
Abstract
Background
Body mass index (BMI) is known to influence the risk of various site-specific cancers, however, dissecting which subcomponents of this heterogenous risk factor are predominantly responsible for driving disease effects has proven difficult to establish. We have leveraged tissue-specific gene expression to separate the effects of distinct phenotypes underlying BMI on the risk of seven site-specific cancers.
Methods
SNP-exposure estimates were weighted in a multivariable Mendelian randomisation analysis by their evidence for colocalization with subcutaneous adipose- and brain-tissue-derived gene expression using a recently developed methodology.
Results
Our results provide evidence that brain-tissue-derived BMI variants are predominantly responsible for driving the genetically predicted effect of BMI on lung cancer (OR: 1.17; 95% CI: 1.01–1.36; P = 0.03). Similar findings were identified when analysing cigarettes per day as an outcome (Beta = 0.44; 95% CI: 0.26–0.61; P = 1.62 × 10−6), highlighting a possible shared aetiology or mediator effect between brain-tissue BMI, smoking and lung cancer. Our results additionally suggest that adipose-tissue-derived BMI variants may predominantly drive the effect of BMI and increased risk for endometrial cancer (OR: 1.71; 95% CI: 1.07–2.74; P = 0.02), highlighting a putatively important role in the aetiology of endometrial cancer.
Conclusions
The study provides valuable insight into the divergent underlying pathways between BMI and the risk of site-specific cancers.
Similar content being viewed by others
Introduction
Body mass index (BMI) is an important risk factor for multiple types of cancer. Mendelian randomisation (MR) [1] studies have been integral in elucidating evidence of causal relationships between variation in BMI and site-specific cancer risk [2], although further granular insight is required to clarify the specific mechanistic and biological pathways which may explain these effects. While BMI is a commonly used proxy for excess adiposity in population studies, it retains a high degree of heterogeneity and therefore captures multiple phenotypes [3].
Recently, we have developed a novel multivariable MR approach to separate the effects of phenotypic subcomponents of BMI on complex traits and disease risk. This is implemented through fractionation of the genetic variants associated with BMI according to whether the BMI signal colocalises with gene expression in the brain or subcutaneous adipose tissue [4]. In this framework, ‘adipose-’ and ‘brain-tissue instrumented BMI’ were analysed as separate exposures in a one-sample multivariable MR analysis using genetic risk scores (GRS) based on subsets of adipose and brain expression colocalizing BMI variants. We found that these distinct tissue-dependent exposures related differentially to measures of fat distribution and visceral adiposity, with brain-tissue colocalizing variants driving the effect of BMI on cardiometabolic disease outcomes and subcutaneous adipose-tissue colocalizing variants predominantly being responsible for the effect of BMI on measures of heart structure [4].
In this study, we sought to adapt this tissue-partitioned MR approach such that it can be applied in a two-sample MR setting, allowing us to leverage findings from large consortia. Next, we applied this approach to investigate the putatively independent effects of adipose- and brain-tissue instrumented BMI on the risk of seven site-specific cancer outcomes. Lastly, we conducted further analyses using additional datasets to evaluate the robustness of potential independent effects highlighted in our primary analysis.
Methods
Tissue-partitioned genetic instruments for adult BMI
Full details on the genetic instruments identified for MR analyses in this study have been reported previously [4] and are explained in further detail in Supplementary Note 1. In brief, we incorporated gene expression data to identify genetic variants whose robust effects on BMI are putatively mediated via the expression of a nearby gene in either subcutaneous adipose and neural tissues. This was assessed by conducting extensive genetic colocalization analyses using the method ‘coloc’ [5] where a posterior probability for colocalization (PPA4) ≥0.8 was applied to formally define instrument sets, as recommended by the authors of the method.
Genetic colocalization analyses were performed systematically at 915 independent loci robustly associated with adult BMI (i.e., P < 5 × 10−08 & r2 < 0.01) from a meta-analysis GWAS from the Genetic Investigation of Anthropometric Traits (GIANT) consortium and the UK Biobank (UKB) (n = 681,275) [6]. To ensure that the highest SNP coverage available was implemented for colocalization analyses, we combined these data with the summary statistics from a BMI GWAS involving participants of European ancestry from the UK Biobank only (n = 463,005) to obtain summary statistics for SNPs not included in the meta-analysis with GIANT. The combined BMI datasets provided summary statistics on a total of 12,322,387 SNPs. To minimise the incorporation of findings which may potentially be influenced by strong regional linkage disequilibrium (LD) structure we omitted variants which reside within the human leukocyte antigen (HLA) region (chr6:25 Mb–35 Mb).
In total, 86 genetic variants provided evidence of colocalization between BMI and proximal subcutaneous adipose-tissue-derived gene expression using meta-analysed expression quantitative loci (eQTL) data derived from subcutaneous adipose (n = 1257). Similarly, 140 genetic variants with evidence of colocalization were found between BMI and proximal brain-tissue-derived gene expression data using meta-analysed brain-tissue samples (n = 1194). These two instrument sets were used as proxies for what we refer to as 'adipose-' and 'brain-tissue instrumented BMI', respectively. These tissues were selected due to their important biological relevance for adiposity and resulting instruments were subject to various robustness evaluations such as ensuring that both resulting instrument sets have very similar average effect estimates on BMI (adipose = 0.0148 and brain = 0.0149 standard deviation change in BMI per effect allele) (Supplementary Note 1). Full details of the genetic variants incorporated into adipose- and brain-tissue instrumented BMI exposures are provided in Supplementary Table S1.
Robust simulation studies in the literature have suggested that sample sizes of eQTL studies over n = 1000 should maintain a high true positive and low false-positive rate for the majority of common variants identified by GWAS [7]. We additionally assessed how the number of instruments may influence exposure strength in the multivariable model. Randomly sampling pools of adipose- and brain-tissue instruments suggested that even when 30 BMI instruments for both tissues are available our multivariable approach is capable of separating these two exposures (Fadipose = 30.6 & Fbrain = 29.9).
