakqualtools package [27]. Normalised strand cross-correlation and relative strand cross-correlation metrics were required to adhere to the ENCODE standards [28].
Next-generation sequencing analysis
For ChIP-Seq analysis, sequence reads were mapped against human reference genome build hg38 using Bowtie2 [25]. Removed reads prior to peak calling include: reads >5 bases with Phred score <30, reads containing undefined bases and reads mapping to DAC ENCODE consensus excluded regions [29]. Peak calling was performed using MACS2 with default settings [30]. To identify significantly dysregulated H3K27ac peaks, we used the Bioconductor package DiffBind with DESeq2 settings [31, 32]. DiffBind was used to analyse ChIP-Seq signal distribution in genomic locations of annotated enhancers obtained from the GeneHancer database [33], incorporating the remove grey list and remove blacklist options. Enhancer occupancy heat maps were generated using the pheatmap R package [34]. Principal component analysis was performed and plotted using DiffBind [35] and ggplot2 [36] packages, respectively. Volcano plots and pathway enrichment plots were also generated using ggplot2. Genome browser plots were generated using Gviz R package [37]. Upset plots were generated using UpSetR package [38]. Venn diagrams were created using Inkscape v0.92 [39]. Pathway analyses were performed using WebGestaltR [40]. For RNA-Seq analysis, sequence reads were mapped against human reference genome build hg38 using STAR [26]. Prior to alignment, low-quality read ends were removed using the sickle window adaptive trimming tool [41]. Gene count tables were generated using the STAR aligner quant mode function.
MAGIC enhancer matrix and transcription factor enrichment
As MAGIC was originally designed to assess enriched TF binding in gene promoter regions, a novel MAGIC matrix was designed to map enhancer locations annotated in the GeneHancer database [33]. We developed a custom R script to generate our matrix, where 1695 BED files from TF ChIP-Seq experiments for both human cell lines and primary samples were downloaded from ENCODE using the ENCODExplorer R package (R version 3.6.3) [42]. With this newly generated matrix, MAGIC was used to identify enriched TF binding within enhancers (FDR < 0.01, K-S test). ‘Find Individual Motif Occurrences’ (FIMO) [43] was used to refine the list of TFs produced by MAGIC, removing TFs with no significant enrichment of motif binding sites (P < 0.001) using a custom R script. Motif sequence logos were generated using the diffLogo R package [44]. Molecular surface render plots of complex members were obtained from Uniprot [45] and UCSF Chimera [46].
Protein complex and compound search
The CORUM DB protein complex database [47] was used to isolate enriched protein components using a custom R script. The STITCH database was used to assess chemicals linked to specific complex coregulators, filtered and ranked using a STITCH combined score (>700) [20]. The ChEMBL database was used to identify chemicals corresponding to drug compounds approved by the FDA or undergoing clinical trial [48]. Bar plots, RPKM complex factor diagrams and pie charts were generated using ggplot2 [36]. Network diagrams were generated using Cytoscape [49].
Compounds, cell culture and cell viability assays
Identified drug compounds were provided by GlaxoSmithKline (i-CBP112, GSK343, GSK503, UNC0646), purchased from Cayman Chemical (MI, US): Seliciclib (CAY10009569), BIX-01294 (CAY13124), SNS-032 (CAY17904) and NU-6102 (CAY113317) or MedChemExpress (NJ, US): AUZ 454 (HY-15004). Patient-derived primary ovarian cancer cells were isolated from ovarian biopsies using a protocol adapted from Sheperd et al. [50]. Human biological samples were sourced ethically and their research use was in accord with the terms of the informed consents under an IRB/EC-approved protocol. Primary cells were maintained in 50% MCDB 105 + 50% M199 media (Merck, DE) supplemented with 20% foetal calf serum (ThermoFisher Scientific, MA, USA). Sample size was determined by availability. Ovarian cancer cell lines were purchased from ATCC® (LGC Ltd, UK). SKOV3 (CVCL_0532) and OVCAR-3 (CVCL_0465) were maintained in RPMI 1640 media (Gibco™, 11875093) supplemented with 20% foetal calf serum (FBS; Gibco™, 10270106) and 10 μg/ml of insulin solution from bovine pancreas (I0516, Merck; DE). UWB1.289 (CVCL_B079) and CAOV3 (CVCL_0201) were maintained in DMEM/F-12+GlutaMAX™ (ThermoFisher Scientific) (10% FBS). All cell lines used within this study were authenticated at NorthGene™ using the PowerPlex® Fusion kit (DC24, Promega; WI, USA) in 2022. Laboratory reports are available upon request. Micoplasma contamination was routinely tested with the MycoAlert Mycoplasma Detection Kit (LZLT07218, Lonza, CH). The RealTime-Glo™ MT Cell Viability Assay (Promega, G9712) was used to assess live cell viability following the manufacturer’s instructions. All samples were tested in triplicates (n = 3). The experiment was replicated three times in the laboratory. Half-inhibitory concentration values (IC50) and heatmaps were generated using Graphpad Prism (V9). Raw luminescence values were transformed into logarithmic values, normalised and fitted to a dose-response curve using a non-linear regression; 100% and 0% values were defined by vehicle control and Staurosporine positive control (Biotechne, MN, USA), respectively.
