
Abstract
Background
EGFR tyrosine kinase inhibitors (TKIs) induce cytolysis and release of tumour proteins, which can stimulate antigen-specific T cells. The safety and efficacy of durvalumab and gefitinib in combination for TKI-naive patients with advanced EGFRm NSCLC was evaluated.
Methods
This Phase 1 open-label, multicentre trial (NCT02088112) was conducted in 56 patients with NSCLC. Dose expansion permitted TKI-naive patients, primarily with activating L858R or Ex19del EGFRm. Arms 1 + 1a received concurrent therapy; Arm 2 received 4 weeks of gefitinib induction followed by concurrent therapy.
Results
From dose escalation, the recommended dose of durvalumab was 10 mg/kg Q2W with 250 mg QD gefitinib. Pharmacokinetics were as expected, consistent with inhibition of soluble PD-L1 and no treatment-emergent immunogenicity. In dose expansion, 35% of patients had elevated liver enzymes leading to drug discontinuation. In Arms 1 + 1a, objective response rate was 63.3% (95% CI: 43.9–80.1), median progression-free survival (PFS) was 10.1 months (95% CI: 5.5–15.2) and median response duration was 9.2 months (95% CI: 3.7–14.0).
Conclusions
Durvalumab and gefitinib in combination had higher toxicity than either agent alone. No significant increase in PFS was detected compared with historical controls. Therefore, concurrent PD-L1 inhibitors with gefitinib should be generally avoided in TKI-naive patients with EGFRm NSCLC.
Similar content being viewed by others

Background
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) are the preferred first-line therapy for patients with metastatic non-small cell lung cancer (NSCLC) harbouring sensitising EGFR mutations.1 Advances in the chemistry of EGFR TKIs offer the potential for improved response and survival.1,2,3,4 However, acquired resistance to EGFR TKIs remains largely inevitable, with disease progression typically occurring within 2 years.1,5,6,7 Furthermore, programmed cell death-1 (PD-1) axis blockade as monotherapy has attenuated efficacy in EGFR mutation-positive lung cancer, with reported response rates of ≤10%.8,9,10,11,12 Therefore, novel strategies to boost the durability of TKI responses are urgently needed. Targeting EGFR may promote an inflamed tumour microenvironment through engagement of Fc-γ receptors on immune cells, thereby boosting T cell cross-priming and antigen presentation.13 EGFR TKIs cause immunogenic apoptosis of tumour cells,14 releasing aberrant intracellular antigens and recruiting T cells via interferon-γ-induced major histocompatibility complex (MHC) class I presentation.15 This phenomenon further promotes expression of T cell chemoattractants, chemokine (C-C motif) ligand 2 (CCL2), CCL5 and chemokine (C-X-C motif) ligand 10.16 Gefitinib treatment has been shown to boost CD8+ T cell recruitment via MHC I upregulation and antigen cross-presentation within the tumour.17,18,19,20,21 Interestingly, programmed cell death ligand-1 (PD-L1)-expressing clones have been identified as EGFR TKI-resistant tumours.22,23 In fact, PD-L1 expression may predict poor response and lower survival rates with EGFR TKI monotherapies for patients with activating EGFR mutations.24,25,26,27,28 Therefore, PD-L1 immune checkpoint inhibition may be an attractive combination to partner with gefitinib in the first-line setting.
Durvalumab is a selective, high-affinity human IgG1κ monoclonal antibody that blocks PD-L1 binding to PD-1 and CD80.29 Objective response rates of approximately 12% have been reported with durvalumab monotherapy in EGFR TKI-resistant tumours with strong PD-L1 expression.30 We hypothesised that the combination of gefitinib with durvalumab would exert therapeutic synergy by inducing differentiation and engraftment of memory T cells immediately after initial TKI treatment, therefore inducing more durable clinical remissions with the EGFR TKI. We performed a Phase 1 study to assess the safety and efficacy of concurrent gefitinib and durvalumab for the treatment of TKI-naive patients with EGFR mutation-positive NSCLC.
Methods
Study design
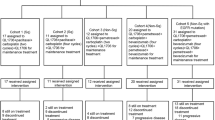
This was an open-label, multicentre Phase 1 trial (NCT02088112) with a modified 3 + 3 dose-escalation phase followed by a multi-arm dose-expansion phase, conducted at seven sites in the US, Japan and Korea. A fixed dose of gefitinib 250 mg daily (QD) was selected for all cohorts, based upon the established maximal biologic activity in vivo.31 In the dose-escalation phase (Fig. 1), patients received gefitinib 250 mg QD plus durvalumab (MEDI4736) at 3 or 10 mg/kg intravenous (IV) every 2 weeks (Q2W). Cohort A received durvalumab at 3 mg/kg IV Q2W. Next, a subsequent Cohort B and a Japan Cohort received durvalumab at 10 mg/kg. Dose-limiting toxicity (DLT) was defined as any possible treatment-related Grade ≥3 adverse event (AE), regardless of duration, within the first treatment cycle of 28 days. This included any Grade 4 immune-mediated AEs that were not attributable to lung cancer.