Tissue-partitioned childhood body-size instruments
In this study, we additionally applied our instrument derivation pipeline as described above to GWAS results from a measure of childhood body size using recall data from the UK Biobank study at age 10 [8]. UKB participants completed recall questionnaires asking if they were ‘thinner’, ‘plumper’ or ‘about average’ when they were aged 10 years old compared to the average. These GWAS results have been previously validated using measured childhood BMI in three independent cohorts which found that they predict BMI at this early stage in the lifecourse more strongly compared to adult BMI genetic variants [9,10,11]. In addition, the childhood BMI phenotype has been shown to directly influence outcomes measured during childhood such as vitamin D levels [12], but have an indirect effect after accounting for genetically predicted adulthood adiposity on the same outcome when measured in adulthood. There were 56 and 53 childhood body-size-associated variants which colocalized with adipose- and brain-tissue eQTL, respectively. A list of these genetic variants can be found in Supplementary Table S2.
Genome-wide association study data on site-specific cancers
GWAS estimates were obtained on the following seven site-specific cancer outcomes: colon, breast, endometrial, lung, ovarian, kidney and prostate cancer [13,14,15,16,–17]. All site-specific cancers investigated in the present analysis have previously been shown to be causally influenced by adiposity in MR analyses [18,19,–20]. If a particular SNP was not present in the outcome summary data extracted from the MRC-IEU OpenGWAS database using MR-base [21, 22], a proxy SNP in LD with the requested SNP was provided by default. LD proxies were determined using the 1000 genomes of European ancestry sample data. To maximise statistical power for cancer outcomes where reported parental history of disease in the UK Biobank study provided a larger number of cases compared to accessible datasets (i.e., colon and lung cancer), we obtained estimates based on a GWAS by proxy approach previously shown to be highly genetically correlated with findings from GWAS of diagnosed cases [23, 24]. A summary of the outcome datasets used in this study is provided in Supplementary Table S3.
Univariable Mendelian randomisation to estimate the total effect of BMI
We firstly estimated the total effect of genetically predicted adult BMI using the full set of 915 instruments (i.e., without considering their tissue-dependent effects on gene expression) on the seven site-specific cancer outcomes which have previously been shown to be influenced by adiposity [18,19,–20]. Analyses were conducted using two-sample Mendelian randomisation (MR) with the inverse-variance weighted (IVW) method [25] and repeated using the MR Egger, weighted median and MR penalised weighed median methods, which are typically more robust to horizontal pleiotropy [26]. All analyses were conducted using the ‘TwoSampleMR’ R package. Estimates for instruments when analysing cancer endpoints based on GWAS of sex-stratified populations (i.e., breast, endometrial and ovarian cancers in female-only populations, prostate cancer in a male-only population) were obtained from previously conducted sex-stratified GWAS analyses of BMI [27]. MR analyses were conducted using exposure and outcome data from non-overlapping samples where possible to avoid overfitting bias [28].
Tissue-partitioned Mendelian randomisation
Next, we used the sets of adipose and brain expression variants which colocalized with BMI based on PPA4 ≥ 0.8 as instrumental variables within the MR framework. When using the adipose colocalized variants as genetic proxies for BMI, we refer to this exposure as ‘adipose-tissue instrumented BMI’ hereafter, whereas when using the subset of brain colocalized variants as instruments, we refer to this exposure as ‘brain-tissue instrumented BMI’. We conducted univariable MR as above to estimate the total effect of adipose and brain-expressed BMI separately on all seven site-specific cancers.
We next employed a multivariable MR (MVMR) approach to estimate the direct effects of these tissue-partitioned exposures on each outcome by simultaneously estimating their effects in the same model. We previously demonstrated the use of MVMR to separate the effects of adipose- and brain-tissue instrumented BMI in a one-sample MR setting for various cardiovascular disease traits [4]. Due to the current limited availability of individual-level data with large numbers of cancer cases, we adapted the methodology to leverage GWAS summary statistics for which there are publicly available data from highly powered meta-analyses studies conducted by consortia. Simulations were conducted using the ‘simulateGP’ R package to evaluate the relative power of this approach across a range of effect sizes (0.1, 0.125 and 0.15), outcome sample sizes (10,000, 25,000, 50,000, 75,000 and 100,000) and proportion of variance explained by tissue-partitioned instruments (0.5%, 1%, 1.5%, 2%, 2.5% and 3%) derived from a simulated GWAS of n = 700,000 with a pool of 915 independent genetic instruments (based the BMI GWAS by Yengo used in our applied analysis).
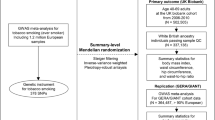
The independent effects of adipose- and brain-tissue instrumented BMI were estimated using MVMR by weighting the beta effect estimates of the SNP-exposure associations by their PPA4 values assessed by colocalization for each tissue, respectively. This weighting scheme was devised to incorporate the evidence that genetic instruments putatively influence BMI due to their expression in either adipose or brain tissues (i.e., SNPs with a very small PPA4 value were down-weighted using our approach as they are unlikely to influence BMI via gene expression in adipose or brain tissue). A schematic diagram of this approach is illustrated in Fig. 1.

The total effect of BMI (a) was estimated using univariable MR analysis. The independent effect of adipose- and brain-tissue instrumented BMI was estimated using a multivariable MR approach (b) by weighting the beta effect estimates of the SNP-exposure associations by their PPA4 values assessed by colocalization for each tissue respectively.
We next applied our novel two-sample MVMR approach to investigate whether the effect of BMI on the seven cancer types is predominantly attributed to genetic variants exerting their effects via subcutaneous adipose- or brain-tissue-related pathways. Importantly, we emphasize that the effect estimates derived using tissue-partitioned instruments should not be interpreted as causal effects in the same manner as more conventional risk factors when analysed using MR. Instead, we have developed this approach to investigate the separate contributions of genetic instruments that relate to different forms of a given trait, applied in this study using BMI and adipose/brain-tissue-derived gene expression as an exemplar. To this end, our framework provides a novel approach to dissect disease pathways between risk factors and endpoints by leveraging genetic instruments under the principles of MR.
