
Abstract
Obesity is a risk factor for at least 13 different types of cancer, many of which are hormonally driven, and is associated with increased cancer incidence and morbidity. Adult obesity rates are steadily increasing and a subsequent increase in cancer burden is anticipated. Obesity-related dysfunction can contribute to cancer pathogenesis and treatment resistance through various mechanisms, including those mediated by insulin, leptin, adipokine, and aromatase signalling pathways, particularly in women. Furthermore, adiposity-related changes can influence tumour vascularity and inflammation in the tumour microenvironment, which can support tumour development and growth. Trials investigating non-pharmacological approaches to target the mechanisms driving obesity-mediated cancer pathogenesis are emerging and are necessary to better appreciate the interplay between malignancy, adiposity, diet and exercise. Diet, exercise and bariatric surgery are potential strategies to reverse the cancer-promoting effects of obesity; trials of these interventions should be conducted in a scientifically rigorous manner with dose escalation and appropriate selection of tumour phenotypes and have cancer-related clinical and mechanistic endpoints. We are only beginning to understand the mechanisms by which obesity effects cell signalling and systemic factors that contribute to oncogenesis. As the rates of obesity and cancer increase, we must promote the development of non-pharmacological lifestyle trials for the treatment and prevention of malignancy.
Similar content being viewed by others
Background
The rates of adult obesity are increasing yearly and have already reached epidemic proportions.1 Obesity—classically defined as a body mass index (BMI) of 30 or greater2—is a well-known contributor to overall mortality and, specifically, to death from cardiometabolic diseases like diabetes and coronary artery disease.3 In the past decade, the links between obesity and rising rates of cancer incidence and cancer-specific death have been increasingly recognised, and current evidence implicates obesity as a risk factor for at least 13 different types of cancer, including oesophageal, gastric, colorectal, breast and endometrial cancers.4,5 The relative risks of oesophageal, gastric and colon cancer for obese individuals are 4.8-, 1.8-, and 1.3-fold greater, respectively, and a staggering 7.1-fold greater for endometrial cancer, than those for non-obese individuals.5 A longer exposure time to obesity is also associated with an increased incidence of many of these cancers.5,6 As the number of young obese and overweight individuals continues to rise, a related acceleration in the global cancer burden is likely to follow. Indeed, this prediction has already been realised by the increased incidence of endometrial, gallbladder, pancreatic and other obesity-related cancers in younger cohorts (25–29 years old).7 In terms of mortality, it is estimated that elevated body weight and excess adiposity negatively impact clinical outcomes in ~20% of all cancer cases.8 In obese women, observational studies indicate a 2.12-fold increase in the relative risk of death from breast cancer and a 6.25-fold increase in the relative risk of death from uterine cancer.9 In men, obesity has been associated with more than quadruple and nearly double the risk of death from liver and colorectal cancers, respectively.9
The recognition of obesity as a leading modifiable risk factor for cancer development and mortality has triggered an active area of investigation and a rationale for testing anti-obesity interventions in oncology. Weight loss strategies targeting overweight or obese individuals account for most of these interventions. Despite multiple completed and ongoing clinical trials, however, it is still unclear whether weight loss reduces the risk of developing cancer and/or cancer-related death.10 Reliance on diagnostics that are useful for the assessment of population health but imprecise at the individual level might, in part, contribute to the challenge of identifying successful interventions for obesity-related cancers. Indeed, nearly all large epidemiology studies use convenient but imprecise surrogates of adiposity (e.g., BMI, waist circumference) to approximate the impact of obesity on cancer. However, such anthropometric measures frequently mischaracterise obesity-related dysfunction and related disease incidence. For example, nearly one-third of women with normal BMI (<25 kg/m2) have subclinical evidence of metabolic obesity.11 Similarly, increased adiposity is associated with a two-fold increase in the risk of invasive breast cancer among postmenopausal women with normal BMI.12 Conversely, up to 30% of obese individuals can be defined as metabolically healthy.13 The developmental paradigm of interventions for obesity-related cancers must therefore incorporate a more precise characterisation of disease phenotype in order to parallel the successes of other cancer therapies that target specific biological pathways (Box 1).
The scope of obesity-related malignancy is vast and varied. In this review, we will discuss the need to characterise obesity via biological targets that are relevant to oncological pathways to facilitate mechanistically driven and precise interventions for obesity-related cancers. We will focus on hormonally driven cancer, such as breast and endometrial cancers, and on the changes in peptide and steroid hormones, including insulin and insulin-like growth factors (IGFs), various adipokines such as leptin and adiponectin, and oestrogen, that link metabolic dysfunction with chronic low-level systemic inflammation. Finally, we will discuss the translation of biological findings into the development of interventions, with a focus on lifestyle modification strategies, that aim to attenuate the drivers of obesity-induced tumorigenesis.
Obesity and dysregulated insulin signalling
Under normal physiological conditions, increases in the levels of systemic glucose induce pancreatic cells to release the hormone insulin, which, in addition to mediating glucose metabolism, stimulates key pathways implicated in cell survival, protein synthesis and replication.14 It does this by binding to insulin receptors (IR) on the surface of cells and activating various signalling cascades, including the extracellular-signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) and the phosphatidylinositol 3-kinase (PI3K) pathways15,16,17 (Fig. 1). Two IR isoforms, IR-A and IR-B, are present among various tissues in different ratios and carry out different functions. IR-A has largely a mitogenic role in early life,18 and its expression in adulthood is linked to insulin resistance and unregulated cell proliferation.19 In comparison, IR-B is expressed in the liver and other differentiated adult tissue and is involved primarily in glucose metabolism.18 High levels of IR-A are implicated in tumorigenesis and are found in various cancers including breast, endometrial, colon and hepatocellular cancer.20,21,22 IR-A also shows an increased affinity for IGFs compared with IR-B.23 IGF-1 and IGF-2 are small peptides synthesised in the liver in response to growth hormone. While insulin circulates mostly in its free form, IGFs circulate largely bound to IGF-binding proteins (IGFBPs), which regulate their levels and biological function.24 By binding to an IGF-1 receptor (IGF-1R), IGF-1 and IGF-2 promote cell growth and proliferation,25 and several studies have demonstrated that the expression of IGF-1R is increased in breast and endometrial cancers.21,26,27 In the setting of obesity, higher levels of IGF-2 stimulate both IGF-1R and IR-A.28

IR-A Insulin Receptor A, IR-B Insulin Receptor B, IGF-1 Insulin Growth Factor-1, IGF-2 Insulin Growth Factor 2, IGF-1R Insulin Growth Factor Receptor 1, SHBG Sex Hormone Binding Globulin, ER Estrogen receptor.
Although genetic mutation of IGF-1R as the primary driver event in tumorigenesis is infrequent, dysregulation of the IGFR axis can occur secondary to other events that influence the expression of ligands and receptors in this pathway.29,30,31,32 Several factors allow transformed malignant cells to heavily depend on the dysregulation of insulin and IGF signalling pathways for proliferation and invasion.32 In a state of energy abundance, such as occurs in obesity, insulin and IGFRs are chronically activated, resulting in increased glucose uptake into cells, cell proliferation, angiogenesis and, ultimately, greater potential for malignant transformation and growth.33,34 Additionally, hyperinsulinaemia and insulin resistance occur in the setting of excess visceral adiposity,35 but are not strongly associated with subcutaneous or total body adiposity;36 this difference is thought to be mediated by a rise in circulating free fatty acids due to increased rates of lipolysis in visceral, but not subcutaneous, fat depots.37 Although the mechanisms through which excess free fatty acids released from visceral adipose tissue cause insulin resistance remain the subject of ongoing investigation, proposed mechanisms include production of lipid metabolites and secretion of pro-inflammatory cytokines that stimulate insulin release.38 Accordingly, in studies that differentiate adipose compartments, visceral adiposity is associated with an increased risk of several cancers and is a stronger predictor of risk than BMI.37 For breast cancer, specifically, elevated insulin levels in non-diabetic patients are associated with worse progression-free survival (PFS) then normal insulin levels, and patients with obesity and diabetes have significantly higher mortality rates compared with non-diabetics.9,39 Hyperactivation of IR and IGFR also promotes downstream signalling through PI3K, which is dysregulated and constitutively activated in various obesity-associated cancers, including breast, endometrial and colorectal cancers.40,41,42 Inhibition of PI3K is a strategy that is currently used in cancer treatment and is associated with on-target hyperglycaemia and hyperinsulinaemia.43 In preclinical models, the subsequent surge in insulin after PI3K inhibition can reactivate this pathway and stimulate further tumour-cell proliferation.44 Thus, insulin resistance, characterised by prolonged periods of hyperinsulinaemia and stimulation of IR-A and IGFR1, is a key means by which obesity promotes the development and growth of cancer.
Targeting the insulin signalling pathway
Strategies that target insulin and IGF signalling for cancer treatment include ligand- or receptor-specific agents, as well as interventions that globally alter glucose homoeostasis.
Receptor-specific agents
The high-affinity binding of IGFs to IR-A and IGF1R offers potentially useful pharmacological targets, and antibodies to IGF1R and IR-A, as well as various tyrosine kinase inhibitors (TKIs), have been tested in early phase clinical trials.45 However, given the ubiquitous nature of both IR and IGF1R in human tissues, the toxicities associated with targeting these receptors pose serious challenges. Furthermore, strategic targeting has proven to be difficult. On the one hand, blocking both IGF1R and IR can result in dose-limiting hyperglycaemia,46,47 but, on the other hand, exclusively inhibiting IGF1R can cause compensatory activation of IR signalling.48,49 Investigation of various targets are ongoing, but for the purposes of this review, we will highlight some that are the most advanced in clinical development.
Figitumumab, an IGF1R monoclonal antibody, was investigated in Phase 3 clinical trials in combination with carboplatin and paclitaxel for the treatment of advanced non-small-cell lung cancer (NSCLC). However, the trial was closed early due to an increased incidence of serious adverse events, including grade 3/4 hyperglycaemia and treatment-related deaths.50 Two other IGF1R monoclonal antibodies, ganitumab and dalotuzumab, were investigated for the treatment of metastatic pancreatic cancer and metastatic colon cancer, respectively, but both trials were also terminated after preplanned futility assessments.51,52 In oestrogen receptor-positive (ER+) breast cancer, ganitumab in combination with the aromatase inhibitor exemestane or the ER downregulator fulvestrant failed to improve PFS and also induced significant rates of grade 3/4 hyperglycaemia.53 Other IGF1R monoclonal antibodies, including cixutumumab, robatumumab and istiratumab, have been investigated in Phase 1 and Phase 2 clinical trials but have shown limited efficacy and poor tolerability.46,54,55,56
Small-molecule TKIs targeting IGF1R, IR-A and IR-B have also been studied in the clinical setting. Although dual targeting of IGF1R and IR circumvents compensatory IR activation, this approach leads to a higher rate of hyperinsulinaemia and hyperglycaemia.57 In a Phase 3 randomised controlled trial (RCT), no difference in overall survival was seen in patients with adrenocortical carcinoma treated with linsitinib, which targets IGF1R and IR, versus those receiving placebo.58 The combination of linsitinib and paclitaxel chemotherapy did not improve survival in ovarian cancer, and linsitinib maintenance with erlotinib, a TKI of the epidermal growth factor receptor (EGFR), did not improve overall survival in patients with NSCLC.59,60
Ligand-specific agents
As well as inhibiting IGF1R and IR, other potential strategies include targeting the IGFs. Dusigitumab, a monoclonal antibody that binds IGF-2, has been explored in a Phase 1 basket trial of advanced solid malignancies and resulted in stable disease at best response, with a favourable toxicity profile.61 However, no further development of this agent is currently being planned. Early phase studies of xentuzumab, a monoclonal antibody that binds IGF-1 and IGF-2, have demonstrated promising anti-tumour activity in patients with breast cancer. No improvement in the overall PFS was reported with the addition of xentuzumab to exemestane and the mammalian target of rapamycin (mTOR) inhibitor everolimus. However, in patients without visceral metastasis, the three-drug regimen had a longer PFS (hazard ratio (HR): 0.21 (0.05–0.98)) compared with the combination of exemestane and everolimus alone.62
Agents that alter glucose homoeostasis
Repurposing medications labelled for the treatment of diabetes is an area of active investigation in cancer therapy. In preclinical models, metformin was shown to downregulate IGF signalling and inhibit proliferation of uterine serous carcinoma cells.63 Metformin also attenuates the expression of IGF1 and the activation of mTOR and Akt (downstream effectors of insulin signalling) in breast, lung and pancreatic cancer cells.64,65,66 However, the clinical response to metformin has been mixed. When combined with other cytotoxic agents during neoadjuvant treatment of breast cancer, metformin improved pathological complete response rates, but did not improve PFS in the metastatic setting.67,68 Similarly, the data supporting metformin in the treatment of endometrial cancer have been mixed. Inhibiting the IGF1 and PI3K signalling pathways with metformin lowers cellular proliferation in endometrial tumours.69,70 In small window-of-opportunity (presurgery) trials, metformin reduced tumour proliferation (as indicated by the marker Ki-67) by 11.75% (P = 0.008) in patients in one trial and 17.2% (P = 0.002) in another trial, but these findings were not replicated in a confirmatory Phase 3 trial.71,72,73
Based on encouraging observational, preclinical and early phase data, several clinical trials testing metformin in the presurgical/neoadjuvant, adjuvant and metastatic settings in combination with standard anti-tumour therapies are ongoing.