d days, EGFR epidermal growth factor receptor, IV intravenous, N number of patients assigned to treatment, NSCLC non-small cell lung cancer, QD once daily, Q2W once every 2 weeks, TKI tyrosine kinase inhibitor.
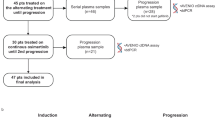
The dose-expansion phase comprised three arms. Patients enrolled in Arm 1 received gefitinib 250 mg QD plus durvalumab 10 mg/kg IV Q2W. Arm 1 was intended to address whether concurrent gefitinib and durvalumab could achieve a more durable response than historical gefitinib monotherapy. Patients enrolled in Arm 2 received gefitinib monotherapy induction for 28 days followed by concurrent gefitinib plus durvalumab. The rationale for the induction Arm 2 was that gefitinib would induce tumour autophagy with MHC class I cross-presentation of tumour antigens and the activation of CD8+ T cells over time, thereby priming T cells for durvalumab at Day 28.32 Arm 1a was later added to the study protocol to further explore the safety and clinical activity of the dosing schedule used in Arm 1. For all cohorts, concurrent therapy was given for up to 12 months; and thereafter patients continued with gefitinib monotherapy until disease progression.
Patients
Screening was conducted between March 2014 and February 2015. Patients were required to have tissue-confirmed metastatic or advanced NSCLC by AJCC seventh edition cancer staging criteria33 that was not amenable to definitive surgery or radiation. The dose-escalation phase permitted patients with any relapsed/refractory NSCLC or those who were intolerant or not eligible for any line of standard treatment. This cohort did not require an activating EGFR mutation, and prior treatment with EGFR TKIs was permitted. The dose-expansion phase permitted only EGFR TKI-naive patients with tumours harbouring a sensitising EGFR mutation. Mandatory tumour biopsies were required at screening and on Day 10 of treatment. Patients in Arm 1a were permitted to submit an archival tissue sample in place of the screening sample, if collected within 90 days prior to the first dose (N = 12). For additional patient eligibility criteria, please see Supplementary Data 1. All patients provided written informed consent; the final protocol was approved by the local ethics committee or Institutional Review Board at each site.
Assessments
In the dose-escalation phase, the primary objective was to assess the safety and tolerability of concurrent gefitinib plus durvalumab and establish a recommended dose of durvalumab for use in the dose-expansion phase. In the dose-expansion phase, the primary objective was to confirm the safety and tolerability of the gefitinib plus durvalumab combination in the intent-to-treat population, for use in future studies. Secondary objectives included pharmacokinetics, durvalumab immunogenicity, durvalumab pharmacodynamics and efficacy.34 Efficacy endpoints included overall response rate (ORR), disease control rate (DCR), DCR at 16 weeks, duration of response and progression-free survival (PFS). Overall survival was added as a protocol amendment later in the course of the study; however, few patients could subsequently consent to this protocol amendment.
EGFR mutation was determined by local site laboratories. Exploratory objectives included correlation of baseline tumour PD-L1 expression with efficacy. Tumour cell (TC) PD-L1 immunohistochemistry (Ventana, clone SP26335) was blindly scored by a pathologist using an established scoring protocol.36 An exploratory cut-point (PD-L1 TC ≥20%) was empirically chosen as it provided more meaningful group numbers for analysis (PD-L1 TC ≥20%: N = 12; PD-L1 TC <20%: N = 24) than the more typical cut-off of PD-L1 TC ≥25% (N = 7; PD-L1 TC: <25%: N = 29). Safety and tolerability were assessed in the safety population of all patients who received at least one dose of study medication. Pharmacokinetics were assessed in all patients who had at least one measurable post-dose pharmacokinetic concentration. Tumour response was assessed in all patients with a baseline tumour assessment who received study medication. Study sample size was based on the desire to obtain adequate tolerability, safety, pharmacokinetic and pharmacodynamic data while exposing as few subjects as possible to the investigational product and procedures. Further details are provided in Supplementary Data 1.
Results
Patient characteristics
Of the 70 patients screened, 56 were eligible and treated (Supplementary Fig. 1). Patient demographics and baseline characteristics are reported in Supplementary Table 1. Overall, in the dose-escalation and dose-expansion phases, patients had a median age of 61 years (range: 27–83) and 55% were female. There was a slightly higher prevalence of Asian (55%) patients, compared with Caucasian (43%) and Black (4%) patients. EGFR mutation status in the dose-expansion phase included 2 patients with exon 18 mutations, 21 with exon 19 deletions, 16 with exon 21 L858R and 1 with L858R/L861Q.
Dose-escalation phase
In the dose-escalation phase, three patients in Cohort A received durvalumab 3 mg/kg IV Q2W and 13 patients in Cohort B and a Japan Cohort received durvalumab 10 mg/kg IV Q2W. No DLTs were reported, and the maximum tolerated dose of durvalumab was not reached. The recommended dose for the dose-expansion phase was thus deemed to be 10 mg/kg IV Q2W. For the subset of patients in the dose-escalation phase with EGFR mutations (11/16), the majority were either EGFRL858R (5/9) or EGFRΔEx19 (4/9) (Supplementary Table 1); some had prior TKI treatment and others did not. AEs are summarised in Supplementary Table 2; common AEs included diarrhoea (69%), transaminitis (44%), fatigue (44%) and nausea (44%). AEs of special interest are shown in Fig. 2. In the dose-escalation phase, no diarrhoea above Grade 2 was reported; one patient reported Grade 3 dermatitis/rash. However, after completion of the 28-day DLT evaluation period, a further 8 hepatic events were reported, including 3 Grade 3/4.