In addition, we assessed the sensitivity of this weighted two-sample MVMR approach by analysing the same cardiovascular disease endpoints and measures of cardiac structure investigated in our previous study [4] (Supplementary Tables S4 and S5). Doing so suggested that the more widely applicable two-sample approach was capable of recapitulating findings from its application in a one-sample MVMR setting (Supplementary Note 2). Instrument strength in MVMR analyses was evaluated using conditional F-statistics as derived with the ‘MVMR’ R package with F > 10 used as an indication that weak instrument bias was not influencing our findings [29] (Supplementary Table S6). Conditional F-statistics were particularly important to evaluate within our MVMR framework to demonstrate that the two molecular forms of BMI being analysed could be instrumented as two separate exposures in our model. In order to maximise the number of reliable instruments incorporated in exposure variables, we demonstrated the effect of varying the PPA threshold for eQTL instrument identification on conditional F-statistics in an analysis on coronary artery disease (Supplementary Fig. S1). Lowering the PPA threshold in our inclusion criteria resulted in weaker instruments as indicated by their conditional F-statistics. We therefore advocate that the recommended PPA4 threshold proposed by the method developers (i.e PPA4 > 0.8) be used when identifying tissue-partitioned instruments.
Results
Univariable MR analyses of BMI effects on cancer outcomes
MR analyses were first carried out to estimate the total effect of genetically predicted BMI using all 915 independent variants and each of the 7 cancer endpoints. The MR estimates reproduced findings from previously published MR studies of the relationship between adiposity and site-specific cancers (i.e., without considering tissue-dependent effects on gene expression) [18,19,–20] (Supplementary Table S7 and Supplementary Fig. 2a). Univariable MR analyses of BMI instrumented with the adipose- and brain-tissue colocalized variants found strong evidence of an effect on the risk of outcomes such as endometrial (adipose: OR = 1.84; 95% CI = 1.35–2.51; P = 9.88 × 10−5; brain: OR = 1.61; 95% CI = 1.32–1.97; P = 3.7 × 10−6) and lung (adipose: OR = 1.07; 95% CI = 0.95–1.19; P = 0.27; brain: OR = 1.17; 95% CI:1.07–1.27; P = 0.0003) cancer. Evidence of a genetically predicted effect using tissue-partitioned BMI was also found on a lower risk of prostate cancer (adipose: OR = 0.79; 95% CI = 0.6–0.98; P = 0.03; brain: OR = 0.77; 95% CI = 0.65–0.91; P = 0.002).

Summary of Mendelian randomisation results for BMI on 7 site-specific cancers based on (a) univariable analyses using the total set of BMI variants and (b) analyses instrumented in a multivariable setting with tissue-partitioned variants. Forest plots illustrating the odds ratios per change in risk factor and 95% confidence intervals (CIs) for each outcome analysed by MR are shown. The effect estimates of BMI instrumented with all 915 BMI SNPs is illustrated in (a) (red), and the independent effect estimates of BMI instrumented by adipose- (blue) and brain (orange)-tissue-derived instruments in the multivariable MR model are illustrated in (b).
Multivariable MR analyses of tissue-partitioned BMI effects on cancer outcomes
Applying our weighted two-sample MVMR approach to separate the ‘independent’ effects of adipose- and brain-tissue instrumented BMI highlighted instances where these tissue-partitioned sets of variants may contribute differentially to site-specific cancer risk (Fig. 2). For example, the independent effect of brain-tissue instrumented BMI on risk of endometrial cancer attenuated when analysed simultaneously with adipose-tissue instrumented BMI (OR = 1.20; 95% CI = 0.81–1.78; P = 0.36), whereas the effect of adipose-tissue instrumented BMI remained strong (OR = 1.71; 95% CI = 1.07–2.74; P = 0.02). Conversely, evidence of an independent effect of adipose-tissue instrumented BMI on lung cancer risk attenuated in the MVMR model (OR = 0.98; 95% CI = 0.82–1.17; P = 0.83) and the effect of brain-tissue instrumented BMI remained strongly positive (OR = 1.17; 95% CI = 1.01–1.36; P = 0.03). A comparison of all univariable and multivariable estimates for each site-specific cancer outcome is provided in Supplementary Table S8.
Weak evidence of an independent effect was detected for both adipose- (OR = 1.03; 95% CI = 0.78–1.37; P = 0.80) and brain-tissue (OR = 0.85; 95% CI = 0.67–1.07; P = 0.17) instrumented BMI and breast cancer. Given the emerging role of childhood obesity in breast cancer risk [9], we re-applied our entire instrument derivation pipeline using results from a large-scale GWAS of childhood body size based at age 10 in the lifecourse. In total, 56 variants provided strong evidence of colocalization with proximal gene expression derived from subcutaneous adipose tissue, and 53 variants using gene expression data from brain-derived tissue (Supplementary Table S2). The mean absolute effect for each subset of these tissue-partitioned instruments on childhood body size were similar (adipose = 0.013, brain = 0.013). Although both adipose- and brain-tissue instrumented childhood body size effects provided evidence of an effect on breast cancer risk in a univariable setting (adipose: OR = 0.59; 95% CI = 0.41–0.87; P = 0.007; brain: OR = 0.58; 95% CI = 0.42–0.81; P = 0.001), only weak evidence of an independent effect was found in the multivariable MR analysis for adipose-tissue instrumented childhood body size (OR = 0.98; 95% CI = 0.55–1.73; P = 0.93). In contrast, the central estimate for genetically predicted childhood body size when instrumented using brain-tissue colocalized variants remained robust in a multivariable setting (OR = 0.57; 95% CI: 0.33–0.98; P = 0.04) (Supplementary Table S9).
Replication and negative control analyses
As a further analysis, we incorporated additional datasets to investigate the replicability of the findings for endometrial and lung cancer, and we advocate similar validation analyses for any future applications of our MVMR approach. The independent effect of adipose-tissue instrumented BMI on endometrial cancer risk is supported by an analysis using data obtained from the UK Biobank (UKB) and the Kaiser Permanente Genetic Epidemiology Research on Adult Health and Aging (GERA) cohorts [30] (OR = 3.03; 95% CI: 1.42–6.47; P = 0.004), despite the small case numbers in this dataset. We were unable to replicate the independent effects of brain-tissue instrumented BMI on lung cancer using an additional case–control GWAS study [31] (OR = 1.10; 95% CI: 0.78–1.55; P = 0.57). However, we were able to provide evidence that brain-tissue instrumented BMI is predominantly responsible for driving the relationship between BMI and ‘cigarettes smoked per day’ when analysed as an outcome (adipose: Beta = 0.03; 95% CI = –0.18–0.24; P = 0.76; brain: Beta = 0.44; 95% CI = 0.26–0.61; P = 1.62 × 10−6) (Supplementary Table S8), which is noteworthy given the strong causal effect that smoking has on lung cancer risk. Detailed results of all replication analyses are provided in Supplementary Table S10.