Obesity and dysregulated adipokine signalling
Dysregulated circulating levels of adipokines—hormones and cytokines secreted by adipose tissue—is a hallmark of hyperadiposity and can promote tumour growth. The primary function of one such adipocyte-secreted hormone and biomarker of adiposity,74,75 leptin, is hypothalamic-mediated regulation of appetite, which modulates feeding behaviour and energy expenditure.76,77 Circulating levels of leptin are elevated in obese individuals and are associated with an increased risk of the development and progression of cancer, such as endometrial, breast, colon, and kidney cancers, among others.78,80,80
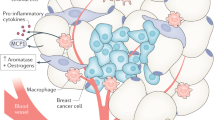
The mechanisms through which leptin promotes cancer growth are best outlined in the setting of breast cancer. Binding of leptin to one of the six isoforms of the leptin receptor induces the activation of various signalling pathways including the Janus kinase/signal transducer and activator of transcription (JAK/STAT), MAPK and PI3K pathways, which ultimately promote cell proliferation.81,82 Leptin signalling through the leptin receptor also activates mammary cancer stem cells and is necessary for mammary stem cell survival and maintenance83 (Fig. 2). Insulin and IGF1 can also increase the expression of leptin and its receptor in mammary epithelial tissues, and this increased expression is associated with worse prognosis in breast cancer.79 Furthermore, the mRNA and protein expression of leptin in breast cancer cells can be regulated by hyperinsulinaemia and hypoxia (through hypoxia-inducible factor (HIF)).84 In turn, leptin can stimulate angiogenesis and create vascular permeability to enable further malignant cell growth.85,86 Leptin is, therefore, an important mediator of interactions between the tumour and the tumour microenvironment (TME).

ObR leptin binding receptor, HIF-1 hypoxia-inducible factor, TNF-alpha tumor necrosis factor alpha, IL-6 interleukin 6, TME tumor microenvironment.
Another adipocyte-secreted hormone is adiponectin, which has anti-diabetic and anti-inflammatory properties. Plasma levels of adiponectin are decreased in obesity and metabolic syndrome, as is the expression of adiponectin receptors (AdipoR1 and AdipoR2), resulting in further reduced adiponectin sensitivity.87 Low adiponectin levels are associated with insulin resistance and an increased risk of obesity-associated malignancies, including breast and endometrial cancer.88,89,90,91 The mechanisms contributing to this relationship are not known; however, adiponectin has been shown to inhibit the growth of several cancer cell lines in vitro.92 Additionally, adiponectin activates the 5’-adenosine monophosphate-activated protein kinase (AMPK) pathway, leading to upregulation of p53 and p21, key regulators of the cell cycle and apoptosis.93 Furthermore, because adiposity increases leptin and decreases adiponectin levels, the leptin/adiponectin ratio has been suggested to be a predictor of breast cancer growth.94,95 The levels of other adipokines, such as resistin and visfatin, are elevated in obesity; these increased levels are markers of inflammation, and are associated with the development and progression of various cancers.96,97,98,99 For example, resistin is thought to promote growth of breast cancer cells through Toll-like receptor 4 (TLR4)-mediated activation of nuclear factor (NF)-κB and STAT3.96
Targeting adipokine signalling and repurposing diabetic and cardiometabolic medications
Diabetic agents that inhibit leptin signalling
Despite the well-established role of leptin in promoting tumour growth, no pharmacological interventions directly targeting leptin signalling are currently approved for the prevention or treatment of cancer. Interestingly, metformin has been shown to decrease leptin levels in patients with either breast or endometrial cancer.100,101 In patients with endometrial cancer, metformin reduces cancer cell proliferation (as measured by Ki-67 staining) and has inhibitory effects on the PI3K–mTOR signalling pathways in the presurgical window.71,72 In a trial of 200 non-diabetic patients with breast cancer, metformin did not significantly decrease breast cancer cell proliferation. However, trends were identified in an unplanned analysis of Ki-67 reduction in overweight women with insulin resistance.102 Metformin is currently being tested for adjuvant breast cancer treatment in the MA.32 trial, a Phase 3 multicentre trial that has completed accrual with results anticipated after maturation of follow-up data.103 A leptin receptor antagonist has been investigated in preclinical models in triple-negative breast cancer.104,105 Pegylated leptin peptide receptor antagonist 2 (PEG-LPrA2) was shown to inhibit leptin signalling pathways and inhibit breast cancer growth both in vitro and in vivo in breast cancer xenograft models.106 These promising preclinical findings warrant further investigation in early phase human trials.
Diabetic agents that increase adiponectin levels
Peroxisome-proliferator-activated receptor γ (PPARγ) synthetic ligands, such as rosiglitazone and pioglitazone, are diabetes drugs that regulate glucose metabolism, reduce hyperinsulinaemia and alter fatty acid metabolism.107 Additionally, PPARγ synthetic ligands have been shown to increase adiponectin levels in preclinical models and in humans.108,109,110,111,112 Based on the observations that low adiponectin levels are associated with cancer progression as discussed above, the propensity of PPARγ agonists to increase adiponectin levels may be beneficial for treating obesity-driven cancers.113,114
Statins
Statins, which are widely used for the management of lipid levels, might also have anticancer properties, and many preclinical studies have suggested a protective role for statins against cancer development and progression.115,116,117,118,119,120 Several mechanisms have been proposed to underlie this anticancer effect: impaired tumour-cell proliferation via inhibition of Ras and Rho activation;121,122 inhibition of cellular proliferation via cell cycle arrest;123 induction of apoptosis;116,124,125 dose-dependent inhibition of angiogenesis;126 and anti-inflammatory properties.118,119,120 Epidemiology data supporting an anticancer effect of statins have been mixed. Several population studies have reported a reduced risk of breast cancer in statin users compared with non-statin users,127,128,129,130,131 but meta-analyses have not confirmed this association.132,133,134,135,136 Notably, high-dose statin consumption might provide a greater anticancer effect.130 Additionally, whether the statin is hydrophobic or lipophilic could differentially affect cancer risk, although data on this point are conflicting.128,131 The use of statins might also be associated with reduced cancer mortality after diagnosis,137 although observational studies do not support this link in breast cancer.138,139,140 Taken together, the epidemiology reports to date provide a signal that statin use might be protective against breast cancer for some, but not all, patients. Identifying this high-risk or statin-responsive population will be critical to developing successful intervention and prevention strategies that use statins.
Oestrogen signalling
It has long been established that oestrogen signalling is a key driver of various cellular processes including cell proliferation and survival, and that removal of the source of oestrogens—predominantly the ovaries in premenopausal women—provides clinical benefit and tumour regression for oestrogen-sensitive cancers.141 Increased levels of oestrogen function to increase cell proliferation and angiogenesis through various mechanisms,142 including binding to the ER and stimulating the IGF1 signalling pathway in breast cancer;143 in endometrial cancer, oestrogen binding to the G-protein-coupled oestrogen receptor (GPER) can result in hyperplasia in endometrial tissue.144 Furthermore, through activation of GPER, oestrogens play a role in hypoxia-induced angiogenesis in breast cancer145 (Fig. 3).

AI aromatase inhibitor, GnRH gonadotropin releasing hormone, SERM selective estrogen receptor modulator (i.e; tamoxifen), SERDselectve receptor degrader (i.e.; fulvestrant), CDK4/6 cyclin dependent kinases 4 and 6 inhibitors, GPER G-coupled estrogen receptor.
After menopause, the main source of systemic oestrogen comes from the peripheral conversion of androgens by the oestrogen biosynthetic enzyme, aromatase, and one of the most well-characterised obesity-related mechanisms for cancer pathogenesis involves the increased activity of aromatase in adipose tissue, consistent with the dysregulation of oestrogens being implicated in the development of obesity-associated ER+ breast and endometrial cancers.11,146,147 Obesity and metabolic syndrome have been linked to increased inflammation and increased expression in breast tissue and adipose stromal cells of the aromatase-encoding gene CYP19A1.148,149 In vitro studies using isolated primary human breast preadipocytes or adipose stromal cells, the main cell type responsible for oestrogen biosynthesis in the breast, have contributed to defining the mechanism by which inflammatory mediators drive aromatase expression in the context of obesity. For example, prostaglandin E2 (PGE2), a crucial inflammatory mediator, has been shown to strongly stimulate the expression of CYP19A1 via activation of PII, the promoter contributing to the majority of aromatase transcripts in breast tissue in both obesity and breast cancer.150,151,152 This increased expression is dependent on the binding and activity of a number of transcription factors and co-regulators,153,154,155,156,157,158,159,160,161 and is regulated by several pathways,152,159,160,162,163,164 some of which, notably, involve regulation by leptin.148,159 Conversely, p53 has been shown to act as a transcriptional repressor of the CYP19A1 gene, but is inhibited by both PGE2 and leptin.148,165 The effects of p53 in tumour suppression therefore go beyond its established role in promoting cell cycle arrest and apoptosis.166 Other inflammatory mediators, such as tumour necrosis factor (TNF) and interleukin (IL)-6, have also been shown to stimulate the expression of the CYP19A1 promoter I.4.167,168,169,170
Targeting oestrogen signalling
Aromatase inhibitors
Metformin has been identified as a potential breast-specific aromatase inhibitor.171,172 Interestingly, the adipokine adiponectin and the hunger hormone ghrelin have also been shown to suppress aromatase expression in a promoter-specific manner, which may help to explain the association between low levels of adiponectin and breast cancer growth in the setting of obesity.159,173,174 It remains to be determined whether these results can be leveraged to improve treatment of obesity-related breast cancer.
Specific steroidal and non-steroidal aromatase inhibitors have demonstrated efficacy for the prevention and treatment of ER+ breast cancers, with an approximate 50% reduction in the risk of ER+ breast cancer development or recurrence.175 Aromatase inhibitors might also have clinical utility in ER+ endometrial cancers, although the efficacy of aromatase inhibitors for the treatment of endometrial cancer is modest.176 Anastrozole, a non-steroidal aromatase inhibitor, has been shown to reduce proliferation in endometrial cancer cells when used in the neoadjuvant setting, and has modest activity for the treatment of recurrent ER+ endometrial cancer.177,178 Letrozole, another non-steroidal aromatase inhibitor, in combination with everolimus, an mTOR inhibitor, is associated with an overall response rate of 32% in an unselected endometrial cancer population.179 Exemestane, an irreversible steroidal aromatase inhibitor, is currently being tested for the treatment of endometrial hyperplasia and low-grade endometrial cancer (NCT03300557). Other ongoing trials are assessing various combinations of aromatase inhibitors with inhibitors of the PI3K–mTOR pathway (NCT02730923, NCT03008408). As well as the use of aromatase inhibitors, targeting the ER is a promising strategy for the treatment of ER+ endometrial cancer. An ongoing clinical trial is assessing fulvestrant, a selective ER downregulator, in combination with abemaciclib, a cyclin-dependent kinase (CDK)4/6 inhibitor, for the treatment of ER+ endometrial cancer (NCT03643510). Whether obesity affects the efficacy of various hormone therapies for endometrial cancer is currently unknown and warrants further investigation. In the setting of breast cancer, however, obesity is associated with reduced efficacy of aromatase inhibitors.180 This observation may be explained in part by increased expression of aromatase in the breast due to obesity-related adipose tissue inflammation, which will be discussed below.
Obesity and the microenvironment
The tumour microenvironment has an established role in tumour formation and metastatic invasion. It consists of various cells including lymphocytes, antigen presenting cells, cancer fibroblasts and the extracellular matrix. Increased adiposity can create chronic inflammation and hypoxic conditions that disrupt the intricate web of connections, and subsequent perturbations contribute to carcinogenesis.