Maximum CTCAE grade is shown for all cohorts, including Cohort A (N = 3), Cohort B (N = 7), Japan Cohort (N = 6), Arm 1 (N = 10), Arm 1a (N = 20) and Arm 2 (N = 10). CTCAE Common Terminology Criteria for Adverse Events, N number of patients assigned to treatment.
Overall, serious AEs were reported in 50.0% of patients and Grade 3/4 AEs were reported in 68.8% of patients. Five patient deaths occurred in the dose-escalation phase. The primary causes of death were NSCLC (N = 3), pericardial effusion (N = 1) and suicide (N = 1) and were not considered to be treatment related by the investigator. Most patients discontinued combination treatment after the DLT evaluation period (94%); AEs were the most common cause for discontinuation. Eight patients (50%) discontinued treatment as a result of AEs, with elevated alanine aminotransferase (ALT) being the most frequently reported AE leading to discontinuation (N = 3). In the dose-escalation phase, response data were not considered meaningful due to the wide heterogeneity of these patients in terms of disease stage, EGFR mutation status and absence/presence of prior TKI therapy.
Safety and tolerability in the dose-expansion phase
In the dose-expansion phase, all patients (N = 40) were TKI naive and had a sensitising EGFR mutation (Supplementary Table 1). Ten patients were each enrolled into Arm 1 and Arm 2. As no patients in Arm 1 had AEs leading to treatment discontinuation, a further 20 patients were enrolled into the concomitant dosing group, Arm 1a (Supplementary Table 2). Common AEs included diarrhoea (78%), elevated ALT (60%) and aspartate aminotransferase (AST) (45%), rash (53%) and pruritus (45%). The most common causally related AEs are summarised in Supplementary Table 3. No deaths were treatment related. Common AEs of special interest were dermatitis/rash, diarrhoea/colitis and hepatic events; 17 patients had high-grade hepatic events (Fig. 2). Management of elevations in ALT and AST primarily consisted of dose interruptions and prompt serial rechecking of AST/ALT levels. If AST/ALT levels continued to increase, despite cessation of both drugs, then administration of a corticosteroid, usually 1 mg/kg oral prednisone, was initiated. Once AST/ALT levels improved, corticosteroid dose was tapered over the course of 3–5 weeks. With this management, hepatic AEs of special interest resolved for most patients (87.1%) but remained a pervasive problem during the trial. The majority of patients discontinued gefitinib plus durvalumab combination treatment before the full 1-year treatment period ended (N = 28; 70.0%). Of these, 17 patients discontinued as a result of AEs, most frequently due to elevated ALT (47% [8/17]) and AST (35% [6/17]). Although these patients stopped combination treatment, they continued on gefitinib monotherapy. Patients with hepatic AEs that led to treatment discontinuation are shown in Table 1.
Pharmacokinetics and pharmacodynamics in the dose-expansion phase
The pharmacokinetics of each compound were similar to those previously reported in gefitinib and durvalumab monotherapy trials,37,38 indicating no drug–drug interaction between gefitinib and durvalumab (Supplementary Table 4 and Supplementary Fig. 2). No treatment-emergent anti-drug antibodies were observed for durvalumab when combined with gefitinib. Complete inhibition of soluble PD-L1, a pharmacodynamic biomarker for durvalumab activity, was observed in all patients (Supplementary Fig. 3), consistent with durvalumab monotherapy at this dose.30
Efficacy in the dose-expansion phase
ORR was 63.3% and 70.0% in Arms 1 + 1a and Arm 2, respectively (Table 2). DCR was 100.0% in Arms 1 + 1a and 90.0% in Arm 2, indicating that almost all patients in the dose-expansion phase achieved disease control.
Median duration of response was 9.2 months (95% confidence interval [CI]: 3.7–14.0) in Arms 1 + 1a and 12.6 months (95% CI 5.5–20.4) in Arm 2, while median PFS was 10.1 (95% CI: 5.5–15.2) in Arms 1 + 1a and 12.0 months (95% CI: 2.7–15.6) in Arm 2 (Fig. 3). However, given the small number of patients in Arm 2, these results should be interpreted with caution. The duration of PFS for individual patients in Arms 1 + 1a and Arm 2 is shown in Fig. 4. In an exploratory analysis, a trend towards favourable PFS was noted in patients expressing baseline PD-L1 TC ≥20% (N = 12 vs. 24; hazard ratio: 0.46; 95% CI: 0.19–1.03); Figs. 3 and 4). It was not possible to compare the median duration of response for patients expressing PD-L1 TC ≥20%, due to small patient numbers.

a Overall and b by PD-L1 expression (tumour response analysis set). PD-L1 TC expression (high: ≥20%; low/negative: <20%) was determined at baseline. CI confidence interval, DoR duration of response, NC not calculable, PD-L1 programmed cell death ligand-1, PFS progression-free survival, TC tumour cell.