As a final sensitivity analysis, we repeated our entire instrument derivation pipeline using gene expression data from tissues which are unlikely to be biologically relevant for the BMI-associated genetic variants (e.g., minor salivary gland and ovary tissues [32, 33] (Supplementary Note 3)). In contrast to findings from our primary analysis, there was very weak evidence to suggest that partitioning variants from these putatively non-causal tissues for BMI leads to robust evidence of an effect on the site-specific cancer endpoints analysed previously (Supplementary Table S11). In addition, we sought to evaluate the effects where adipose- and brain-tissue instruments provided evidence of an independent effect on cancer outcomes using data from the eQTLGen consortium [34] (n = 31,684) to assess the sensitivity of the findings to gene expression data derived from whole blood that may not capture tissue-specific effects (Supplementary Note 3). The independent effect of brain-instrumented BMI on lung cancer risk remained robust when analysed simultaneously with whole blood instrumented BMI (OR = 1.14; 95% CI = 1.00:1.30; P = 0.04), while the whole blood BMI effect attenuated (OR = 1.09; 95% CI = 0.96:1.23; P = 0.18). Similarly, the independent effect of adipose-instrumented BMI on endometrial cancer replicated when analysed simultaneously with whole blood instrumented BMI (OR = 2.33; 95% CI = 1.06:5.12; P = 0.03), while weak evidence of an independent effect was obtained for whole blood instrumented BMI (OR = 1.07; 95% CI = 0.58:1.96; P = 0.83) (Supplementary Table S12).
Lastly, we attempted to partition genetic instruments using the adipose- and brain-tissue-derived datasets used in our primary analysis for a phenotype where these tissues are unlikely to be functionally important. We selected a GWAS of psoriasis for this purpose previously conducted in the UKB (n = 462,933) which further reinforced that tissue types need to be carefully selected for our approach to produce meaningful results, given that we identified only 2 variants with evidence of colocalization with adipose-tissue-derived gene expression and only 1 variant with brain-derived gene expression (Supplementary Note 3).
Discussion
The role of obesity in cancer aetiology is highly complex. In this study, we have applied the principles of MR to estimate the effects of separate tissue-partitioned subcomponents of BMI on the risk of seven site-specific cancer outcomes which have previously been shown to be influenced by adiposity [18,19,–20]. We firstly demonstrated that the results generated using our two-sample MVMR approach to detect distinct adipose- and brain-tissue BMI-mediated effects provide concordant results with a recently conducted one-sample multivariable MR analysis on cardiovascular disease and cardiac structure phenotypes [4]. Application of this novel extension of multivariable MR to cancer outcomes provides mechanistic insight into the distinct pathways underlying variation in BMI and risk of developing certain cancer types, particularly endometrial, lung and breast cancer.
Endometrial cancer is more strongly associated with obesity than any other cancer [35, 36]. Adipose-tissue accumulation is an important driver of endometrial cancer progression via three main mechanisms: excess oestrogen exposure [37, 38], insulin resistance [39], and the induction of pro-inflammatory phenotypes as a result of hypoxia following adipose-tissue expansion [40, 41]. The variation in gene expression captured by the subcutaneous adipose-tissue instrumented BMI exposure in this study may have several molecular consequences which can be postulated to differentially influence disease aetiology. For instance, adipose tissue is a major source of multipotent mesenchymal stem cells (MSCs) which have significant proliferative capacity [42, 43]. The characteristic migration of MSCs towards sites of injury [44, 45] includes the sites of several tumour types and has been shown to contribute to cell growth in tumour microenvironments [46,47,–48]. Regional differences in proliferation and differentiation may favourably impact metabolic phenotypes, explaining the attenuated effect observed between adipose-BMI on common obesity comorbidities such as T2D [4, 49, 50]. A plausible explanation for this has been attributed to variation in fat distribution, whereby adiposity-increasing alleles are associated with a greater capacity to store fat subcutaneously as opposed to viscerally are protective [50].
On the other hand, our results suggest that adipose-tissue BMI may capture a particular phenotype which is more susceptible to inducing endometrial tumorigenicity. Genetic loci incorporated into the adipose-tissue BMI exposure included several regulators of adipogenesis which may be of prognostic importance for endometrial cancer. For example, FST encodes the adipokine follistatin which has been shown to regulate adipocyte differentiation [51,52,–53] and is also a marker of polycystic ovary syndrome (PCOS) [54]. The developmental transcription factor TBX15 influences adipogenesis [55,56,–57] and has recently been identified as a key regulator of co-expression networks regulating central adiposity [58]. Furthermore, the expression of CADM1 has a role in extracellular matrix adhesion [59] and has been shown to promote endometrial cancer progression [60,61,–62]. These findings help to establish an important link between the regulation of body composition and endometrial cancer risk and warrant further functional investigation.
The epidemiological evidence for the relationship between obesity and lung cancer is complicated, with some studies reporting seemingly paradoxical findings [63,64,65,–66]. This can most likely be attributed to the strong potential for confounding caused by smoking status and the effect of smoking on body weight. Our results are consistent with a positive causal effect of genetically predicted higher BMI on lung cancer risk, which has been reported in earlier MR studies [18, 19]. The results of the multivariable MR analysis suggest that BMI when instrumented using the brain-tissue BMI exposure independently increases risk of lung cancer when the effect of adipose-tissue BMI is accounted for. This is further supported by the independent effect observed for brain-tissue BMI on cigarette smoking shown in our sensitivity analysis. A potential limitation of the analysis to detect supporting evidence of the brain-tissue instrumented BMI effect in our replication dataset may have been introduced by covariate adjustment amongst the contributing consortia for variables including alcohol dependence. Investigating the effect of our tissue-partitioned instruments on alcohol intake frequency suggests that the brain-tissue-derived variants relate more strongly to this behavioural trait (Supplementary Table S8). As such, this finding therefore requires further investigation by future studies.