Changes in vascularity
In the context of a tumour, it is well established that the increasing mass resulting from rapidly dividing cells generates hypoxic areas; HIF-1α mediates the adaptive response to the low availability of oxygen, with higher levels of HIF-1α promoting angiogenesis, thereby supporting further tumour growth and metastasis181,182 (Fig. 1). Indeed, higher levels of HIF-1α have been associated with recurrence, metastasis and reduced survival in several tumour types.183,184 The mechanisms underlying these observations and potential opportunities to intervene have been reviewed elsewhere by Pouysségur and colleagues.185 Hypoxia also induces the expression of vascular endothelial growth factor (VEGF), which further promotes angiogenesis and tumour growth.185 Obesity is also associated with an increase in tissue hypoxia due to expansion of adipose tissue beyond its vascular supply,186 which also promotes neovascularisation.187 Furthermore, hypoxia-induced VEGF expression can promote adipose tissue expansion, as well as inflammation, and this can generate a microenvironment that is supportive of tumour growth (discussed below).186,188 In endometrial cancer, VEGF is upregulated in the visceral adipose tissue of obese women and drives endometrial hyperplasia and endometrial cell growth through the PI3K–Akt–mTOR pathway.189 Anti-VEGF therapies used to target breast cancer have failed to improve overall survival, and preclinical evidence suggests that this might be related to obesity-induced resistance to anti-VEGF therapy by the production of inflammatory factors such as IL-6, which, as alluded to above, can promote a favourable TME.190 Small retrospective studies in ovarian and colorectal cancer have suggested that increased adiposity is associated with decreased efficacy of bevacizumab.191,192
Targeting angiogenesis
Inhibitors of HIF are currently under investigation for the treatment of various types of cancer in early phase clinical trials. For example, Phase 1 trials of EZN-2968 (a HIF-1 inhibitor) and PT2977 (a HIF-2 inhibitor) demonstrated some clinical activity, suggested by prolonged stable disease (>24 weeks) in one patient with a duodenal neuroendocrine tumour and five responses (one partial response, four stable disease) in six patients with clear cell renal cell carcinoma.193,194 Inhibitors of VEGF signalling have progressed further than HIF inhibitors in clinical development. The anti-VEGF monoclonal antibody bevacizumab is currently used for the treatment of lung, colon, cervical and ovarian cancers,195,196,197,198 while ramucirumab, an anti-VEGF receptor antibody, is also approved for the treatment of gastric, colorectal and hepatocellular cancers.199,200,201 Finally, small-molecule TKIs of VEGF signalling, such as sorafenib, sunitinib, pazopanib, lenvatinib and others, have demonstrated efficacy for the treatment of kidney, thyroid and hepatocellular cancers.202,203,204 In endometrial cancer, lenvatinib in combination with the checkpoint inhibitor pembrolizumab has FDA breakthrough designation and is undergoing confirmatory Phase 3 investigation.205
Chronic inflammation
Obesity is associated with a chronic state of subclinical inflammation that is characterised by white adipose tissue inflammation. Such inflammation can be histologically detected by the presence of crown-like structures (CLS),206 in which dead or dying adipocytes are surrounded by activated macrophages. These macrophages are associated with the production of several pro-inflammatory mediators, the expression of aromatase, and the presence of a fibrotic extracellular matrix.207,208 In humans, adipose inflammation in the breast is present in many overweight/obese individuals and is associated with postmenopausal status.11 In preclinical models of postmenopausal obesity, inflammation of mammary adipose tissue is associated with increased levels of TNF-α, IL-1β, IL-6 and cyclo-oxygenase (COX)-2 and an increased risk of developing breast cancer and reduced distant disease-free survival after breast cancer diagnosis.209,210 These chronic inflammatory changes associated with dysfunctional adipose tissue contribute to a microenvironment that is rich in tumour growth factors.211,212 We have previously reviewed the mechanisms through which this pro-inflammatory microenvironment promotes tumour growth.213 Interventions that reduce adipose inflammation, such as diet and exercise, might therefore reduce breast cancer risk and/or mortality, and clinical trials investigating the effects of diet and exercise on cancer-related outcomes are currently underway.
Non-pharmacological/lifestyle interventions
We have so far outlined the various mechanisms through which increased adiposity drives changes in insulin signalling, adipokine signalling, oestrogen signalling and in the TME, including angiogenesis and chronic inflammation. We have also briefly addressed the current landscape of pharmacological interventions in the context of cancer treatment for targets that are dysregulated by obesity. However, as well as such targeted approaches, the pleiotropic effects of lifestyle interventions offer a promising strategy to reverse the cancer-promoting effects of obesity. Furthermore, combining lifestyle interventions with pharmacological therapies could further augment the efficacy of anticancer therapies.
Dietary interventions
Although the biological mechanisms through which the modulation of specific macro- and micro-nutrients impact tumour biology are beyond the scope of this discussion and have been reviewed elsewhere,214,215 we outline here the key findings from RCTs that have tested strategies to shift overall dietary patterns in cancer populations. In the case of breast cancer, several trials have established that dietary modification as well as exercise are achievable and safe after diagnosis.216,217 Subsequent trials have examined the effects of diet and exercise interventions on weight loss, breast cancer outcomes, and circulating blood factors (Table 1). Two large RCTs that tested dietary interventions to improve breast cancer outcomes have been completed, but the results are conflicting. The Women’s Intervention Nutrition Study (WINS) demonstrated a 24% reduction in the recurrence of breast cancer in patients randomly assigned to a low-fat diet group versus control patients.218 Conversely, however, the Women’s Healthy Eating and Living (WHEL) trial did not show any improvement in the risk of recurrence for women randomised to a low-fat, high-fibre diet;219 diets high in fibre are known to increase microbial biodiversity (see below) and decrease insulin resistance.220,221 The long-term results of another RCT, the Women’s Health Initiative (WHI), were reported in 2019 and demonstrated a 21% reduction in mortality after breast cancer diagnosis in patients randomised to a low-fat diet intervention compared with a usual diet.222 Although large-scale clinical trial data are still lacking in this area, several RCTs testing the efficacy of diet and/or exercise interventions are ongoing (Table 2).
For patients diagnosed with endometrial or breast cancer, preclinical evidence suggests that a ketogenic diet (KD; a diet of high fats, moderate proteins, and very low carbohydrates) might improve the efficacy of PI3K inhibitors by inhibiting insulin signalling.44,223 It has recently been shown in murine KPC tumour models that treatment with PI3K inhibitors causes a transient hyperglycaemia and hyperinsulinaemia. This resultant hyperinsulinaemia can partially reactivate PI3K signalling, and following PI3K inhibition, can reactivate PI3K signalling in both normal and tumour tissues.44 A ketogenic diet, which is deficient in carbohydrate, prevents hyperinsulinaemia and can thereby reduce the paradoxical reactivation of PI3K by PI3K inhibitor-associated hyperglycaemia.44
RCTs of a KD have demonstrated reductions in visceral adiposity and serum insulin levels without adversely affecting blood lipid levels despite elevated dietary fat intake.224,225 Notably, in xenograft models of pancreatic cancer, a KD also increased sensitivity to radiation—putatively by reducing oxidative stress; however, the diet was poorly tolerated in a pilot study of nine people.226
In obese patients without malignancy, a very-low-calorie KD reduces visceral adiposity and obesity-related metabolic dysfunction, restores leptin and resistin levels to normal, and reduces the expression of inflammatory markers.227,228,229 This approach might therefore be particularly valuable for cancer populations where weight loss is a critical priority. The definition of a KD varies among clinical trials—all KDs include low carbohydrates, but varying cut-offs for daily calories and lipid targets exist.230 Thus, the tolerability and durability of a KD intervention requires further testing, which would be aided by standardisation of KD parameters. Finally, it is important to note that, although a KD might be beneficial for certain established tumour phenotypes (e.g., PIK3CA-mutated tumours), this approach might not be effective—and could potentially be detrimenta—in certain other tumour types and in the preventive setting. For example, high dietary fat intake has been associated with an increased risk of developing breast cancer.231,232 Accordingly, the selection of an appropriate KD protocol (e.g., low-calorie, carbohydrate-restricted, and/or limited-fat) will be important for the development of this approach for use in cancer populations. However, it is important to note that a KD might not be beneficial in all circumstances or cancer histologies, as other groups have noted that changing to a high protein intake can increase insulin signalling through IGF-1.220,221 There remains ambiguity regarding which diets can effectively reverse tumorigenesis mechanisms, and future studies should aim to identify the appropriate populations and tumour phenotypes for rational dietary intervention.
The gut microbiome
Investigations carried out over the past decade have demonstrated that particular gut microbiome signatures are associated with the development of cancer,233,234 and that alterations in the gut microbiota can promote chronic inflammation and immunological changes that facilitate carcinogenesis.235,236 As obesity and diet alter the health and diversity of the gut microbiome, research on the role of the gut microbiome in contributing to obesity-associated cancers is active and ongoing. Although there is less data regarding hormonally driven cancers such as breast and endometrial cancers, an individual’s metabolic profile and oestrogen status can affect their microbiome. In breast cancer, there is a growing interest in the ‘oestrobolome’, which includes genes that encode bacterial enzymes such as β-glucuronidases, which are involved in the processing of endogenous oestrogens,237 and understanding how changes in oestrogen-dependent pathways influence the gut microbiome.238 Obesity can disrupt this oestrobolome, resulting in increased levels of oestrogen and its metabolites, which could affect the development and treatment of breast and endometrial cancers.239,240
Exercise interventions
A substantial body of observational data suggests that post-diagnosis exercise could prevent cancer progression and improve cancer-related mortality. In a seminal study by Holmes et al.,241 9–14.9 MET (metabolic equivalent of task) hours·per week (equivalent to ~150–250 min of moderate-intensity exercise per week) was associated with an adjusted 50% reduction in breast cancer death compared with <3 MET-hours per.week among 2987 patients with primary breast cancer. In another systematic review, post-diagnosis exercise was associated with, on average, a 37% reduction (95% confidence interval (CI) 0.54–0.73) in the risk of cancer-specific mortality in the most- versus least-active patients.242 Collectively, observational data support the hypothesis that exercise confers anti-tumour effects for several cancer types.
Although data from investigations into the effect of post-diagnosis exercise on cancer progression from prospective RCTs are not yet available, such trials are underway and outlined in Table 2. The Colon Health and Life-Long Exercise Change (CHALLENGE) trial is an international, multicentre, Phase 3 trial investigating the impact of exercise on recurrence and cancer-specific mortality in patients with resected high-risk stage II or stage III colorectal cancer.243 Another international, multicentre Phase 3 trial, the INTense Exercise foR survival (INTERVAL) trial, is investigating the effects of high-intensity aerobic and resistance training on disease outcomes in 866 patients with metastatic castrate-resistant prostate cancer (NCT02730338). Data from these, and other, Phase 3 trials of exercise in cancer populations are eagerly awaited, but it is important to note that the ‘dose’ of exercise that confers optimal anticancer efficacy or predictors of favourable response to exercise has not yet been identified. Early phase dose-finding trials of exercise are needed, and a Phase 1a/1b trial of exercise in ER+ metastatic breast cancer is currently ongoing (NCT03988595).
Combination diet and exercise interventions
Several RCTs have demonstrated that combining diet and exercise interventions provides an effective approach for inducing weight loss in patients who have survived obesity-related breast cancer244,245,246 (Table 1). Several of these weight loss interventions have also demonstrated improvements in circulating metabolic and inflammatory factors.246,247,248,249,250 For example, in the Lifestyle, Exercise, and Nutrition (LEAN) study, breast cancer survivors with a BMI ≥25 randomly assigned to diet and physical activity counselling experienced reductions in the level of circulating C-reactive protein (CRP) and body fat percentage compared with usual care.246 Participants who achieved a 5% or greater weight loss by caloric restriction and increased physical activity were also found to have reductions in their levels of circulating insulin, leptin and IL-6.246 Several other studies have established that weight loss is an effective method for reducing circulating levels of CRP, insulin, glucose and lipids.251,252,253,254,255
Diet and exercise interventions can also influence the levels of circulating hormones in individuals with or without malignancy. In the Nutrition and Exercise for Women (NEW) trial, circulating levels of estrone and oestradiol in overweight and obese postmenopausal women were reduced with energy-restricted diet, exercise, or combined diet plus exercise, versus control.256 The interventions also increased the circulating levels of sex-hormone-binding globulin (SHBG) and decreased free oestradiol and testosterone levels, which could inhibit the recurrence or growth of hormone-sensitive tumours. The magnitude of effect on SHBG and oestrogens was greatest in the diet plus exercise arm. Encouraging findings from these trials collectively support the further development of diet and exercise interventions in the prevention and treatment of cancer.257
Bariatric surgery
Given the various mechanisms by which obesity contributes to carcinogenesis, weight loss mediated by bariatric surgery has been investigated as a strategy for adjunct cancer treatment and prevention. The Swedish Obesity Study demonstrated that, especially for women, bariatric surgery reduced the incidence of cancer with a HR of 0.67 (95% CI 0.53–0.85);258 this risk reduction was confirmed in a large multicentre retrospective study in the USA in obesity-related cancers including breast cancer (HR 0.58; 95% CI 0.44–0.77) and in endometrial cancer (HR 0.50; 95% CI 0.37–0.67).259 A prospective trial is investigating the efficacy of bariatric surgery in reducing recurrence in breast cancer patients (NCT03946423).260 Although additional randomised prospective data are needed, it seems that weight loss modulates many of the effects of obesity on carcinogenesis.
Future directions
As increasing data elucidate the mechanisms by which obesity can alter cancer cell signalling, the prospective TME and systemic factors, additional targets that can be therapeutically exploited to improve obesity-related cancer risk and outcomes are likely to be identified. Figure 4 provides a summary of the pathways and mechanisms through which obesity promotes tumour growth, which establishes the paradigm for interventions. A number of pharmacologic agents could be repurposed for the prevention and treatment of obesity-related cancers, and obesity might be associated with a differential response to existing and novel anticancer therapies. Lifestyle interventions, including dietary modification and exercise, also demonstrate potential anticancer efficacy; however, the identification of appropriate ‘dose’, populations and tumour phenotypes is needed to leverage the promise of this approach. Significant progress has been made in elucidating the mechanisms through which obesity promotes cancer risk and mortality. Interestingly, a number of pathways that are dysregulated in obesity are also key drivers of oestrogen production, cancer growth and angiogenesis. Targeting these pathways would therefore potentially lead to a multifaceted approach to tumour suppression through both direct and indirect mechanisms. Translating these findings into effective clinical strategies is urgently needed to halt the accelerating global burden of obesity-related cancer.