Flow cytometry
All flow cytometry experiments were undertaken using a CellStream® (Luminex; TX, US). Cell cycle staining was performed using DAPI ready-made solution (1 µl/mL, MBD0015; Merck, DE) following the manufacturer’s instructions. Prior to staining, cells were fixed in 70% ethanol. Cell cycle profiles were analysed using FlowJo™ 10 using the Watson (Pragmatic) model; all data met the assumptions of the test. Schematic representations of cell percentages in different cell cycle phases were graphed using Graphpad Prism (V9). Values shown are defined as mean; error bars represent the coefficient of variation. Apoptosis staining was performed using Annexin V-FITC (640906, Biolegend; CA, US) and Propidium iodide (PI, 421301, Biolegend) as per manufacturer’s instructions, which included the use of Annexin V Binding Buffer (422201, Biolegend) and Cell Staining Buffer (420201, Biolegend). Unstained cells and cells stained with Annexin V/PI only were used to calculate the compensation matrix that was applied to all the data to adjust for signal overlap between channels of the emission spectra. All data met the assumptions of the test.
Results
Epigenomic landscapes reveal novel therapeutic avenues
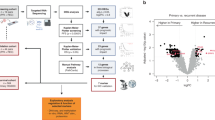
Deep epigenomic data mining offers an exciting alternative approach to current computational drug repurposing pipelines that typically use genomic or transcriptomic datasets [51, 52]. In this study, we implemented a novel computational approach to identify potential candidates for the treatment of OC subtypes (Fig. 1) using both cell line and primary sample H3K27ac ChIP-Seq datasets as discovery cohorts. Specifically, five primary OC sample datasets per subtype (HGSC, CC, MC 1–5) and three HGSC (Kuramochi, OAW42, UWB1.289), CC (JHOC5, RMG-II, ES2) and MC (MCAS, GTFR230, EFO27) cell line datasets were explored (Supplementary Table S1). Datasets from patient-derived primary ovarian samples (IOE4 and IOE11) were used as non-cancerous controls [21].

The diagram was created with BioRender.com.
Prediction of regulatory elements in ovarian cancer ChIP samples
Extensive quality control was performed on NGS data as samples were derived from different sources (Supplementary Table S1). Based on standard quality check parameters [53], all samples were deemed acceptable to pursue further analysis. At first, we undertook peak calling using MACS2, which resulted in the identification of an average of 65,000 peaks per sample. To facilitate downstream analyses with MAGIC [19], enriched regions were also identified using annotated enhancers obtained from the GeneHancer database [33]. The GeneHancer repository contains ~400,000 human regulatory elements divided into proximal (promoter), distal (enhancer) and proximal with distal enhancer functions. Using this approach, we identified 11,485–70,139 predicted regulatory elements per sample (Supplementary Table S1). Signal enrichment of proximal elements displayed a bimodal genomic distribution, while distal and proximal/distal elements displayed a unimodal genomic distribution (Fig. 2a), as expected [54, 55]. Due to H3K27ac being considered primarily as an active enhancer mark [10], we decided to concentrate on the examination of distal and proximal/distal enhancer elements, henceforth simply labelled as enhancers.

a Genome wide coverage of H3K27ac ChIP signal around promoter and enhancer sites in HGSC primary samples. b Counts per million of H3K27ac ChIP signal in GH19J009069 MUC16 interacting enhancer, HNF1B GH17J0377409 interacting enhancer and GH19J041709 CEACAM5 interacting enhancer in representative HGSC, CC and MC primary cells (red), cell lines (blue) and healthy samples (green). Red highlighted areas indicate the extent of enhancer regions. c, d Principal component analysis of ChIP signal in GeneHancer enhancer sites of healthy cells and ovarian cancer cell lines or primary cells. e, f Overlaps of H3K27ac peaks in replicates of UWB1.289 and GTFR230 cells. g Intersections of H3K27ac occupied GeneHancer enhancer regions for all HGSC samples analysed. See also Supplementary Fig. S1 for intersections of CC and MC samples.
To validate predicted enhancer locations at genomic level, we evaluated specific enrichment at the sites of well-known biomarkers of OC. MUC16 is the most widely used biomarker for OC [56] and we found enhancers linked with its expression in all HGSC samples and 50% of CC (Fig. 2b). HNF1B is commonly regarded as a CC-specific biomarker [57]. In accordance, an enhancer located proximally to its transcriptional start site was enriched in most CC samples but not in HGSC (Fig. 2b). CEACAM5 is a known tumour marker specific for MC [58] and analysis identified that corresponding enhancers were enriched in most MC samples (Fig. 2b).
Prior to differential analysis, we evaluated broad enhancer landscape similarities and differences, and discovered a clear contrast between cell line profiles of diseased and healthy samples (Fig. 2c). Separate clustering was also observed when analysing profiles of primary cells of all subtypes (Fig. 2d). In most instances replicates clustered together, confirming the high reproducibility of samples (Fig. 2c–f). To compare the persistence of enhancer regions across subtypes, we searched for MACS2 peaks which overlapped with enhancer regions in the GeneHancer database. Comparison of overlapping regions in HGSC revealed a total of 112,256 enriched enhancers across all samples, of which 12.8% were present in all primary samples and 9.6% were present in all cell line samples (Fig. 2g). In the case of CC (6.8% primaries, 15.5% cell lines) and MC (9.5% primaries, 20.5% cell lines), the overlap is higher in cell lines compared to primary samples (Supplementary Fig. S1). The largest intersection of enhancers occurred when comparing all primary samples and cell lines, suggesting a recurrent but still highly privative and sample-dependent enhancer landscape, as has been noted in cancer cells of colon, gastric and ovarian origin [59,60,61].
Differential analysis reveals subtype-specific target enhancer landscapes
Next, we performed differential analysis comparing the peak strength of H3K27ac-enriched regions between cancerous and healthy samples (Fig. 3a). We identified a total of 88,245 enhancer regions that were significantly enriched across the three HGSC cell lines (Fig. 3b), whereas 86,904 were identified when comparing HGSC primary cells and healthy samples (Fig. 3c). Differential analysis of CC and MC cell lines and primary samples rendered analogous peak representation, while primary enriched peaks were significantly greater than active enhancers in cell line samples (Fig. 3d, e). Given that enhancer enrichment, as determined by elevated levels of H3K27ac, signifies an open chromatin architecture, we were keen to understand what this effect could have on TF and concomitant coregulator recruitment. We therefore focused only on those enhancer regions that were enriched, and common to both the cell line and primary sample datasets.