Previously, we postulated that the brain-tissue BMI exposure may relate to a molecular phenotype which predominantly influences BMI through a genetic predisposition for increased adiposity, likely arising from variation in appetite and energy intake [4]. In addition, our findings contribute to our understanding of the positive relationship between BMI and smoking. BMI has been shown to bi-directionally associate with smoking [67]; whereby having a higher BMI is positively associated with smoking [67,68,–69], and smoking heaviness inversely effects BMI [70,71,–72]. The overall positive relationship between BMI and smoking behaviour has been well replicated, likely influenced both by behavioural [73,74,75,–76] and physiological factors [77], while the inverse relationship between smoking heaviness and BMI may be mediated by the effect of nicotine on energy balance [2]. The brain-tissue BMI exposure reflects the BMI phenotype leading to smoking, suggesting that smoking may be a mediator between BMI and lung cancer, or may be partly influenced by a shared aetiology for BMI and smoking [78, 79]. For example, adiposity genes incorporated into the brain-tissue BMI exposure such as BDNF and OPRL1, have each been shown to contribute to energy intake [80,81,–82], binge eating [83,84,–85], and smoking initiation [86,87,88,–89]. Furthermore, association studies have identified a positive relationship between sensitivity to sweet-tasting stimuli and impulsive behaviour [90]. Among the loci incorporated into the brain-tissue BMI exposure are several genes which are highly represented within the sweet taste signalling pathway (e.g., KCNK3, PLCD4, PRKCD) [91]. Taken together, these findings highlight compelling parallels between the impact of variation in neuroregulatory pathways on energy intake, smoking behaviour, and lifetime risk of lung cancer which will be important to delineate further.
Our univariable MR results align with studies which have established strong evidence indicating that a larger body size in childhood is protective against breast cancer risk [9, 92, 93]. The results of the multivariable MR suggest that the independent effect of childhood body size when instrumented using the brain-tissue BMI exposure may contribute to the protective effect on breast cancer. Nutritional status and higher adiposity in childhood are important drivers of earlier pubertal onset [94, 95], which is a demonstrated risk for breast cancer [96, 97]. Further exploration of the molecular characteristics of higher childhood BMI phenotypes on key developmental stages, such as age of menarche, may provide important insight on potential preventative measures. Previous MR studies [18, 93], including the results presented here, have also reported an inverse relationship between lifetime BMI and breast cancer risk. However, observational studies have suggested that higher adiposity is an important driver of breast cancer susceptibility in post-menopausal women [36, 98, 99]. As such, additional analyses stratified by pre- and post-menopause are needed to further investigate the independent effects of BMI via distinct tissue types on breast cancer risk.
This study has noteworthy limitations. In all MR analyses, a null effect was observed for the relationship between BMI and colon cancer (based on a GWAS by proxy study). While GWAS ascertained from a family history of the disease has demonstrated utility [23, 100], these resources are liable to have attenuated effect sizes and reduced statistical power relative to conventional GWAS datasets. Repeated analyses will be needed to determine these effects should the summary statistics from large-scale cohort studies on colon cancer become publicly available. Similarly, we do not report evidence for independent effects of adipose- or brain-tissue BMI on kidney cancer. Simulations suggest that our approach is adequately powered as long as tissue-partitioned instruments explained at least 1% of the variance in the exposure trait, as well as analysing outcome GWAS datasets based on at least 75,000 participants (Supplementary Fig. S2). As such, our approach should be repeated to evaluate the independent effects of tissue-partitioned instruments on kidney cancer once sufficient sample sizes become accessible. Furthermore, another important aspect to address in future studies will be the different aetiological subtypes of several of the cancer types assessed in the present study. For example, BMI is heterogeneously associated with the development of the histological subtypes of renal cell carcinoma (RCC) [101, 102], which may potentially influence attenuation of the observed associations between the tissue-stratified BMI exposures and kidney cancer. Lastly, the present study is focused on the effects of BMI mediated predominantly by neural and subcutaneous adipose gene expression, due to both sample size availability and biological relevance to BMI. Future analyses incorporating gene expression data from additional tissue types will likely yield further insight on important aetiological effects for site-specific cancers once sufficiently powered datasets are available.
In summary, we have demonstrated a novel application of multivariable MR which allowed us to investigate the genetically predicted effects of distinct molecular subcomponents of BMI on the risk of site-specific cancers. By extending this approach into a two-sample setting, we envisage that this will have wide applicability on a spectrum of disease outcomes where individual-level data obtained in highly powered cohort studies is not currently publicly available. Furthermore, our findings provide important insight into the divergent underlying pathways between body mass index and risk of site-specific cancers.
Data availability
All summary-level data analysed in this study is publicly available, with the exception of the kidney and lung cancer GWAS data used for replication analysis. These can be accessed via a request to Dr Paul Brennan and Dr Chris Amos, respectively.
References
Davey Smith G, Ebrahim S. Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease?*. Int J Epidemiol. 2003;32:1–22.
Fang Z, Song M, Lee D, Giovannucci EL. The role of Mendelian randomization studies in deciphering the effect of obesity on cancer. J Natl Cancer Inst. 2021;114:361–71.
Sulc J, Winkler TW, Heid IM, Kutalik Z. Heterogeneity in obesity: genetic basis and metabolic consequences. Curr Diab Rep. 2020;20:1.
Leyden GM, Shapland CY, Davey Smith G, Sanderson E, Greenwood MP, Murphy D, et al. Harnessing tissue-specific genetic variation to dissect putative causal pathways between body mass index and cardiometabolic phenotypes. Am J Hum Genet. 2022;109:240–52.
Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383.
Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet. 2018;27:3641–9.
Huang QQ, Ritchie SC, Brozynska M, Inouye M. Power, false discovery rate and Winner’s Curse in eQTL studies. Nucleic Acids Res. 2018;46:e133.
Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–9.
Richardson TG, Sanderson E, Elsworth B, Tilling K, Davey Smith G. Use of genetic variation to separate the effects of early and later life adiposity on disease risk: mendelian randomisation study. BMJ. 2020;369:m1203.
Brandkvist M, Bjørngaard JH, Ødegård RA, Åsvold BO, Smith GD, Brumpton B, et al. Separating the genetics of childhood and adult obesity: a validation study of genetic scores for body mass index in adolescence and adulthood in the HUNT Study. Hum Mol Genet. 2021;29:3966–73.
Richardson TG, Mykkänen J, Pahkala K, Ala-Korpela M, Bell JA, Taylor K, et al. Evaluating the direct effects of childhood adiposity on adult systemic metabolism: a multivariable Mendelian randomization analysis. Int J Epidemiol. 2021;50:1580–92.