HIF1α hypoxia- inducible factor 1- alpha, VEGF vascular endothelial growth factor, TNFα tumor necrosis factor alpha, IL-1β interleukin 1 beta, IL- 6 interleukin 6, COX-2 cyclooxygenase isoenzyme 2, IGFR- insuline-like growth factor receptor, JAK-STAT Janus kinases-signal transducer and activtor of transcription proteins, MAPK mitogen-activated protein kinase.
References
Hales, C. M., Carroll, M. D., Fryar, C. D. & Ogden, C. L. Prevalence of obesity among adults and youth: United States, 2015-2016. NCHS Data Brief 1–8 (2017).
world health association. Global Database on Body Mass Index. https://apps.who.int/bmi/index (2020).
Poirier, P., Giles, T. D., Bray, G. A., Hong, Y., Stern, J. S., Pi-Sunyer, F. X. et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 113, 898–918 (2006).
Vainio, H., Kaaks, R. & Bianchini, F. Weight control and physical activity in cancer prevention: international evaluation of the evidence. Eur. J. Cancer Prev. 11, S94–S100 (2002).
Lauby-Secretan, B., Scoccianti, C., Loomis, D., Grosse, Y., Bianchini, F. & Straif, K. Body fatness and cancer— viewpoint of the IARC working group. New Engl. J. Med. 375, 794–798 (2016).
Arnold, M., Jiang, L., Stefanick, M. L., Johnson, K. C., Lane, D. S., LeBlanc, E. S. et al. Duration of adulthood overweight, obesity, and cancer risk in the women’s health initiative: a longitudinal study from the United States. PLoS Med. 13, e1002081 (2016).
Sung, H., Siegel, R. L., Rosenberg, P. S. & Jemal, A. Emerging cancer trends among young adults in the USA: analysis of a population-based cancer registry. Lancet Public Health 4, e137–e147 (2019).
Wolin, K. Y., Carson, K. & Colditz, G. A. Obesity and cancer. The oncologist 15, 556–565 (2010).
Calle, E. E., Rodriguez, C., Walker-Thurmond, K. & Thun, M. J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. New Engl. J. Med. 348, 1625–1638 (2003).
Chlebowski, R. T., Aragaki, A. K., Anderson, G. L., Thomson, C. A., Manson, J. E., Simon, M. S. et al. Low-fat dietary pattern and breast cancer mortality in the women’s health initiative randomized controlled trial. New Engl. J. Med. 35, 2919–2926 (2017).
Iyengar, N. M., Brown, K. A., Zhou, X. K., Gucalp, A., Subbaramaiah, K., Giri, D. D. et al. Metabolic obesity, adipose inflammation and elevated breast aromatase in women with normal body mass index. Cancer Prev. Res. 10, 235–243 (2017).
Iyengar, N. M., Arthur, R., Manson, J. E., Chlebowski, R. T., Kroenke, C. H., Peterson, L. et al. Association of body fat and risk of breast cancer in postmenopausal women with normal body mass index: a secondary analysis of a randomized clinical trial and observational study. JAMA Oncol. 5, 155–163 (2019).
Karelis, A. D. Metabolically healthy but obese individuals. Lancet 372, 1281–1283 (2008).
Poloz, Y. & Stambolic, V. Obesity and cancer, a case for insulin signaling. Cell Death Dis. 6, e2037 (2015).
Sadagurski, M. & White, M. F. Integrating metabolism and longevity through insulin and IGF1 signaling. Endocrinol. Metab. Clinics North Am. 42, 127–148 (2013).
Ruderman, N. B., Kapeller, R., White, M. F. & Cantley, L. C. Activation of phosphatidylinositol 3-kinase by insulin. Proc. Natl Acad. Sci. USA 87, 1411–1415 (1990).
Avruch, J. Insulin signal transduction through protein kinase cascades. Mol. Cell. Biochem. 182, 31–48 (1998).
Belfiore, A., Malaguarnera, R., Vella, V., Lawrence, M. C., Sciacca, L., Frasca, F. et al. Insulin receptor isoforms in physiology and disease: an updated view. Endocr. Rev. 38, 379–431 (2017).
Belfiore, A., Frasca, F., Pandini, G., Sciacca, L. & Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 30, 586–623 (2009).
Sciacca, L., Costantino, A., Pandini, G., Mineo, R., Frasca, F., Scalia, P. et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene 18, 2471–2479 (1999).
Flannery, C. A., Saleh, F. L., Choe, G. H., Selen, D. J., Kodaman, P. H., Kliman, H. J. et al. Differential expression of IR-A, IR-B and IGF-1R in endometrial physiology and distinct signature in adenocarcinoma. J. Clin. Endocrinol. Metab. 101, 2883–2891 (2016).
Benabou, E., Salame, Z., Wendum, D., Lequoy, M., Tahraoui, S., Merabtene, F. et al. Insulin receptor isoform A favors tumor progression in human hepatocellular carcinoma by increasing stem/progenitor cell features. Cancer Lett. 450, 155–168 (2019).
Frasca, F., Pandini, G., Scalia, P., Sciacca, L., Mineo, R., Costantino, A. et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 19, 3278–3288 (1999).
Allard, J. B. & Duan, C. IGF-binding proteins: why do they exist and why are there so many? Front. Endocrinol. 9, 117 (2018).
Lawrence, M. C., McKern, N. M. & Ward, C. W. Insulin receptor structure and its implications for the IGF-1 receptor. Curr. Opin. Struct. Biol. 17, 699–705 (2007).
Farabaugh, S. M., Boone, D. N. & Lee, A. V. Role of IGF1R in breast cancer subtypes, stemness, and lineage differentiation. Front. Endocrinol. 6, 59 (2015).
Hirano, S., Ito, N., Takahashi, S. & Tamaya, T. Clinical implications of insulin-like growth factors through the presence of their binding proteins and receptors expressed in gynecological cancers. Eur. J. Gynaecol. Oncol. 25, 187–191 (2004).
O’Dell, S. D. & Day, I. N. Insulin-like growth factor II (IGF-II). Int. J. Biochem. Cell Biol. 30, 767–771 (1998).
Berns, E. M., Klijn, J. G., van Staveren, I. L., Portengen, H. & Foekens, J. A. Sporadic amplification of the insulin-like growth factor 1 receptor gene in human breast tumors. Cancer Res. 52, 1036–1039 (1992).
Tran, T. N., Selinger, C. I., Yu, B., Ng, C. C., Kohonen-Corish, M. R., McCaughan, B. et al. Alterations of insulin-like growth factor-1 receptor gene copy number and protein expression are common in non-small cell lung cancer. J. Clin. Pathol. 67, 985–991 (2014).
Badzio, A., Wynes, M. W., Dziadziuszko, R., Merrick, D. T., Pardo, M., Rzyman, W. et al. Increased insulin-like growth factor 1 receptor protein expression and gene copy number in small cell lung cancer. J. Thoracic Oncol. 5, 1905–1911 (2010).
Werner, H. Tumor suppressors govern insulin-like growth factor signaling pathways: implications in metabolism and cancer. Oncogene 31, 2703 (2011).
Wang, C. F., Zhang, G., Zhao, L. J., Qi, W. J., Li, X. P., Wang, J. L. et al. Overexpression of the insulin receptor isoform A promotes endometrial carcinoma cell growth. PLoS ONE 8, e69001 (2013).
Lu, C. C., Chu, P. Y., Hsia, S. M., Wu, C. H., Tung, Y. T. & Yen, G. C. Insulin induction instigates cell proliferation and metastasis in human colorectal cancer cells. Int. J. Oncol. 50, 736–744 (2017).
Calle, E. E. & Kaaks, R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 4, 579–591 (2004).
Kissebah, A. H., Vydelingum, N., Murray, R., Evans, D. J., Hartz, A. J., Kalkhoff, R. K. et al. Relation of body fat distribution to metabolic complications of obesity. J. Clin. Endocrinol. Metab. 54, 254–260 (1982).
Donohoe, C. L., Doyle, S. L. & Reynolds, J. V. Visceral adiposity, insulin resistance and cancer risk. Diabetol. Metab. Syndr. 3, 12 (2011).
Boden, G. Obesity, insulin resistance and free fatty acids. Curr. Opin. Endocrinol. Diabetes Obes. 18, 139–143 (2011).
Ferroni, P., Riondino, S., Laudisi, A., Portarena, I., Formica, V., Alessandroni, J. et al. Pretreatment insulin levels as a prognostic factor for breast cancer progression. Oncologist 21, 1041–1049 (2016).
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Levine, D. A., The Cancer Genome Atlas Research N, Getz, G., Gabriel, S. B., Cibulskis, K., Lander, E. et al. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67 (2013).
Samuels, Y., Wang, Z., Bardelli, A., Silliman, N., Ptak, J., Szabo, S. et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554 (2004).
Rubinstein, M. M., Hyman, D. M., Caird, I., Won, H., Soldan, K., Seier, K. et al. Phase 2 study of LY3023414 in patients with advanced endometrial cancer harboring activating mutations in the PI3K pathway. Cancer 126, 1274–1282 (2019).
Hopkins, B. D., Pauli, C., Du, X., Wang, D. G., Li, X., Wu, D. et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503 (2018).
Simpson, A., Petnga, W., Macaulay, V. M., Weyer-Czernilofsky, U. & Bogenrieder, T. Insulin-like growth factor (IGF) pathway targeting in cancer: role of the IGF axis and opportunities for future combination studies. Target. Oncol. 12, 571–597 (2017).
Abou-Alfa, G. K., Capanu, M., O’Reilly, E. M., Ma, J., Chou, J. F., Gansukh, B. et al. A phase II study of cixutumumab (IMC-A12, NSC742460) in advanced hepatocellular carcinoma. J. Hepatol. 60, 319–324 (2014).
Lerario, A. M., Worden, F. P., Ramm, C. A., Hesseltine, E. A., Stadler, W. M., Else, T. et al. The combination of insulin-like growth factor receptor 1 (IGF1R) antibody cixutumumab and mitotane as a first-line therapy for patients with recurrent/metastatic adrenocortical carcinoma: a multi-institutional NCI-sponsored trial. Hormones Cancer 5, 232–239 (2014).
Buck, E., Gokhale, P. C., Koujak, S., Brown, E., Eyzaguirre, A., Tao, N. et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol. Cancer Ther. 9, 2652–2664 (2010).
Forest, A., Amatulli, M., Ludwig, D. L., Damoci, C. B., Wang, Y., Burns, C. A. et al. Intrinsic resistance to cixutumumab is conferred by distinct isoforms of the insulin receptor. Mol. Cancer Res. 13, 1615–1626 (2015).
Langer, C. J., Novello, S., Park, K., Krzakowski, M., Karp, D. D., Mok, T. et al. Randomized, phase III trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non–small-cell lung cancer. J. Clin. Oncol. 32, 2059–2066 (2014).
Fuchs, C. S., Azevedo, S., Okusaka, T., Van Laethem, J. L., Lipton, L. R., Riess, H. et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: the GAMMA trial. Ann. Oncol. 26, 921–927 (2015).
Sclafani, F., Kim, T. Y., Cunningham, D., Kim, T. W., Tabernero, J., Schmoll, H. J. et al. A randomized phase II/III study of dalotuzumab in combination with cetuximab and irinotecan in chemorefractory, KRAS wild-type, metastatic colorectal cancer. J. Natl Cancer Inst. 107, https://doi.org/10.1093/jnci/djv258 (2015).
Robertson, J. F., Ferrero, J. M., Bourgeois, H., Kennecke, H., de Boer, R. H., Jacot, W. et al. Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, phase 2 trial. Lancet Oncol. 14, 228–235 (2013).
Lin, E. H., Lenz, H.-J., Saleh, M. N., Mackenzie, M. J., Knost, J. A., Pathiraja, K. et al. A randomized, phase II study of the anti-insulin-like growth factor receptor type 1 (IGF-1R) monoclonal antibody robatumumab (SCH 717454) in patients with advanced colorectal cancer. Cancer Med. 3, 988–997 (2014).
Ko, A. H., Cubillo, A., Kundranda, M., Zafar, S. F., Meiri, E., Bendell, J. et al. LBA29CARRIE: a randomized, double-blind, placebo-controlled phase II study of istiratumab (MM-141) plus nab-paclitaxel and gemcitabine versus nab-paclitaxel and gemcitabine in front-line metastatic pancreatic cancer. Ann. Oncol. 29, https://doi.org/10.1093/annonc/mdy424.031 (2018).