a Reads per kilobase of transcript, per million mapped reads (RPKM) normalised H3K27ac ChIP signal for GeneHancer enhancers with significantly enriched H3K27ac binding in OC subtypes. b, c Volcano plots showing dysregulation of H3K27ac ChIP signal in healthy cells versus HGSC cell lines and HGSC primary samples. d, e Number of enriched and under-represented GeneHancer enhancer regions in HGSC, CC and MC cell line and primary samples extracted from the differential analysis results (FDR < 0.05). f Intersection of enriched GeneHancer enhancer sites in cell line and primary samples for all HGSC, CC and MC samples. g Intersection of final lists of enriched enhancers for all HGSC, CC and MC samples. h Examples of significantly enriched pathways for genes associated with specific GeneHancer enhancers in OC samples.
In the case of HGSC, we found an overlap of 23.2% between cell lines and primary samples, while we observed 12.5% and 20.1% in CC and MC, respectively (Fig. 3f). Interestingly, we found a global overlap of 4.7% between the intersected enriched enhancer regions of the different subtypes. A relatively high percentage of cell type-specific enhancers were also observed in HGSC (34.3%), CC (16.7%) and MC (25.6%) (Fig. 3g). Lastly, we assigned enriched enhancer locations to putative target genes based on GeneHancer annotations and performed pathway analyses using WebGestaltR [40]. Subtype-specific regulatory networks were found to be strongly associated with predicted enriched enhancer regions (FDR < 0.05), including NOTCH1 signalling pathways, known to be altered in HGSC [62] and the ERBB signalling pathway, linked with CC [57] (Fig. 3h). These results suggest that there is a predominance of subtype-specific enhancer landscapes, corroborating earlier observations [12, 13, 61].
Identification of enriched transcription factor binding
Subsets of aberrantly over-represented H3K27ac peaks were analysed using MAGIC and our custom enhancer matrix, with 534 TFs exhibiting significant enrichment (P < 0.001). This list was cross-referenced with publicly available RNA-Seq data to filter out non-expressed TFs (GSE121103) [12], resulting in the identification of 164 TFs that displayed significant enrichment scores across the OC subtypes (Fig. 4a and Supplementary Table S2). Of these, 100 (61%) were shared amongst all subtypes, 28 between two subtypes (17%), and 36 (22%) were subtype-specific (Fig. 4b). ESR1 and NR3C1were the predominant TFs (Fig. 4c, d), both of which have been previously associated with ovarian carcinogenesis [63, 64]. ESR1 was also the top enriched TF for the HGSC subtype (Fig. 4c), suggesting a major role for this TF in regulating aberrant epigenomic landscapes that lead to HGSC tumour development. As well as hormone receptors, we identified other major general TFs, like MYC (Fig. 4c, d), a TF that has been associated with all stages and types of ovarian tumorigenesis [65].

a Binding motifs identified by FIMO for top three predicted enriched TFs in enriched HGSC, CC and MC enhancer sites. b Subtype unique TFs with enriched binding in enriched enhancer sites. c MAGIC score of top ten TF binding sites for enriched GeneHancer enhancer sites in HGSC, CC and MC subtypes after filtering using MOTIF analysis and RNA-Seq expression levels. d Subtype-specific significant enrichment of TFs for the top twenty significantly enriched TFs from each subtype. e RPKM score from RNA-Seq HGSC and MC primaries for complexes only identified in HGSC and MC samples, respectively. Green borders indicate TFs identified in the pipeline and blue borders indicate complex coregulators. f Number of complexes where each coregulator is a component for HGSC samples, including only coregulators present in more than ten complexes. See also Supplementary Fig. S2 for coregulators of CC and MC samples. g E2F-1-DP-1-Cyclin A-CDK2 complex identified uniquely in HGSC. Created with BioRender.com.
Identification of enriched complex coregulators
Transcriptional regulation involves the recruitment of dynamic complexes to regulatory regions either subsequently to or together with the direct binding of TFs [66]. Accordingly, we interrogated the CORUM database to identify coregulators that are known to associate with each of the TFs linked with the deregulation of OC enhancer landscapes [47]. After removing non-expressed coregulators using RNA-Seq data, analysis resulted in the identification of 201 regulatory complexes, of which 175 (87%) were shared amongst all subtypes, 15 between two subtypes (7.5%), and interestingly 11 (5.5%) were subtype-specific for either HGSC or MC, suggesting that OC subtype-specific regulatory complexes could exist (Fig. 4e and Supplementary Table S3). HGSC subtype-specific complexes include E2F-1-DP-1-cyclin A-CDK2 and CTNNB1-FERMT2-TCF7L2, while MC-specific complexes are mainly formed by interacting SMAD coregulators (Fig. 4e–g and Supplementary Table S3). Many of the coregulators identified are known to be present in several of the 201 protein complexes and were extensively shared amongst the subtypes (259 out of 290), while only a small percentage were privative of specific subtypes (8 HGSC, 4 CC and 19 shared between 2 different subtypes) (Fig. 4e and Supplementary Table S3). Amongst the more prevalent cofactors, HDAC1 and HDAC2 were present in approximately 20-30% of the identified complexes across OC subtypes (Fig. 4f and Supplementary Fig. S2).