Richardson TG, Power GM, Davey Smith G. Adiposity may confound the association between vitamin D and disease risk—a lifecourse Mendelian randomization study. Elife. 2022;11:e79798.
Michailidou K, Lindström S, Dennis J, Beesley J, Hui S, Kar S, et al. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551:92–4.
Schumacher FR, Al Olama AA, Berndt SI, Benlloch S, Ahmed M, Saunders EJ, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet. 2018;50:928–36.
Phelan CM, Kuchenbaecker KB, Tyrer JP, Kar SP, Lawrenson K, Winham SJ, et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat Genet. 2017;49:680–91.
O’Mara TA, Glubb DM, Amant F, Annibali D, Ashton K, Attia J, et al. Identification of nine new susceptibility loci for endometrial cancer. Nat Commun. 2018;9:3166.
Richardson TG, Wang Q, Sanderson E, Mahajan A, McCarthy MI, Frayling TM, et al. Effects of apolipoprotein B on lifespan and risks of major diseases including type 2 diabetes: a mendelian randomisation analysis using outcomes in first-degree relatives. Lancet Healthy Longev. 2021;2:e317–e26.
Gao C, Patel CJ, Michailidou K, Peters U, Gong J, Schildkraut J, et al. Mendelian randomization study of adiposity-related traits and risk of breast, ovarian, prostate, lung and colorectal cancer. Int J Epidemiol. 2016;45:896–908.
Carreras-Torres R, Johansson M, Haycock PC, Wade KH, Relton CL, Martin RM, et al. Obesity, metabolic factors and risk of different histological types of lung cancer: a Mendelian randomization study. PLoS ONE. 2017;12:e0177875.
Painter JN, O’Mara TA, Marquart L, Webb PM, Attia J, Medland SE, et al. Genetic risk score Mendelian randomization shows that obesity measured as body mass index, but not waist:hip ratio, is causal for endometrial cancer. Cancer Epidemiol Biomark Prev. 2016;25:1503–10.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife. 2018;7:e34408.
Elsworth B, Lyon M, Alexander T, Liu Y, Matthews P, Hallett J, et al. The MRC IEU OpenGWAS data infrastructure. bioRxiv 2020:244293. https://doi.org/10.1101/2020.08.10.244293.
Liu JZ, Erlich Y, Pickrell JK. Case-control association mapping by proxy using family history of disease. Nat Genet. 2017;49:325–31.
DeBoever C, Tanigawa Y, Aguirre M, McInnes G, Lavertu A, Rivas MA. Assessing digital phenotyping to enhance genetic studies of human diseases. Am J Hum Genet. 2020;106:611–22.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–65.
Hemani G, Bowden J, Davey Smith G. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum Mol Genet. 2018;27:R195–R208.
Pulit SL, Stoneman C, Morris AP, Wood AR, Glastonbury CA, Tyrrell J, et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet. 2019;28:166–74.
Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol. 2016;40:597–608.
Sanderson E, Davey Smith G, Windmeijer F, Bowden J. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int J Epidemiol. 2019;48:713–27.
Rashkin SR, Graff RE, Kachuri L, Thai KK, Alexeeff SE, Blatchins MA, et al. Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat Commun. 2020;11:4423.
McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet. 2017;49:1126–32.
Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826.
Consortium G. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–30.
Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, Zeng B, et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. 2021;53:1300–10.
Reeves GK, Pirie K, Beral V, Green J, Spencer E, Bull D, et al. Cancer incidence and mortality in relation to body mass index in the million women study: cohort study. BMJ. 2007;335:1134.
Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–78.
Grodin JM, Siiteri PK, MacDonald PC. Source of estrogen production in postmenopausal women. J Clin Endocrinol Metab. 1973;36:207–14.
Rodriguez AC, Blanchard Z, Maurer KA, Gertz J. Estrogen signaling in endometrial cancer: a key oncogenic pathway with several open questions. Horm Cancer. 2019;10:51–63.
Mu N, Zhu Y, Wang Y, Zhang H, Xue F. Insulin resistance: a significant risk factor of endometrial cancer. Gynecol Oncol. 2012;125:751–7.
Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56:901–11.
Sahoo SS, Lombard JM, Ius Y, O’Sullivan R, Wood LG, Nahar P, et al. Adipose-derived VEGF-mTOR signaling promotes endometrial hyperplasia and cancer: implications for obese women. Mol Cancer Res. 2018;16:309–21.
Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, et al. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell. 2002;13:4279–95.
Minteer D, Marra KG, Rubin JP. Adipose-derived mesenchymal stem cells: biology and potential applications. Adv Biochem Eng Biotechnol. 2013;129:59–71.
Chapel A, Bertho JM, Bensidhoum M, Fouillard L, Young RG, Frick J, et al. Mesenchymal stem cells home to injured tissues when co-infused with hematopoietic cells to treat a radiation-induced multi-organ failure syndrome. J Gene Med. 2003;5:1028–38.
Caplan AI. Why are MSCs therapeutic? New data: new insight. J Pathol. 2009;217:318–24.
Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–63.
Prantl L, Muehlberg F, Navone NM, Song YH, Vykoukal J, Logothetis CJ, et al. Adipose tissue-derived stem cells promote prostate tumor growth. Prostate. 2010;70:1709–15.
Kucerova L, Matuskova M, Hlubinova K, Altanerova V, Altaner C. Tumor cell behaviour modulation by mesenchymal stromal cells. Mol Cancer. 2010;9:129.
Martin S, Tyrrell J, Thomas EL, Bown MJ, Wood AR, Beaumont RN, et al. Disease consequences of higher adiposity uncoupled from its adverse metabolic effects using Mendelian randomisation. Elife. 2022;11:e72452.
Loos RJF, Kilpeläinen TO. Genes that make you fat, but keep you healthy. J Intern Med. 2018;284:450–63.
Flanagan JN, Linder K, Mejhert N, Dungner E, Wahlen K, Decaunes P, et al. Role of follistatin in promoting adipogenesis in women. J Clin Endocrinol Metab. 2009;94:3003–9.
Brown ML, Bonomi L, Ungerleider N, Zina J, Kimura F, Mukherjee A, et al. Follistatin and follistatin like-3 differentially regulate adiposity and glucose homeostasis. Obesity. 2011;19:1940–9.