Gradishar, W. J., Yardley, D. A., Layman, R., Sparano, J. A., Chuang, E., Northfelt, D. W. et al. Clinical and translational results of a phase II, randomized trial of an anti-IGF-1R (cixutumumab) in women with breast cancer that progressed on endocrine therapy. Clin. Cancer Res. 22, 301–309 (2016).
Weroha, S. J. & Haluska, P. IGF-1 receptor inhibitors in clinical trials–early lessons. J. Mammary Gland Biol. Neoplasia 13, 471–483 (2008).
Fassnacht, M., Berruti, A., Baudin, E., Demeure, M. J., Gilbert, J., Haak, H. et al. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: a double-blind, randomised, phase 3 study. Lancet Oncol. 16, 426–435 (2015).
Oza, A., Kaye, S., Van Tornout, J., Sessa, C., Gore, M., Naumann, R. W. et al. Phase 2 study evaluating intermittent and continuous linsitinib and weekly paclitaxel in patients with recurrent platinum resistant ovarian epithelial cancer. Gynecologic Oncol. 149, 275–282 (2018).
Ciuleanu, T.-E., Ahmed, S., Kim, J.-H., Mezger, J., Park, K., Thomas, M. et al. Randomised phase 2 study of maintenance linsitinib (OSI-906) in combination with erlotinib compared with placebo plus erlotinib after platinum-based chemotherapy in patients with advanced non-small cell lung cancer. Br. J. Cancer 117, 757–766 (2017).
Haluska, P., Menefee, M., Plimack, E. R., Rosenberg, J., Northfelt, D., LaVallee, T. et al. Phase I dose-escalation study of MEDI-573, a bispecific, antiligand monoclonal antibody against IGFI and IGFII, in patients with advanced solid tumors. Clin. Cancer Res. 20, 4747–4757 (2014).
Crown, J., Sablin, M.-P., Cortés, J., Bergh, J., Im, S.-A., Lu, Y.-S. et al. Abstract P6-21-01: xentuzumab (BI 836845), an insulin-like growth factor (IGF)-neutralizing antibody (Ab), combined with exemestane and everolimus in hormone receptor-positive (HR+) locally advanced/metastatic breast cancer (LA/mBC): randomized phase 2 results. Cancer Res. 79, P6-21-01–P26-21-01 (2019).
Sarfstein, R., Friedman, Y., Attias-Geva, Z., Fishman, A., Bruchim, I. & Werner, H. Metformin downregulates the insulin/IGF-I signaling pathway and inhibits different uterine serous carcinoma (USC) cells proliferation and migration in p53-dependent or -independent manners. PLoS ONE 8, e61537 (2013).
Mohammed, A., Janakiram, N. B., Brewer, M., Ritchie, R. L., Marya, A., Lightfoot, S. et al. Antidiabetic drug metformin prevents progression of pancreatic cancer by targeting in part cancer stem cells and mTOR signaling. Transl. Oncol. 6, 649–647 (2013).
Dowling, R. J., Zakikhani, M., Fantus, I. G., Pollak, M. & Sonenberg, N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 67, 10804–10812 (2007).
Memmott, R. M., Mercado, J. R., Maier, C. R., Kawabata, S., Fox, S. D. & Dennis, P. A. Metformin prevents tobacco carcinogen–induced lung tumorigenesis. Cancer Prev. Res. 3, 1066–1076 (2010).
Martin-Castillo, B., Pernas, S., Dorca, J., Álvarez, I., Martínez, S., Pérez-Garcia, J. M. et al. A phase 2 trial of neoadjuvant metformin in combination with trastuzumab and chemotherapy in women with early HER2-positive breast cancer: the METTEN study. Oncotarget 9, 35687–35704 (2018).
Goodwin, P., Ennis, M., Cescon, D., Elser, C., Haq, R., Hamm, C. et al. Abstract P1-16-03: phase II randomized clinical trial (RCT) of metformin (MET) vs placebo (PLAC) in combination with chemotherapy (CXT) in refractory locally advanced (LABC) or metastatic breast cancer (MBC). Cancer Res. 79, P1-16-03–P11-16-03 (2019).
Zhao, Y., Sun, H., Feng, M., Zhao, J., Zhao, X., Wan, Q. et al. Metformin is associated with reduced cell proliferation in human endometrial cancer by inbibiting PI3K/AKT/mTOR signaling. Gynecol. Endocrinol. 34, 428–432 (2018).
Cai, D., Sun, H., Qi, Y., Zhao, X., Feng, M. & Wu, X. Insulin-like growth factor 1/mammalian target of rapamycin and AMP-activated protein kinase signaling involved in the effects of metformin in the human endometrial cancer. Int. J. Gynecol. Cancer 26, 1667–1672 (2016).
Schuler, K. M., Rambally, B. S., DiFurio, M. J., Sampey, B. P., Gehrig, P. A., Makowski, L. et al. Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med. 4, 161–173 (2015).
Sivalingam, V. N., Kitson, S., McVey, R., Roberts, C., Pemberton, P., Gilmour, K. et al. Measuring the biological effect of presurgical metformin treatment in endometrial cancer. Br. J. Cancer 114, 281 (2016).
Kitson, S. J., Maskell, Z., Sivalingam, V. N., Allen, J. L., Ali, S., Burns, S. et al. PRE-surgical metformin in uterine malignancy (PREMIUM): a multi-center, randomized double-blind, placebo-controlled phase III trial. Clin. Cancer Res. 25, 2424–2432 (2019).
Thomas, T., Burguera, B., Melton, L. J. 3rd, Atkinson, E. J., O’Fallon, W. M., Riggs, B. L. et al. Relationship of serum leptin levels with body composition and sex steroid and insulin levels in men and women. Metab.: Clin. Exp. 49, 1278–1284 (2000).
Leroy, P., Dessolin, S., Villageois, P., Moon, B. C., Friedman, J. M., Ailhaud, G. et al. Expression of ob gene in adipose cells: regulation by insulin. J. Biol. Chem. 271, 2365–2368 (1996).
Surmacz, E. Obesity hormone leptin: a new target in breast cancer? Breast Cancer Res. 9, 301 (2007).
Kolaczynski, J. W., Considine, R. V., Ohannesian, J., Marco, C., Opentanova, I., Nyce, M. R. et al. Responses of leptin to short-term fasting and refeeding in humans: a link with ketogenesis but not ketones themselves. Diabetes 45, 1511–1515 (1996).
Yoon, Y. S., Kwon, A. R., Lee, Y. K. & Oh, S. W. Circulating adipokines and risk of obesity related cancers: a systematic review and meta-analysis. Obesity Res. Clin. Pract. 13, 329–339 (2019).
Garofalo, C., Koda, M., Cascio, S., Sulkowska, M., Kanczuga-Koda, L., Golaszewska, J. et al. Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: possible role of obesity-related stimuli. Clin. Cancer Res. 12, 1447–1453 (2006).
Stattin, P., Lukanova, A., Biessy, C., Soderberg, S., Palmqvist, R., Kaaks, R. et al. Obesity and colon cancer: does leptin provide a link? Int. J. Cancer 109, 149–152 (2004).
Bjorbaek, C., Uotani, S., da Silva, B. & Flier, J. S. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J. Biol. Chem. 272, 32686–32695 (1997).
Donato, J. Jr., Frazao, R. & Elias, C. F. The PI3K signaling pathway mediates the biological effects of leptin. Arquivos brasileiros de endocrinologia e metabologia 54, 591–602 (2010).
Crean-Tate, K. K. & Reizes, O. Leptin regulation of cancer stem cells in breast and gynecologic cancer. Endocrinology 159, 3069–3080 (2018).
Cascio, S., Bartella, V., Auriemma, A., Johannes, G. J., Russo, A., Giordano, A. et al. Mechanism of leptin expression in breast cancer cells: role of hypoxia-inducible factor-1α. Oncogene 27, 540 (2007).
Cao, R., Brakenhielm, E., Wahlestedt, C., Thyberg, J. & Cao, Y. Leptin induces vascular permeability and synergistically stimulates angiogenesis with FGF-2 and VEGF. Proc. Natl Acad. Sci. USA 98, 6390–6395 (2001).
Barone, I., Catalano, S., Gelsomino, L., Marsico, S., Giordano, C., Panza, S. et al. Leptin mediates tumor-stromal interactions that promote the invasive growth of breast cancer cells. Cancer Res. 72, 1416–1427 (2012).
Yamauchi, T., Nio, Y., Maki, T., Kobayashi, M., Takazawa, T., Iwabu, M. et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 13, 332–339 (2007).
Soliman, P. T., Wu, D., Tortolero-Luna, G., Schmeler, K. M., Slomovitz, B. M., Bray, M. S. et al. Association between adiponectin, insulin resistance, and endometrial cancer. Cancer 106, 2376–2381 (2006).
Cust, A. E., Kaaks, R., Friedenreich, C., Bonnet, F., Laville, M., Lukanova, A. et al. Plasma adiponectin levels and endometrial cancer risk in pre- and postmenopausal women. J. Clin. Endocrinol. Metab. 92, 255–263 (2007).
Körner, A., Pazaitou-Panayiotou, K., Kelesidis, T., Kelesidis, I., Williams, C. J., Kaprara, A. et al. Total and high-molecular-weight adiponectin in breast cancer: in vitro and in vivo studies. J. Clin. Endocrinol. Metab. 92, 1041–1048 (2007).
Mantzoros, C., Petridou, E., Dessypris, N., Chavelas, C., Dalamaga, M., Alexe, D. M. et al. Adiponectin and breast cancer risk. J. Clin. Endocrinol. Metabo. 89, 1102–1107 (2004).
Grossmann, M. E., Nkhata, K. J., Mizuno, N. K., Ray, A. & Cleary, M. P. Effects of adiponectin on breast cancer cell growth and signaling. Br. J. Cancer 98, 370–379 (2008).
van Kruijsdijk, R. C., van der Wall, E. & Visseren, F. L. Obesity and cancer: the role of dysfunctional adipose tissue. Cancer Epidemiol. Biomark. Prev. 18, 2569–2578 (2009).
Duggan, C., Irwin, M. L., Xiao, L., Henderson, K. D., Smith, A. W., Baumgartner, R. N. et al. Associations of insulin resistance and adiponectin with mortality in women with breast cancer. J. Clin. Oncol. 29, 32–39 (2010).
Grossmann, M. E., Ray, A., Dogan, S., Mizuno, N. K. & Cleary, M. P. Balance of adiponectin and leptin modulates breast cancer cell growth. Cell Res. 18, 1154–1156 (2008).
Wang, C. H., Wang, P. J., Hsieh, Y. C., Lo, S., Lee, Y. C., Chen, Y. C. et al. Resistin facilitates breast cancer progression via TLR4-mediated induction of mesenchymal phenotypes and stemness properties. Oncogene 37, 589–600 (2018).
Avtanski, D., Garcia, A., Caraballo, B., Thangeswaran, P., Marin, S., Bianco, J. et al. Resistin induces breast cancer cells epithelial to mesenchymal transition (EMT) and stemness through both adenylyl cyclase-associated protein 1 (CAP1)-dependent and CAP1-independent mechanisms. Cytokine 120, 155–164 (2019).
Nakayama, S., Miyoshi, Y., Ishihara, H. & Noguchi, S. Growth-inhibitory effect of adiponectin via adiponectin receptor 1 on human breast cancer cells through inhibition of S-phase entry without inducing apoptosis. Breast Cancer Res. Treat. 112, 405–410 (2008).
Cabia, B., Andrade, S., Carreira, M. C., Casanueva, F. F. & Crujeiras, A. B. A role for novel adipose tissue-secreted factors in obesity-related carcinogenesis. Obes. Rev. 17, 361–376 (2016).
Rahmani, J., Manzari, N., Thompson, J., Gudi, S. K., Chhabra, M., Naik, G. et al. The effect of metformin on biomarkers associated with breast cancer outcomes: a systematic review, meta-analysis, and dose-response of randomized clinical trials. Clin. Transl. Oncol. 22, 37–49 (2019).
Soliman, P. T., Zhang, Q., Broaddus, R. R., Westin, S. N., Iglesias, D., Munsell, M. F. et al. Prospective evaluation of the molecular effects of metformin on the endometrium in women with newly diagnosed endometrial cancer: a window of opportunity study. Gynecologic Oncol. 143, 466–471 (2016).
Bonanni, B., Puntoni, M., Cazzaniga, M., Pruneri, G., Serrano, D., Guerrieri-Gonzaga, A. et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J. Clin. Oncol. 30, 2593–2600 (2012).
Goodwin, P. J., Parulekar, W. R., Gelmon, K. A., Shepherd, L. E., Ligibel, J. A., Hershman, D. L. et al. Effect of metformin vs placebo on and metabolic factors in NCIC CTG MA.32. J. Natl Cancer Inst. 107, djv006 (2015).
Beccari, S., Kovalszky, I., Wade, J. D., Otvos, L. Jr. & Surmacz, E. Designer peptide antagonist of the leptin receptor with peripheral antineoplastic activity. Peptides 44, 127–134 (2013).