Coregulator interactions reveal potential drug candidates
Having identified TFs and coregulators associated with aberrant enhancer switching, we next sought to identify chemical compounds that could be used to target these regulatory proteins by assigning chemical/protein interaction scores via STITCH [20]. Despite differences in subtype-specific enhancer landscapes, potential drug candidates were largely shared between subtypes, with an 87.9% overlap likely due to shared TFs and coregulators. The top ten ranked drug candidates for all subtypes were monopolised by those directly targeting ESR1 and NR3C1 (Table 1 and Supplementary Table S4). Interestingly, Sulindac, a non-steroidal anti-inflammatory identified in previous in silico repositioning studies [51], was ranked among the top compounds for all subtypes (Table 1).
Screening identified seliciclib/CYC202 and SNS-032/BMS-387032 as HGSC-specific molecules that inhibit CDK2, 7 and 9 [67, 68] (Fig. 5a and Table 1). Both drug compounds have been investigated for the treatment of solid malignancies in Phase I clinical trials [69, 70] but they did not progress to Phase II. In addition, we broadened for potential therapeutic candidates search, to identify drugs that have not yet been included in clinical trials for any cancer setting by exploring the CHEMBL database [48] (Supplementary Table S4). Using this approach we identified BIX-01294, a molecule that inhibits histone methyltransferase EHMT2/G9A, involved in anti-tumour immune response and chemoresistance in HGSC [71] (Fig. 5a and Table 1).

a Network diagram of HGSC targets (CDKs and EHMT2) and targeting compounds (STITCH score >0.95). Edge thickness represents combined STITCH confidence score. Round nodes and diamond nodes represent compounds that have undergone some phase of clinical trials and have not undergone clinical trials, respectively. Compound node colours represent compound mechanism of action (MOA). Factor node outline colour indicates the number of the expressed complexes which the factor is a component of. Red and green highlight circles indicate CDK and EHMT-related protein groups respectively. b Heatmap representing the effect of drug compounds on patient-derived clinical samples treated for 72 h. Each 4 rows of the heatmap represent the top concentrations used to derive IC50 values (10 μM, 1 μM, 100 nM and 10 nM). Heatmap values were calculated using relative viability compared to the vehicle control (DMSO). Red colour indicates low viability following treatment. c Table displaying calculated best fit IC50 values after 72 h of treatment with varying concentrations (10 pM-10 μM) of displayed drug compounds. DMSO was used as vehicle control and staurosporine was used as positive control (+). d Flow cytometry cell cycle analyses of CAOV3 and OVCAR-3 cells treated with selected compounds for 24 h. Bar chart error represents coefficients of variation (cv). Blue peaks represent cells in G0/G1, while green peaks represent cells in G2/M phase. The area depicted as yellow represents cells in the S phase. e Flow cytometry apoptosis analysis of CAOV3 and OVCAR-3 cells treated with selected compounds for 24 h. Cells were stained with Propidium iodide and Annexin V-FITC, rendering 4 populations: viable (−, −), early apoptotic (−,+), late apoptotic (+, +) and dead (+, −), three of which are highlighted in the panels. Graphs display cell densities, whereby red, green and blue colours indicate high, medium and low cell densities, respectively.
Validation of predicted HGSC-specific therapeutics
The efficacy of selected molecules was evaluated in primary cells derived from late-stage HGSC clinical samples and cell line models (Fig. 5b, Supplementary Table S5 and Supplementary Fig. S3). The histone acetyltransferase inhibitor i-CBP112 ranked poorly (~400th) and was selected as a negative control. Overall, SNS-032 exhibited potent and clinically achievable anti-tumour activity across the panel of patient samples and cell lines; seliciclib and i-CBP112 showed little to no activity (IC50 > 10 μM) (Fig. 5b, c and Supplementary Fig. S3). Although SNS-032 was primarily selected based on its activity against CDK2, it is also sensitive to CDK7/9 (IC50 in cell-free assays: 4 nM [CDK9], 48 nM [CDK2] and 62 nM [CDK7]) [67]. To validate the specific vulnerability of HGSC towards CDK2 inhibition, we tested the CDK1/2 inhibitor NU-6102 and the CDK2-specific inhibitor AUZ 454. Whilst NU-6102 showed no activity (IC50 > 10 μM), probably due to its higher sensitivity towards CDK1, AUZ 454 exhibited considerable levels of anti-tumour activity in the range of μM (Supplementary Fig. S3), suggesting an abnormal behaviour of CDK2 in HGSC. Next, we performed qRT-PCR and western blot analyses to confirm predicted modes of action. The expression of known CDK2 targets was significantly upregulated following SNS-032 treatment (Supplementary Fig. S4) [72]. At the same time, CDK2-specific target Rb-Thr821 was de-phosphorylated after exposure with SNS-032 and AUZ 454 [73], indicating the presence of a CDK2-specific mechanism of action. In addition, we performed cell cycle and apoptosis assays which, as expected, led to apoptotic cell death coupled with cell cycle arrest (Fig. 5d, e and Supplementary Fig. S5).
We also tested compounds BIX-01294 and UNC0646 targeting EHMT2 as well as GSK503 and GSK343 targeting methyltransferase EZH2 as means of comparison. After 72 h, both EHMT2 inhibitors exhibited potent anti-tumour in vitro activity in all models (Fig. 5b, c and Supplementary Fig. S3). Neither of the EZH2 inhibitors was able to match the in vitro efficacy observed with BIX-01294 or UNC0646 (Fig. 5b, c and Supplementary Fig. S3). The expression of known EHMT2 targets was significantly up and/or downregulated following treatment [74], and EHMT2 inhibition led to a significant decrease in the levels of H3K9me2, the main histone methylation target of EHMT2 (Supplementary Fig. S4); these results point to a specific EHMT2-driven effect. EHMT2 inhibition led to increased levels of cellular apoptosis, whereas no significant changes were observed in the DNA profile (Fig. 5d, e and Supplementary Fig. S5). Overall, the results of in vitro validation studies in patient-derived cells and cell lines further confirmed the efficacy of our approach, supporting the concept of epigenomic profiling as means to guide therapeutic strategies.