Braga M, Reddy ST, Vergnes L, Pervin S, Grijalva V, Stout D, et al. Follistatin promotes adipocyte differentiation, browning, and energy metabolism. J Lipid Res. 2014;55:375–84.
Raeisi T, Rezaie H, Darand M, Taheri A, Garousi N, Razi B, et al. Circulating resistin and follistatin levels in obese and non-obese women with polycystic ovary syndrome: a systematic review and meta-analysis. PLoS ONE. 2021;16:e0246200.
Gesta S, Bezy O, Mori MA, Macotela Y, Lee KY, Kahn CR. Mesodermal developmental gene Tbx15 impairs adipocyte differentiation and mitochondrial respiration. Proc Natl Acad Sci USA. 2011;108:2771–6.
Sun W, Zhao X, Wang Z, Chu Y, Mao L, Lin S, et al. Tbx15 is required for adipocyte browning induced by adrenergic signaling pathway. Mol Metab. 2019;28:48–57.
Gburcik V, Cawthorn WP, Nedergaard J, Timmons JA, Cannon B. An essential role for Tbx15 in the differentiation of brown and “brite” but not white adipocytes. Am J Physiol Endocrinol Metab. 2012;303:E1053–60.
Pan DZ, Miao Z, Comenho C, Rajkumar S, Koka A, Lee SHT, et al. Identification of TBX15 as an adipose master trans regulator of abdominal obesity genes. Genome Med. 2021;13:123.
Moiseeva EP, Straatman KR, Leyland ML, Bradding P. CADM1 controls actin cytoskeleton assembly and regulates extracellular matrix adhesion in human mast cells. PLoS ONE. 2014;9:e85980.
De Strooper LM, van Zummeren M, Steenbergen RD, Bleeker MC, Hesselink AT, Wisman GB, et al. CADM1, MAL and miR124-2 methylation analysis in cervical scrapes to detect cervical and endometrial cancer. J Clin Pathol. 2014;67:1067–71.
Yadav VK, Lee TY, Hsu JB, Huang HD, Yang WV, Chang TH. Computational analysis for identification of the extracellular matrix molecules involved in endometrial cancer progression. PLoS ONE. 2020;15:e0231594.
Wang J, Lei C, Shi P, Teng H, Lu L, Guo H, et al. LncRNA DCST1-AS1 promotes endometrial cancer progression by modulating the MiR-665/HOXB5 and MiR-873-5p/CADM1 pathways. Front Oncol. 2021;11:714652.
Bhaskaran K, Douglas I, Forbes H, dos-Santos-Silva I, Leon DA, Smeeth L. Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5·24 million UK adults. Lancet. 2014;384:755–65.
Yang Y, Dong J, Sun K, Zhao L, Zhao F, Wang L, et al. Obesity and incidence of lung cancer: a meta-analysis. Int J Cancer. 2013;132:1162–9.
Mavridis K, Michaelidou K. The obesity paradox in lung cancer: is there a missing biological link? J Thorac Dis. 2019;11:S363–S6.
Ardesch FH, Ruiter R, Mulder M, Lahousse L, Stricker BHC, Kiefte-de Jong JC. The obesity paradox in lung cancer: associations with body size versus body shape. Front Oncol. 2020;10:591110.
Taylor AE, Richmond RC, Palviainen T, Loukola A, Wootton RE, Kaprio J, et al. The effect of body mass index on smoking behaviour and nicotine metabolism: a Mendelian randomization study. Hum Mol Genet. 2019;28:1322–30.
Clark MM, Decker PA, Offord KP, Patten CA, Vickers KS, Croghan IT, et al. Weight concerns among male smokers. Addict Behav. 2004;29:1637–41.
Farley AC, Hajek P, Lycett D, Aveyard P. Interventions for preventing weight gain after smoking cessation. Cochrane Database Syst Rev. 2012;1:CD006219.
Freathy RM, Kazeem GR, Morris RW, Johnson PC, Paternoster L, Ebrahim S, et al. Genetic variation at CHRNA5-CHRNA3-CHRNB4 interacts with smoking status to influence body mass index. Int J Epidemiol. 2011;40:1617–28.
Åsvold BO, Bjørngaard JH, Carslake D, Gabrielsen ME, Skorpen F, Smith GD, et al. Causal associations of tobacco smoking with cardiovascular risk factors: a Mendelian randomization analysis of the HUNT Study in Norway. Int J Epidemiol. 2014;43:1458–70.
Morris RW, Taylor AE, Fluharty ME, Bjørngaard JH, Åsvold BO, Elvestad Gabrielsen M, et al. Heavier smoking may lead to a relative increase in waist circumference: evidence for a causal relationship from a Mendelian randomisation meta-analysis. The CARTA consortium. BMJ Open. 2015;5:e008808.
Winter AL, de Guia NA, Ferrence R, Cohen JE. The relationship between body weight perceptions, weight control behaviours and smoking status among adolescents. Can J Public Health. 2002;93:362–5.
Tomeo CA, Field AE, Berkey CS, Colditz GA, Frazier AL. Weight concerns, weight control behaviors, and smoking initiation. Pediatrics. 1999;104:918–24. 4 Pt 1
Calzo JP, Sonneville KR, Haines J, Blood EA, Field AE, Austin SB. The development of associations among body mass index, body dissatisfaction, and weight and shape concern in adolescent boys and girls. J Adolesc Health. 2012;51:517–23.
Howe LJ, Trela-Larsen L, Taylor M, Heron J, Munafò MR, Taylor AE. Body mass index, body dissatisfaction and adolescent smoking initiation. Drug Alcohol Depend. 2017;178:143–9.
Jain RB, Bernert JT. Effect of body mass index and total blood volume on serum cotinine levels among cigarette smokers: NHANES 1999-2008. Clin Chim Acta. 2010;411:1063–8.
Criscitelli K, Avena NM. The neurobiological and behavioral overlaps of nicotine and food addiction. Prev Med. 2016;92:82–9.
Carreras-Torres R, Johansson M, Haycock PC, Relton CL, Davey Smith G, Brennan P, et al. Role of obesity in smoking behaviour: Mendelian randomisation study in UK Biobank. BMJ. 2018;361:k1767.
Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–42.