Otvos, L., Jr., Kovalszky, I., Riolfi, M., Ferla, R., Olah, J., Sztodola, A. et al. Efficacy of a leptin receptor antagonist peptide in a mouse model of triple-negative breast cancer. Eur. J. Cancer 47, 1578–1584 (2011).
Catalano, S., Leggio, A., Barone, I., De Marco, R., Gelsomino, L., Campana, A. et al. A novel leptin antagonist peptide inhibits breast cancer growth in vitro and in vivo. J. Cell. Mol. Med. 19, 1122–1132 (2015).
Miyazaki, Y., Glass, L., Triplitt, C., Matsuda, M., Cusi, K., Mahankali, A. et al. Effect of rosiglitazone on glucose and non-esterified fatty acid metabolism in type II diabetic patients. Diabetologia 44, 2210–2219 (2001).
Majuri, A., Santaniemi, M., Rautio, K., Kunnari, A., Vartiainen, J., Ruokonen, A. et al. Rosiglitazone treatment increases plasma levels of adiponectin and decreases levels of resistin in overweight women with PCOS: a randomized placebo-controlled study. Eur. J. Endocrinol. 156, 263–269 (2007).
Wong Wing, T., Tian Xiao, Y., Xu, A., Yu, J., Lau Chi, W., Hoo Ruby, L. C. et al. Adiponectin is required for PPARγ-mediated improvement of endothelial function in diabetic mice. Cell Metab. 14, 104–115 (2011).
Tonelli, J., Li, W., Kishore, P., Pajvani, U. B., Kwon, E., Weaver, C. et al. Mechanisms of early insulin-sensitizing effects of thiazolidinediones in type 2. Diabetes. Diabetes 53, 1621–1629 (2004).
Yee, L. D., Williams, N., Wen, P., Young, D. C., Lester, J., Johnson, M. V. et al. Pilot study of rosiglitazone therapy in women with breast cancer: effects of short-term therapy on tumor tissue and serum markers. Clin. Cancer Res. 13, 246–252 (2007).
Maeda, N., Takahashi, M., Funahashi, T., Kihara, S., Nishizawa, H., Kishida, K. et al. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 50, 2094–2099 (2001).
Augimeri, G., Giordano, C., Gelsomino, L., Plastina, P., Barone, I., Catalano, S. et al. The role of PPARγ ligands in breast cancer: from basic research to clinical studies. Cancers 12, 2623 (2020).
Kelesidis, I., Kelesidis, T. & Mantzoros, C. S. Adiponectin and cancer: a systematic review. Br. J. Cancer 94, 1221–1225 (2006).
Shellman, Y. G., Ribble, D., Miller, L., Gendall, J., Vanbuskirk, K., Kelly, D. et al. Lovastatin-induced apoptosis in human melanoma cell lines. Melanoma Res. 15, 83–89 (2005).
Koyuturk, M., Ersoz, M. & Altiok, N. Simvastatin induces proliferation inhibition and apoptosis in C6 glioma cells via c-jun N-terminal kinase. Neurosci. Lett. 370, 212–217 (2004).
Girgert, R., Vogt, Y., Becke, D., Bruchelt, G. & Schweizer, P. Growth inhibition of neuroblastoma cells by lovastatin and L-ascorbic acid is based on different mechanisms. Cancer Lett. 137, 167–172 (1999).
Demierre, M. F., Higgins, P. D., Gruber, S. B., Hawk, E. & Lippman, S. M. Statins and cancer prevention. Nat. Rev. Cancer 5, 930–942 (2005).
Graaf, M. R., Richel, D. J., van Noorden, C. J. & Guchelaar, H. J. Effects of statins and farnesyltransferase inhibitors on the development and progression of cancer. Cancer Treat. Rev. 30, 609–641 (2004).
Chan, K. K., Oza, A. M. & Siu, L. L. The statins as anticancer agents. Clin. Cancer Res. 9, 10–19 (2003).
Kang, S., Kim, E. S. & Moon, A. Simvastatin and lovastatin inhibit breast cell invasion induced by H-Ras. Oncol. Rep. 21, 1317–1322 (2009).
Laezza, C., Malfitano, A. M., Proto, M. C., Esposito, I., Gazzerro, P., Formisano, P. et al. Inhibition of 3-hydroxy-3-methylglutaryl-coenzyme A reductase activity and of Ras farnesylation mediate antitumor effects of anandamide in human breast cancer cells. Endocrine-related Cancer 17, 495–503 (2010).
Jakobisiak, M., Bruno, S., Skierski, J. S. & Darzynkiewicz, Z. Cell cycle-specific effects of lovastatin. Proc. Natl Acad. Sci. USA 88, 3628–3632 (1991).
Wong, W. W., Dimitroulakos, J., Minden, M. D. & Penn, L. Z. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia 16, 508–519 (2002).
Wu, J., Wong, W. W., Khosravi, F., Minden, M. D. & Penn, L. Z. Blocking the Raf/MEK/ERK pathway sensitizes acute myelogenous leukemia cells to lovastatin-induced apoptosis. Cancer Res. 64, 6461–6468 (2004).
Skaletz-Rorowski, A. & Walsh, K. Statin therapy and angiogenesis. Curr. Opin. Lipidol. 14, 599–603 (2003).
Ahern, T. P., Pedersen, L., Tarp, M., Cronin-Fenton, D. P., Garne, J. P., Silliman, R. A. et al. Statin prescriptions and breast cancer recurrence risk: a Danish nationwide prospective cohort study. J. Natl Cancer Inst. 103, 1461–1468 (2011).
Cauley, J. A., McTiernan, A., Rodabough, R. J., LaCroix, A., Bauer, D. C., Margolis, K. L. et al. Statin use and breast cancer: prospective results from the Women’s Health Initiative. J. Natl Cancer Inst. 98, 700–707 (2006).
Graaf, M. R., Beiderbeck, A. B., Egberts, A. C., Richel, D. J. & Guchelaar, H. J. The risk of cancer in users of statins. J. Clin. Oncol. 22, 2388–2394 (2004).
Karp, I., Behlouli, H., Lelorier, J. & Pilote, L. Statins and cancer risk. Am. J. Med. 121, 302–309 (2008).
Kumar, A. S., Benz, C. C., Shim, V., Minami, C. A., Moore, D. H. & Esserman, L. J. Estrogen receptor-negative breast cancer is less likely to arise among lipophilic statin users. Cancer Epidemiol. Biomark. Prev. 17, 1028–1033 (2008).
Bonovas, S., Filioussi, K., Tsavaris, N. & Sitaras, N. M. Use of statins and breast cancer: a meta-analysis of seven randomized clinical trials and nine observational studies. J. Clin. Oncol. 23, 8606–8612 (2005).
Browning, D. R. & Martin, R. M. Statins and risk of cancer: a systematic review and metaanalysis. Int. J. Cancer 120, 833–843 (2007).
Cholesterol Treatment Trialists C, Emberson, J. R., Kearney, P. M., Blackwell, L., Newman, C., Reith, C. et al. Lack of effect of lowering LDL cholesterol on cancer: meta-analysis of individual data from 175,000 people in 27 randomised trials of statin therapy. PLoS ONE 7, e29849 (2012).
Dale, K. M., Coleman, C. I., Henyan, N. N., Kluger, J. & White, C. M. Statins and cancer risk: a meta-analysis. J. Am. Med. Assoc. 295, 74–80 (2006).
Undela, K., Srikanth, V. & Bansal, D. Statin use and risk of breast cancer: a meta-analysis of observational studies. Breast Cancer Res. Treat. 135, 261–269 (2012).
Nielsen, S. F., Nordestgaard, B. G. & Bojesen, S. E. Statin use and reduced cancer-related mortality. New Engl. J. Med. 367, 1792–1802 (2012).
Boudreau, D. M., Yu, O., Miglioretti, D. L., Buist, D. S., Heckbert, S. R. & Daling, J. R. Statin use and breast cancer risk in a large population-based setting. Cancer Epidemiol. Biomark. Prev. 16, 416–421 (2007).
Coogan, P. F., Rosenberg, L. & Strom, B. L. Statin use and the risk of 10 cancers. Epidemiology 18, 213–219 (2007).
Eliassen, A. H., Colditz, G. A., Rosner, B., Willett, W. C. & Hankinson, S. E. Serum lipids, lipid-lowering drugs, and the risk of breast cancer. Arch. Intern. Med. 165, 2264–2271 (2005).
Beatson, G. T. On the treatment of inoperable cases of carcinoma of the mamma: suggestions for a new method of treatment, with illustrative cases. Trans. Med. Chir. Soc. Edinb. 15, 153–179 (1896).
Deroo, B. J. & Korach, K. S. Estrogen receptors and human disease. J. Clin. Invest. 116, 561–570 (2006).
Martin, M. B. & Stoica, A. Insulin-like growth factor-i and estrogen interactions in breast cancer. J. Nutr. 132, 3799S–3801S (2002).
Petrie, W. K., Dennis, M. K., Hu, C., Dai, D., Arterburn, J. B., Smith, H. O. et al. G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obstetr. Gynecology Int. 2013, 472720 (2013).
De Francesco, E. M., Pellegrino, M., Santolla, M. F., Lappano, R., Ricchio, E., Abonante, S. et al. GPER mediates activation of HIF1alpha/VEGF signaling by estrogens. Cancer Res. 74, 4053–4064 (2014).
Bulun, S. E., Chen, D., Moy, I. & Brooks, D. C. Zhao H. Aromatase, breast cancer and obesity: a complex interaction. Trends Endocrinol. Metab. 23, 83–89 (2012).
Onstad, M. A., Schmandt, R. E. & Lu, K. H. Addressing the role of obesity in endometrial cancer risk, prevention, and treatment. J. Clin. Oncol. 34, 4225–4230 (2016).
Zahid, H., Subbaramaiah, K., Iyengar, N. M., Zhou, X. K., Chen, I. C., Bhardwaj, P. et al. Leptin regulation of the p53-HIF1alpha/PKM2-aromatase axis in breast adipose stromal cells: a novel mechanism for the obesity-breast cancer link. Int. J. Obes. 42, 711–720 (2018).
Morris, P. G., Hudis, C. A., Giri, D., Morrow, M., Falcone, D. J., Zhou, X. K. et al. Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev. Res. 4, 1021–1029 (2011).
Agarwal, V. R., Bulun, S. E., Leitch, M., Rohrich, R. & Simpson, E. R. Use of alternative promoters to express the aromatase cytochrome P450 (CYP19) gene in breast adipose tissues of cancer-free and breast cancer patients. J. Clin. Endocrinol. Metab. 81, 3843–3849 (1996).
Mahendroo, M. S., Mendelson, C. R. & Simpson, E. R. Tissue-specific and hormonally controlled alternative promoters regulate aromatase cytochrome P450 gene expression in human adipose tissue. J. Biol. Chem. 268, 19463–19470 (1993).
Zhao, Y., Agarwal, V. R., Mendelson, C. R. & Simpson, E. R. Estrogen biosynthesis proximal to a breast tumor is stimulated by PGE2 via cyclic AMP, leading to activation of promoter II of the CYP19 (aromatase) gene. Endocrinology 137, 5739–5742 (1996).
Clyne, C. D., Speed, C. J., Zhou, J. & Simpson, E. R. Liver receptor homologue-1 (LRH-1) regulates expression of aromatase in preadipocytes. J. Biol. Chem. 277, 20591–20597 (2002).
Safi, R., Kovacic, A., Gaillard, S., Murata, Y., Simpson, E. R., McDonnell, D. P. et al. Coactivation of liver receptor homologue-1 by peroxisome proliferator-activated receptor gamma coactivator-1alpha on aromatase promoter II and its inhibition by activated retinoid X receptor suggest a novel target for breast-specific antiestrogen therapy. Cancer Res. 65, 11762–11770 (2005).
Michael, M. D., Michael, L. F. & Simpson, E. R. A CRE-like sequence that binds CREB and contributes to cAMP-dependent regulation of the proximal promoter of the human aromatase P450 (CYP19) gene. Mol. Cell. Endocrinol. 134, 147–156 (1997).
Means, G. D., Mahendroo, M. S., Corbin, C. J., Mathis, J. M., Powell, F. E., Mendelson, C. R. et al. Structural analysis of the gene encoding human aromatase cytochrome P-450, the enzyme responsible for estrogen biosynthesis. J. Biol. Chem. 264, 19385–19391 (1989).
Mendelson, C. R., Cleland, W. H., Smith, M. E. & Simpson, E. R. Regulation of aromatase activity of stromal cells derived from human adipose tissue. Endocrinology 111, 1077–1085 (1982).
Samarajeewa, N. U., Docanto, M. M., Simpson, E. R. & Brown, K. A. CREB-regulated transcription co-activator family stimulates promoter II-driven aromatase expression in preadipocytes. Horm. Cancer 4, 233–241 (2013).
Brown, K. A., McInnes, K. J., Hunger, N. I., Oakhill, J. S., Steinberg, G. R. & Simpson, E. R. Subcellular localization of cyclic AMP-responsive element binding protein-regulated transcription coactivator 2 provides a link between obesity and breast cancer in postmenopausal women. Cancer Res. 69, 5392–5399 (2009).