Discussion
In 2012, the ENCODE project proposed that there were 400,000 regions with enhancer-like features in the human genome [75]. Since then, numerous studies have investigated the epigenomic landscape of cancer, labelling active enhancers in the regions of tens of thousands per cell type. For example, Lidschreiber et al identified 58,457 putative enhancers across 7 cancer cell lines from different origins [76], Mack et al. profiled more than 30,000 enhancers in ependymoma primary cells [77] and Ooi et al. detected 36,973 predicted distal enhancer regions in gastric cancer cell lines [60]. In ovarian cancer, Shang et al identified 36,388 regulatory elements across four cell line models [61]. We have refined this approach to define cancer-associated enhancer landscapes and have identified aberrant enhancer activity associated exclusively to specific OC subtypes. Linking broad epigenomic signatures, in this case H3K27ac, we were subsequently able to map enhancer locations to enriched TF binding sites and associated coregulator proteins. Interestingly, through this process, we observed a notable disparity between the subtype-specific nature of most abnormal cis-regulatory elements and the fact that the TFs, and more pronouncedly, the cofactors governing such networks display substantial homogeneity across the range of studied OC subtypes.
To discover novel therapeutic agents for OC, we sought to identify chemicals that targeted the TFs and coregulators uniquely enriched at enhancers. To validate our findings and the usefulness of our approach, we tested the in vitro efficacy of a subset of pipeline drug outputs in patient-derived primary samples. SNS-032 is a potent and selective CDK2, 7 and 9 inhibitor [67], whose in vitro efficacy has been proven in OC cell lines, and their sensitivity towards this compound linked with cyclin E1 overexpression, relatively common in HGSC patients (15–20%) [78]. SNS-032 was investigated for the treatment of haematologic and solid malignancies in two Phase I clinical trials [69, 70] but no further developments have been undertaken. We extended previous investigations on the effect of SNS-032 in OC using patient-derived primary samples and cell lines, although unfortunately, we could not confirm sample CCNE1 status. Our study suggests that SNS-032 represents a good therapeutic option for HGSC patients, given its outstanding efficacy in vitro. In contraposition, seliciclib or NU-6102 displayed overall lower in vitro efficacy levels, which correlates with known IC50 values obtained in cell-free assays as well as previous in vitro observations [79]. Importantly, CDK2 was one of the few cofactors uniquely identified in HGSC, as part of the protein complex E2F-1-DP-1-cyclin A-CDK2 [80]. Our findings highlight the critical role of this cofactor modulating broad epigenomic landscapes and demonstrate a potential for further research into the clinical effectiveness of targeting CDK2 in HGSC.
In our effort to put forward new therapeutic targets against HGSC, we performed further in vitro testing of compounds targeting EZH2 and EHMT2 molecules. Preclinical studies have previously highlighted the potential of targeting EZH2, although no clinical trial is currently investigating the use of EZH2 inhibitors, specifically in OC subjects [81]. The EHMT1/2 inhibitor BIX-01294 is known to block the proliferation of cancer cell lines but has shown considerable toxicity problems in preclinical studies [82]. We have shown that while EZH2 inhibitors mainly displayed poor in vitro efficacy results, BIX-01294 analogue UNC0646 exhibited an improved response, with IC50 values in the range of ~1–3 μM across the studied samples.
Epigenomic deregulation continues to emerge as a key driver in ovarian tumorigenesis, and more specifically in the differentiation of OC subtypes [23]. Such epigenomic deficiencies have the potential to be targeted with existing therapies already in clinical or preclinical trials for other diseases. Here we present the first attempt to investigate subtype-specific enhancer re-programming aimed at drug discovery and repurposing in OC. Overall, in silico and in vitro investigations suggest that EHMT2, together with CDK2, represent the best candidates to pursue further preclinical studies aimed at improving HGSC treatment strategies. The effectiveness of this approach, identifying potential candidate molecules based on the evaluation of one histone mark alone, is extremely encouraging. Combining enhancer-localised epigenomic datasets for H3K27ac and other histone marks could lead to further subtype stratification using this approach.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Code availability
Code and usage instructions are available at https://github.com/RBGO-Lab/EnhancerDrugTargetPipeline.
References
McGuire AL, Gabriel S, Tishkoff SA, Wonkam A, Chakravarti A, Furlong EEM, et al. The road ahead in genetics and genomics. Nat Rev Genet. 2020;21:581–96. https://doi.org/10.1038/s41576-020-0272-6
Brogi S, Ramalho TC, Kuca K, Medina-Franco JL, Valko M. Editorial: in silico methods for drug design and discovery. Front Chem. 2020;8:612. https://doi.org/10.3389/fchem.2020.00612
Brunak S, Bjerre Collin C, Eva Ó Cathaoir K, Golebiewski M, Kirschner M, Kockum I, et al. Towards standardization guidelines for in silico approaches in personalized medicine. J Integr Bioinforma. 2020;17. https://doi.org/10.1515/JIB-2020-0006.