Baseline PD-L1 TC ≥20% was indicative of high PD-L1 expression. AE adverse event, EGFR epidermal growth factor receptor, H high (PD-L1 TC ≥20%), L low/negative (PD-L1 TC <20%), NE not evaluable (no sample or <100 TCs), PD-L1 programmed cell death ligand-1, PFS progression-free survival, TC tumour cell.
Of note, in a post hoc analysis, patient baseline data were assessed for evidence of central nervous system (CNS) metastases, which were reported as present in 14/40 patients at baseline. At least 23.1% of patients who had not reported CNS metastases at baseline had progression to new CNS lesions. Furthermore, for patients with baseline CNS metastases, 50% progressed due to CNS lesions. Unfortunately, few patients consented to long-term survival follow-up, yielding insufficient events at the time of data cut-off to provide an interpretable Kaplan–Meier overall survival estimate.
Discussion
In this trial combining gefitinib with durvalumab immunotherapy, no synergistic efficacy signal was detected, and the incidence of AEs was higher than expected. The incidence of hepatic AEs with concurrent gefitinib plus durvalumab was notably higher than previously reported for gefitinib (2.4%) and durvalumab (2.8%) monotherapy.5,30,39 The observed transaminitis led to treatment discontinuation in >25% of patients, potentially compromising the dose intensity of the EGFR inhibitor. Although some patients were successfully managed with dose interruption or corticosteroids, this remained a significant concern during the course of the trial. Owing to the small numbers of patients in the study, it is difficult to draw definite conclusions around the impact of AEs on PFS, which was no better than historical reports of PFS with gefitinib monotherapy (Supplementary Fig. 4). It is possible that PFS was reduced due to toxicity; however, when considering the width of the CIs, the PFS in each of the groups was not dissimilar despite the observed differences in the number of patients discontinuing treatment due to AEs. This hepatic phenomenon suggests a potential synergy in liver toxicity. Interestingly, treatment-related AEs associated with elevations in ALT and AST have been observed in other first-generation EGFR TKI/immunotherapy combinations, such as erlotinib plus atezolizumab (14.3%),40 erlotinib plus nivolumab (14.3%)11 and erlotinib plus pembrolizumab (25.0%),41 and particularly gefitinib plus pembrolizumab (71.4%).41 Recruitment to the latter trial was stopped after seven patients enrolled, due to frequency and severity of transaminitis. Hepatotoxicity may be due to the formation of reactive gefitinib metabolites in the liver,42 leading to inflammation when combined with an immune checkpoint inhibitor.
Since this study was initiated, the third-generation EGFR TKI osimertinib is now available for first-line treatment of patients with EGFR-mutant metastatic NSCLC.1,43 In contrast to gefitinib, osimertinib had a high incidence of interstitial lung disease when combined with durvalumab (13/34; 38%).44 This resulted in early termination of the subsequent Phase 3 CAURAL combination trial.45 In the first 13 ALK+ patients treated with nivolumab plus crizotinib, 5 developed severe hepatic toxicities leading to drug discontinuation.46 Of these, two patients died and the presence of severe hepatic toxicities may have contributed to death. Taken together, the safety profiles associated with EGFR/ALK TKI plus PD-(L)1 inhibitor combinations have generally shown somewhat higher toxicity than expected, reflecting the potential exacerbation of intrinsic but typically minimal toxicities of various TKIs.
In this Phase 1 trial, there was no improvement in PFS or ORR compared to that previously reported with gefitinib monotherapy in similar patient populations.5,39 Similar trials of TKI plus PD-1 axis inhibitors have also had no clear evidence of therapeutic synergy, compared with EGFR TKI monotherapy.40,41 In a small Phase 1b study of erlotinib plus atezolizumab, response rate of 75% and median PFS of 15 months was observed; likewise erlotinib plus either pembrolizumab or nivolumab had modestly favourable median PFS and ORR.11 Perhaps due to this unfavourable efficacy-to-toxicity ratio, the clinical investigation of EGFR TKI plus PD-1 axis inhibitor combinations has largely curtailed in the past 2 years, particularly for TKI-naive patients. To the best of our knowledge, no Phase 3 trials with an EGFR TKI plus PD-1 inhibitor for EGFR TKI-naive patients are currently planned or actively accruing.
Although our trial did observe numerically greater improvement in median PFS in patients with baseline PD-L1 TC ≥20%, this finding needs to be interpreted with caution due to the small sample size. Similarly, an association between PD-L1 expression and improved efficacy was suggested with another EGFR TKI/immunotherapy combination in KEYNOTE-021, in which partial response was reported in all patients with baseline PD-L1 tumour proportion scores ≥50%.41 Although PD-L1 TC ≥25% was associated with efficacy of durvalumab monotherapy in patients with EGFR-mutant NSCLC with acquired TKI resistance,30 a recent Phase 2 study found that pembrolizumab monotherapy was ineffective for the treatment of EGFR TKI-naive patients with PD-L1 TC ≥1%, in which many were ≥50%.47 Although higher PD-L1 and tumour mutational burden may also be predictive for early relapse for EGFR-mutant patients while receiving EGFR TKI monotherapy, these reports remain largely exploratory and inconclusive.24,27,48
In summary, results from this Phase 1 study do not support the combination of TKIs and anti-PD-L1 in the EGFR TKI treatment-naive setting. Given the diverse array of resistance mutations and clonal heterogeneity for EGFR TKI-resistant patients, it is possible that the relapsed/refractory setting may be a more opportune setting for T cell or immune checkpoint-based therapy. Further trials are warranted to elucidate the role of anti-PD-1/PD-L1 agents in the treatment paradigm for patients with EGFR-mutant NSCLC and determine whether baseline tumour PD-L1 expression is predictive of improved durability of response.