Chen, D., Reierstad, S., Lin, Z., Lu, M., Brooks, C., Li, N. et al. Prostaglandin E(2) induces breast cancer related aromatase promoters via activation of p38 and c-Jun NH(2)-terminal kinase in adipose fibroblasts. Cancer Res. 67, 8914–8922 (2007).
Samarajeewa, N. U., Yang, F., Docanto, M. M., Sakurai, M., McNamara, K. M., Sasano, H. et al. HIF-1alpha stimulates aromatase expression driven by prostaglandin E2 in breast adipose stroma. Breast Cancer Res. 15, R30 (2013).
Richards, J. A. & Brueggemeier, R. W. Prostaglandin E2 regulates aromatase activity and expression in human adipose stromal cells via two distinct receptor subtypes. J. Clin. Endocrinol. Metab. 88, 2810–2816 (2003).
Schonwasser, D. C., Marais, R. M., Marshall, C. J. & Parker, P. J. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol. Cell. Biol. 18, 790–798 (1998).
Zhang, B., Perpetua, M., Fulmer, M. & Harbrecht, B. G. JNK signaling involved in the effects of cyclic AMP on IL-1beta plus IFNgamma-induced inducible nitric oxide synthase expression in hepatocytes. Cell. Signal. 16, 837–846 (2004).
Wang, X., Docanto, M. M., Sasano, H., Kathleen Cuningham Foundation Consortium for Research into Familial Breast C, Lo, C., Simpson, E. R. et al. Prostaglandin E2 inhibits p53 in human breast adipose stromal cells: a novel mechanism for the regulation of aromatase in obesity and breast cancer. Cancer Res. 75, 645–655 (2015).
Wang, X., Simpson, E. R. & Brown, K. A. p53: protection against tumor growth beyond effects on cell cycle and apoptosis. Cancer Res. 75, 5001–5007 (2015).
To S. Q., Simpson E. R., Knower K. C., Clyne C. D. Involvement of early growth response factors in TNFalpha-induced aromatase expression in breast adipose. Breast Cancer Res Treat (2013).
Zhao, Y., Nichols, J. E., Valdez, R., Mendelson, C. R. & Simpson, E. R. Tumor necrosis factor-alpha stimulates aromatase gene expression in human adipose stromal cells through use of an activating protein-1 binding site upstream of promoter 1.4. Mol. Endocrinol. 10, 1350–1357 (1996).
Macdiarmid, F., Wang, D., Duncan, L. J., Purohit, A., Ghilchick, M. W. & Reed, M. J. Stimulation of aromatase activity in breast fibroblasts by tumor necrosis factor alpha. Mol. Cell. Endocrinol. 106, 17–21 (1994).
Singh, A., Purohit, A., Wang, D. Y., Duncan, L. J., Ghilchik, M. W. & Reed, M. J. IL-6sR: release from MCF-7 breast cancer cells and role in regulating peripheral oestrogen synthesis. J. Endocrinol. 147, R9–R12 (1995).
Brown, K. A., Hunger, N. I., Docanto, M. & Simpson, E. R. Metformin inhibits aromatase expression in human breast adipose stromal cells via stimulation of AMP-activated protein kinase. Breast Cancer Res. Treat. 123, 591–596 (2010).
Samarajeewa, N. U., Ham, S., Yang, F., Simpson, E. R. & Brown, K. A. Promoter-specific effects of metformin on aromatase transcript expression. Steroids 76, 768–771 (2011).
Docanto, M. M., Yang, F., Callaghan, B., Au, C. C., Ragavan, R., Wang, X. et al. Ghrelin and des-acyl ghrelin inhibit aromatase expression and activity in human adipose stromal cells: suppression of cAMP as a possible mechanism. Breast Cancer Res. Treat. 147, 193–201 (2014).
Au, C. C., Docanto, M. M., Zahid, H., Raffaelli, F. M., Ferrero, R. L., Furness, J. B. et al. Des-acyl ghrelin inhibits the capacity of macrophages to stimulate the expression of aromatase in breast adipose stromal cells. J. Steroid Biochem. Mol. Biol. 170, 49–53 (2017).
Smith, I. E. & Dowsett, M. Aromatase inhibitors in breast cancer. New Engl. J. Med. 348, 2431–2442 (2003).
Kawahara, T., Okamoto, N., Takae, S., Kashiwagi, M., Nakajima, M., Uekawa, A. et al. Aromatase inhibitor use during ovarian stimulation suppresses growth of uterine endometrial cancer in xenograft mouse model. Hum. Reprod. 33, 303–310 (2018).
Mileshkin, L., Edmondson, R., O’Connell, R. L., Sjoquist, K. M., Andrews, J., Jyothirmayi, R. et al. Phase 2 study of anastrozole in recurrent estrogen (ER)/progesterone (PR) positive endometrial cancer: the PARAGON trial—ANZGOG 0903. Gynecologic Oncol. 154, 29–37 (2019).
Thangavelu, A., Hewitt, M. J., Quinton, N. D. & Duffy, S. R. Neoadjuvant treatment of endometrial cancer using anastrozole: a randomised pilot study. Gynecologic Oncol. 131, 613–618 (2013).
Slomovitz, B. M., Jiang, Y., Yates, M. S., Soliman, P. T., Johnston, T., Nowakowski, M. et al. Phase II study of everolimus and letrozole in patients with recurrent endometrial carcinoma. J. Clin. Oncol. 33, 930–936 (2015).
Sestak, I., Distler, W., Forbes, J. F., Dowsett, M., Howell, A. & Cuzick, J. Effect of body mass index on recurrences in tamoxifen and anastrozole treated women: an exploratory analysis from the ATAC trial. J. Clin. Oncol. 28, 3411–3415 (2010).
Semenza, G. L. HIF-1 and human disease: one highly involved factor. Genes Dev. 14, 1983–1991 (2000).
Masoud, G. N. & Li, W. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B 5, 378–389 (2015).
Baba, Y., Nosho, K., Shima, K., Irahara, N., Chan, A. T., Meyerhardt, J. A. et al. HIF1A overexpression is associated with poor prognosis in a cohort of 731 colorectal cancers. Am. J. Pathol. 176, 2292–2301 (2010).
Theodoropoulos, V. E., Lazaris, A., Sofras, F., Gerzelis, I., Tsoukala, V., Ghikonti, I. et al. Hypoxia-inducible factor 1 alpha expression correlates with angiogenesis and unfavorable prognosis in bladder cancer. Eur. Urol. 46, 200–208 (2004).
Pouysségur, J., Dayan, F. & Mazure, N. M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441, 437–443 (2006).
Hosogai, N., Fukuhara, A., Oshima, K., Miyata, Y., Tanaka, S., Segawa, K. et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56, 901–911 (2007).
Gonzalez, F. J., Xie, C. & Jiang, C. The role of hypoxia-inducible factors in metabolic diseases. Nat. Rev. Endocrinol. 15, 21–32 (2019).
Costa, C., Incio, J. & Soares, R. Angiogenesis and chronic inflammation: cause or consequence? Angiogenesis 10, 149–166 (2007).
Sahoo, S. S., Lombard, J. M., Ius, Y., O’Sullivan, R., Wood, L. G., Nahar, P. et al. Adipose-derived VEGF-mTOR signaling promotes endometrial hyperplasia and cancer: implications for obese women. Mol. Cancer Res. 16, 309–321 (2018).
Incio, J., Ligibel, J. A., McManus, D. T., Suboj, P., Jung, K., Kawaguchi, K. et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci. Transl. Med. 10, 432 (2018).
Guiu, B., Petit, J. M., Bonnetain, F., Ladoire, S., Guiu, S., Cercueil, J. P. et al. Visceral fat area is an independent predictive biomarker of outcome after first-line bevacizumab-based treatment in metastatic colorectal cancer. Gut 59, 341–347 (2010).
Slaughter, K. N., Thai, T., Penaroza, S., Benbrook, D. M., Thavathiru, E., Ding, K. et al. Measurements of adiposity as clinical biomarkers for first-line bevacizumab-based chemotherapy in epithelial ovarian cancer. Gynecologic Oncol. 133, 11–15 (2014).
Jeong, W., Rapisarda, A., Park, S. R., Kinders, R. J., Chen, A., Melillo, G. et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1alpha), in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 73, 343–348 (2014).
Papadopoulos, K. P., Jonasch, E., Zojwalla, N. J., Wang, K. & Bauer, T. M. A first-in-human phase 1 dose-escalation trial of the oral HIF-2a inhibitor PT2977 in patients with advanced solid tumors. J. Clin. Oncol. 36, 2508 (2018).
Rossi, L., Verrico, M., Zaccarelli, E., Papa, A., Colonna, M., Strudel, M. et al. Bevacizumab in ovarian cancer: a critical review of phase III studies. Oncotarget 8, 12389–12405 (2017).
Tewari, K. S., Sill, M. W., Long, H. J. 3rd, Penson, R. T., Huang, H., Ramondetta, L. M. et al. Improved survival with bevacizumab in advanced cervical cancer. New Engl. J. Med. 370, 734–743 (2014).
Crino, L., Dansin, E., Garrido, P., Griesinger, F., Laskin, J., Pavlakis, N. et al. Safety and efficacy of first-line bevacizumab-based therapy in advanced non-squamous non-small-cell lung cancer (SAiL, MO19390): a phase 4 study. Lancet Oncol. 11, 733–740 (2010).
Hurwitz, H., Fehrenbacher, L., Novotny, W., Cartwright, T., Hainsworth, J., Heim, W. et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. New Engl. J. Med. 350, 2335–2342 (2004).
Wilke, H., Muro, K., Van Cutsem, E., Oh, S.-C., Bodoky, G., Shimada, Y. et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 15, 1224–1235 (2014).
Garon, E. B., Ciuleanu, T.-E., Arrieta, O., Prabhash, K., Syrigos, K. N., Goksel, T. et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet 384, 665–673 (2014).
Zhu, A. X., Kang, Y.-K., Yen, C.-J., Finn, R. S., Galle, P. R., Llovet, J. M. et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 20, 282–296 (2019).
Haas, N. B., Manola, J., Uzzo, R. G., Flaherty, K. T., Wood, C. G., Kane, C. et al. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial. Lancet 387, 2008–2016 (2016).
Kudo, M., Finn, R. S., Qin, S., Han, K. H., Ikeda, K., Piscaglia, F. et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 391, 1163–1173 (2018).
Nair, A., Lemery, S. J., Yang, J., Marathe, A., Zhao, L., Zhao, H. et al. FDA approval summary: lenvatinib for progressive, radio-iodine-refractory differentiated thyroid cancer. Clin. Cancer Res. 21, 5205–5208 (2015).
Makker, V., Rasco, D. W., Vogelzang, N. J., Messing, M., Brose, M. S., Cohn, A. L. et al. Lenvatinib + pembrolizumab in patients with advanced endometrial cancer: updated results. J. Clin. Oncol. 36, 5596–5596 (2018).
Olefsky, J. M. & Glass, C. K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 72, 219–246 (2010).
Cinti, S., Mitchell, G., Barbatelli, G., Murano, I., Ceresi, E., Faloia, E. et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 46, 2347–2355 (2005).
Vaysse, C., Lømo, J., Garred, Ø., Fjeldheim, F., Lofteroed, T., Schlichting, E. et al. Inflammation of mammary adipose tissue occurs in overweight and obese patients exhibiting early-stage breast cancer. npj Breast Cancer 3, 19 (2017).
Carter, J. M., Hoskin, T. L., Pena, M. A., Brahmbhatt, R., Winham, S. J., Frost, M. H. et al. Macrophagic “crown-like structures” are associated with an increased risk of breast cancer in benign breast disease. Cancer Prev. Res. 11, 113–119 (2018).
Iyengar, N. M., Zhou, X. K., Gucalp, A., Morris, P. G., Howe, L. R., Giri, D. D. et al. Systemic correlates of white adipose tissue inflammation in early-stage breast cancer. Clin. Cancer Res. 22, 2283–2289 (2016).
Quail, D. F. & Dannenberg, A. J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 15, 139–154 (2019).
Crujeiras, A. B., Cabia, B., Carreira, M. C., Amil, M., Cueva, J., Andrade, S. et al. Secreted factors derived from obese visceral adipose tissue regulate the expression of breast malignant transformation genes. Int. J. Obes. 40, 514–523 (2016).
Iyengar, N. M., Gucalp, A., Dannenberg, A. J. & Hudis, C. A. Obesity and cancer mechanisms: tumor microenvironment and inflammation. J. Clin. Oncol. 34, 4270–4276 (2016).
Franceschi, S., Favero, A., Decarli, A., Negri, E., La Vecchia, C., Ferraroni, M. et al. Intake of macronutrients and risk of breast cancer. Lancet 347, 1351–1356 (1996).
Lucenteforte, E., Talamini, R., Montella, M., Dal Maso, L., Tavani, A., Deandrea, S. et al. Macronutrients, fatty acids and cholesterol intake and endometrial cancer. Ann. Oncol. 19, 168–172 (2008).