Zhang Y, Qazi S, Raza K. Differential expression analysis in ovarian cancer: a functional genomics and systems biology approach. Saudi J Biol Sci. 2021;28:4069–81. https://doi.org/10.1016/J.SJBS.2021.04.022
Lu W, Zhang R, Jiang H, Zhang H, Luo C. Computer-aided drug design in epigenetics. Front Chem. 2018;6:57. https://doi.org/10.3389/fchem.2018.00057
Qazi S, Raza K. In silico approach to understand epigenetics of POTEE in ovarian cancer. J Integr Bioinform. 2021;18:20210028
Dawson MA. The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science. 2017;355:1147–52. https://doi.org/10.1126/science.aam7304
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46. https://doi.org/10.1158/2159-8290.CD-21-1059
Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu Rev Genomics Hum Genet. 2006;7:29–59. https://doi.org/10.1146/annurev.genom.7.080505.115623
Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107:21931–6. https://doi.org/10.1073/pnas.1016071107
Okabe A, Kaneda A. Transcriptional dysregulation by aberrant enhancer activation and rewiring in cancer. Cancer Sci. 2021;112:2081. https://doi.org/10.1111/CAS.14884
Corona RI, Seo JH, Lin X, Hazelett DJ, Reddy J, Fonseca MAS, et al. Non-coding somatic mutations converge on the PAX8 pathway in ovarian cancer. Nat Commun. 2020;11:1–11. https://doi.org/10.1038/s41467-020-15951-0
Lawrenson K, Fonseca MAS, Liu AY, Freedman ML, Gayther SA, Noushmehr H. A study of high-grade serous ovarian cancer origins implicates the SOX18 transcription factor in tumor development. Cell Rep. 2019;29:3726–35.
Ravindran F, Choudhary B. Ovarian cancer: molecular classification and targeted therapy. Ovarian Cancer Updat Tumour Biol Ther. 2021. https://doi.org/10.5772/intechopen.95967.
Chandra A, Pius C, Nabeel M, Nair M, Vishwanatha JK, Ahmad S, et al. Ovarian cancer: current status and strategies for improving therapeutic outcomes. Cancer Med. 2019;8:7018–31. https://doi.org/10.1002/cam4.2560
Marchetti C, De Felice F, Romito A, Iacobelli V, Sassu CM, Corrado G, et al. Chemotherapy resistance in epithelial ovarian cancer: mechanisms and emerging treatments. Semin Cancer Biol. 2021;77:144–66. https://doi.org/10.1016/j.semcancer.2021.08.011
Horikawa N, Abiko K, Matsumura N, Baba T, Hamanishi J, Yamaguchi K, et al. Anti-VEGF therapy resistance in ovarian cancer is caused by GM-CSF-induced myeloid-derived suppressor cell recruitment. Br J Cancer. 2020;122:778–88. https://doi.org/10.1038/s41416-019-0725-x
Dias MP, Moser SC, Ganesan S, Jonkers J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat Rev Clin Oncol. 2021;18:773–91. https://doi.org/10.1038/s41571-021-00532-x
Roopra A. MAGIC: A tool for predicting transcription factors and cofactors driving gene sets using ENCODE data. PLoS Comput Biol. 2020;16:e1007800. https://doi.org/10.1371/journal.pcbi.1007800
Szklarczyk D, Santos A, Von Mering C, Jensen LJ, Bork P, Kuhn M. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016;44:D380–4. https://doi.org/10.1093/nar/gkv1277
Coetzee SG, Shen HC, Hazelett DJ, Lawrenson K, Kuchenbaecker K, Tyrer J, et al. Cell-type-specific enrichment of risk-associated regulatory elements at ovarian cancer susceptibility loci. Hum Mol Genet. 2015;24:3595–607. https://doi.org/10.1093/HMG/DDV101
Lin X, Spindler TJ, de Souza Fonseca MA, Corona RI, Seo JH, Dezem FS, et al. Super-enhancer-associated LncRNA UCA1 interacts directly with AMOT to activate YAP target genes in epithelial ovarian cancer. IScience. 2019;17:242–55. https://doi.org/10.1016/j.isci.2019.06.025
Jones MR, Peng PC, Coetzee SG, Tyrer J, Reyes ALP, Corona RI, et al. Ovarian cancer risk variants are enriched in histotype-specific enhancers and disrupt transcription factor binding sites. Am J Hum Genet. 2020;107:622–35. https://doi.org/10.1016/j.ajhg.2020.08.021
Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. https://doi.org/10.1038/nmeth.1923
Dobin A, Gingeras TR, Spring C, Flores R, Sampson J, Knight R, et al. Mapping RNA-seq with STAR. Curr Protoc Bioinforma. 2016;51:586–97. https://doi.org/10.1002/0471250953.bi1114s51.Mapping
Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol. 2008;26:1351–9. https://doi.org/10.1038/nbt.1508
Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. 2012:1813–31. https://doi.org/10.1101/gr.136184.111.
Carroll TS, Liang Z, Salama R, Stark R, de Santiago I. Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front Genet. 2014;5:1–11. https://doi.org/10.3389/fgene.2014.00075
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:1–9. https://doi.org/10.1186/GB-2008-9-9-R137
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. https://doi.org/10.1186/s13059-014-0550-8
Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, et al. {O}rchestrating high-throughput genomic analysis with {B}ioconductor. Nat Methods. 2015;12:115–21.
Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database. 2017;2017. https://doi.org/10.1093/database/bax028.
Kolde R. pheatmap: Pretty Heatmaps. R package version 1.0.12 (2019). https://CRAN.R-project.org/package=pheatmap
Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–93. https://doi.org/10.1038/nature10730
Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag New York; 2016.
Hahne F, Ivanek R. Visualizing genomic data using Gviz and bioconductor. Methods Mol Biol. 2016;1418:335–51. https://doi.org/10.1007/978-1-4939-3578-9_16
Gehlenborg N. UpSetR: a more scalable alternative to Venn and Euler diagrams for visualizing intersecting sets. R package version 1.4.0 (2019). https://CRAN.R-project.org/package=UpSetR
Inkscape Project. Inkscape [Internet]. 2020. Available from https://inkscape.org.
Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47:W199–205. https://doi.org/10.1093/nar/gkz401
Joshi NA, Fass JN. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]. Available at https://github.com/najoshi/sickle
Beauparlant CJ, Lemacon A, Fournier E, Droit A. ENCODExplorer: a compilation of ENCODE metadata. R package version 2.12.1 (2019). https://rdrr.io/bioc/ENCODExplorer/
Grant CE, Bailey TL, Noble WS. FIMO: Scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–8. https://doi.org/10.1093/bioinformatics/btr064
Nettling M, Treutler H, Grau J, Keilwagen J, Posch S, Grosse I. DiffLogo: a comparative visualization of sequence motifs. BMC Bioinforma. 2015;16:1–9. https://doi.org/10.1186/S12859-015-0767-X
Bateman A, Martin MJ, Orchard S, Magrane M, Agivetova R, Ahmad S, et al. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–9. https://doi.org/10.1093/nar/gkaa1100
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. https://doi.org/10.1002/jcc.20084
Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, et al. CORUM: the comprehensive resource of mammalian protein complexes—2019. Nucleic Acids Res. 2019;47:D559–63. https://doi.org/10.1093/nar/gky973
Gaulton A, Hersey A, Nowotka ML, Patricia Bento A, Chambers J, Mendez D, et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017;45:D945–54. https://doi.org/10.1093/nar/gkw1074
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Shepherd TG, Thériault BL, Campbell EJ, Nachtigal MW. Primary culture of ovarian surface epithelial cells and ascites-derived ovarian cancer cells from patients. Nat Protoc. 2007;1:2643–9. https://doi.org/10.1038/nprot.2006.328
Nagaraj AB, Wang QQ, Joseph P, Zheng C, Chen Y, Kovalenko O, et al. Using a novel computational drug-repositioning approach (DrugPredict) to rapidly identify potent drug candidates for cancer treatment. Oncogene. 2018;37:403–14. https://doi.org/10.1038/onc.2017.328
Qazi S, Raza K. Integrative analysis of ovarian serious adenocarcinoma to understand disease network biology. Lect Notes Comput Sci. 2022;13347 LNBI:3–17. https://doi.org/10.1007/978-3-031-07802-6_1/FIGURES/6
Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE. 2012;7. https://doi.org/10.1371/journal.pone.0030619.
Bae S, Lesch BJ. H3K4me1 distribution predicts transcription state and poising at promoters. Front Cell Dev Biol. 2020;8:289. https://doi.org/10.3389/fcell.2020.00289
Cai W, Huang J, Zhu Q, Li BE, Seruggia D, Zhou P, et al. Enhancer dependence of cell-type–specific gene expression increases with developmental age. Proc Natl Acad Sci USA. 2020;117:21450–8. https://doi.org/10.1073/pnas.2008672117
Høgdall EVS, Christensen L, Kjaer SK, Blaakaer J, Kjærbye-Thygesen A, Gayther S, et al. CA125 expression pattern, prognosis and correlation with serum CA125 in ovarian tumor patients: from The Danish “MALOVA” Ovarian Cancer Study. Gynecol Oncol. 2007;104:508–15. https://doi.org/10.1016/j.ygyno.2006.09.028
Iida Y, Okamoto A, Hollis RL, Gourley C, Herrington CS. Clear cell carcinoma of the ovary: a clinical and molecular perspective. Int J Gynecol Cancer. 2021;31:605–16. https://doi.org/10.1136/IJGC-2020-001656
Babaier A, Ghatage P. Mucinous cancer of the ovary: overview and current status. Diagnostics. 2020;10. https://doi.org/10.3390/diagnostics10010052.
Akhtar-Zaidi B, Cowper-Sallari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012;336:736–9. https://doi.org/10.1126/science.1217277
Ooi WF, Xing M, Xu C, Yao X, Ramlee MK, Lim MC, et al. Epigenomic profiling of primary gastric adenocarcinoma reveals super-enhancer heterogeneity. Nat Commun. 2016;7. https://doi.org/10.1038/ncomms12983.
Shang S, Yang J, Jazaeri AA, Duval AJ, Tufan T, Fischer NL, et al. Chemotherapy-induced distal enhancers drive transcriptional programs to maintain the chemoresistant state in ovarian cancer. Cancer Res. 2019;79:4599–611. https://doi.org/10.1158/0008-5472.CAN-19-0215
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. https://doi.org/10.1038/nature10166
Simpkins F, Garcia-Soto A, Slingerland J. New insights on the role of hormonal therapy in ovarian cancer. Steroids. 2013;78:530. https://doi.org/10.1016/J.STEROIDS.2013.01.008
Veneris JT, Huang L, Churpek JE, Conzen SD, Fleming GF. Glucocorticoid receptor expression is associated with inferior overall survival independent of BRCA mutation status in ovarian cancer. Int J Gynecol Cancer. 2019;29:357–64. https://doi.org/10.1136/IJGC-2018-000101
Li C, Bonazzoli E, Bellone S, Choi J, Dong W, Menderes G, et al. Mutational landscape of primary, metastatic, and recurrent ovarian cancer reveals c-MYC gains as potential target for BET inhibitors. Proc Natl Acad Sci USA. 2019;116:619–24. https://doi.org/10.1073/pnas.1814027116
Gonzalez D, Hamidi N, Del Sol R, Benschop JJ, Nancy T, Li C, et al. Suppression of mediator is regulated by Cdk8-dependent Grr1 turnover of the Med3 coactivator. Proc Natl Acad Sci USA. 2014;111:2500–5. https://doi.org/10.1073/pnas.1307525111
Misra RN, Xiao HY, Kim KS, Lu S, Han WC, Barbosa SA, et al. N-(Cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-dependent kinase 2. N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor agent. J Med Chem. 2004;47:1719–28.