References
Soria, J. C., Ohe, Y., Vansteenkiste, J., Reungwetwattana, T., Chewaskulyong, B., Lee, K. H. et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 378, 113–125 (2018).
Schuler, M., Yang, J. C., Park, K., Kim, J. H., Bennouna, J., Chen, Y. M. et al. Afatinib beyond progression in patients with non-small-cell lung cancer following chemotherapy, erlotinib/gefitinib and afatinib: Phase III randomized LUX-Lung 5 trial. Ann. Oncol. 27, 417–423 (2016).
Yang, J. C., Wu, Y. L., Schuler, M., Sebastian, M., Popat, S., Yamamoto, N. et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 16, 141–151 (2015).
Creelan, B., Gray, J., Lima, D., Antonia, S., Chiappori, A., Tanvetyanon, T. et al. Abstract CT060. Efficacy, safety and tolerability of dasatinib combined with afatinib: a phase I trial in patients with epidermal growth factor receptor mutant (EGFRm) advanced non-small-cell lung cancer (NSCLC) after acquired tyrosine kinase inhibitor (TKI) resistance. Cancer Res. 76, CT060 (2016).
Mok, T. S., Wu, Y. L., Thongprasert, S., Yang, C. H., Chu, D. T., Saijo, N. et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 361, 947–957 (2009).
Rosell, R., Gervais, R., Vergnenegre, A., Massuti, B., Felip, E., Cardenal, F. et al. Erlotinib versus chemotherapy (CT) in advanced non-small cell lung cancer (NSCLC) patients (p) with epidermal growth factor receptor (EGFR) mutations: interim results of the European Erlotinib Versus Chemotherapy (EURTAC) phase III randomized trial. J. Clin. Oncol. 29, 7503 (2011).
Maemondo, M., Inoue, A., Kobayashi, K., Sugawara, S., Oizumi, S., Isobe, H. et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 362, 2380–2388 (2010).
Borghaei, H., Paz-Ares, L., Horn, L., Spigel, D. R., Steins, M., Ready, N. E. et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 373, 1627–1639 (2015).
Herbst, R. S., Baas, P., Kim, D. W., Felip, E., Pérez-Gracia, J. L., Han, J. Y. et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–1550 (2016).
Rittmeyer, A., Barlesi, F., Waterkamp, D., Park, K., Ciardiello, F., von Pawel, J. et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 389, 255–265 (2017).
Gettinger, S., Hellmann, M., Chow, L., Borghaei, H., Antonia, S., Brahmer, J. et al. Nivolumab plus erlotinib in patients with EGFR-mutant advanced NSCLC. J. Thorac. Oncol. 13, 1363–1372 (2018).
Hellmann, M. D., Rizvi, N. A., Goldman, J. W., Gettinger, S. N., Borghaei, H., Brahmer, J. R. et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 18, 31–41 (2017).
Li, X., Lian, Z., Wang, S., Xing, L. & Yu, J. Interactions between EGFR and PD-1/PD-L1 pathway: implications for treatment of NSCLC. Cancer Lett. 418, 1–9 (2018).
Jia, J., Li, X., Jiang, T., Zhao, S., Zhao, C., Zhang, L. et al. EGFR-targeted therapy alters the tumor microenvironment in EGFR-driven lung tumors: implications for combination therapies. Int. J. Cancer 145, 1432–1444 (2019).
Lizotte, P. H., Hong, R.-L., Luster, T. A., Cavanaugh, M. E., Taus, L. J., Wang, S. et al. A high-throughput immune-oncology screen identifies EGFR inhibitors as potent enhancers of antigen-specific cytotoxic T-lymphocyte tumor cell killing. Cancer Immunol. Res. 6, 1511–1523 (2018).
Lulli, D., Carbone, M. L. & Pastore, S. Epidermal growth factor receptor inhibitors trigger a type I interferon response in human skin. Oncotarget 7, 47777–47793 (2016).
Thress, K., Jacobs, V., Angell, H., Yang, J., Sequist, L., Blackhall, F. et al. Modulation of biomarker expression by osimertinib: results of the paired tumor biopsy cohorts of the AURA phase I trial. J. Thorac. Oncol. 12, 1588–1594 (2017).
Pollack, B. P., Sapkota, B. & Cartee, T. V. Epidermal growth factor receptor inhibition augments the expression of MHC class I and II genes. Clin. Cancer Res. 17, 4400–4413 (2011).