Dieli-Conwright, C. M., Lee, K. & Kiwata, J. L. Reducing the risk of breast cancer recurrence: an evaluation of the effects and mechanisms of diet and exercise. Curr. Breast Cancer Rep. 8, 139–150 (2016).
Reeves, M. M., Terranova, C. O., Eakin, E. G. & Demark-Wahnefried, W. Weight loss intervention trials in women with breast cancer: a systematic review. Obes. Rev. 15, 749–768 (2014).
Chlebowski, R. T. B. G. Final survival analysis from the randomized Women’s Intervention Nutrition Study (WINS) evaluating dietary intervention as adjuvant breast cancer therapy. J. Clin. Oncol. Abstract 26, S5-08 (2014).
Pierce, J. P., Natarajan, L., Caan, B. J., Parker, B. A., Greenberg, E. R., Flatt, S. W. et al. Influence of a diet very high in vegetables, fruit, and fiber and low in fat on prognosis following treatment for breast cancer: the Women’s Healthy Eating and Living (WHEL) randomized trial. J. Am. Med. Assoc. 298, 289–298 (2007).
Levine Morgan, E., Suarez Jorge, A., Brandhorst, S., Balasubramanian, P., Cheng, C.-W., Madia, F. et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 19, 407–417 (2014).
Farvid, M. S., Spence, N. D., Holmes, M. D. & Barnett, J. B. Fiber consumption and breast cancer incidence: a systematic review and meta-analysis of prospective studies. Cancer 126, 3061–3075 (2020).
Chlebowski, R. T., Anderson, G. L., Manson, J. E., Prentice, R. L., Aragaki, A. K., Snetselaar, L. et al. Low-fat dietary pattern and cancer mortality in the women’s health initiative (WHI) randomized controlled trial. JNCI Cancer Spectrum 2, pky065 (2019).
Bandera-Merchan, B., Boughanem, H., Crujeiras, A. B., Macias-Gonzalez, M. & Tinahones, F. J. Ketotherapy as an epigenetic modifier in cancer. Rev. Endocr. Metab. Disord. 21, 509–519 (2020).
Cohen, C. W., Fontaine, K. R., Arend, R. C., Gower, B. A. A ketogenic diet is acceptable in women with ovarian and endometrial cancer and has no adverse effects on blood lipids: a randomized, controlled trial. Nutr. Cancer 72, 584–594 (2019).
Cohen, C. W., Fontaine, K. R., Arend, R. C., Alvarez, R. D., Leath, C. A. III, Huh, W. K. et al. A ketogenic diet reduces central obesity and serum insulin in women with ovarian or endometrial cancer. J. Nutr. 148, 1253–1260 (2018).
Zahra, A., Fath, M. A., Opat, E., Mapuskar, K. A., Bhatia, S. K., Ma, D. C. et al. Consuming a ketogenic diet while receiving radiation and chemotherapy for locally advanced lung cancer and pancreatic cancer: the university of iowa experience of two phase 1 clinical trials. Radiation Res. 187, 743–754 (2017).
de Luis, D., Domingo, J. C., Izaola, O., Casanueva, F. F., Bellido, D. & Sajoux, I. Effect of DHA supplementation in a very low-calorie ketogenic diet in the treatment of obesity: a randomized clinical trial. Endocrine 54, 111–122 (2016).
Gomez-Arbelaez, D., Bellido, D., Castro, A. I., Ordoñez-Mayan, L., Carreira, J., Galban, C. et al. Body composition changes after very-low-calorie ketogenic diet in obesity evaluated by 3 standardized methods. J. Clin. Endocrinol. Metab. 102, 488–498 (2017).
Sajoux, I., Lorenzo, P. M., Gomez-Arbelaez, D., Zulet, M. A., Abete, I., Castro, A. I. et al. Effect of a very-low-calorie ketogenic diet on circulating myokine levels compared with the effect of bariatric surgery or a low-calorie diet in patients with obesity. Nutrients 11, 2368 (2019).
Trimboli, P., Castellana, M., Bellido, D. & Casanueva, F. F. Confusion in the nomenclature of ketogenic diets blurs evidence. Rev. Endocr. Metab. Disord. 21, 1–3 (2020).
Linos, E., Willett, W. C., Cho, E. & Frazier, L. Adolescent diet in relation to breast cancer risk among premenopausal women. Cancer Epidemiol. Biomark. Prev. 19, 689–696 (2010).
Sieri, S., Chiodini, P., Agnoli, C., Pala, V., Berrino, F., Trichopoulou, A. et al. Dietary fat intake and development of specific breast cancer subtypes. J. Natl Cancer Inst. 106, 5 (2014).
Ternes, D., Karta, J., Tsenkova, M., Wilmes, P., Haan, S. & Letellier, E. Microbiome in colorectal cancer: how to get from meta-omics to mechanism? Trends Microbiol. 28, 401–423 (2020).
Gopalakrishnan, V., Helmink, B. A., Spencer, C. N., Reuben, A. & Wargo, J. A. The Influence Of The Gut Microbiome On Cancer, Immunity, And Cancer Immunotherapy. Cancer Cell 33, 570–580 (2018).
Gopalakrishnan, V., Spencer, C. N., Nezi, L., Reuben, A., Andrews, M. C., Karpinets, T. V. et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018).
Zitvogel, L., Galluzzi, L., Viaud, S., Vétizou, M., Daillère, R., Merad, M. et al. Cancer and the gut microbiota: an unexpected link. Sci. Transl. Med. 7, 271ps271–271ps271 (2015).
Ervin, S. M., Li, H., Lim, L., Roberts, L. R., Liang, X., Mani, S. et al. Gut microbial β-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens. J. Biol. Chem. 294, 18586–18599 (2019).
Kwa, M., Plottel, C. S., Blaser, M. J. & Adams, S. The intestinal microbiome and estrogen receptor-positive female breast cancer. J. Natl Cancer Inst. 108, djw029 (2016).
Goedert, J. J., Jones, G., Hua, X., Xu, X., Yu, G., Flores, R. et al. Investigation of the association between the fecal microbiota and breast cancer in postmenopausal women: a population-based case-control pilot study. J. Natl Cancer Inst. 107, djv147 (2015).
Łaniewski, P., Ilhan, Z. E. & Herbst-Kralovetz, M. M. The microbiome and gynaecological cancer development, prevention and therapy. Nat. Rev. Urol. 17, 232–250 (2020).
Holmes, M. D., Chen, W. Y., Feskanich, D., Kroenke, C. H. & Colditz, G. A. Physical activity and survival after breast cancer diagnosis. J. Am. Med. Assoc. 293, 2479–2486 (2005).
Friedenreich, C. M., Neilson, H. K., Farris, M. S. & Courneya, K. S. Physical activity and cancer outcomes: a precision medicine approach. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.CCR-16-0067 (2016).
Courneya, K. S., Booth, C. M., Gill, S., O’Brien, P., Vardy, J., Friedenreich, C. M. et al. The colon health and life-long exercise change trial: a randomized trial of the National Cancer Institute of Canada Clinical Trials Group. Curr. Oncol. 15, 279–285 (2008).
Demark-Wahnefried, W., Jones, L. W., Snyder, D. C., Sloane, R. J., Kimmick, G. G., Hughes, D. C. et al. Daughters and mothers against breast cancer (DAMES): main outcomes of a randomized controlled trial of weight loss in overweight mothers with breast cancer and their overweight daughters. Cancer 120, 2522–2534 (2014).
Rock, C. L., Flatt, S. W., Byers, T. E., Colditz, G. A., Demark-Wahnefried, W., Ganz, P. A. et al. Results of the exercise and nutrition to enhance recovery and good health for you (ENERGY) trial: a behavioral weight loss intervention in overweight or obese breast cancer survivors. J. Clin. Oncol. 33, 3169–3176 (2015).
Harrigan, M., Cartmel, B., Loftfield, E., Sanft, T., Chagpar, A. B., Zhou, Y. et al. Randomized trial comparing telephone versus in-person weight loss counseling on body composition and circulating biomarkers in women treated for breast cancer: the lifestyle, exercise, and nutrition (LEAN) study. J. Clin. Oncol. 34, 669–676 (2016).
Villarini, A., Pasanisi, P., Traina, A., Mano, M. P., Bonanni, B., Panico, S. et al. Lifestyle and breast cancer recurrences: the DIANA-5 trial. Tumori 98, 1–18 (2012).
Rack, B., Andergassen, U., Neugebauer, J., Salmen, J., Hepp, P., Sommer, H. et al. The German SUCCESS C Study—the first european lifestyle study on breast cancer. Breast Care 5, 395–400 (2010).
ClinicalTrials.gov. Prevention of breast cancer recurrence through weight control, diet, and physical activity intervention (PREDICOP). https://clinicaltrialsgov/show/NCT02035631. (2014)
ClinicalTrials.gov. Randomized phase III trial evaluating the role of weight loss in adjuvant treatment of overweight and obese women with early breast caner (BWEL study). https://clinicaltrialsgov/show/NCT02750826 (2021).
Nicklas, J. M., Sacks, F. M., Smith, S. R., LeBoff, M. S., Rood, J. C., Bray, G. A. et al. Effect of dietary composition of weight loss diets on high-sensitivity c-reactive protein: the Randomized POUNDS LOST trial. Obesity 21, 681–689 (2013).
Tchernof, A., Nolan, A., Sites, C. K., Ades, P. A. & Poehlman, E. T. Weight loss reduces C-reactive protein levels in obese postmenopausal women. Circulation 105, 564–569 (2002).
Ross, R., Hudson, R., Stotz, P. J. & Lam, M. Effects of exercise amount and intensity on abdominal obesity and glucose tolerance in obese adults: a randomized trial. Ann. Intern. Med. 162, 325–334 (2015).
Shai, I., Schwarzfuchs, D., Henkin, Y., Shahar, D. R., Witkow, S., Greenberg, I. et al. Weight loss with a low-carbohydrate, Mediterranean, or low-fat diet. New Engl. J. Med. 359, 229–241 (2008).
Friedenreich, C. M., Neilson, H. K., Woolcott, C. G., McTiernan, A., Wang, Q., Ballard-Barbash, R. et al. Changes in insulin resistance indicators, IGFs, and adipokines in a year-long trial of aerobic exercise in postmenopausal women. Endocrine-related Cancer 18, 357–369 (2011).
Campbell, K. L., Foster-Schubert, K. E., Alfano, C. M., Wang, C. C., Wang, C. Y., Duggan, C. R. et al. Reduced-calorie dietary weight loss, exercise, and sex hormones in postmenopausal women: randomized controlled trial. J. Clin. Oncol. 30, 2314–2326 (2012).
Chlebowski, R. T. & Reeves, M. M. Weight loss randomized intervention trials in female cancer survivors. J. Clin. Oncol. 34, 4238–4248 (2016).
Sjöström, L., Gummesson, A., Sjöström, C. D., Narbro, K., Peltonen, M., Wedel, H. et al. Effects of bariatric surgery on cancer incidence in obese patients in Sweden (Swedish Obese Subjects Study): a prospective, controlled intervention trial. Lancet Oncol. 10, 653–662 (2009).
Schauer, D. P., Feigelson, H. S., Koebnick, C., Caan, B., Weinmann, S., Leonard, A. C. et al. Bariatric surgery and the risk of cancer in a large multisite cohort. Ann. Surg. 269, 95 (2019).
Zhang, S., Ikramuddin, S., Beckwith, H. C., Sheka, A. C., Wirth, K. M. & Blaes, A. H. The impact of bariatric surgery on breast cancer recurrence: case series and review of literature. Obes. Surg. 30, 780–785 (2020).
Author information
Authors and Affiliations
Contributions
M.M.R., K.A.B. and N.M.I. contributed to the conception, design, writing and editing of this review.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Data availability
Not applicable.
Competing interests
M.M.R. and K.A.B. report no competing interests. N.M.I. reports consulting and speaking fees from Novartis and Seattle Genetics unrelated to this work.
Funding information
This research was supported in part through the National Institutes of Health/National Cancer Institute (NIH/NCI) Cancer Center Support Grant P30 CA008748. M.M.R. is supported through the Conquer Cancer ASCO YIA award. K.A.B. is supported through NIH R01 CA215797 grant. N.M.I. is supported through the NIH 1 R01CA235711 grant, the Breast Cancer Research Foundation, American Cancer Society, and the Kat’s Ribbon of Hope Foundation.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rubinstein, M.M., Brown, K.A. & Iyengar, N.M. Targeting obesity-related dysfunction in hormonally driven cancers. Br J Cancer 125, 495–509 (2021). https://doi.org/10.1038/s41416-021-01393-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01393-y
This article is cited by
-
The Impact of Bariatric Surgery on the Incidence of Colorectal Cancer in Patients with Obesity—a Systematic Review and Meta-analysis of Registry Data
Obesity Surgery (2023)
-
Obesity, cancer risk, and time-restricted eating
Cancer and Metastasis Reviews (2022)
-
Inflammatory biomarkers and risk of breast cancer among young women in Latin America: a case-control study
BMC Cancer (2022)