De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M, Kim SH. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur J Biochem. 1997;243:518–26. https://doi.org/10.1111/J.1432-1033.1997.0518A.X
Tong WG, Chen R, Plunkett W, Siegel D, Sinha R, Harvey RD, et al. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J Clin Oncol. 2010;28:3015 https://doi.org/10.1200/JCO.2009.26.1347
Heath EI, Bible K, Martell RE, Adelman DC, LoRusso PM. A phase 1 study of SNS-032 (formerly BMS-387032), a potent inhibitor of cyclin-dependent kinases 2, 7 and 9 administered as a single oral dose and weekly infusion in patients with metastatic refractory solid tumors. Invest N. Drugs. 2008;26:59–65. https://doi.org/10.1007/S10637-007-9090-3
Spiliopoulou P, Spear S, Mirza H, Garner I, McGarry L, Grundland-Freile F, et al. Dual G9A and EZH2 inhibition stimulates an anti-tumour immune response in ovarian high-grade serous carcinoma. Mol Cancer Ther. 2022;21:522–34. https://doi.org/10.1158/1535-7163.MCT-21-0743
Han Y, Wei Y, Yao J, Chu Y-Y, Li C-W, Hsu JL, et al. Inhibition of CDK2 reduces EZH2 phosphorylation and reactivates ERα expression in high-grade serous ovarian carcinoma. Am J Cancer Res. 2020;10:1194.
Lentine B, Antonucci L, Hunce R, Edwards J, Marallano V, Krucher NA. Dephosphorylation of threonine-821 of the retinoblastoma tumor suppressor protein (Rb) is required for apoptosis induced by UV and Cdk inhibition. Cell Cycle. 2012;11:3324. https://doi.org/10.4161/CC.21693
Hua K-T, Wang M-Y, Chen M-W, Wei L-H, Chen C-K, Ko C-H, et al. The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol Cancer. 2014;13. https://doi.org/10.1186/1476-4598-13-189.
Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. https://doi.org/10.1038/nature11247
Lidschreiber K, Jung LA, Emde H von der, Dave K, Taipale J, Cramer P, et al. Transcriptionally active enhancers in human cancer cells. Mol Syst Biol. 2021;17. https://doi.org/10.15252/MSB.20209873.
Mack SC, Pajtler KW, Chavez L, Okonechnikov K, Bertrand KC, Wang X, et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature. 2017;553::101–5. https://doi.org/10.1038/nature25169
Yang L, Fang D, Chen H, Lu Y, Dong Z, Ding HF, et al. Cyclin-dependent kinase 2 is an ideal target for ovary tumors with elevated cyclin E1 expression. Oncotarget. 2015;6:20801 https://doi.org/10.18632/oncotarget.4600
Chen R, Wierda WG, Chubb S, Hawtin RE, Fox JA, Keating MJ, et al. Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood. 2009;113:4637–45. https://doi.org/10.1182/blood-2008-12-190256
Xu M, Sheppard KA, Peng CY, Yee AS, Piwnica-Worms H. Cyclin A/CDK2 binds directly to E2F-1 and inhibits the DNA-binding activity of E2F-1/DP-1 by phosphorylation. Mol Cell Biol. 1994;14:8420–31. https://doi.org/10.1128/MCB.14.12.8420-8431.1994
Jones BA, Varambally S, Arend RC. Histone methyltransferase EZH2: a therapeutic target for ovarian cancer. Mol Cancer Ther. 2018;17:591. https://doi.org/10.1158/1535-7163.MCT-17-0437
Jan S, Dar MI, Wani R, Sandey J, Mushtaq I, Lateef S, et al. Targeting EHMT2/ G9a for cancer therapy: progress and perspective. Eur J Pharm. 2021;893:173827. https://doi.org/10.1016/j.ejphar.2020.173827
Acknowledgements
Special thanks to Dr Avtar Roopra for his help in adapting the original MAGIC algorithm to our specific enhancer search. Part of this work was supported by the MRC IAA 2021 Swansea University AccelerateAI facility (MR/X502686/1), part-funded by the European Regional Development Fund through the Welsh Government via Ser Cymru.
Funding
The research conducted herein was funded by SMARTExpertise programmes CEAT and RISE via the Welsh Government and the European Regional Development Fund (2017/COL/004; 2017/COL/001).
Author information
Authors and Affiliations
Contributions
Conceptualisation: MQ, DWJ, DG, RSC and LWF; methodology and software: DWJ, JG and KE; validation, formal analysis and investigation: MQ and DWJ; resources: JG, KE, LM, ND and KLS; writing—original draft: MQ and DWJ; writing—major reviews & editing: MQ, NRH, DG, RSC and LWF; supervision: LM, NRH, ALB, DG, RSC and LWF; project administration and funding acquisition: ALB, IR, RKP, NRH, DG, RSC and LWF. Manuscript revision and final approval: all authors. Accountable for all aspects of the work: all authors.
Corresponding author
Ethics declarations
Competing interests
RKP and NRH are employees and shareholders of GlaxosSmithKline. IR was an employee and shareholder of GlaxoSmithKline when the study was carried out. The remaining authors declare no competing interests.
Ethics approval and consent to participate
Ethical approval for processing ovarian patient samples has been obtained through Local Research Ethics Committee LREC Wales (ref 15/wa/0065) for the collection of biopsies from consented patients. Formal written consent was obtained from all patients at the time of recruitment into the study. The study was performed in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Quintela, M., James, D.W., Garcia, J. et al. In silico enhancer mining reveals SNS-032 and EHMT2 inhibitors as therapeutic candidates in high-grade serous ovarian cancer. Br J Cancer 129, 163–174 (2023). https://doi.org/10.1038/s41416-023-02274-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-023-02274-2