Kumai, T., Matsuda, Y., Oikawa, K., Aoki, N., Kimura, S., Harabuchi, Y. et al. EGFR inhibitors augment antitumour helper T-cell responses of HER family-specific immunotherapy. Br. J. Cancer 109, 2155–2166 (2013).
Champiat, S., Ileana, E., Giaccone, G., Besse, B., Mountzios, G., Eggermont, A. et al. Incorporating immune-checkpoint inhibitors into systemic therapy of NSCLC. J. Thorac. Oncol. 9, 144–153 (2014).
Gurule, N. J. & Heasley, L. E. Linking tyrosine kinase inhibitor-mediated inflammation with normal epithelial cell homeostasis and tumor therapeutic responses. Cancer Drug Resist. 1, 118–125 (2018).
Kunimasa, K., Nakamura, H., Sakai, K., Kimura, M., Inoue, T., Tamiya, M. et al. Heterogeneity of EGFR-mutant clones and PD-L1 highly expressing clones affects treatment efficacy of EGFR-TKI and PD-1 inhibitor. Ann. Oncol. 29, 2145–2147 (2018).
Gainor, J. F., Shaw, A. T., Sequist, L. V., Fu, X., Azzoli, C. G., Piotrowska, Z. et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin. Cancer Res. 22, 4585–4593 (2016).
D’Incecco, A., Andreozzi, M., Ludovini, V., Rossi, E., Capodanno, A., Landi, L. et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br. J. Cancer 112, 95–102 (2015).
Schmidt, L., Kümmel, A., Görlich, D., Mohr, M., Bröckling, S., Mikesch, J. et al. PD-1 and PD-L1 expression in NSCLC indicate a favorable prognosis in defined subgroups. PLoS ONE 10, e0136023 (2015).
Tang, Y., Fang, W., Zhang, Y., Hong, S., Kang, S., Yan, Y. et al. The association between PD-L1 and EGFR status and the prognostic value of PD-L1 in advanced non-small cell lung cancer patients treated with EGFR-TKIs. Oncotarget 6, 14209–14219 (2015).
Su, S., Dong, Z.-Y., Xie, Z., Yan, L.-X., Li, Y.-F., Su, J. et al. Strong programmed death ligand 1 expression predicts poor response and de novo resistance to EGFR tyrosine kinase inhibitors among NSCLC patients with EGFR mutation. J. Thorac. Oncol. 13, 1668–1675 (2018).
Offin, M., Rizvi, H., Tenet, M., Ni, A., Sanchez-Vega, F., Li, B. T. et al. Tumor mutation burden and efficacy of EGFR-tyrosine kinase inhibitors in patients with EGFR-mutant lung cancers. Clin. Cancer Res. 25, 1063–1069 (2019).
Stewart, R., Morrow, M., Hammond, S. A., Mulgrew, K., Marcus, D., Poon, E. et al. Identification and characterization of MEDI4736, an antagonistic anti-PD-L1 monoclonal antibody. Cancer Immunol. Res. 3, 1052–1062 (2015).
Garassino, M. C., Cho, B. C., Kim, J. H., Mazières, J., Vansteenkiste, J., Lena, H. et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): an open-label, single-arm, phase 2 study. Lancet Oncol. 19, 521–536 (2018).
Wolf, M., Swaisland, H. & Averbuch, S. Development of the novel biologically targeted anticancer agent gefitinib: determining the optimum dose for clinical efficacy. Clin. Cancer Res. 10, 4607–4613 (2004).
He, S., Yin, T., Li, D., Gao, X., Wan, Y., Ma, X. et al. Enhanced interaction between natural killer cells and lung cancer cells: involvement in gefitinib-mediated immunoregulation. J. Transl. Med. 11, 1–11 (2013).
Edge, S., Byrd, D. R., Compton, C. C., Fritz, A. G., Greene, F. L. & Trotti, A. AJCC Cancer Staging Manual 7th edn (Springer, New York, NY, 2010).
Eisenhauer, E. A., Therasse, P., Bogaerts, J., Schwartz, L. H., Sargent, D., Ford, R. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Rebelatto, M., Midha, A., Mistry, A., Sabalos, C., Schechter, N., Li, X. et al. Development of a programmed cell death ligand-1 immunohistochemical assay validated for analysis of non-small cell lung cancer and head and neck squamous cell carcinoma. Diagn. Pathol. 11, 95 (2016).
Roche. VENTANA PD-L1 (SP263) assay (CE IVD). https://diagnostics.roche.com/global/en/products/tests/ventana-pd-l1-_sp263-assay2.html (2020).
Baverel, P., Dubois, V., Jin, C., Zheng, Y., Song, X., Jin, X. et al. Population pharmacokinetics of durvalumab in cancer patients and association with longitudinal biomarkers of disease status. Clin. Pharmacol. Ther. 103, 631–642 (2018).
Swaisland, H. C., Smith, R. P., Laight, A., Kerr, D. J., Ranson, M., Wilder-Smith, C. H. et al. Single-dose clinical pharmacokinetic studies of gefitinib. Clin. Pharmacokinet. 44, 1165–1177 (2005).