Rios M. BDNF and the central control of feeding: accidental bystander or essential player? Trends Neurosci. 2013;36:83–90.
Farhang B, Pietruszewski L, Lutfy K, Wagner EJ. The role of the NOP receptor in regulating food intake, meal pattern, and the excitability of proopiomelanocortin neurons. Neuropharmacology. 2010;59:190–200.
Akkermann K, Hiio K, Villa I, Harro J. Food restriction leads to binge eating dependent upon the effect of the brain-derived neurotrophic factor Val66Met polymorphism. Psychiatry Res. 2011;185:39–43.
Statnick MA, Chen Y, Ansonoff M, Witkin JM, Rorick-Kehn L, Suter TM, et al. A novel nociceptin receptor antagonist LY2940094 inhibits excessive feeding behavior in rodents: a possible mechanism for the treatment of binge eating disorder. J Pharm Exp Ther. 2016;356:493–502.
Hardaway JA, Jensen J, Kim M, Mazzone CM, Sugam JA, Diberto JF, et al. Nociceptin receptor antagonist SB 612111 decreases high fat diet binge eating. Behav Brain Res. 2016;307:25–34.
Lang UE, Sander T, Lohoff FW, Hellweg R, Bajbouj M, Winterer G, et al. Association of the met66 allele of brain-derived neurotrophic factor (BDNF) with smoking. Psychopharmacology. 2007;190:433–9.
Ohmoto M, Takahashi T. Effect of genetic polymorphism of brain-derived neurotrophic factor and serotonin transporter on smoking phenotypes: a pilot study of Japanese participants. Heliyon. 2019;5:e01234.
Korhonen T, Loukola A, Hällfors J, Salomaa V, Kaprio J. Is brain-derived neurotrophic factor associated with smoking initiation? Replication using a large Finnish population sample. Nicotine Tob Res. 2020;22:293–6.
Kasai S, Nishizawa D, Hasegawa J, Sato N, Tanioka F, Sugimura H, et al. Nociceptin/orphanin FQ receptor gene variation is associated with smoking status in Japanese. Pharmacogenomics. 2016;17:1441–51.
Weafer J, Burkhardt A, de Wit H. Sweet taste liking is associated with impulsive behaviors in humans. Front Behav Neurosci. 2014;8:228.
Belinky F, Nativ N, Stelzer G, Zimmerman S, Iny Stein T, Safran M, et al. PathCards: multi-source consolidation of human biological pathways. Database. 2015;2015:bav006. https://doi.org/10.1093/database/bav006.
Shawon MSR, Eriksson M, Li J. Body size in early life and risk of breast cancer. Breast Cancer Res. 2017;19:84.
Guo Y, Warren Andersen S, Shu XO, Michailidou K, Bolla MK, Wang Q, et al. Genetically predicted body mass index and breast cancer risk: mendelian randomization analyses of data from 145,000 women of European descent. PLoS Med. 2016;13:e1002105.
Kaplowitz PB. Link between body fat and the timing of puberty. Pediatrics. 2008;121:S208–17.
Freedman DS, Khan LK, Serdula MK, Dietz WH, Srinivasan SR, Berenson GS. Relation of age at menarche to race, time period, and anthropometric dimensions: the Bogalusa Heart Study. Pediatrics 2002;110:e43.
Cancer CGoHFiB. Menarche, menopause, and breast cancer risk: individual participant meta-analysis, including 118 964 women with breast cancer from 117 epidemiological studies. Lancet Oncol. 2012;13:1141–51.
Ritte R, Tikk K, Lukanova A, Tjønneland A, Olsen A, Overvad K, et al. Reproductive factors and risk of hormone receptor positive and negative breast cancer: a cohort study. BMC Cancer. 2013;13:584.
Guo W, Key TJ, Reeves GK. Adiposity and breast cancer risk in postmenopausal women: results from the UK Biobank prospective cohort. Int J Cancer. 2018;143:1037–46.
van den Brandt PA, Ziegler RG, Wang M, Hou T, Li R, Adami HO, et al. Body size and weight change over adulthood and risk of breast cancer by menopausal and hormone receptor status: a pooled analysis of 20 prospective cohort studies. Eur J Epidemiol. 2021;36:37–55.
Purcell S, Sham P, Daly MJ. Parental phenotypes in family-based association analysis. Am J Hum Genet. 2005;76:249–59.
van de Pol JAA, George L, van den Brandt PA, Baldewijns MMLL, Schouten LJ. Etiologic heterogeneity of clear-cell and papillary renal cell carcinoma in the Netherlands Cohort Study. Int J Cancer. 2021;148:67–76.
Callahan CL, Hofmann JN, Corley DA, Zhao WK, Shuch B, Chow WH, et al. Obesity and renal cell carcinoma risk by histologic subtype: a nested case-control study and meta-analysis. Cancer Epidemiol. 2018;56:31–7.
Acknowledgements
We would like to thank the authors of all the GWAS who made their summary statistics available for the benefit of this study, including the following cancer consortia: the Breast Cancer Association Consortium (BCAC), the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL), the Ovarian Cancer Association Consortium (OCAC), the Endometrial Cancer Association Consortium (OCAC), the Transdisciplinary Research of Cancer in Lung of the International Lung Cancer Consortium (TRICL-ILCCO) and the Lung Cancer Cohort Consortium (LC3). We also thank Dr Michael Love for helpful discussion regarding simulation studies of expression quantitative trait loci.
Funding
GML is supported by a grant from the British Heart Foundation (FS/17/60/33474). This work was supported by the Integrative Epidemiology Unit, which receives funding from the UK Medical Research Council and the University of Bristol (MC_UU_00011/1). The funder had no role in the study design, analysis or interpretation of data; the funder had no role in the manuscript or the decision to submit the study for publication.
Author information
Authors and Affiliations
Contributions
Conceptualisation: TGR; formal analysis: GML; investigation: GML; supervision: TGR and GDS; methodology: TGR, GDS and GML; resources: VG, TH, CIA and PB; writing—original draft: GML; all authors contributed to the review and editing of the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leyden, G.M., Greenwood, M.P., Gaborieau, V. et al. Disentangling the aetiological pathways between body mass index and site-specific cancer risk using tissue-partitioned Mendelian randomisation. Br J Cancer 128, 618–625 (2023). https://doi.org/10.1038/s41416-022-02060-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-022-02060-6