Douillard, J.-Y., Ostoros, G., Cobo, M., Ciuleanu, T., McCormack, R., Webster, A. et al. First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: a phase-IV, open label, single arm study. Br. J. Cancer 110, 55–62 (2014).
Rudin, C., Cervantes, A., Dowlati, A., Besse, B., Ma, B., Costa, D. et al. Abstract MA15.02 Long-term safety and clinical activity results from a Phase Ib study of erlotinib plus atezolizumab in advanced NSCLC. J. Thorac. Oncol. 13, MA15.02 (2018).
Yang, J., Gadgeel, S., Sequist, L., Wu, C., Papadimitrakopoulou, V., Su, W. et al. Pembrolizumab in combination with erlotinib or gefitinib as first-line therapy for advanced NSCLC with sensitizing EGFR mutation. J. Thorac. Oncol. 14, 553–559 (2019).
Teo, Y., Ho, H. & Chan, A. Formation of reactive metabolites and management of tyrosine kinase inhibitor-induced hepatotoxicity: a literature review. Expert Opin. Drug Metab. Toxicol. 11, 231–242 (2015).
US Food and Drug Administration. Osimertinib highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208065s008lbl.pdf (2018).
Ahn, M.-J., Yang, J., Yu, H., Saka, H., Ramalingam, S., Goto, K. et al. Abstract 136O: Osimertinib combined with durvalumab in EGFR-mutant non-small cell lung cancer: results from the TATTON phase Ib trial. J. Thorac. Oncol. 11, 136O (2016).
Chih-Hsin Yang, J., Shepherd, F., Kim, D., Lee, G., Lee, J., Chang, G. et al. Osimertinib plus durvalumab versus osimertinib monotherapy in EGFR T790M-positive NSCLC following previous EGFR-TKI therapy: CAURAL brief report. J. Thorac. Oncol. 14, 933–939 (2019).
Spigel, D. R., Reynolds, C., Waterhouse, D., Garon, E. B., Chandler, J., Babu, S. et al. Phase 1/2 study of the safety and tolerability of nivolumab plus crizotinib for the first-line treatment of anaplastic lymphoma kinase translocation - positive advanced non-small cell lung cancer (CheckMate 370). J. Thorac. Oncol. 13, 682–688 (2018).
Lisberg, A., Cummings, A., Goldman, J. W., Bornazyan, K., Reese, N., Wang, T. et al. A Phase II study of pembrolizumab in EGFR-mutant, PD-L1+, tyrosine kinase inhibitor naïve patients with advanced NSCLC. J. Thorac. Oncol. 13, 1138–1145 (2018).
Brown, H., Vansteenkiste, J., Nakagawa, K., Cobo Dols, M., John, T., Barker, C. et al. MA15.03 PD-L1 expression in untreated EGFRm advanced NSCLC and response to osimertinib and SoC EGFR-TKIs in the FLAURA trial. J. Thorac. Oncol. 13, MA15.03 (2018).
Acknowledgements
The authors would like to thank the study participants and their families. Medical writing support, under the direction of the authors, was provided by Lauren McNally, MSc, CMC Connect, McCann Health Medical Communications, funded by AstraZeneca, Cambridge, UK in accordance with Good Publication Practice (GPP3) guidelines.
Author information
Authors and Affiliations
Contributions
B.C.C. (Site Principal Investigator) contributed to the study design, recruitment and management of patients and interpretation of data. T.C.Y. (Translational Sciences Lead) contributed to the development and implementation of the biomarker strategy; was responsible for the delivery and oversight of pharmacokinetics, soluble PD-L1, anti-drug antibodies and exploratory biomarker data (i.e. PD-L1) and contributed to the interpretation of data. S.-W.K. (Site Principal Investigator) contributed to the study design, recruitment and management of patients and interpretation of data. N.N. (Site Principal Investigator) contributed to the recruitment and management of patients, assisted with the provision of study materials/patients, and was responsible for the collection and assembly of data. D.-W.K. and S.K. (Site Principal Investigators) assisted with the provision of study materials/patients, were responsible for the collection and assembly of data and contributed to the interpretation of data. L.Q.M.C. (Site Principal Investigator) contributed to the study design, assisted with the provision of study materials/patients, was responsible for the collection and assembly of data and contributed to the interpretation of data. R.T. contributed to the study design, oversaw the analysis and contributed to the interpretation of data. W.T. (Clinical Pharmacologist) was responsible for the analysis and interpretation of the gefitinib pharmacokinetics data. M.T. (Clinical Pharmacologist) was responsible for the analysis and interpretation of the durvalumab pharmacokinetics, soluble PD-L1 and anti-drug antibodies data. H.K.A. was responsible for the analysis of PD-L1 in tumour samples and contributed to the interpretation of data. M.P.R. was responsible for the assessment of tumour PD-L1 expression in tissue sections. M.M. (Clinical Lead) contributed to the study design, development of the protocol and interpretation of data. D.L.G. (Site Principal Investigator) contributed to the study design, recruitment and management of patients, development of the protocol and interpretation of data. All authors contributed to the development of the manuscript and have given final approval for the final version to be published.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was performed in accordance with ethical principles that have their origin in the Declaration of Helsinki and are consistent with the International Conference on Harmonisation (ICH)/Good Clinical Practice (GCP) guidelines, applicable regulatory requirements and the AstraZeneca policy on Bioethics and Human Biological Samples. All patients provided written informed consent, including for exploratory biomarker research. The study protocol, and all other written information and/or materials provided to patients, were approved by the following Ethics Committees/Institutional Review Boards: National Cancer Center Hospital, Chuo-ku, Japan; National Hospital Organization Shikoku Cancer Center, Matsuyama-shi, Japan; Seoul National University Hospital, Seoul, South Korea; Asan Medical Center, Seoul, South Korea; Liberty Institutional Review Board, DeLand, USA; MD Anderson Cancer Center, Houston, USA; Western Institutional Review Board, Puyallup, USA.
Data availability
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Competing interests
B.C.C. has received institutional research grants/supplies from Biodesix, Boehringer Ingelheim, Bristol-Myers Squibb, Iovance Biotherapeutics, Neogenomics and Prometheus; participated in speaker bureaus for Achilles, AstraZeneca, Bristol-Myers Squibb, Foundation Medicine, F. Hoffmann-La Roche AG, Gilead and Takeda; and has participated in advisory boards for AbbVie, BergenBio, Bristol-Myers Squibb and GlaxoSmithKline. T.C.Y., R.T., W.T. and H.K.A. are employees or contracted employees of AstraZeneca and may be shareholders of AstraZeneca. S.-W.K. has received clinical research support from AstraZeneca, Boehringer Ingelheim and Eli Lilly. N.N. has received research grants from AstraZeneca, Boehringer Ingelheim, Chugai Pharmaceutical, Kyowa Hakko Kirin, ONO Pharmaceutical and Taiho Pharmaceutical and personal fees from AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Chugai Pharmaceutical, Eli Lilly Japan, Kyowa Hakko Kirin, Meiji Seika Pharma, Merck Sharp & Dohme, Nikkei Business Publications, ONO Pharmaceutical, Pfizer Japan, Reno Medical and Taiho Pharmaceutical. D.-W.K’s institution has received research funding from Alpha Biopharma, AstraZeneca/MedImmune, Hanmi, Janssen, Merus, Mirati Therapeutics, MSD, Novartis, ONO Pharmaceutical, Pfizer Inc., Roche/Genentech, Takeda, TP Therapeutics, Xcovery and Yuhan. L.Q.M.C. is an employee of the University of Texas, Austin and a former employee of the University of Washington/Seattle Cancer Care Alliance. L.Q.M.C’s institution has received research funding from Alkermes, AstraZeneca/MedImmune, Bristol-Myers Squibb, Dynavax, Eli Lilly, Genentech, Incyte, Merck, Novartis, Pfizer Inc., Seattle Genetics and VentiRx; and the University of Washington/Seattle Cancer Care Alliance received institutional funding from AstraZeneca for this study. L.Q.M.C. has received honoraria from Amgen and has participated in advisory boards for Alkermes, Amgen, AstraZeneca, Bristol-Myers Squibb, Dynavax, Genentech, Merck, Novartis, Pfizer Inc., Sanofi Genzyme, Seattle Genetics, Synthorx and Takeda. S.K. has received research grant funding from AbbVie, AstraZeneca and ONO Pharmaceutical; honoraria from AstraZeneca, Bristol-Myers Squibb, Chugai Pharmaceutical, Novartis and ONO Pharmaceutical; and has participated in advisory boards for AstraZeneca. M.T. is an employee of Astellas Pharma US and a former employee of AstraZeneca. M.P.R. is an employee of the Institute for Prostate Cancer Research and a former employee of AstraZeneca. M.M. is a former employee of AstraZeneca. D.L.G. has received research grants from AstraZeneca, Janssen Research & Development and Takeda; has participated in advisory boards for AstraZeneca, GlaxoSmithKline and Sanofi; and has received travel expenses from AstraZeneca. D.L.G’s institution has received compensation for conducting the study.
Funding information
This study was sponsored by AstraZeneca PLC.
Additional information
Note This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International (CC BY 4.0).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Creelan, B.C., Yeh, T.C., Kim, SW. et al. A Phase 1 study of gefitinib combined with durvalumab in EGFR TKI-naive patients with EGFR mutation-positive locally advanced/metastatic non-small-cell lung cancer. Br J Cancer 124, 383–390 (2021). https://doi.org/10.1038/s41416-020-01099-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-020-01099-7
This article is cited by
-
At the crossroads of immunotherapy for oncogene-addicted subsets of NSCLC
Nature Reviews Clinical Oncology (2023)
-
Treatment of advanced non-small cell lung cancer with driver mutations: current applications and future directions
Frontiers of Medicine (2023)
-
Third-generation EGFR and ALK inhibitors: mechanisms of resistance and management
Nature Reviews Clinical Oncology (2022)
-
EGFR and HER2 exon 20 insertions in solid tumours: from biology to treatment
Nature Reviews Clinical Oncology (2022)
-
Non-small Cell Lung Cancer with EGFR or HER2 Exon 20 Insertion Mutations: Diagnosis and Treatment Options
BioDrugs (2022)
